

ABSTRACT
Objective: To verify the diagnosis of channelopathies in two families and explore the mechanism of the overlap between periodic paralysis (PP) and paramyotonia congenita (PMC). Methods: We have studied two cases with overlapping symptoms of episodic weakness and stiffness in our clinical center using a series of assessment including detailed medical history, careful physical examination, laboratory analyses, muscle biopsy, electrophysiological evaluation, and genetic analysis. Results: The first proband and part of his family with the overlap of PMC and hyperkalemic periodic paralysis (HyperPP) has been identified as c.2111C > T (T704M) substitution of the gene SCN4A. The second proband and part of his family with the overlap of PMC and hypokalemic periodic paralysis type 2 (HypoPP2) has been identified as c.4343G > A (R1448H) substitution of the gene SCN4A. In addition, one member of the second family with overlapping symptoms has been identified as a novel mutation c.2111C > T without the mutation c.4343G > A. Conclusions: SCN4A gene mutations can cause the overlap of PMC and PP (especially the HypoPP2). The clinical symptoms of episodic weakness and stiffness could happen at a different time or temperature. Based on diagnosis assessments such as medical history and muscle biopsy, further evaluations on long-time exercise test, genetic analysis, and patch clamp electrophysiology test need to be done in order to verify the specific subtype of channelopathies. Furthermore, the improvement of one member in the pregnancy period can be used as a reference for the other female in the child-bearing period with T704M.
KEYWORDS: SCN4A gene, paramyotonia congenita, hyperkalemic periodic paralysis, hypokalemic periodic paralysis type 2
Introduction
The voltage-gated sodium channel NaV1.4 encoded by SCN4A gene is a transmembrane complex mainly expressed in skeletal muscle and participated in contraction. The activation of the channel generates action potential (AP) and the fast inactivation after AP can prevent repetitive discharge, which insures the physiological excitability changes of sarcolemma and the normal skeletal muscle contraction.
SCN4A gene mutations lead to abnormal changes of skeletal muscle excitability, which is connected with the activation or inactivation speed of muscle ion channels. Two main clinical phenotypes are non-dystrophic myotonia with improved excitability, including paramyotonia congenita (PMC) and sodium channel myotonia (SCM), and episodic flaccid weakness with reduced excitability, including hyperkalemic periodic paralysis (HyperPP) and hypokalemic periodic paralysis type 2 (HypoPP2).
We have studied two cases in our clinical center, one with a three-generation Chinese family and another one with a four-generation Chinese family. The diagnosis relies on the clinical symptoms, laboratory tests, electrophysiological examinations including needle electromyography and the exercise test at different status, muscle biopsy, and genetic analysis. In this paper, we present a report of the two families and a literature review on SCN4A gene mutations with overlapping symptoms.
Methods
Muscle biopsy is the gold standard for many muscle diseases, and there are characteristic features in biopsy specimen. In channelopathies, tubular aggregates or vacuoles can present in HE, NADH, MGT and other routine histological or histochemical staining of muscle sections. In addition, nonspecific pathological changes have been found including increased fiber size variation, diffused necrosis and regeneration, some atrophic or hypertrophic fibers, preferential central nucleation and so on.
Electrophysiological examinations are widely used to identify different types of channelopathies. Needle electromyography can show the myotonic discharge, and it is evidence for supporting the diagnosis of PMC or SCM in channelopathies. In electrophysiological exercise tests, several specific compound muscle action potential (CMAP) amplitude change patterns have been found correlated with different clinical types [1–4]. The long-time exercise test (ET) method has been proposed by McManis initially[1]. In our center, we use the ET to verify the subtypes of channelopathies following the method and evaluation criteria in China [5–7] Our subjects were instructed to try their best to abduce their right or left abductor digiti minimi (ADM) (exercise 45 s – relax 15 s/repeat 5 times continuously). The CMAP amplitude was recorded with the supramaximal stimulation of the ulnar nerve before and after exercise at specific times. There are different patterns in CMAP amplitude change curves, and we choose extreme muscles like ADM to avoid general paralysis or myotonia, which may cause discomfort or panic. In normal subjects, CMAP amplitude fluctuates from −20% to 10% immediately, and returns to baseline within 60 s[2]. In China, the normal reference range is −33%-0% based on the 100 healthy subjects (all from China) in 2008[5]. Deviations larger than −33% on the CMAP amplitude are considered abnormal and widely used as the standard in China[6]. There are following studies based on subjects from China to explore ET reference value, one of the studies from the same neuroelectrophysiology center concluded that CMAP amplitude decrease after exercise of greater than 35.50% is abnormal in 2014[7], but the −33% standard is still the most commonly used in China. With PMC patients, the CMAP amplitude shows a conspicuous decrease immediately after exercise, followed by a continued decrease within 5 min and a slow recovery after 30 min. This agrees with the pattern I in Fournier guides[2]. For some patients with p.R1448C, the decrease even reached −95%, usually followed by a persistent weakness. For HypoPP patients, there is a steady decrease of CMAP amplitude after exercise, and the amplitude often falls back to baseline after a few hours. This agrees with pattern V in Fournier guides[2]. For HyperPP patients, the CMAP amplitude shows a slight transient increase within seconds after exercise, then decreases in 30 min and falls back to baseline after a few hours. This agrees with pattern IV in Fournier guides[2].
Gene tests with certain directionality can be used for further diagnoses. Gene analysis usually includes Sanger test and NGS panel test (including SCN4A, CLCN1, CACNA1S, KCNJ2 gene).
Case report
Case 1
The proband (Figure 1A III 8), an 18-year-old Chinese male, complained of overlap between periodic limbs flaccid weakness and stiffness since the toddler period, was hospitalized in December 2017. When he was two years old, he was found crying weaker than others and having limbs stiffness, which would last for several hours and recover after feeding. The frequency was 2 times per year approximately. The stiffness was found in hands and eyelids with cold exposure, apparently more frequent and susceptible in winter than summer. The severity and frequency of the similar symptoms were aggravated with age. In his school age, he often developed episodic weakness such as having difficulties climbing and squatting each month, which would last one or two days and recover after rest. The symptoms were triggered or exacerbated by cold circumstance, movement, starvation or fatigue and alleviated by rest, warming or eating. The past medical history was negative. The social history was negative for smoking or alcohol. The family history was positive, and his mother (Figure 1A II 5), uncle (Figure 1A II 7) older sister (Figure 1A III 7) and maternal grandmother (Figure 1A I 2) presented similar overlapping characteristics from a very young age. His mother and uncle relieved in their 30s and 40 s, and the symptoms of the former relieved during her pregnancy period. Physical examinations showed flaccid weakness of limbs (4/5, distal > proximal, upper > lower) and cervical flexors (4+/5), without sensory, reflexes, muscle tension impairment or muscle atrophy. There was no “warm-up effect” or percussion myotonia. Laboratory analyses indicated that serum potassium was lower at 3.2mmol/L (reference interval (RI), 3.5–5.5) and serum creatine kinase (CK) was elevated at 848U/L (RI, 50–310). The assay indexes reflected nutritional status like lipid, protein, and vitamins were lower than average. Other investigations were basically normal. EMG showed diffuse myotonia discharge at rest (Figure 2D), and muscle biopsy showed occasional tubular aggregates and vacuoles (Figure 3). We then applied ET on him, and the change of CMAP amplitude reflected probable HyperPP pattern at room temperature and PMC pattern at cold temperature (Figure 2A–C). We further completed gene analysis using Sanger test and NGS panel test (including SCN4A, CLCN1, CACNA1S, KCNJ2 gene) according to the outcomes of ET and found c.2111C > T chr17-62,034,787 (p.T704M) mutation of SCN4A gene (Figure 1B). Considering the family history, we also tested the mutation c.2111C > T in his family. The segregation analysis reflected c.2111C > T mutation of SCN4A gene as the disease-causing gene, which was maternally inherited in this family. Six members (including the proband) out of 25 presented similar clinical overlapping symptoms and one in the six was not exact. We then identified p.T704M in four members (Figure 1A II 5, II 7, III 7, III 8) of the six with positive symptoms and the two residuals rejected the gene test. We also tested two family members without clinical symptoms (Figure 1A II 6, II 9) and they were normal in chr17-62034787 (Figure 1C).
Figure 1.
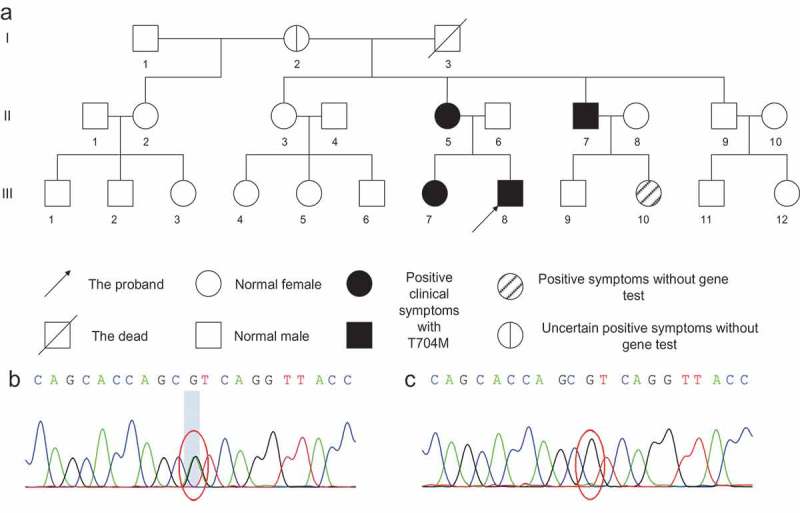
Pedigree and genetic analysis of Case 1.
(A) Pedigree of Case 1. B/C. Genetic analysis by Sanger test and NGS panel test (containing SCN4A, CLCN1, CACNA1S, KCNJ2 gene). (B) c.2111C > T chr17-62034787 (p.T704M) gene mutation. (C) Normal in chr17-62034787.
Figure 2.
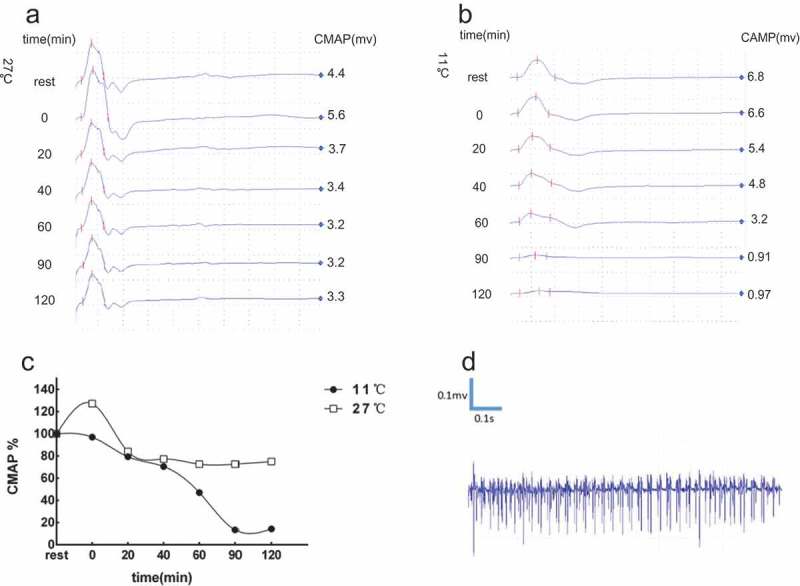
Electrophysiological evaluations of Case 1.
(A) At room temperature (27°C), the CMAP amplitudes increased (+27%) immediately after exercise. Then, the CMAP amplitudes decreased gradually in 20 min-60 min (−27% at most) and started to back to baseline from 90 min. (B) At cold status (11°C), the CMAP amplitudes decreased after exercise immediately, following a continued remarkable decrease after 60min-90min (−87%), and recovered gradually after 90 min. (C) The change of CMAP amplitudes tended to be HyperPP pattern but not reached the abnormal standard (>−33%) in room temperature, and it is PMC pattern in cold temperature. (D) Needle electromyography showed diffuse myotonia discharge in the rest state.
Figure 3.
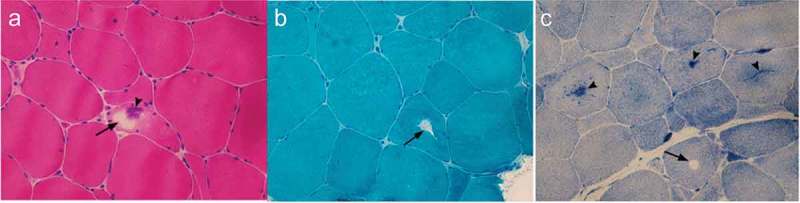
Muscle biopsy of Case 1.
HE (A), MGT (B) and NADH-TR (C) demonstrates tubular aggregates (arrowhead) and vacuoles (arrow) under the light microscope.
Case 2
The proband (Figure 4A III 3), a 24-year-old Chinese male, complained of episodic limbs stiffness since the age of 3 and overlapping episodic weakness since the age of 20. From the age of 3, he presented hands stiffness in cold circumstances, and the symptom would relieve in 10 h after keeping his hands in a warm environment. When he was 20 years old, he began to present episodic flaccid weakness in the morning, such as having difficulties in squatting and climbing, approximately 3 times per month. The weakness progressed in several years and persisted after his age of 23. The symptom of weakness was more obvious and frequent in early spring, which would relieve after taking potassium supplement or receiving nutrition support treatment, but the proband never recovered completely. There was no significant correlations to fatigue, movement, starvation, satiation, emotional fluctuation, or cold or warm circumstances. The past medical history was negative and the social history was positive for smoking and alcohol. Family history was positive for his grandfather (Figure 4A I 1), father (Figure 4A II 1), uncle (Figure 4A II 5), aunt (Figure 4A II 7), elder male cousin (Figure 4A III 11) and niece (Figure 4A IV 1). His father (Figure 4A II 1) and niece (Figure 4A IV 1) had similar overlapping characteristics from a young age. His grandfather (Figure 4A I 1), aunt (Figure 4A II 7) and elder male cousin (Figure 4A III 11) presented stiffness in cold circumstances and his uncle (Figure 4A II 5) presented episodic weakness. Physical examinations showed flaccid weakness of proximal limbs (4/5), without sensory, reflexes, muscle tension impairment or muscle atrophy. There was no “warm-up effect” or percussion myotonia. Laboratory analyses indicated that the serum potassium was 3.3mmol/L. The serum CK was elevated at 402U/L. The ALT and AST were slightly elevated. The level of serum lipid was lower than average. Other investigations were basically normal. EMG showed diffuse myotonia discharge at rest (Figure 5D), and muscle biopsy showed occasional tubular aggregates (Figure 6). The ET reflected approximately normal at room temperature and HypoPP pattern at cold temperature (Figure 5A–C). We further completed gene analysis (including SCN4A, CLCN1, CACNA1S, KCNJ2 gene) and found c.4343G > A chr17-62019299 (p.R1448H) mutation of SCN4A gene (Figure 4B). Considering the family history, we also tested the mutation c.4343G > A in his family and the segregation analysis reflected that the c.4343G > A allele was paternally inherited. Seven members (Figure 4A I 1, II 1, II 5, II 7, III 3, III 11, IV 1) out of 24 (including the proband) presented similar clinical symptoms. We then identified p.R1448H mutation in four members (Figure 4A II 1, II 5, II 7, III 3) of the seven with positive symptoms. One member (Figure 4A III 11) rejected the gene test and another one (Figure 4A I 1) passed away in the seven with positive symptoms. We also tested four family members without clinical symptoms (Figure 4A II 2, III 1, III 5, III 6) and they were normal in chr17-62019299 (Figure 4C). Interestingly, one of the seven with positive symptoms, the proband’s niece (Figure 4A IV 1), presented similar overlapping symptoms with the proband without p.R1448H. We further tested the genes of his niece (Figure 4A IV 1) using Sanger test and NGS Panel capture test, and we identified the c.2111C > T chr17-62034787 (p.T704M) gene mutation as the disease-causing gene. We also verified that father (Figure 4A III 5) and mother (Figure 4A III 6) of his niece were normal in chr17-62034787, and we concluded that it is a novel mutation in his niece (Figure 4A IV 1).
Figure 4.
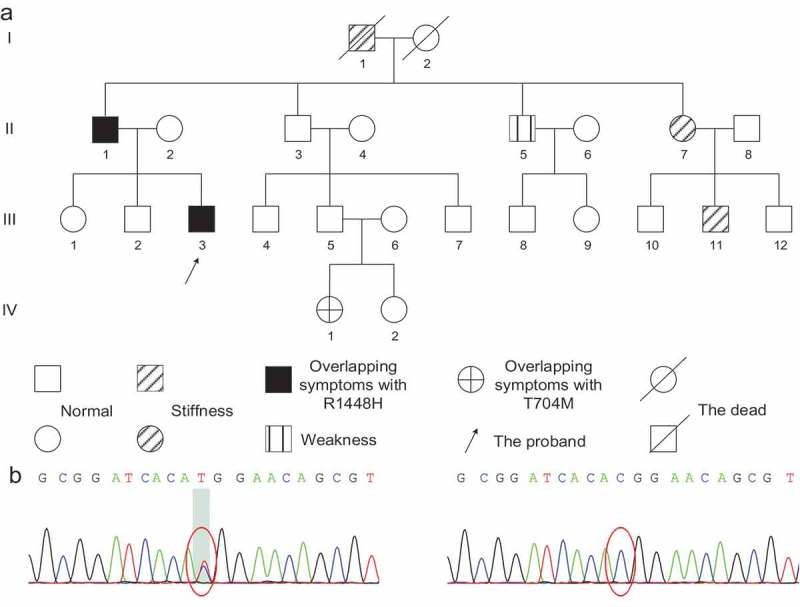
Pedigree and genetic analysis of Case 2.
(A) Pedigree of Case 2. B/C. Genetic analysis by Sanger test and NGS panel test (containing SCN4A, CLCN1, CACNA1S, KCNJ2 gene): (B) c.4343G > A chr17-62019299 (p.R1448H) gene mutation. (C) Normal in chr17-62019299.
Figure 5.
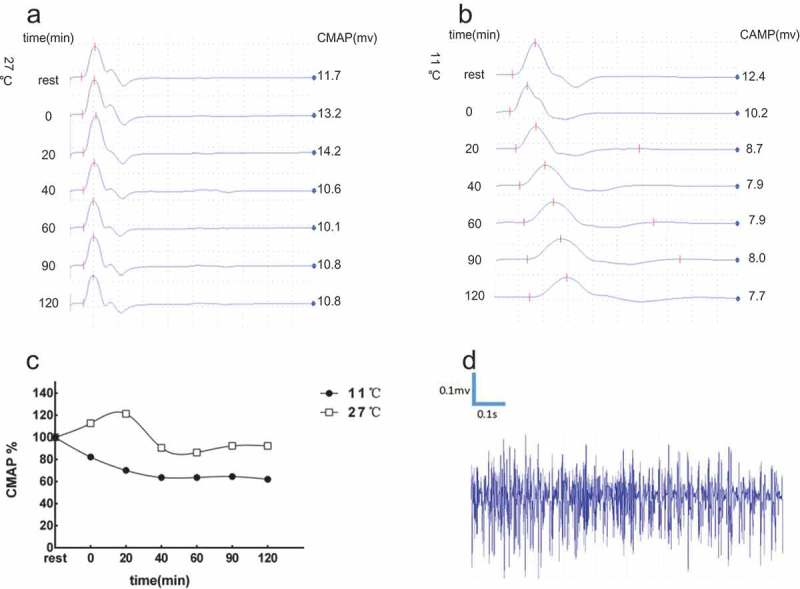
Electrophysiological evaluations of Case 2.
(A) At room temperature (27°C), the CMAP amplitudes showed a slight transient increase (+13%) immediately after exercise, then there was a slowly decrease in 20 min-60 min (−25% at most) and backed to baseline from 90 min. (B) At cold status (11°C), the CMAP amplitudes decreased gradually after exercise (−36% in 40 min), maintaining the level after 40min-120min, and recovery after few hours. (C) Though the change was not reached the abnormal standard (>−33%), it tended to be PMC pattern in room temperature. The change of CMAP amplitudes was HypoPP pattern in cold status. (D) Needle electromyography showed diffuse myotonia discharge in the rest state.
Figure 6.
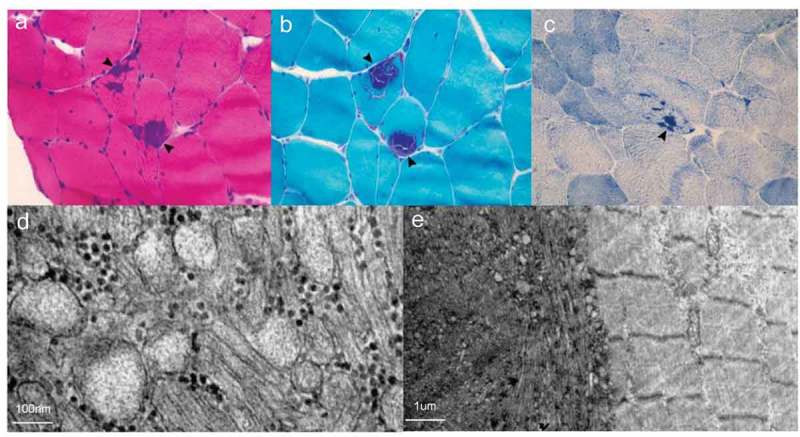
Muscle biopsy of Case 2.
HE (A), MGT (B) and NADH-TR (C) demonstrates tubular aggregates (arrowhead) and vacuoles (arrow) under the light microscope. D. Tubular aggregates under electron microscopy. E. The right area shows relatively normal skeletal muscle structure and the left area shows tubular aggregates under electron microscopy.
Discussion
Nav1.4 channel introduction and mechanisms of mutation in SCN4A
Skeletal muscle ion channelopathies are rare diseases including sodium channel disorders, calcium channel disorders, chloride channel disorders, and potassium channel disorders with fluctuating stiffness or episodic weakness usually. Several clinical phenotypes have been found in recent studies comprising myotonia congenita (MC), PMC, SCM, HyperPP, HypoPP, Andersen-Tawil syndrome (ATS) and so on.
Sodium channels have many subtypes, and the NaV1.4 is mainly presented in skeletal muscles. NaV1.4 consists of an α subunit and an auxiliary β subunit. The α subunit consists of four homologous domains (I -IV), and each domain is comprised of six α-helical transmembrane segments (S1-S6). The α subunit is encoded by the SCN4A gene (c.17q23) [8] and the β subunit is encoded by the SCN1B gene (c.19q13.11)[9]. The mutations in SCN4A may cause to some subtypes of sodium channel disorders. Six allelic diseases are connected to those mutations, including PMC, HyperPP, HypoPP, SCM, congenital myopathy, and myasthenia, while the mutations in SCN1B are linked to other disorders such as ataxia, epilepsy or cardiac arrhythmia[10].
NaV1.4 carries the majority of the inward Na+ current. The activation of the channel generates AP and the fast inactivation of the channel after AP helps the membrane potential to back to rest potential (RP) rapidly. The whole process maintains normal skeletal muscle contraction. Mutations of NaV1.4 channel always act on the process of activation or inactivation, leading to abnormal changes of skeletal muscle excitability. There are different pathophysiological mechanisms corresponding to different phenotypes: gain-of-function corresponding to HyperPP, PMC, and SCM, and anomalous gating pore current connected to loss-of-function corresponding to HypoPP[11].
Gain-of-function mechanism enhances inward Na+ current of NaV1.4, which will lead to the impairment of fast inactivation, or sometimes the improvement of activation. Furthermore, there is enhanced excitability in slightly Na+ influx (PMC) or impaired excitability in plentiful Na+ influx (HyperPP). This pathophysiological mechanism can lead to an initial burst of myotonia discharges and result in the symptom of stiffness. However, following the increasing level of discharge, most of NaV1.4 will turn to inactivation and hold back immediately stimulating again, which can result in the symptom of paralysis. Due to the sequential process of the gain-of-function mechanism, the overlap of HyperPP and PMC is not very rare [12–14]. In our first case, we found the c.2111C > T chr17-62034787 (p.T704M) mutation of SCN4A gene, which is related to HyperPP according to the Human Gene Mutation Database (HGMD). It is not very rare to see the overlap of HyperPP and PMC in this mutation for the same pathophysiological mechanism as mentioned earlier, and we conclude that it is the disease-causing mutation in the first family.
The mechanism of HypoPP mutations is the loss-of-function related to anomalous depolarizing gating pore current, which can cause intermittent paralysis[15]. Normally, there is a gating charge transfer center in voltage sensor domains-arginine residues in S4 segment, which will form a hydrophobic narrow waist averted the current. Most HypoPP mutations site at S4 segment, which can cause that hydrophobic amino acid residue replaces the first arginine in S4 segment. Different from the conventional alpha current, this pathophysiological mechanism can trigger the omega current (gating pore current) through the cation pore domain and cause intermittent paralysis [4,16,17].
Generally, we think that the symptom of stiffness does not show in HypoPP, and it is an exclusion criterion for HypoPP. Many PMC mutations has been found in the DIVS4 of the α subunit. However, definite HypoPP mutations has not been found in the domain IV until now[17]. The resistance to the gating pore current in DIVS4 may be caused by the fact that the gating charge transfer center extends a large distance[18]. We have found a few reports on the overlap of PMC and HypoPP [19,20]. However, there is still no further electrophysiological function verification [21,22]. In the second case, we conclude that the R1448H mutation is the disease-causing mutation that leads to the overlap of PMC and HypoPP. Hydrophobic residue replaces the first arginine in S4 segment in the R1448H mutation. Though the mutation is in the relatively distant DIV, we conclude that it can cause pore current connected to HypoPP, and there may be some undiscovered mechanisms in DIVS4 different from S4 segment in other domains. Electrophysiological function tests can be used in these rare cases to study the anomalous gating pore current or other new mechanisms.
Exercise test verify the subtype of channelopathies
In our first case, the CMAP amplitudes increased (+27%) immediately after exercise. The CMAP amplitudes then decreased gradually within 60 min (−27% at most) and started to fall back to the baseline from 90 min at room temperature (27°CSSS). The CMAP amplitudes decreased after exercise immediately, followed by a remarkable decrease after 60 min (−87% at most) and a slow recovery after 90 min at cold temperature (11°C). The change of the CMAP amplitudes tended to be the HyperPP pattern but not reached the abnormal standard (>−33%) at room temperature, and tended to be the PMC pattern at cold temperature. In the second case, the CMAP amplitudes showed a slight transient increase (+13%) immediately after exercise, then a slow decrease within 60 min (−25% at most) and went back to the baseline from 90 min at room temperature (27°C). The CMAP amplitudes decreased gradually after exercise (−36% at most), maintaining the level after 40 min, and recovered after several hours at cold temperature. The change of CMAP amplitudes was HypoPP pattern at cold temperature. Though the change did not reach the abnormal standard (>−33%) at room temperature, it tended to be PMC pattern in consideration of the myotonia charge in EMG and the p.R1448H mutation is related to PMC according to the HGMD.
Inherited mutation or novel mutation
Interestingly, one member of the family (Figure 4A IV 1) was normal in chr17-62019299, who presented the similar overlapping symptoms with the proband of the second family, with clinical episodic weakness and diffuse myotonia discharge in EMG. We then tested his genes using Sanger test and NGS Panel capture test and found the c.2111C > T chr17-62034787 (T704M) gene mutation. We also verified that his father (Figure 4A III 5) and mother (Figure 4A III 6) were normal in chr17-62034787, and we conclude that it is a novel disease-causing gene mutation in him (Figure 4A IV 1). The mutation T704M was disease-causing gene of the first family and their clinical symptoms were similar. This has confirmed that the mutation T704M is a relatively common mutation of channelopathies with the overlap of episodic weakness and stiffness. The novel mutation is occasional or related to undiscovered SCN4A gene variation or other mechanisms, which needs further explorations.
Improvement during pregnancy in patients with channelopathies
Episodic weakness in HyperPP can generally be aggravating during pregnancy [23,24]. However, there have been a few reports on the improvement in HyperPP in pregnancy period[25]. In the first family, the proband’s mother (Figure 4A II 5) was pregnant for the first time when she was 21 years old and the second time when she was 29 years old. During her first pregnant period, the episodic weakness and myotonia did not aggravate. In the second pregnant period, the symptoms alleviated significantly. The cause for the improvement in those patient’s pregnant period may be hyperglycemia for hormonal changes or a low level of potassium during that period. Some studies have also shown that gestagen may have a directive effect on sodium channels. Furthermore, the changes in diet, fluid retention, progesterone, and other aspect during pregnant can also contribute to the improvement in that special period[26]. The unusual improvement can provide some references for the other female in child-bearing period with channelopathies, especially in p.T704M mutation.
Prospect of patch clamp electrophysiology technology
Neurophysiological assessments are widely used to study the function of ion channels, and the most common method is patch clamp electrophysiology technology. Whole-cell patch clamp recording is a conventional method[27]. There are also new patch clamp techniques in channelopathies. These technologies can be used to diagnosis different types of channelopathies, but also could apply to drug research and development for channelopathies[28]. Further function tests are still needed to study, and we want to observe the activation or inactivation kinetics and alterations of the curves recorded by patch clamp electrophysiology technology in myoblasts or myotubes from muscle biopsy in our future research.
Funding Statement
No funding was received.
Glossary
- PP
= periodic paralysis;
- PMC
= paramyotonia congenita;
- HyperPP
= hyperkalemic periodic paralysis;
- HypoPP2
= hypokalemic periodic paralysis type 2;
- AP
= action potential;
- SCM
= sodium channel myotonia;
- ET
= long-time exercise test;
- CMAP
= compound muscle action potential;
- ADM
= abductor digiti minimi;
- RI
= reference interval;
- CK
= creatine kinase;
- MC
= myotonia congenita;
- ATS
= Andersen-Tawil syndrome;
- RP
= rest potential.
Note
1.DlNG ZY, LIU MS, CUI LY. Reference value of long-time exercise test in the diagnosis of primary periodic paralysis. Chin Med J (Engl). 2014;127(18):3219-23PMID: 25266516. A PDF copy of the translated paper will be provided on request to the Corresponding Author.
Acknowledgments
We thank Pathology Lab of our Department and Electron Microscope Room of Shanxi Medical University for supporting the pathological data. We thank Electrophysiology Center of our Department for supporting the electrophysiological data. We thank Huaxing Meng and Jing Zhang for data analysis and their valuable suggestions in the study.
Disclosure statement
The authors have no competing interests.
Financial disclosures
Shan Huang, Wei Zhang, Xueli Chang, and Junhong Guo report no disclosures
Availability of data and materials
All data related to this case report are contained within the manuscript.
Authors’ contributions
JHG, SH, WZ, and XLC participated in the design of the study. SH collected literature and integrated related data and drafted the manuscript. JHG made substantial contribution to manuscript revision. JHG and SH were responsible for data acquisition and analysis. All authors participated in review the manuscript and agreed to be accountable for all aspects of the work. All authors read and approved the final manuscript.
References
- [1].McManis PG, Lambert EH, Daube JR.. The exercise test in periodic paralysis. Muscle Nerve. 1986;9:704–710. [DOI] [PubMed] [Google Scholar]
- [2].Fournier E, Arzel M, Sternberg D, et al. Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol. 2004;56:650–661. [DOI] [PubMed] [Google Scholar]
- [3].Kuntzer T, Flocard F, Vial C, et al. Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve. 2000;23:1089–1094. [DOI] [PubMed] [Google Scholar]
- [4].Cannon SC. Channelopathies of skeletal muscle excitability. Compr Physiol. 2015;5:761–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ding ZY. The value of exercise test in the diagnosis of primary periodic paralysis [Doctoral dissertation]. Peking Union Medical College; 2008. Chinese. [Google Scholar]
- [6].Ding ZY, Cui LY. Value of exercise test in the diagnosis of periodic paralysis. Chin J Neuroimmunol Neurol. 2008;1:69–71. Chinese. [Google Scholar]
- [7].Ding ZY, Liu MS, Cui LY. Reference value of long-time exercise test in the diagnosis of primary periodic paralysis. Chin Med J (Engl). 2014;127(18):3219–3223. [PubMed] [Google Scholar]
- [8].George AJ, Komisarof J, Kallen RG, et al. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Ann Neurol. 1992;31:131–137. [DOI] [PubMed] [Google Scholar]
- [9].McClatchey AI, Cannon SC, Slaugenhaupt SA, et al. The cloning and expression of a sodium channel beta 1-subunit cDNA from human brain. Hum Mol Genet. 1993;2:745–749. [DOI] [PubMed] [Google Scholar]
- [10].Calhoun JD, Isom LL. The role of non-pore-forming beta subunits in physiology and pathophysiology of voltage-gated sodium channels. Handb Exp Pharmacol. 2014;221:51–89. [DOI] [PubMed] [Google Scholar]
- [11].Cannon SC. Sodium channelopathies of skeletal muscle. Handb Exp Pharmacol. 2018;246:309–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brancati F, Valente EM, Davies NP, et al. Severe infantile hyperkalaemic periodic paralysis and paramyotonia congenita: broadening the clinical spectrum associated with the T704M mutation in SCN4A. J Neurol Neurosurg Psychiatry. 2003;74:1339–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kelly P, Yang WS, Costigan D, et al. Paramyotonia congenita and hyperkalemic periodic paralysis associated with a Met 1592 Val substitution in the skeletal muscle sodium channel alpha subunit – a large kindred with a novel phenotype. Neuromuscul Disord. 1997;7:105–111. [DOI] [PubMed] [Google Scholar]
- [14].McClatchey AI, McKenna-Yasek D, Cros D, et al. Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel. Nat Genet. 1992;2:148–152. [DOI] [PubMed] [Google Scholar]
- [15].Cannon SC. Voltage-sensor mutations in channelopathies of skeletal muscle. J Physiol. 2010;588:1887–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moreau A, Gosselin-Badaroudine P, Chahine M. Molecular biology and biophysical properties of ion channel gating pores. Q Rev Biophys. 2014;47:364–388. [DOI] [PubMed] [Google Scholar]
- [17].Matthews E, Labrum R, Sweeney MG, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. 2009;72:1544–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gosselin-Badaroudine P, Delemotte L, Moreau A, et al. Gating pore currents and the resting state of NaV1.4 voltage sensor domains. Proc Natl Acad Sci U S A. 2012;109:19250–19255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Delwaide PJ, Penders CA. Familial paramyotonia and paretic crises with hypokalemia. Rev Neurol (Paris). 1971;125:287–297. [PubMed] [Google Scholar]
- [20].Streib EW. Hypokalemic paralysis in two patients with paramyotonia congenita (PC) and known hyperkalemic/exercise-induced weakness. Muscle Nerve. 1989;12:936–937. [DOI] [PubMed] [Google Scholar]
- [21].Van Osch T, Stunnenberg BC, Sternberg D, et al. Prolonged attacks of weakness with hypokalemia in SCN4A-related paramyotonia congenita. Muscle Nerve. 2018;58:27–28. [DOI] [PubMed] [Google Scholar]
- [22].Mohammadi B, Mitrovic N, Lehmann-Horn F, et al. Mechanisms of cold sensitivity of paramyotonia congenita mutation R1448H and overlap syndrome mutation M1360V. J Physiol. 2003;547:691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lehmann-Horn F, Jurkat-Rott K, Rudel R. Periodic paralysis: understanding channelopathies. Curr Neurol Neurosci Rep. 2002;2:61–69. [DOI] [PubMed] [Google Scholar]
- [24].Finsterer J. Primary periodic paralyses. Acta Neurol Scand. 2008;117:145–158. [DOI] [PubMed] [Google Scholar]
- [25].Finsterer J. Disappearance of episodic weakness during pregnancy in hyperkalemic periodic paralysis from the SCNA4 mutation T704M. Neurologist. 2009;15:289–290. [DOI] [PubMed] [Google Scholar]
- [26].Finsterer J, Wakil SM, Laccone F. Pregnancy reduces severity and frequency of attacks in hyperkalemic periodic paralysis due to the mutation c.2111C>T in the SCN4A gene. Ann Indian Acad Neurol. 2017;20:75–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sakmann B, Neher E. Patch clamp techniques for studying ionic channels in excitable membranes. Annu Rev Physiol. 1984;46:455–472. [DOI] [PubMed] [Google Scholar]
- [28].Li T, Lu G, Chiang EY, et al. High-throughput electrophysiological assays for voltage gated ion channels using SyncroPatch 768PE. PLoS One. 2017;12(7):e0180154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data related to this case report are contained within the manuscript.