

Abstract
Hypertension has emerged as a leading cause of age-related cognitive impairment. Long known to be associated with dementia caused by vascular factors, hypertension has more recently been linked also to Alzheimer’s disease, the major cause of dementia in older people. Thus, while midlife hypertension is a risk factor for late-life dementia, hypertension may also promote the neurodegenerative pathology underlying Alzheimer’s disease. The mechanistic bases of these harmful effects remain to be established. Hypertension is well known to alter in the structure and function of cerebral blood vessels, but how these cerebrovascular effects lead to cognitive impairment and promote Alzheimer disease pathology is not well understood. Furthermore, critical questions also concern whether treatment of hypertension prevents cognitive impairment, the blood pressure threshold for treatment, and the anti-hypertensive agents to be used. Recent advances in neurovascular biology, epidemiology, brain imaging, and biomarker development have started to provide new insights into these critical issues. In this review, we will examine the progress made to date and, after a critical evaluation of the evidence, we will highlight questions still outstanding and seek to provide a path forward for future studies.
Keywords: Dementia, Alzheimer’s disease, neurovascular dysfunction, small vessel disease, cognitive impairment, Hypertension, cerebrovascular disease/stroke
Introduction
Elevated arterial blood pressure (BP) contributes significantly to the global burden of disease, its harmful effects on the brain being a major culprit.1Well established as the leading risk factor for stroke, hypertension has also emerged as a pathogenic factor in cognitive impairment with a vascular basis as well as Alzheimer’s disease (AD), the major causes of dementia in the elderly.2 The harmful effects of hypertension on cognitive function were already noted at the end of the 19th century, in patients who later developed massive cerebral hemorrhages, but systematic investigations on the phenomenon did not appear until the 1950-60s. In particular, pioneering observations on the neuropsychological performance of air traffic controllers with hypertension by Spieth3 and the longitudinal study on cognitive function in hypertensives of Wilkie and Eisdorfer4 provided initial evidence that hypertension deteriorates cognition. The introduction of methods to measure cerebral blood flow (CBF) let to the discovery that individuals with hypertension have increased cerebrovascular resistance, which correlated with the degree of hypertensive retinopathy5, substantiating earlier neuropathological findings linking cerebrovascular damage with the effects of hypertension on brain health. However, the prevailing belief at the time was that the elevation in BP was a beneficial compensatory mechanism aimed at maintaining normal cerebral perfusion, and attempts to lower BP were considered dangerous.6
Subsequent population and prospective studies clearly established the relationship between elevated BP and cognitive impairment,7 and unveiled an unanticipated link between hypertension and AD8. With the development of safe and effective antihypertensive drugs and their widespread use, over the past several decades a dramatic reduction in the morbidity and mortality caused by cardiovascular pathologies, stroke in particular, was observed in a number of large clinical trials9. Surprisingly, however, similar beneficial effects on cognition were not uniformly noted10. Owing to the rapid rise in the prevalence of age-related dementia worldwide, hypertension has received increasing attention as a treatable condition that, if properly controlled, could potentially stave off the onset of the cognitive deficits. Consequently, an increasing number of studies have focused on the mechanisms by which hypertension impairs cognitive function, on the temporal relationships between hypertension and cognitive deficits over the life course, and on the ideal BP targets to achieve in order to minimize its impact on the brain. While this body of new knowledge has provided a deeper insight into the relationship between hypertension and the brain, new questions have emerged that need to be addressed.
The present review seeks to provide an assessment of the state-of-the-art of the effects of hypertension on cognitive function, on the underlying structural and functional alterations and their mechanisms, and on potential preventive and therapeutic strategies to minimize them. After a critical review of the evidence, we will attempt to identify outstanding questions and knowledge gaps that remain to be addressed.
Impact of hypertension on cognition
A growing body of evidence supports the role of hypertension as a risk factor for adverse cognitive outcomes. This evidence primarily comes from epidemiologic studies, which consider hypertension in communities as a risk factor for a range of adverse outcomes including cognitive decline (worsening of cognitive function over years to decades, steeper than expected due to age alone), mild cognitive impairment (MCI) (reduced function in memory, thinking, and other cognitive domains, but not impacting daily functioning), and dementia (impairments in cognition, including memory and other cognitive domains, but with adverse impacts on daily functioning). Studies considering cognitive decline allow evaluation of change, which captures more subtle cases of cognitive dysfunction, without requiring frank thresholds for dementia or MCI to be met, but also may be a less confounded measure of cognitive status, allowing a more pure evaluation of the association between hypertension and cognition.11 In contrast, studies considering MCI and dementia as outcomes have a stronger public health message, as these concepts are easier to define and understand, and cases of preventable disease are easily embraced, as opposed to changes in steepness of cognitive slopes. For hypertension, evidence from epidemiologic studies supports all of these outcomes: hypertension is clearly associated with steeper cognitive decline,12, 13 poor cognitive performance,14 as well as incident MCI15 and dementia.16–19 Furthermore, among adults with MCI, elevated BP is associated with increased progression and worsening of cognition.20 Prehypertension, previously considered as a systolic BP (SBP) <140mmHg, has even been reported as a risk factor for dementia in several studies, including the Atherosclerosis Risk in Communities (ARIC) cohort,18 in which both midlife hypertension and prehypertension conferred a similar risk for dementia, at about 40% higher than among individuals with normotension. New definitions of hypertension now consider SBP in this range consistent with stage 1 hypertension.21
Although epidemiologic data emphasizes the importance of hypertension in the development of stroke, the impact on cognitive outcomes appears to be independent of stroke. This is the case when stroke is defined clinically, with similar associations when individuals with incident stroke are excluded from analyses on cognition,7, 18 but fewer studies have systematically excluded subclinical stroke or small vessel disease (SVD)(including white matter lesions, lacunes and enlarged perivascular spaces), important mediators of the impact of hypertension on cognitive outcomes.
The primary focus of this review is hypertension and its components, systolic (SBP) and diastolic blood pressure (DBP), but different components or aspects of BP may also play an important role in dementia. Although pulse pressure was not significantly associated with dementia in the Honolulu-Asia Aging Study (HAAS),17 it was associated with cognitive decline among APOE ε4 carriers in the Framingham Offspring Study.22 Most human studies do not differentiate between primary and secondary causes of hypertension; although most studies primarily enroll individuals with essential hypertension, rodent models considering several mechanisms of hypertension (including modeling renovascular hypertension along with spontaneously hypertensive rats) have demonstrated similar cognitive deficits and white matter hyperintensities. Orthostatic hypotension, which occurs more frequently in individuals with hypertension, was associated with a 54% higher risk (95% CI 1.20-1.97), with relationships persisting after censoring for ischemic stroke.23 In Uppsala, Sweden, using ambulatory BP monitoring, nondipping circadian BP patterns as well as persistently high 24-hour BP (at age 70) were associated with worse cognitive performance.24 BP variability has also been described in several studies as an important risk factor for reduced cognitive function.25
Age-dependent risk (midlife vs late-life):
Hypertension’s impact on late-life cognitive outcomes appears greatest when considered in middle age. Several studies have demonstrated cognitive decline in the 70’s or older when BP is elevated in the 4th or 5th decade of life. In the HAAS, risk of reduced cognitive function in late-life increased as midlife SBP increased,14 and elevated SBP (>140 mm Hg) in midlife was associated with a 1.77-times higher risk of dementia at ages 71-93.17 Furthermore in HAAS, elevated midlife BP (DBP, especially) and decrease in plasma amyloid-β (Aβ) 1-40 interacted to increase risk of late-life dementia.26 In the National Heart, Lung, and Blood Institute (NHLBI) Twin study, higher midlife SBP was associated with steeper 10-year cognitive decline.13 In the ARIC cohort, hypertension in midlife was associated with steeper 20-year cognitive decline on three repeated cognitive tests, without similar associations with late-life hypertension, which was not associated with cognitive decline over the preceding 20 years.12 Other studies have failed to find an association between BP and dementia or cognitive decline,27 but this may be because they primarily focused on BP in older adults and with a shorter followup period, without evaluation of hypertension during its maximal risk period (middle age).
Some studies have suggested a U-shaped relationship with BP and cognition, but these studies seem isolated to studies of older adults, further reflecting the potential detrimental effect of midlife hypertension but perhaps even a protective effect of late-life hypertension. There is no convincing evidence that low BP, in younger people, is associated with bad cognitive outcomes, but several studies do suggest worse outcomes in older individuals with low BP. In one study, cognitive performance was worst among individuals with either SBP<130 mm Hg or with elevated SBP (≥160 mm Hg), compared to persons at 130-139 mm Hg, but this cohort consisted of adults aged 65-102.28 In an analysis including data from the Rotterdam study and the Göteborg H-70 study, hypertension in old age appeared protective: elderly adults had an inverse association between BP and dementia, with a reduced relative risk for dementia of 0.93 (95% CI 0.88-0.99) per 10 mm Hg higher SBP – but only among elderly adults taking antihypertensive medications.29 In the Bronx Aging Study, adults over age 75 with SBP between 140 and 179 mm Hg had a lower risk for AD (HR 0.55, 95% CI 0.32-0.96), and risk for dementia was especially high in individuals with low DBP at baseline (age>75).30 A U-shaped relationship between BP and cognition was also reported in the Baltimore Longitudinal Study on Aging, among adults over age 80 who had decline at both extremes of BP.31
Although most observed associations are with dementia broadly, several studies have attempted to capture dementia subtype. Given the lack of gold standard for definitively subtyping dementia, it is certainly plausible that these etiologic classifications actually represent mixed etiology, so still may point to a role of hypertension in dementias of other types. In the Hisayama study, midlife (more so than late-life) hypertension was associated with vascular dementia, but not AD,32 with similar findings in the Adult Health Study in Hiroshima, which evaluated SBP in association with subsequent 25-30 year development of vascular dementia.33 In a large Finnish study, midlife SBP≥160 mm Hg was associated with an over two-fold higher risk of AD, independent of other vascular and demographic risk factors.19
Studies using imaging surrogates for cognitive or dementia status also support the importance of midlife hypertension. In the NHLBI Twin study, midlife SBP was associated with not only more white matter hyperintensities in later life but also smaller brain parenchymal volumes.13 Furthermore, BP appears particularly related to atrophy in regions highly relevant for AD (the amygdala and the hippocampus, in this case).34 Other studies have also reported strong associations with hypertension for SVD, including white matter hyperintensities35 and their progression.36
Hypertension over the life course:
Although the above studies emphasize the impact of midlife hypertension as a risk factor for cognitive decline and dementia, change in BP may be relevant as well. As discussed later in this review, BP changes are particularly important when considering shifts that occur in an individual’s autoregulatory curve in response to that individual’s baseline value. In the HAAS study, men who developed dementia had a gradual increase in SBP from mid- to late-life, with a subsequent decrease of 1.36 mm Hg/year in late life; in addition, of the 109 participants in this subcohort who developed dementia, the majority (58%) had had a recent fall in SBP over the preceding 6 years.37 Similar patterns were shown in the Göteborg Longitudinal Population Study, where 70 year olds with elevated BP had more dementia at age 79-85, but many also experienced decline in BP in the years more proximal to a dementia diagnosis.38 In the Framingham Offspring Cohort, elevated midlife BP was an important risk factor for dementia, but a steep decline in BP from mid-to- late-life was associated with a 2.4-times higher risk of dementia in later-life (95% CI 1.4-4.2).39
One complicating factor in studies of BP trajectories is the fact that BP often falls in response to incident cognitive impairment and dementia, making it difficult to determine whether the apparent relationship with recent decline in BP is actually due to reverse causation. Studies support lower and declining BP in elderly individuals with lower cognitive performance, some of whom went on to develop dementia,40 consistent with this concept of reverse causation possibly playing a role. This may also explain the above-cited Rotterdam/ Göteborg H-70 result,29 where hypertension in old age appeared “protective”. Studies considering brain volumes as an outcome have also found evidence that a late-life decline in BP may be a marker for brain volume loss, with more cortical atrophy in individuals with a drop in DBP of more than 10 mm Hg over 20 years in one study.41 In the ARIC study, the pattern of midlife hypertension with subsequent late-life hypotension was associated with smaller brain volumes in regions typically affected in AD.42
Persistently elevated BP, from midlife and continuing through later life, does appear to be consistently associated with worse cognitive outcomes: Yaffe et al., evaluated cumulative SBP and DBP level over 25 years, finding worse cognitive performance on several tests among individuals with higher area-under-the-curves based on cumulative BP.43 However, using a Mendelian Randomization design, genetically-predicted systolic hypertension was associated with lower risk of AD, possibly indicative of subsequent changes in lifestyle or management in response to this elevated risk.44
Cognitive domains affected:
Although several studies have suggested that the impact of hypertension on cognition is global, this is mostly the case because not all studies broadly consider several distinct cognitive domains, or because the most impacted domains in hypertension-associated brain disease are also represented in global cognitive measures. When more detailed neuropsychological batteries are performed and hypertension-specific effects are considered and compared, the biggest impact of hypertension appears to be on executive function,12 motor speed, and attention,45 which are classically considered domains involved with subcortical disease, such as typical vascular disease or pure vascular dementia.46 Even scores on the Mini-Mental State Examination (MMSE) and Montreal Cognitive Assessment (MoCA), two global cognitive measures, although lower in individuals with more vascular risk factors, including hypertension, are primarily driven by impairments in items related to attention (for the MMSE) and visuo-executive function (for lower scoring individuals on the MoCA).47
The role of memory impairments in hypertension-related dementias is complicated by the likely overlap between etiologies and the high prevalence of mixed dementias. Although memory is more typically involved in AD-related cognitive impairments than in vascular types of dementia, it has been noted to have impairments in individuals with a combination of hypertension and elevated brain amyloid by PiB-PET.48 However, many of these studies of domain-specific cognitive deficits are influenced by misdiagnosis or difficulty determining etiology during life. Using a study neuropathologic data, domains did not differ by etiology as might be expected: individuals with pure AD neuropathology had reduced memory scores, more so than executive function, but individuals with cerebrovascular disease had similarly reduced premorbid cognitive function in executive function and verbal and nonverbal memory.49
In summary, epidemiologic data strongly support that hypertension is associated with worse cognitive outcomes, especially when BP is considered in midlife. A pattern of midlife hypertension followed by later-life hypo-or normotension may be especially problematic, and consideration of BP variability not only over the course of a day but over years and decades may be important in understanding what components of BP are most important in increasing risk of cognitive decline, MCI, and dementia. Although hypertension may impact all aspects of cognition, its impact is greatest on motor speed, attention, and executive functioning. The likely overlap with AD pathology, however, in the large group of individuals with mixed pathology of MCI and dementia, makes differentiation of dementia subtype on the basis of patterns of impaired cognitive domains challenging.
Pathobiology of the effects of hypertension on the brain
The cerebral vasculature is the prime target of the deleterious effects of hypertension on the brain. In this section we will briefly review basic concepts on the blood supply of the brain and its regulation, and examine how chronic hypertension affects the structure and function of cerebral blood vessels.
The blood supply of the brain
The brain’s blood supply is provided by the internal carotid arteries and the vertebral arteries. These extracranial vessels enter the skull and merge the base of the brain to form the circle of Willis, from which the major intracranial vessels supplying the brain originate (anterior, middle and posterior cerebral arteries)(Figure 1).50 Branches of these arteries travel along the brain surface giving rise to a highly anastomotic vascular network (pial arteries)50. Branches of the pial arteries penetrate the substance of the brain (penetrating arterioles) surrounded by a virtual space, the perivascular space or Virchow-Robin space50, separating the vascular basal lamina from the astrocytic processes enveloping the brain parenchyma (glial limitans). As the vessels get smaller, the perivascular space disappears and the vascular basal lamina enters in direct contact with the astrocytic foot processes, which completely envelop the vessel’s outer surface50. At this level, the vessel wall includes a single layer of smooth muscle cells resting on the endothelial basement membrane50. As the vessels transition into capillaries, the smooth muscle cell layer becomes discontinuous and is gradually replaced by pericytes covering ≈30% of the abluminal capillary surface.51 Venules are endowed with a discontinuous smooth muscle cell layer, which becomes more pronounced in veins, although not as marked as in the media of arteries. Recent single cell RNA sequencing studies have started to unveil the molecular bases of the diversity of the cellular composition of the cerebrovascular tree52. These efforts, still in an initial stage, will provide critical insight into the cellular, molecular and signaling mechanisms underlying the segmental susceptibility to hypertension of the cerebral vasculature.
Figure 1:

Cerebrovascular anatomy and segmental pathology induced by hypertension. Large extracranial cerebral arteries (internal carotid arteries and vertebral arteries) enter the skull and converge at the base of the brain to form the circle of Willis from which the major intracranial cerebral arteries emanate. Branches of these arteries form a dense anastomotic network (pial arteries ad arterioles) which give rise to arterioles that dive into the substance of the brain (penetrating arterioles). Some vessels, like the lenticulostriate arteries, arise from the circle of Willis and proximal branches and vascularize the deep brain structures including the white matter. The predominant pathology caused by hypertension in the different segment of the cerebrovascular tree is indicated. Due to its precarious blood supply from terminal arterioles particularly vulnerable to hypertensive damage, the subcortical white matter is highly likely to be harmed by hypoxia-ischemia. Abbreviations: ICA, internal carotid artery; MCA, middle cerebral artery; WM, white matter.
Effects of hypertension on cerebrovascular structure
Chronic hypertension exerts a profound influence on the structure of the cerebral vasculature. These structural changes reflect an adaptive response to protect downstream microvessels from the increased transmural pressure. However, this process over time becomes maladaptive resulting in different pathologies depending on the segment of the cerebrovascular tree involved (Figure 1).
Large cerebral arteries:
Hypertension is a leading risk factor for extracranial and intracranial atherosclerosis.53 Extracranial lesions are characterized by lipid accumulation in carotid and vertebral arteries, often associated with ulceration and atheroma instability and protrusions, all linked to artery-to-artery embolism54. Intracranial lesions, affecting the circle of Willis and its major branches, have a more fibrous component, more likely to result in local vascular occlusions and focal ischemic stroke55.
Remodeling is an important structural change induced by hypertension and other vascular risk factors in extracranial and intracranial cerebral arteries56. Hypertrophic remodeling leads to increased wall thickness and reduction in vascular lumen, with an increase in media-to-lumen ratio and in the total volume of wall tissue56. Eutrophic (inward) remodeling consists of an increased media thickness, a reduced lumen and external diameter, without changes in the total amount of wall tissue56. In a recent study assessing remodeling in the carotid arteries using MRI, the pattern of remodeling varied according to the arterial segment57. Whereas the common carotid artery demonstrated hypertrophy, the carotid bifurcation exhibited eutrophic remodeling. In contrast, the internal carotid artery exhibited a mixed pattern of inward and outer remodeling57. Sex and comorbidities had a significant effect on the pattern of remodeling57.
Hypertension is also associated with stiffening of large arteries. In humans as in animal models, stiffening precedes the development of hypertension, raising the possibility that it may contribute to the BP elevation by reducing vascular compliance and increasing pulse pressure58. At the same time, stiffening may also reduce CBF59, deplete cerebrovascular reserves60, and increase the hydrodynamic impact of the elevated pressure (pulsatility) on the cerebral microvasculature61. Perhaps due to these deleterious microvascular actions, arterial stiffening is a sensitive predictor of subsequent white matter lesions and cognitive impairment.
Pial and penetrating arteries and arterioles:
These vessels are responsible for a sizable component of the pressure drop observed between large arteries and the cerebral capillaries50, 62, and, as such, are a prime target of the effects of chronic elevations in BP. Animal studies suggest that hypertrophic and eutrophic remodeling are the predominant alterations in pial and penetrating vessels, stiffening being less common than in large arteries.56 These vessels are often surrounded by enlarged perivascular spaces, a hallmark of SVD.63 Particularly vulnerable are small arteries and arterioles of the deep cortical, periventricular and basal ganglia white matter. These vessels arise either from the first segment of the middle cerebral artery (MCA), e.g., lenticulostriate arteries, or from terminal branches of the pial arteries, and converge on the white matter deep into the hemisphere (Figure 2).64 Due to the limited anastomosis and collateralization among penetrating arterioles, their dysfunction or occlusion is particularly damaging to the white matter.62, 65, 66 Hypertension also induces degenerative changes of the vessel wall, including accumulation of an amorphous material consisting of degenerated smooth muscle and collagen (lipohyalinosis) or infiltration of fibrin and fibrin degradation products (fibrinoid necrosis).67 Possibly due to their shorter length and higher pressures, fibrinoid necrosis is more common in basal ganglia arterioles, and is frequently associated with cerebral hemorrhage.67 Microatheromas can also be observed in larger arterioles, which can lead to vascular occlusion64. Alterations are also observed in capillaries, with reduction in their number, swelling and loss of endothelial cells and pericytes (string vessels), tortuosity, increased thickness of the basal lamina, and fibrin deposition.68
Figure 2:
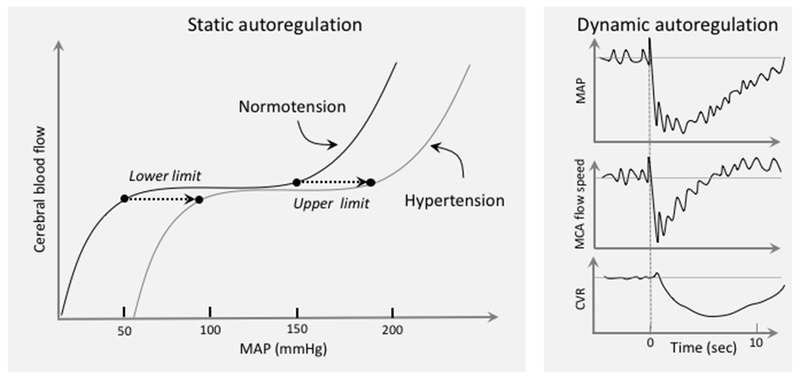
Cerebrovascular autoregulation and hypertension. Autoregulation keeps CBF relatively stable during fluctuations in BP. Autoregulation was first examined by measuring CBF during stepwise changes in BP, once a steady state is reached (static autoregulation) (left). Based largely on animal data, chronic hypertension was found to shift the pressure-flow curve to the right, such that the autoregulated range moves towards higher BP. The development of methods to assess CBF dynamically like transcranial doppler flowmetry, enabled to study the flow velocity changes in large cerebral arteries during BP transients induced by different maneuvers, e.g., standing from a sitting position. As illustrated in the right panel, MCA flow drops in sync with AP. But, owing to an autoregulatory drop in cerebrovascular resistance (CVR), it recovers faster. Evidence suggests that hypertension does not impair dynamic autoregulation, except in severe hypertension (see text for further details). Right panel data from Aaslid et al. 195
What causes remodeling and stiffening?
Remodeling and stiffening result from interacting mechanical, humoral and cellular factors including hemodynamic stress, endothelial dysfunction, vascular inflammation, immune cell infiltration, and calcium deposition69, 70. Both processes are driven by overlapping mediators including inflammatory cytokines, sex hormones, angiotensin-II (AngII), endothelin, aldosterone, and oxidative stress71. In addition, modifications of the extracellular matrix plays an essential role in stiffening. Accumulation of collagen and fibronectin, elastin fragmentation by matrix metalloproteases, and the pro-fibrotic effects of transforming growth factor-β are critical factors in the process69. A deficit in endothelial nitric oxide (NO) and free radicals may play an important role in remodeling.56, 71 In the aorta perivascular macrophages (PVM) expressing the lymphatic hyaluronan receptor LYVE1 safeguard the elasticity of the vascular wall by governing collagen content in smooth muscle cells through hyaluronan and pericellular matrix metalloprotease-9.72 Although LYVE1+ PVM are present also in cerebral arteries,72, 73 it remains unclear if they serve a similar role in the homeostasis of the cerebrovascular wall.
In summary, stiffening, hypertrophic and eutrophic remodeling result from the concerted action of mechanical, cellular and molecular factors that converge on the vascular wall to alter its structure and composition, as well as the balance between collagen and elastin. Intended to protect the downstream microvessels from the mechanical stress associated with elevated pressure, these responses become maladaptive and, in concert with the functional alterations described in the next section, contribute to the harmful effects of hypertension on the brain.
Impact of hypertension on cerebrovascular function
Endowed with limited energy reserves, the brain is highly dependent on the timely delivery of oxygen and glucose through blood flow to maintain its functional and structural integrity50. To this end, sophisticated neurovascular regulatory mechanisms assure that the blood supply is well matched to the brain’s regionally and temporally diverse metabolic requirements, balancing energy delivery with removal of unwanted byproducts of brain activity50. In this section, we will first review these mechanisms and then examine their disruption by hypertension.
Neurovascular coupling and endothelial regulation:
Neurons, astrocytes and vascular cells work as a single functional unit (neurovascular unit) to maintain the homeostasis of the brain’s internal milieu50. Brain activity is a major regulator of local cerebral perfusion, such that increased synaptic activity leads to increases in CBF highly localized to the activated area50. This fundamental property of the cerebral circulation (neurovascular coupling) results from the close interaction among cells of the neurovascular unit at all levels of the cerebrovascular tree. Although the mechanistic bases of the hemodynamic response remain to be completely elucidated, it is now clear that neurons, astrocytes, endothelial cells, vascular smooth muscle cells, and pericytes all participate in this carefully orchestrated process through multiple diffusible mediators (NO, prostanoids, adenosine, ions, etc.), ion channels and segment-specific signaling mechanisms50. Thus, activation of neurons deep in the substance of the brain initiates a series of highly coordinated vascular changes that start at the level of the capillary endothelium and are transmitted to upstream arterioles through intramural vascular signaling resulting in their relaxation. The retrograde propagation of the vasodilation ultimately involves larger vessels at the brain’s surface, which is required to induce a vigorous increase in flow to the activated area50, 74. Endothelial cells are important regulator of cerebral perfusion by producing powerful vasodilators, e.g., NO, prostacylin, or vasoconstrictors, e.g., endothelin-1.75 These vasoactive mediators are released in response to mechanical or chemical stimuli resulting in increased or decreased in blood flow (endothelium-dependent vasodilatation or vasoconstriction).75 The endothelium is also the site of the blood-brain barrier (BBB) governing the bidirectional molecular exchange between brain and blood.76 Furthermore, the endothelium exerts vital trophic effects on brain cells and contribute to maintain the health of neurons, glia, and oligodendrocytes77, 78.
Hypertension reduces resting CBF, suppresses neurovascular coupling and endothelium dependent responses. Contrary to early observations5, several studies have shown focal or global reductions in resting CBF in individuals with hypertension79, 80. In a large scale longitudinal study of hypertensives with ischemic lesions and other vascular risk factors, a reduction in global CBF was observed with uncontrolled or untreated hypertension, but not with satisfactory BP control81. In cognitively normal people with hypertension, but no other risk factors or evidence of brain damage, focal reductions in CBF were observed in the prefrontal, cingulate, temporal, and occipital cortex82, suggesting that the reduction in CBF may precede the development of brain pathology. It remains unclear if these reductions in CBF are a consequence of reduced brain energy metabolism or of a direct effect of hypertension on the cerebral vasculature. Although reductions in glucose utilization, an index of energy metabolism, have been reported in neocortical and subcortical brain regions of patients with well-controlled longstanding hypertension83, it has not been established whether CBF is also reduced in these regions. However, in patients with severe hypertension oxygen consumption is reduced in regions with reduced CBF80, suggesting that with disease progression the vascular effect could be related to reduced metabolic demands possibly related to brain dysfunction and damage.
Relatively few studies have investigated neurovascular coupling in patients with hypertension. The cerebrocortical CBF response produced by cognitive tasks (memory and attention), assessed by O15 positron emission tomography (PET), was reduced in patients with untreated hypertension.84 Furthermore, in patients with repaired aortic coarctation, in whom only pulse pressure was significantly elevated, the flow velocity increase produced by photic stimulation in the posterior cerebral artery, which supplies the visual cortex, was suppressed.85 Consistent with altered neurovascular coupling, the increase in retinal blood flow induced by light flickering was absent in individuals with hypertension.86 These findings, albeit limited, support the view that neurovascular uncoupling may compromise substrate delivery to the brain and render the brain more vulnerable to vascular insufficiency.
Endothelial dysfunction has been studied extensively in peripheral arteries and was found to precede the elevation in BP and, once developed, to correlate with its severity87. Although direct assessment of cerebral endothelial function in humans is not feasible, inhibition of NO synthesis does not reduce retinal flow in patients with hypertension, indicating a deficit of NO and endothelial dysfunction86. Furthermore, post-mortem analysis of arterioles of patients with SVD, most often caused by hypertension, showed a reduced response to the endothelium-dependent vasodilator acetylcholine88. Independent evidence for endothelial dysfunction is also provided by the observation that hypertension alters the BBB89, a property of the cerebral microvasculature attributable to endothelial cells.76
The implications of cerebral endothelial dysfunction for the effects of hypertension on cognition are paramount. Endothelial dysfunction is a prelude to atherosclerosis75 and, as mentioned above, contributes to vascular remodeling and stiffening. Failure of endothelium-dependent vasodilation in peripheral arteries correlates with white matter lesion burden90 and microbleeds91, whereas in a population of elderly hypertensives with subjective memory complaints, white matter lesion burden was associated with circulating levels of von Willebrand factor, an index of endothelial dysfunction.92 The BBB dysfunction is also likely to have a devastating effect on the brain especially in the white matter, wherein increased BBB permeability has been implicated in the damage produced by hypertension.89
Cerebrovascular autoregulation:
Cerebrovascular autoregulation refers to the ability of brain vessels to maintain a relatively stable CBF despite changes in BP within a certain range.93 Owing to cerebrovascular autoregulation, CBF does not follow passively the BP swings that occur during activities of daily living, protecting the brain from excessive CBF fluctuations. Autoregulation has traditionally been assessed using stepwise changes in BP and measuring the corresponding changes in CBF when a steady state is reached (static autoregulation)94, 95. In the normal state, plotting the CBF change as a function of the BP changes leads to an italic S-shaped curve with a plateau in which CBF remains relatively stable despite changes in BP (the autoregulated range: 60-150 mmHg mean BP) (Figure 2)94. More recently, methods have been developed to assess cerebrovascular perfusion dynamics in response to fast changes in BP (dynamic autoregulation)(Figure 2)93. These studies have shown that during pressure changes on a time scale of seconds, blood flow does not remain stable, but follows BP until the autoregulatory adjustments in vascular resistance counteract the BP change (Figure 2). In general, the faster the change in BP the less efficient is autoregulation96.
The cellular bases of autoregulation rest in part on the intrinsic property of the smooth muscle cells to constrict in response to increases in transmural pressure (the myogenic response of Bayliss)95. Accordingly, if BP increases, the smooth muscle cells constrict the vessels and increase vascular resistance, thereby preventing the CBF increase resulting from the increased pressure. Conversely, decreases in BP relaxes the smooth muscle, reducing vascular resistance and counteracting the anticipated CBF decrease. Multiple redundant mechanisms underlie the myogenic response including: (a) mechanosensors that modulate intracellular Ca2+ levels in response to changes in transmural pressure; selected candidates include: transient receptor potential channels97 and G-coupled receptors, including AngII type-1 receptors (AT1R)98 and, possibly, tissue necrosis factor receptors99; (b) amplification of the Ca2+ signal by depolarization and opening of voltage-gated Ca2+ channels leading to further Ca2+ entry, as well as intracellular Ca2+ release100; (c) modulation of the Ca2+ sensitivity of the contractile apparatus by regulating the balance between kinases and phosphatases responsible for myosin light chain phosphorylation and involving Rho kinase and protein kinase-C101. The mechanisms regulating the vasodilatation that occurs during BP lowering are less well understood. It has been suggested that the vasodilatation occurring with reduced BP may be an active phenomenon driven by metabolic byproducts, such as adenosine, which accumulates as a result of the reduced tissue oxygen tension caused by the hypoperfusion102.
Hypertension results in the chronic adaptation of the cerebral circulation to higher levels of BP, such that the autoregulated range of CBF is higher than in normotensive individuals103. The implication of this right shift of the curve is that, if the BP is lowered to a level that would be safe in non-hypertensive individuals, the brain would be more susceptible to hypoperfusion94. The shift of the autoregulatory curve, first described in a baboon model104 and verified in a limited number of hypertensive individuals, has been attributed to the increase in vascular resistance induced by remodeling in cerebral resistance vessels.94 More recent studies of dynamic autoregulation have failed to report major alterations in the moment-to-moment adjustments of CBF to BP changes.105–107 Rather, in patients with moderate hypertension (mean systolic BP: 163±11mmHg), an enhancement of dynamic autoregulation was found.108 Furthermore, lowering BP with anti-hypertensive therapy did not reduce flow velocity,107, 108 but improved carotid artery distensibility after 6 months of treatment.107 However, in patients with malignant hypertension (SBP: 180-260 mmHg) dynamic autoregulation was altered and flow velocity followed passively the reduction in BP during treatment with sodium nitroprusside,109 suggesting that severe disease can have a profound impact on both static and dynamic autoregulation.
These observations, collectively, indicate that while hypertension may shift the autoregulatory range towards higher BPs, the compensatory adjustments taking place during BP variations within this range are not impaired, except in malignant hypertension. In addition, in moderate hypertension, lowering BP does not seem to lead to cerebral hypoperfusion. However, autoregulation data over the life course and in the presence of vascular comorbidities are not available. Therefore, it remains unclear how magnitude and duration of the BP elevation influence dynamic autoregulation and what is the impact of antihypertensive therapy on CBF.
Perivascular spaces, clearance, neuroimmune trafficking:
The brain vascular system and surrounding structures are also responsible for the clearance of waste products generated by brain activity, including toxic proteins such as amyloid-β and tau.110 A transvascular pathway utilizes abluminal transporters that carry solutes through the vascular wall and drain into the circulating blood.76 Other pathways are thought to use the perivascular space, a site of exchange between the interstitial fluid (ISF) and the cerebrospinal fluid (CSF), as a conduit to carry solutes to the brain surface. In particular, a perivascular pathway has been proposed to travel retrogradely within the arterial wall111 whereas a paravascular “glymphatic” pathway, involving aquaporin-4 in astrocytic end-feet, reaches the brain surface through perivenous spaces.112 However, this field is still in evolution and additional evidence is needed to assess the relative weight of these clearance pathways in health and disease. In the end, solutes drain into the cervical lymphnodes, probably through dural lymphatics.113 Cerebral blood vessels and perivascular spaces are also critical for the trafficking of immune cells in and out of the brain required to maintain immune homeostasis and protect the brain from invading pathogens.114 Innate and adaptive immune cells enter the brain through transvascular, meningeal, choroid plexus routes and play a critical role in immune surveillance and in a wide variety of pathologies.114 In particular, as described in the next section PVM located in the perivascular space have emerged as a critical source of reactive oxygen species (ROS) in hypertension with a profound impact on neurovascular function.115
How does hypertension cause neurovascular dysfunction?
Several animal models have been used to investigate the neurovascular effects of acute or chronic elevations in BP.116 Better-studied models involve administration of pressor doses of AngII, which induce acute elevations in BP, or administration of sub-pressor doses (“slow pressor” AngII hypertension) which produce delayed elevations in BP over several days. The slow pressor model has gained popularity since is thought to reflect the progressive increase in BP observed in essential hypertension, although with a more compressed time scale (2-4 weeks)117. In hypertension induced by acute or chronic AngII administration there is attenuation of neurovascular coupling and endothelium-dependent responses.118–120 Interestingly, the mechanical effects of the elevation in transmural pressure are not required for the cerebrovascular dysfunction, at least in the short term.118, 119 Neurovascular dysfunction and cognitive deficits have also been reported in other models of hypertension, including life-long hypertension in spontaneously hypertensive rats (SHR) or mice (BPH mice)73, intermittent hypoxia (a model of sleep apnea),121 hypertension induced by administration of deoxycorticosterone acetate+salt (DOCA-salt)122, eNOS inhibition, etc. (see also116, 123).
The pathways mediating the neurovascular effects of hypertension induced by slow-pressor AngII administration have been studied in some detail and involve both central effects on the brain and peripheral effects on cerebral blood vessels. The evidence suggests that circulating AngII acts on AT1R in the subfornical organs, one of the circumventricular organs, leading to ROS-mediated production of vasopressin from the hypothalamic paraventricular nucleus. Vasopressin, in turn, acts on cerebral blood vessels to trigger local endothelin-1 production which, in concert with circulating AngII, contributes to the neurovascular dysfunction by inducing vascular oxidative stress.124 Evidence from systemic vessels also indicates a critical role of vascular oxidative stress.125 However, how oxidative stress alters vascular function remains to be defined. In AngII hypertension, peroxynitrite, the reaction product of NO and superoxide, is required for the neurovascular dysfunction,126 but whether the effect depends on reduction of NO bioavailability by the reaction with superoxide, or on downstream effects of peroxynitrite, remains to be established. Evidence from the AngII hypertension model indicates that the latter possibility is more likely, since a peroxynitrite decomposition catalyst rescues the neurovascular dysfunction.126 Peroxynitrite could alter vascular function through several mechanisms.127 In the neurovascular dysfunction produced by Aβ, peroxynitrite-induced DNA damage activates the DNA repair enzyme poly-ADP ribose polymerase and leads polyADP ribose production, which, in turn, opens transient receptor potential melastatin-2 channels resulting in endothelial Ca2+ overload and dysfunction128. However, it remains to be established if this mechanism is also involved in the neurovascular effects of AngII hypertension.
There are several potential enzymatic sources of vascular oxidative stress, including mainly mitochondrial complexes, xanthine oxidase, and eNOS uncoupling, a condition in which the enzyme generates ROS instead of NO.129, 130 However, a NOX2-containing NADPH oxidase has emerged as a critical source of the ROS involved in neurovascular dysfunction especially in AngII hypertension. NOX2 is present in vascular and perivascular cells in close association with AT1R.73, 119 Furthermore, pharmacological inhibition or genetic deletion of NOX2 rescues the neurovascular dysfunction in models of hypertension.119, 120, 124 Other isoforms of NOX, particularly NOX1 and NOX4, may also present in the cerebral vasculature, but their cellular localization and role in neurovascular dysfunction have not been elucidated.129
As for the cellular source of ROS, endothelial cells, smooth muscle cells, and pericytes have the potential to produce ROS. However, recent data suggest that PVM are a critical source of the ROS initiating the vascular dysfunction in AngII hypertension. PVM are endowed with AT1R and, as phagocytic cells, express large amounts of NOX2 and have a greater potential for ROS production than vascular cells115. In slow pressor AngII hypertension and in BPH mice, a genetic model of life-long hypertension, PVM depletion prevents vascular oxidative stress, neurovascular dysfunction and cognitive impairment, as well as MCA remodeling in hypertensive rats73, 131. Using bone marrow transplantation to replace PVM with bone marrow-derived macrophages lacking the AT1R or NOX2 abrogates vascular oxidative stress and neurovascular dysfunction, pointing to PVM as the cellular site of the AT1R and NOX2.73 Since in BPH mice and in the slow pressor model AngII is elevated in the circulation but not in brain, circulating AngII was found to cross the BBB and gain access to PVM on the abluminal side of the vessels (Figure 3).73 Although PVM are unlikely to be the sole source of vascular oxidative stress, the observation that their elimination or genetic modification blocks vascular ROS production, suggests that they are the primary source, which may trigger oxidative chain reactions in adjacent vascular cells. Whether PVM are also involved in the neurovascular dysfunction in human hypertension remain to be established. However, as discussed in the next sections, the observation of perivascular space abnormalities, vascular oxidative stress and inflammation in SVD is consistent with findings emerged from animal models implicating PVM.
Figure 3:

Putative mechanisms of neurovascular dysfunction in slow pressor AngII hypertension, BPH mice and DOCA-salt hypertension. Circulating or brain AngII reaches AT1R in PVM wherein it activates Nox2 leading the vascular oxidative stress and neurovascular dysfunction. AT1R and Nox2 are also present in other vascular cells, but they do not seem to play a primary role in these hypertension models. The cartoon depicts a penetrating arteriole, but brain macrophages are also present in the meninges, wherein they may play a role in pial and meningeal vessel vasomotor function and permeability. Whether PVM have a similar role in models of AngII-independent hypertension remains to be established. Abbreviations: AT1R, AngII type 1 receptor; PVM, perivascular macrophages; SMC, smooth muscle cells. (Illustration Credit: Ben Smith).
In summary, experimental studies and human data indicate that oxidative stress and inflammation are critical factor in the alterations in neurovascular and endothelial function produced by hypertension. Although other enzymatic sources cannot be excluded, NOX2 has been identified as a major source of ROS in hypertension involving AngII. Furthermore, innate immune cells, the PVM in particular, have emerged as a powerful source of vascular ROS production.
Brain lesions induced by hypertension and their impact on cognition
The alterations in cerebrovascular structure and function induced by hypertension described in the previous sections predispose the brain to dysfunction and damage, which, in turn, alters cognition. Chronic hypertension is the major risk factor for ischemic and hemorrhagic stroke, which is associated with a 3-6 fold increase in cognitive impairment, especially when multiple strokes are involved (multi-infarct dementia).66 Additional lesions associated with hypertension include microinfarcts and microhemorrhages, microscopic ischemic or hemorrhagic lesions most common in the cerebral cortex, which portend cognitive deficits.132–134 Hypertension is also associated with enlarged perivascular spaces, which also correlate with cognitive impairment.63 The expansion of the perivascular spaces may be due to fluid stagnation and protein accumulation reflecting impaired ISF/CSF clearance, as well as the mechanical stresses on the perivascular tissue by the increased pulsatility and stiffening63. Of note, enlarged perivascular spaces can be confused with small cavitated lesions (lacunae) on conventional brain imaging, and it has been proposed that lacunes rather than enlarged perivascular spaces correlate with cognitive impairment.135 The mechanisms of the brain atrophy induced by hypertension remain unclear, but (a) chronic hypoperfusion leading neuronal loss79, (b) retrograde neurodegeneration from deafferentation due to lesions in other brain regions136, or (c) vascular oxidative stress and inflammation,137 leading to neurovascular trophic failure138 are potential contributing factors.
The most common brain lesions associated with hypertension are white matter lesions, especially in the frontal cortex, which appear as areas of hyperintensity on T2-weighted MRI.139 SVD is considered the major cause of white matter disease, but the mechanisms have not been completely elucidated.140 Cerebral hypoxia-ischemia due to stenosis or occlusion of vessels feeding the white matter, which are particularly susceptible to the effect of hypertension, is considered to be a major contributor.66 In support of this hypothesis, reduction in CBF in normal appearing white matter and expression of hypoxic markers have been reported.141, 142 Other studies have not supported this view143, 144, and have suggested that hypoperfusion is not the cause but the consequence of the white matter lesion.145 Inflammation, upregulation of metalloproteases, and increased BBB permeability in the white matter may lead to water shift and local edema resulting in microvascular compression and occlusion.145, 146 A link between vascular health and white matter maintenance and repair by oligodendrocyte has also been established. A population of oligodendrocytes associated specifically with cerebral blood vessels has been identified147 and oligodendrocyte proliferation and survival requires vascular growth factors.148 These observations have suggested an “oligovascular niche” essential for the maintenance of white matter health. Indeed, arrested development of oligodendrocyte precursors has been observed human white matter in SVD resulting in faulty remyelination of damaged white matter (Figure 4).149
Figure 4:

Potential mechanisms of WM damage by hypertension. Vascular oxidative stress and inflammation disrupt the BBB, induce NVU dysfunction and damage, and impair oligodendrocyte development and function. The resulting alterations in tissue homeostasis, hypoxia-ischemia, reduced brain clearance and impaired remyelination lead to WM damage. Abbreviations: NVU, neurovascular unit.
In summary, oxidative stress, hypoxia-ischemia, inflammation and BBB dysfunction are critical vascular factors threatening the health of the subcortical white matter. There might be regional differences on the impact of hypertension on white matter, and the white matter of the frontal lobe may be more susceptible.133, 139 However, the mechanistic bases for such predilection remain to be established.
How does hypertension cause cognitive dysfunction?
Atrophy, brain damage caused by macro- and micro-infarcts and hemorrhages are important determinants of cognitive impairment. Furthermore, infarcts strategically placed in brain regions involved in cognition, e.g., hippocampus, medial thalamus, frontal lobe, etc., can produced cognitive dysfunction despite relatively small volume of damage,66, 133 and the estimated functional impact of microinfarcts is greater than anticipated by the volume of injury.150 The volume and location of white matter lesions also correlate with the degree and temporal evolution of cognitive dysfunction.133 These white matter lesions may affect cognition by impairing the connectivity between the anterior thalamus to the frontal cortex.133, 151 Indeed, studies using white matter tractography and resting state MR imaging have provided evidence for loss of connectivity and network efficiency degradation, which correlate with a reduction in processing speed, a typical feature of vascular cognitive impairment.151 In addition, brain atrophy could also be a contributing factor by reducing gray matter in critical brain regions such as hippocampus and prefrontal cortex.152
The impact of enlarged perivascular spaces on cognitive impairment of hypertension remains uncertain. The morphological alterations of the perivascular space raise the possibility that perivascular and paravascular clearance systems may be altered and have a role in the white matter damage63, 140. Perivascular spaces are enlarged and distorted in hypertension63, an alteration that could hamper the disposal of potentially toxic byproducts of brain activity. Hypertension is associated with amyloid-β and tau pathology (see section on overlap with AD), suggesting that clearance of these proteins may be reduced. Hypertension-induced large artery stiffening increases pulsatility in microvessels, which has been proposed to alter perivascular spaces and reduce ISF/CSF clearance63. However, a transient increase in pulsatility induced by administration of dobutamine increases CSF clearance in mice153, suggesting that hypertension could increase ISF/CSF clearance. In support of this hypothesis ISF flow is increased in spontaneously hypertensive rats, but such increase is associated with greater retention of tracers at the ISF/CSF interface, indicating a reduction in clearance154. On the other hand, carotid ligation reduced CSF clearance153, suggesting a negative impact of cerebral hypoperfusion and/or reduced pulsatility, which is relevant to large artery stenosis or occlusion often associated with hypertension. Therefore, direct evidence linking hypertension to brain clearance pathways has not been provided, in humans as in animal models, and additional work is needed in this area.
These observations, collectively, suggest that hypertension may lead to cognitive impairment through several pathogenic factors (Figure 5). Gray matter loss, loss of connectivity and network efficiency from white matter lesions, reduced perivascular clearance and neurovascular dysfunction all have the potential to impair brain function, but their relative impact on cognitive processes remain to be established.
Figure 5:
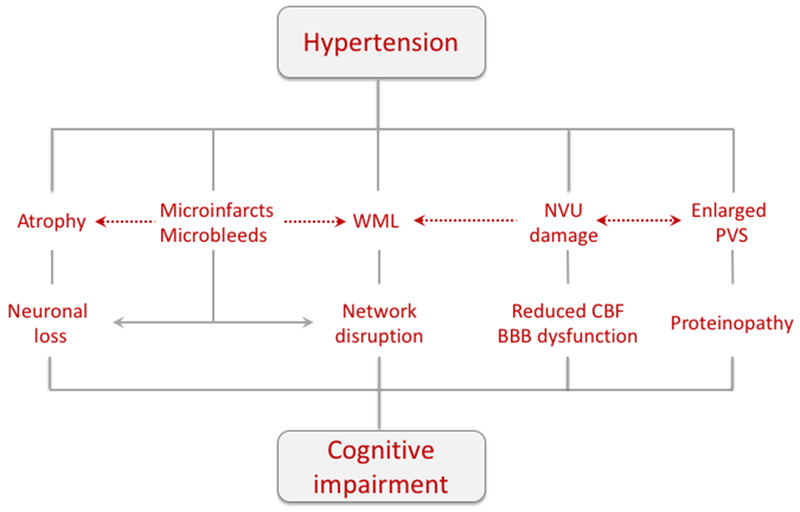
Brain lesions produced by hypertension that underlie cognitive impairment. Brain atrophy, microinfarcts and microbleeds cause neuronal loss and brain dysfunction. In addition, microinfarcts and microbleeds disrupt brain connectivity and reduce network efficiency Damage to WM tracts (WML) also degrades connectivity, especially in thalamo-cortico networks. Alteration in PVS impair brain clearance and may promote protein accumulation in brain and vessels, and dysfunction of the NVU leads to vascular insufficiency and BBB damage. Potential interactions between different pathogenic mechanisms are indicated by the dashed arrows. Abbreviations: NVU, neurovascular unit; PVS, perivascular spaces; WML, white matter lesions.
Hypertension and Alzheimer’s pathology
A large body of evidence indicates that hypertension is a risk factor for AD, a condition once considered purely a neurodegenerative disease. AD is characterized neuropathologically by extracellular accumulation of amyloid-β (amyloid plaques) and intraneuronal aggregates of hyperphosphorylated tau (neurofibrillary tangles).2 Aβ is derived from the proteolytic cleavage of the amyloid precursor protein (APP) by β- and γ-secretase enzymes.2 Vascular brain lesions similar to those associated with vascular cognitive impairment are also present in AD, such that mixed AD-vascular pathology is present in the majority of AD cases diagnosed clinically.155 The recent availability of imaging and biochemical biomarkers of AD has provided the opportunity to investigate the relationship between AD pathology and hypertension in vivo, in an attempt to explore the nature, causal or incidental, of their association.
Clinical-pathological biomarkers of AD in hypertension:
Although hypertension’s impact on cognition is fairly clearly established, whether this acts directly on AD neuropathology or simply as a parallel contributor to cognitive impairment and dementia is still somewhat controversial.8
In the ARIC-PET study, nondemented participants with a greater number of vascular risk factors in middle age, including hypertension, had more elevated brain amyloid in late-life, suggesting a possible direct impact of these risk factors on amyloid; hypertension by itself, however, was not significantly associated with elevated amyloid so may not have an independent role.156 Other studies have failed to find strong relationships between hypertension and brain amyloid, although many of these studied hypertension in older age. Elevated vascular risk, including hypertension, was associated more with atrophy and neurodegeneration than with brain amyloid, among 430 adults studied at >60 years of age with both tau and amyloid PET as well as brain MRI.157 In a neuropathologic sample, late-life BP was not associated with brain amyloid, but SBP was associated with neurofibrillary tangles.158
Using CSF biomarkers, the potential interaction of BP and APOE genotype was identified on tau levels, in one memory clinic cohort, in which hypertension was not associated with amyloid but rather was related to tau among APOE ε4 homozygotes.159 Although not specific for AD, associations between hypertension and regional brain atrophy also support a potential role of elevated BP in Alzheimer’s neurodegeneration. In addition to the above cited studies of elevated BP and brain atrophy,42 midlife BP has also been associated with hippocampal atrophy by MRI, with strongest associations among untreated hypertensives.160
Association between hypertension and AD pathology: experimental studies
Several studies have suggested that experimental hypertension promotes Aβ accumulation and tau phosphorylation. Hypertension, induced either by administration of AngII161, aortic coarctation162, 163, renal artery stenosis162, or in genetic models164, increases microvascular deposition of Aβ or neuronal tau phosphorylation. At the same time, administration of RAS blockers or AT1R genetic deletion ameliorates amyloid deposition and behavioral dysfunction in APP-overexpressing mice.165, 166 There are several mechanisms by which hypertension could worsen AD pathology (Figure 6). As discussed in a previous section, Aβ is cleared in part through vascular and perivascular pathways, and, consequently, the vascular and perivascular dysfunction and damage induced by hypertension could impair Aβ disposal leading to its accumulation. On the other hand, the loss of NO and prostacyclin bioavailability associated with vascular dysfunction may also contribute to promote amyloidogenic APP processing and increase Aβ. Thus, NO reductions upregulate APP and β-secretase, while reduction in prostacyclin may favor amyloidogenic APP processing.167 Mediators of hypertension may also promote amyloidogenic APP processing. For example, AngII increases APP cleavage by β- and γ-secretase and increases Aβ production in vitro and in vivo161, 168. Less is known about a potential effect of hypertension on tau phosphorylation. Endothelial NO prevents tau phosphorylation by inhibiting Cdk5, one of the main kinases phosphorylating tau,169 and consequently the NO deficit associated with hypertension could promote tau phosphorylation. Central administration of AngII induces tau phosphorylation by activation of the tau kinase GSK3-β via AT1R dependent mechanisms.170
Figure 6:

Potential mechanisms underlying the relationship between hypertension and AD. The vascular damage produced by hypertension leads to brain dysfunction thought hypoxia-ischemia, as well as increase in Aβ production due to increased APP processing by secretase enzymes and reduced clearance of AD-related proteins. In turn, Aβ and tau alter vascular function amplifying the deleterious vascular impact of hypertension (reproduced from ref.8 with permission). (Illustration Credit: Ben Smith).
These clinical and experimental observations, collectively, suggest that hypertension has the potential to promote AD pathology by acting at different levels. However, it remains unclear if hypertension is a pathogenic factor in AD, and conversely, if the AD pathology associated with hypertension is a contributor to the cognitive dysfunction.8
Prevention and therapy
Given the strong and consistent associations seen in the epidemiologic literature between hypertension and dementia, and given the strong biological plausibility for a link between the two, the inevitable next question is whether treatment of hypertension reduces risk of dementia. Certainly, this is the major reason why the study of hypertension as a risk factor for dementia is of particular interest: in the absence of other ways to treat or prevent dementia and AD specifically, hypertension is an especially appealing target.
Does treating hypertension reduce dementia risk?
Several epidemiologic studies have considered the role of antihypertensive medication treatment, although all remain susceptible to some indication bias: individuals who are prescribed and take antihypertensives are different than people who don’t, in many ways beyond which can be adjusted statistically. Thus, clinical trials would be the ideal forum in which to test this question, but given the above reviewed evidence that relationships between hypertension and dementia are strongest when hypertension is considered in middle age, decades before the development of dementia, and that clinical trials cannot randomize and follow participants for that long duration, some reliance on longer-term observational study designs is needed to consider these lifespan considerations. These studies also allow consideration of age of treatment or duration of treatment which can extend beyond the windows allowed by clinical trials.
In several observational studies, antihypertensive medication use is associated with less cognitive decline; in the ARIC study, participants taking antihypertensive medications had 20-year cognitive decline equivalent to a prehypertensive group (higher than the normotensives but lower than untreated hypertensives).12 In the Epidemiology of Vascular Aging (EVA) Study Group, treated hypertension was associated with less cognitive decline over only 4 years compared to untreated hypertension.171 Furthermore, duration of antihypertensive therapy appears important in risk reduction. In the HAAS study, per additional year of antihypertensive treatment, dementia risk was lower (HR 0.94, 95% CI 0.89-0.99), with the greatest reduction for individuals with treatment over 12 years in duration, with overall risk nearing that of normotensives.172 In the Rotterdam study, each additional year of antihypertensive use, before age 75, was associated with an 8% reduced risk of dementia; after age 75 the additional risk was reduced and was no longer statistically significant.173
Trial data consists of several randomized clinical trials focusing primarily on antihypertensive therapy, with most trials to date showing negative results, with the exception of Syst-Eur,174 and the most recent SPRINT-MIND trial.175 These negative trials include those in individuals with a history of stroke, in which a Cochrane review yielded a pooled RR of 0.88 (NS) for the use of antihypertensives to reduce dementia,176 as well as studies of individuals without prior known cerebrovascular disease.177
As one of the few positive clinical trials, the Syst-Eur trial showed that dementia-free hypertensive individuals (SBP 160-219 mm Hg and DBP<95 mm Hg) aged ≥60 years who received medications (a dihydropyridine calcium channel blocker plus other antihypertensive drugs as needed) to lower BP to <150 mm Hg systolic, compared to placebo, had less incident dementia over a median followup of 2 years.174 The preliminary SPRINT-MIND data,175 not yet published at the time of preparation of this review, do suggest a benefit in progression of cerebral SVD for individuals treated with intensive BP control vs standard control, as well as for cognitive outcomes, but only for the combination of MCI and dementia, and not for dementia itself.
Importance of other comorbid vascular disease and risk factors:
Although the focus of this review is on hypertension, elevated BP as a risk factor rarely occurs in isolation, and several risk factors co-occurring in the same individuals may further increase accelerated cognitive decline. In the Framingham Heart Study, hypertension was associated with worse decline in the presence of diabetes compared to its effect in nondiabetics,178 and in the Framingham Offspring Study, cognitive outcomes were worst among participants with hypertension and elevated waist-hip ratio.179 In 1449 adults aged 65-79 yo in Finland, the combination of SBP at 160 mm Hg or higher and elevated serum cholesterol, each in midlife, was associated with a markedly increased risk of AD (OR 3.5, 95% CI 1.6-7.9), when compared to the presence of either risk factor alone.19
Similar relationships are seen when consideration is made of any additional risk factor: in 8,945 participants of the Kaiser HMO, an increasing number of midlife vascular risk factors was associated with elevated risk of dementia in late-life, with a HR of 1.27 for individuals with only one midlife vascular risk factor and 2.37 for four midlife vascular risk factors (considering smoking, hypertension, hypercholesterolemia, and diabetes).16 The CAIDE score, which considers several vascular risk factors as a dementia risk score, including hypertension, has been reliably associated with dementia,180 with reasonably strong predictive power (area-under-the-curve 0.74).181 In parallel, better Life’s Simple 7 scores, which consider optimal control of vascular (including hypertension) and lifestyle risk factors, were associated with less cognitive decline, in the ARIC study.182
As the primary genetic risk factor for AD, carriage of the APOE ε4 allele independently increases risk for AD, but the combination of the ε4 genotype and hypertension may further increase risk of AD.183 In the PATH Through Life study, the combination of an APOE ε4 allele with hypertension was associated with greater decline over 8 years in 1,474 cognitively normal adults aged 60-64.184 In ARIC-PET, as cited above, although no statistically significant difference was found among APOE ε4 carriers vs noncarriers for risk of elevated BP on late-life brain amyloid, there was a suggestion of more brain amyloid in individuals with at least one ε4 allele, as midlife BP increased.156 This potential interaction was further supported by a study by Rodrigue et al in which the combination of hypertension and at least one APOE ε4 allele was associated with greater brain amyloid, by PiB PET,185 with a similar pattern with greater cortical thinning among individuals with both hypertension and at least one ε4 allele.186
Class-specific effects of antihypertensive drugs:
There are conflicting data as to the potential role of distinct antihypertensive medications: in HAAS, the only antihypertensive drug category when considered at mean age 77 which was associated with reduced cognitive impairment was β-blockers (incidence rate ratio 0.69, 95% CI 0.50-0.94).187 Syst-Eur primarily had a calcium channel-blocker (CCB) based intervention, and showed reduction in dementia rates,174 and the Perindopril Protection Against Recurrent Stroke Study (PROGRESS) showed a benefit in dementia reduction among individuals with a prior history of stroke or transient ischemic attack who were treated with active therapy (perindopril (an ACE inhibitor) or indapamide (a diuretic)) over 3.9 years, but only among individuals who also experienced recurrent stroke.188 In the 3-City cohort study, participants who took a combination of selective serotonin reuptake inhibitors (SSRI’s) with CCBs, compared with SSRI’s combined with other antihypertensives, had improved cognitive performance, as well as improved depression scores, at 2-year followup,189 with a similar beneficial effect on cognitive decline from CCBs noted in the Newcastle 85+ Study of elderly adults, without a similar benefit from other antihypertensives.190 This is in direct contrast to findings from the Canadian Study of Health and Aging, in which Canadians aged 65 and older who took CCBs had steeper decline, measured by a modified mini-mental state examination repeated serially over 5 years, than did their counterparts who used other antihypertensive drugs (75% on CCBs declined, versus 59% on other antihypertensive drugs).191
A growing body of evidence supports the biological importance of the RAS, and points to the potential importance of drugs within this family for dementia prevention in individuals with hypertension. Intervention trials in spontaneously hypertensive rats have shown reduced post-stroke cognitive impairment in rodents treated with RAS modifiers (specifically candesartan, an AT1R blocker).192 The ongoing HEART phase 1B randomized controlled trial will evaluate the RAS system specifically, in nondemented adults at risk for AD, in which participants will be randomized to placebo vs one of two doses of telmisartan.193
The optimal treatment regimen to prevent future cognitive impairment or dementia is difficult to ascertain, since nearly all antihypertensives have studies supporting their potential benefit. Again, these are still likely confounded by indication bias, similar to the overall issue of indication bias regarding any antihypertensive use vs none, although to a lesser degree (considering one antihypertensive versus another may be influenced by comorbidities or demographic characteristics, but are less profoundly impacted by socioeconomic or other factors influencing access to medical care more broadly.
Outstanding Questions and Future Prospects
Although the body of knowledge relating hypertension to cognitive outcomes, dementia, and AD has expanded tremendously in recent years, several key questions remain. Answers to some of these questions, listed below, will be critical in order to gain a better insight into how hypertension impacts cognitive function and to best recommend prevention and management strategies.
-
Is the neurovascular dysfunction induced by hypertension sufficient to cause cognitive impairment?
Hypertension induces alterations in neurovascular function, which are thought to induce brain lesions associated with cognitive impairment (Figure 5). However, it remains to be established if the neurovascular dysfunction alone is sufficient to induced cognitive impairment. Reduced cerebral perfusion, alterations in brain clearance and BBB, as well as vascular growth factor deficiency have the potential to alter neuronal function in metabolically-active brain regions involved in cognitive function, such as the hippocampus, entorinal cortex and prefrontal cortex. A better understanding of the natural history of the impact of hypertension on cerebrovascular function, network degradation and cognition would be needed to address this question, which is relevant to the initiation of anti-hypertensive therapy. In parallel, a more nuanced understanding of the signaling mechanisms by which the neurovascular dysfunction interferes with neuronal function would also be desirable. Is CBF insufficiency the major factor? Or, are there other aspect of cerebrovascular biology, such as trophic support by endothelial factors or perivascular clearance, also at play?
-
When does antihypertensive therapy need to be initiated in order to minimize risk of cognitive decline and dementia?
As summarized above, the benefits of antihypertensive therapy are likely to be greatest when initiated in midlife, and continued over decades, although preliminary results from the SPRINT-MIND trial suggest that benefits may still be possible with a shorter duration of antihypertensive therapy. In this regard, it would be important to assess if the appearance of brain lesions diminish the impact of hypertensive therapy on cognitive health. Although difficult to study in clinical trials, further consideration will need to be made of optimal age and duration of antihypertensive therapy, and correlation with cardiovascular and structural-functional imaging biomarkers, in order to best prevent cognitive decline, MCI, and dementia.
-
Are there subgroups of individuals who are especially susceptible to the cognitive effects of hypertension and in whom more aggressive antihypertensive therapy might be especially warranted?
Individuals with more vascular risk factors appear especially vulnerable to hypertension, with worse outcomes among individuals with several vascular risk factors. This suggests that individuals with other known risk factors, such as diabetes, may need better screening of BP with a lower threshold for initiation of antihypertensive therapy. This is consistent with the management of vascular risk factors as they relate to other cardiovascular outcomes. Beyond vascular risk, there may be genetic susceptibilities, such as has been seen with the APOE data, leading to greater risk, and therefore greater potential benefit from antihypertensive therapy, in individuals with a known risk allele, or even of a particular race or sex. Focused trials with enrollment of these higher-risk groups could identify a subgroup in whom prevention might be especially effective.
-
What class of antihypertensive is the most effective for prevention of cognitive decline, MCI, and dementia?
Although ongoing studies are directly addressing the potential benefit of particular antihypertensives, at least on surrogate endpoints, at the time of this manuscript preparation no convincing data point to a clinical difference with the use of particular antihypertensive medications. This question—whether any antihypertensive is as effective as the next—will be critical as evidence is translated into practice. Furthermore, it is possible that the same recommended therapeutics may not be ideal for all individuals. Race-based differences in therapeutic effect for distinct antihypertensives has been noted for other cardiovascular outcomes,194 and similar differences by race, or other demographic or genetic factors, may be found for reduction of adverse cognitive outcomes.
-
What are the best surrogate endpoints or biomarkers for measuring a cerebral or cognitive benefit in treating hypertension?
Biomarkers or surrogate endpoints are valuable in studying long-term relationships as seen between hypertension and dementia because they allow for earlier assessment of therapeutic effect, both for research purposes as well as clinically, to evaluate benefits of treatment. Ongoing studies and consortia, including the MarkVCID consortium, are evaluating biomarkers for the vascular contribution to cognitive impairment and dementia, which likely would be candidates for the specific effect of hypertension on the brain as well, and may include serum, CSF, or imaging markers to examine the contribution from AD pathology.
-
Is there a synergistic relationship between hypertension and AD pathology?
As discussed in a previous section, there is biological plausibility that hypertension and other vascular risk factors may promote AD pathology, and vice-versa. However, the clinical-pathological evidence remains contradictory. The recent development and validation of amyloid and tau PET imaging provides the opportunity to examine in vivo the reciprocal interaction between markers of AD and hypertensive neuropathology and their relative contribution to cognitive impairment. These findings would have important implications for targeting treatments to the relevant pathology responsible for the cognitive deficits in a particular individual. At the same time, in depth investigations on the impact of AD and hypertension on vessel-associated clearance pathways would be invaluable to provide insight into the role of failure of Aβ and tau disposal on disease onset and progression.
These are a few of the outstanding questions that remain to be addressed. Answering these questions will require engaging both the pre-clinical and clinical scientific communities in a concerted effort to advance the understanding of how hypertension affects brain function leading to cognitive impairment. Thus, epidemiological, biomarker, and clinical-pathological studies should be closely related to basic science efforts to unravel mechanisms and provide new targets to be tested in clinical trials. Such cooperation will be essential for the development of new diagnostic tools and treatment strategies for one of the most devastating health challenges of our times.
Acknowledgments
Sources of funding: Supported by National Institutes of Health grants R37-NS089323 (Dr Iadecola), R01-NS100447 (Dr Iadecola), R01-NS09544 (Dr Iadecola), R01-NS/HL037853 (Dr Iadecola), K24-AG052573 (R.F. Gottesman), RF1-AG050745 (R.F. Gottesman), and R01-AG040282 (R.F. Gottesman).
Non-Standard Abbreviations and Acronyms
- Aβ:
amyloid-β
- ACE:
angiotensin converting enzyme
- AD:
Alzheimer’s disease
- ACA:
anterior cerebral artery
- AngII:
angiotensin-II
- ApoE:
apolipoprotein-E
- APP:
amyloid precursor protein
- ARIC:
Atherosclerosis Risk in Communities
- AT1R:
angiotensin type-1receptor
- BBB:
blood-brain barrier
- BP:
blood pressure
- BPH:
blood pressure high
- CBF:
cerebral blood flow
- CCBs:
calcium channel blockers
- Cdk5:
cyclin dependent kinase-5
- CSF:
cerebrospinal fluid
- DBP:
diastolic blood pressure
- EVA:
Epidemiology of Vascular Aging
- GSK3:
glycogen synthase kinase-3
- HAAS:
Honolulu Asia Aging Study
- ISF:
interstitial fluid
- LYVE1:
Lymphatic vessel endothelial hyaluronan receptor-1
- MCA:
middle cerebral artery
- MCI:
mild cognitive impairment
- MMSE:
mini-mental state examination
- MoCA:
Montreal cognitive assessment
- MRI:
magnetic resonance imaging
- NHLBI:
National Heart Lung and Blood Institute
- NO:
nitric oxide
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NOX:
NADPH oxidase
- PET:
positron emission tomography
- PiB:
Pittsburgh compound-B
- PVM:
perivascular macrophages
- RAS:
renin angiotensin system
- ROS:
reactive oxygen species
- SBP:
systolic blood pressure
- SSRI:
selective serotonin reuptake inhibitors
- SVD:
small vessel disease
Footnotes
Disclosures: C. Iadecola serves on the Scientific Advisory Board of Broadview Ventures. R.F. Gottesman is Associate Editor for the journal Neurology.
Contributor Information
Costantino Iadecola, Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, New York, NY 10021
Rebecca F. Gottesman, Departments of Neurology and Epidemiology, Johns Hopkins University, Baltimore, MD 21287
References
- 1.Forouzanfar MH, Liu P, Roth GA, Ng M, Biryukov S, Marczak L, Alexander L, Estep K, Hassen Abate K, Akinyemiju TF, Ali R, Alvis-Guzman N, Azzopardi P, Banerjee A, Bärnighausen T, Basu A, Bekele T, Bennett DA, Biadgilign S, Catalá-López F, Feigin VL, Fernandes JC, Fischer F, Gebru AA, Gona P, Gupta R, Hankey GJ, Jonas JB, Judd SE, Khang Y-H, Khosravi A, Kim YJ, Kimokoti RW, Kokubo Y, Kolte D, Lopez A, Lotufo PA, Malekzadeh R, Melaku YA, Mensah GA, Misganaw A, Mokdad AH, Moran AE, Nawaz H, Neal B, Ngalesoni FN, Ohkubo T, Pourmalek F, Rafay A, Rai RK, Rojas-Rueda D, Sampson UK, Santos IS, Sawhney M, Schutte AE, Sepanlou SG, Shifa GT, Shiue I, Tedla BA, Thrift AG, Tonelli M, Truelsen T, Tsilimparis N, Ukwaja KN, Uthman OA, Vasankari T, Venketasubramanian N, Vlassov VV, Vos T, Westerman R, Yan LL, Yano Y, Yonemoto N, Zaki MES, Murray CJL. Global burden of hypertension and systolic blood pressure of at least 110 to 115 mm hg, 1990-2015. JAMA. 2017;317:165–118 [DOI] [PubMed] [Google Scholar]
- 2.Scheltens P, Blennow K, Breteler MMB, De Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer’s disease. Lancet. 2016;388:505–517 [DOI] [PubMed] [Google Scholar]
- 3.Spieth W Cardiovascular health status, age, and psychological performance. J Gerontol 1964;19:277–284 [DOI] [PubMed] [Google Scholar]
- 4.Wilkie F, Eisdorfer C. Intelligence and blood pressure in the aged. Science. 1971;172:959–962 [DOI] [PubMed] [Google Scholar]
- 5.Kety S, Hafkenschiel J, Jeffers W, Leopold I, Shenkin H. The blood flow, vascular resistance, and oxygen consumption of the brain in essential hypertension. J. CLin. Invest 1948;27:511–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saklayen MG, Deshpande NV. Timeline of history of hypertension treatment. Front. Cardiovasc. Med 2016;3:621–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elias MF, Wolf PA, D’Agostino RB, Cobb J, White LR. Untreated blood pressure level is inversely related to cognitive functioning: The framingham study. Am J Epidemiol 1993;138:353–364 [DOI] [PubMed] [Google Scholar]
- 8.Iadecola C, Gottesman RF. Cerebrovascular alterations in alzheimer disease: Incidental or pathogenic? Circ Res 2018;123:406–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser M, Roccella EJ. The treatment of hypertension: A remarkable success story. J Clin Hypertens (Greenwich). 2013;15:88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iadecola C, Yaffe K, Biller J, Bratzke LC, Faraci FM, Gorelick PB, Gulati M, Kamel H, Knopman DS, Launer LJ, Saczynski JS, Seshadri S, Zeki Al Hazzouri A. Impact of hypertension on cognitive function: A scientific statement from the american heart association. Hypertension. 2016;68:e67–e94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottesman RF, Rawlings AM, Sharrett AR, Albert M, Alonso A, Bandeen-Roche K, Coker LH, Coresh J, Couper DJ, Griswold ME, Heiss G, Knopman DS, Patel MD, Penman AD, Power MC, Selnes OA, Schneider ALC, Wagenknecht LE, Windham BG, Wruck LM, Mosley TH Jr.. Impact of differential attrition in the association of education with cognitive change over 20 years of follow-up: The aric-neurocognitive study. American Journal of Epidemiology. 2014;179:956–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottesman RF, Schneider AL, Albert M, Alonso A, Bandeen-Roche K, Coker L, Coresh J, Knopman D, Power MC, Rawlings A, Sharrett AR, Wruck LM, Mosley TH. Midlife hypertension and 20-year cognitive change: The atherosclerosis risk in communities neurocognitive study. JAMA neurology. 2014;71:1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swan GE, DeCarli C, Miller BL, Reed T, Wolf PA, Jack LM, Carmelli D. Association of midlife blood pressure to late-life cognitive decline and brain morphology. Neurology. 1998;51:986–993 [DOI] [PubMed] [Google Scholar]
- 14.Launer LJ, Masaki K, Petrovitch H, Foley D, Havlik RJ. The association between midlife blood pressure levels and late-life cognitive function. The honolulu-asia aging study. JAMA. 1995;274:1846–1851 [PubMed] [Google Scholar]
- 15.Kivipelto M, Helkala EL, Hanninen T, Laakso MP, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A. Midlife vascular risk factors and late-life mild cognitive impairment: A population-based study. Neurology. 2001;56:1683–1689 [DOI] [PubMed] [Google Scholar]
- 16.Whitmer RA, Sidney S, Selby J, Johnston SC, Yaffe K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005;64:362–381 [DOI] [PubMed] [Google Scholar]
- 17.Freitag MH, Peila R, Masaki K, Petrovitch H, Ross GW, White LR, Launer LJ. Midlife pulse pressure and incidence of dementia: The honolulu-asia aging study. Stroke. 2006;37:33–37 [DOI] [PubMed] [Google Scholar]
- 18.Gottesman RF, Albert MS, Alonso A, Coker LH, Coresh J, Davis SM, Deal JA, McKhann GM, Mosley TH, Sharrett AR, Schneider ALC, Windham BG, Wruck LM, Knopman DS. Associations between midlife vascular risk factors and 25-year incident dementia in the atherosclerosis risk in communities (aric) cohort. JAMA neurology. 2017;74:1246–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A. Midlife vascular risk factors and alzheimer’s disease in later life: Longitudinal, population based study. British Medical Journal. 2001;322:1447–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein FC, Levey AI, Steenland NK. High blood pressure and cognitive decline in mild cognitive impairment. Journal of the American Geriatrics Society. 2013;61:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC Jr, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA Sr, Williamson JD, Wright JT Jr.. 2017 acc/aha/aapa/abc/acpm/ags/apha/ash/aspc/nma/pcna guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: A report of the american college of cardiology/ american heart association task force on clinical practice guidelines. Hypertension. 2018;71:1269–1324 [DOI] [PubMed] [Google Scholar]
- 22.Nation DA, Preis SR, Beiser A, Bangen KJ, Delano-Wood L, Lamar M, Libon DJ, Seshadri S, Wolf PA, Au R. Pulse pressure is associated with early brain atrophy and cognitive decline: Modifying effects of apoe-e4. Alzheimer Disease and Associated Disorders. 2016;30:210–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rawlings AM, Juraschek SP, Heiss G, Hughes T, Meyer ML, Selvin E, Sharrett AR, Windham BG, Gottesman RF. Association of orthostatic hypotension with incident dementia, stroke, and cognitive decline. Neurology. 2018;91:e759–e768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kilander L, Nyman H, Boberg M, Hansson L, Lithell H. Hypertension is related to cognitive impairment: A 20-year follow-up of 999 men. Hypertension. 1998;31:780–786 [DOI] [PubMed] [Google Scholar]
- 25.Sabayan B, Wijsman L, Foster-Dingley JC, Stott DJ, Ford I, Buckley BM, Sattar N, Jukema JW, van Osch MJ, van der Grond J, van Buchem MA, Westendorp RG, De Craen AJ, Mooijaart SP. Association of visit-to-visit variability in blood pressure with cognitive function in old age: A prospective cohort study. British Medical Journal. 2013;347:1–11 [DOI] [PubMed] [Google Scholar]
- 26.Shah NS, Vidal JS, Masaki K, Petrovitch H, Ross GW, Tilley C, DeMattos RB, Tracy RP, White LR, Launer LJ. Midlife blood pressure, plasma b amyloid and the risk for alzheimer’s disease: The honolulu asia aging study. Hypertension. 2012;59:780–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah RC, Wilson RS, Bienias JL, Arvanitakis Z, Evans DA, Bennett DA. Relation of blood pressure to risk of incident alzheimer’s disease and change in global cognitive function in older persons. Neuroepidemiology. 2006;26:30–36 [DOI] [PubMed] [Google Scholar]
- 28.Glynn RJ, Beckett LA, Hebert LE, Morris MC, Scherr PA, Evans DA. Current and remote blood pressure and cognitive decline. JAMA. 1999;281:438–445 [DOI] [PubMed] [Google Scholar]
- 29.Ruitenberg A, Skoog I, Ott A, Aevarsson O, Lernfelt B, van Harskamp F, Hofman A, Breteler MM. Blood pressure and risk of dementia: Results from the rotterdam study and the gothenburg h-70 study. Dementia and Geriatric Cognitive Disorders. 2001;12:33–39 [DOI] [PubMed] [Google Scholar]
- 30.Verghese J, Lipton RB, Hall CB, Kuslansky G, Katz MJ. Low blood pressure and the risk of dementia in very old individuals. Neurology. 2003;61:1667–1672 [DOI] [PubMed] [Google Scholar]
- 31.Waldstein SR, Giggey PP, Thayer JF, Zonderman AB. Nonlinear relations of blood pressure to cognitive function: The baltimore longitudinal study of aging. Hypertension. 2005;45:374–379 [DOI] [PubMed] [Google Scholar]
- 32.Ninomiya T, Ohara T, Hirakawa Y, Yoshida D, Doi Y, Hata J, Kanba S, Iwaki T, Kiyohara Y. Midlife and late-life blood pressure and dementia in japanese elderly: The hisayama study. Hypertension. 2011;58:22–28 [DOI] [PubMed] [Google Scholar]
- 33.Yamada M, Kasagi F, Sasaki H, Masunari N, Mimori Y, Suzuki G. Association between dementia and midlife risk factors: The radiation effects research foundation adult health study. Journal of the American Geriatrics Society. 2003;51:410–414 [DOI] [PubMed] [Google Scholar]
- 34.den Heijer T, Launer LJ, Prins ND, van Dijk EJ, Vermeer SE, Hofman A, Koudstaal PJ, Breteler MM. Association between blood pressure, white matter lesions, and atrophy of the medial temporal lobe. Neurology. 2005;64:263–267 [DOI] [PubMed] [Google Scholar]
- 35.Dufouil C, de Kersaint-Gilly A, Besancon V, Levy C, Auffray E, Brunnereau L, Alperovitch A, Tzourio C. Longitudinal study of blood pressure and white matter hyperintensities: The eva mri cohort. Neurology. 2001;56:921–926 [DOI] [PubMed] [Google Scholar]
- 36.Gottesman RF, Coresh J, Catellier DJ, Sharrett AR, Rose KM, Coker LH, Shibata DK, Knopman DS, Jack CR, Mosley TH Jr. Blood pressure and white-matter disease progression in a biethnic cohort: Atherosclerosis risk in communities (aric) study. Stroke. 2010;41:3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart R, Xue QL, Masaki K, Petrovitch H, Ross GW, White LR, Launer LJ. Change in blood pressure and incident dementia: A 32-year prospective study. Hypertension. 2009;54:233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, Persson G, Oden A, Svanborg A. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–1145 [DOI] [PubMed] [Google Scholar]
- 39.McGrath ER, Beiser AS, DeCarli C, Plourde KL, Vasan RS, Greenberg SM, Seshadri S. Blood pressure from mid- to late life and risk of incident dementia. Neurology. 2017;89:2447–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo Z, Viitanen M, Winblad B, Fratiglioni L. Low blood pressure and incidence of dementia in a very old sample: Dependent on initial cognition. Journal of the American Geriatrics Society. 1999;47:723–726 [DOI] [PubMed] [Google Scholar]
- 41.den Heijer T, Skoog I, Oudkerk M, de Leeuw F-E, de Groot JC, Hofman A, Breteler MMB. Association between blood pressure levels over time and brain atrophy in the elderly. Neurobiology of Aging. 2003;24:307–313 [DOI] [PubMed] [Google Scholar]
- 42.Power MC, Schneider AL, Wruck L, Griswold M, Coker LH, Alonso A, Jack CR Jr, Knopman D, Mosley TH Jr., Gottesman RF. Life-course blood pressure in relation to brain volumes. Alzheimer’s and Dementia. 2016;12:890–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yaffe K, Vittinghoff E, Pletcher MJ, Hoang TD, Launer LJ, Whitmer R, Coker LH, Sidney S. Early adult to midlife cardiovascular risk factors and cognitive function. Circulation. 2014;129:1560–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ostergaard SD, Mukherjee S, Sharp SJ, Proitsi P, Lotta LA, Day F, Perry JR, Boehme KL, Walter S, Kauwe JS, Gibbons LE, Alzheimer’s Disease Genetics Consortium, GERAD1 Consortium, EPIC-InterAct Consortium, Larson EB, Powell JF, Langenberg C, Crane PK, Wareham NJ, Scott RA. Associations between potentially modifiable risk factors in alzheimer disease: A mendelian randomization study. PLoS Medicine. 2015;12:e1001841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldstein FC, Hajjar IM, Dunn CB, Levey AI, Wharton W. The relationship between cognitive functioning and the jnc-8 guidelines for hypertension in older adults. Journal of Gerontology A: Biological Science and Medical Science. 2017;72:121–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smits LL, van Harten AC, Pijnenburg YA, Koedam EL, Bouwman FH, Sistermans N, Reuling IE, Prins ND, Lemstra AW, Scheltens P, van der Flier WM. Trajectories of cognitive decline in different types of dementia. Psychological Medicine. 2015;45:1051–1059 [DOI] [PubMed] [Google Scholar]
- 47.Sikaroodi H, Yadegari S, Miri SR. Cognitive impairments in patients with cerebrovascular risk factors: A comparison of mini mental status exam and montreal cognitive assessment. Clinical Neurology and Neurosurgery. 2013;115:1276–1280 [DOI] [PubMed] [Google Scholar]
- 48.Smith EE, Muzikansky A, McCreary CR, Batool S, Viswanathan A, Dickerson BC, Johnson K, Greenberg SM, Blacker D. Impaired memory is more closely associated with brain beta-amyloid than leukoaraiosis in hypertensive patients with cognitive symptoms. PLoS One. 2018;13:e0191345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reed BR, Mungas DM, Kramer JH, Ellis W, Vinters HV, Zarow C, Jagust WJ, Chui HC. Profiles of neuropsychological impairment in autopsy-defined alzheimer’s disease and cerebrovascular disease. Brain. 2007;130:731–739 [DOI] [PubMed] [Google Scholar]
- 50.Iadecola C The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron. 2017;96:17–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Armulik A, Genove G, Betsholtz C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215 [DOI] [PubMed] [Google Scholar]
- 52.Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;80:844–480 [DOI] [PubMed] [Google Scholar]
- 53.Hollander W, Prusty S, Kemper T, Rosene DL, Moss MB. The effects of hypertension on cerebral atherosclerosis in the cynomolgus monkey. Stroke. 1993;24:1218–1226; discussion 1226-1217 [DOI] [PubMed] [Google Scholar]
- 54.Agmon Y, Khandheria BK, Meissner I, Schwartz GL, Petterson TM, O’Fallon WM, Gentile F, Whisnant JP, Wiebers DO, Seward JB. Independent association of high blood pressure and aortic atherosclerosis: A population-based study. Circulation. 2000;102:2087–2093 [DOI] [PubMed] [Google Scholar]
- 55.Qureshi AI, Caplan LR. Intracranial atherosclerosis. Lancet. 2014;383:984–998 [DOI] [PubMed] [Google Scholar]
- 56.Hu X, De Silva TM, Chen J, Faraci FM. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ Res 2017;120:449–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watase H, Sun J, Hippe DS, Balu N, Li F, Zhao X, Mani V, Fuster V, Hatsukami TS, Yuan C. Carotid artery remodeling is segment specific: An in vivo study by vessel wall magnetic resonance imaging. Arterioscler Thromb Vasc Biol 2018;38:927–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA. 2012;308:875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jefferson AL, Cambronero FE, Liu D, Moore EE, Neal JE, Terry JG, Nair S, Gifford KA, Hohman TJ, Bell SP, Wang TJ, Beckman JA, Carr JJ. Higher aortic stiffness is related to lower cerebral blood flow and preserved cerebrovascular reactivity in older adults. Circulation. 2018;in press:CIRCULATIONAHA.118.032410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DuBose LE, Boles Ponto LL, Moser DJ, Harlynn E, Reierson L, Pierce GL. Higher aortic stiffness is associated with lower global cerebrovascular reserve among older humans. Hypertension. 2018;72:476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schnerr RS, Jansen JFA, Uludag K, Hofman PAM, Wildberger JE, van Oostenbrugge RJ, Backes WH. Pulsatility of lenticulostriate arteries assessed by 7 tesla flow mri-measurement, reproducibility, and applicability to aging effect. Front Physiol 2017;8:961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blanco PJ, Müller LO, Spence JD. Blood pressure gradients in cerebral arteries: A clue to pathogenesis of cerebral small vessel disease. Stroke Vasc Neurol 2017;2:108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown R, Benveniste H, Black SE, Charpak S, Dichgans M, Joutel A, Nedergaard M, Smith KJ, Zlokovic BV, Wardlaw JM. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res 2018;114:1462–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marinkovic S, Milisavljevic M, Puskas L. Microvascular anatomy of the hippocampal formation. Surg Neurol 1992;37:339–349 [DOI] [PubMed] [Google Scholar]
- 65.Blinder P, Tsai PS, Kaufhold JP, Knutsen PM, Suhl H, Kleinfeld D. The cortical angiome: An interconnected vascular network with noncolumnar patterns of blood flow. Nat Neurosci 2013;16:889–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iadecola C The pathobiology of vascular dementia. Neuron. 2013;80:844–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenblum WI. Fibrinoid necrosis of small brain arteries and arterioles and miliary aneurysms as causes of hypertensive hemorrhage: A critical reappraisal. Acta Neuropathol 2008;116:361–369 [DOI] [PubMed] [Google Scholar]
- 68.Brown W, Moody D, Thore C, Challa V, Anstrom J. Vascular dementia in leukoaraiosis may be a consequence of capillary loss not only in the lesions, but in normal-appearing white matter and cortex as well. J Neurol Sci 2007;257:62–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iulita MF, Noriega de la Colina A, Girouard H. Arterial stiffness, cognitive impairment and dementia: Confounding factor or real risk? J Neurochem 2017;144:527–548 [DOI] [PubMed] [Google Scholar]
- 70.Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med 2018;215:21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ogola BO, Zimmerman MA, Clark GL, Abshire CM, Gentry KM, Miller KS, Lindsey SH. New insights into arterial stiffening: Does sex matter? Am J Physiol Heart Circ Physiol 2018;6:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, Chew SHS, See P, Chakarov S, Wang XN, Lim LH, Johnson LA, Lum J, Fong CY, Bongso A, Biswas A, Goh C, Evrard M, Yeo KP, Basu R, Wang JK, Tan Y, Jain R, Tikoo S, Choong C, Weninger W, Poidinger M, Stanley RE, Collin M, Tan NS, Ng LG, Jackson DG, Ginhoux F, Angeli V. Hyaluronan receptor lyve-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity. 2018;49:326–341 e327 [DOI] [PubMed] [Google Scholar]
- 73.Faraco G, Sugiyama Y, Lane D, Garcia-Bonilla L, Chang H, Santisteban MM, Racchumi G, Murphy M, van Rooijen N, Anrather J, Iadecola C. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J. CLin. Invest 2016;126:4674–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D, Nelson MT. Capillary k(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 2017;20:717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vanhoutte PM, Shimokawa H, Feletou M, Tang EHC. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol (Oxf) 2017;219:22–96 [DOI] [PubMed] [Google Scholar]
- 76.Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: From physiology to disease and back. Physiol Rev 2019;99:21–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marie C, Pedard M, Quirié A, Tessier A, Garnier P, Totoson P, Demougeot C. Brain-derived neurotrophic factor secreted by the cerebral endothelium: A new actor of brain function? J Cereb Blood Flow Metab 2018;38: 935–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tran KA, Zhang X, Predescu D, Huang X, Machado RF, Göthert JR, Malik AB, Valyi-Nagy T, Zhao Y-Y. Endothelial β-catenin signaling is required for maintaining adult blood-brain barrier integrity and central nervous system homeostasis. Circulation. 2016;133:177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dai W, Lopez OL, Carmichael OT, Becker JT, Kuller LH, Gach HM. Abnormal regional cerebral blood flow in cognitively normal elderly subjects with hypertension. Stroke. 2008;39:349–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fujishima M, Ibayashi S, Fujii K, Mori S. Cerebral blood flow and brain function in hypertension. Hypertens Res 1995;18:111–117 [DOI] [PubMed] [Google Scholar]
- 81.Muller M, van der Graaf Y, Visseren FL, Mali WPTM, Geerlings MI, Group SS. Hypertension and longitudinal changes in cerebral blood flow: The smart-mr study. Ann Neurol 2012;71:825–833 [DOI] [PubMed] [Google Scholar]
- 82.Beason-Held LL, Moghekar A, Zonderman AB, Kraut MA, Resnick SM. Longitudinal changes in cerebral blood flow in the older hypertensive brain. Stroke. 2007;38:1766–1773 [DOI] [PubMed] [Google Scholar]
- 83.Mentis MJ, Salerno J, Horwitz B, Grady C, Schapiro MB, Murphy DG, Rapoport SI. Reduction of functional neuronal connectivity in long-term treated hypertension. Stroke. 1994;25:601–607 [DOI] [PubMed] [Google Scholar]
- 84.Jennings JR, Muldoon MF, Ryan C, Price JC, Greer P, Sutton-Tyrrell K, van der Veen FM, Meltzer CC. Reduced cerebral blood flow response and compensation among patients with untreated hypertension. Neurology. 2005;64:1358–1365 [DOI] [PubMed] [Google Scholar]
- 85.Wong R, Ahmad W, Davies A, Spratt N, Boyle A, Levi C, Howe P, Collins N. Assessment of cerebral blood flow in adult patients with aortic coarctation. Cardiology in the young. 2017;27:1606–1613 [DOI] [PubMed] [Google Scholar]
- 86.Delles C, Michelson G, Harazny J, Oehmer S, Hilgers KF, Schmieder RE. Impaired endothelial function of the retinal vasculature in hypertensive patients. Stroke. 2004;35:1289–1293 [DOI] [PubMed] [Google Scholar]
- 87.Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I, Li H, Lamas S, Munzel T. Targeting vascular (endothelial) dysfunction. Br J Pharmacol 2017;174:1591–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bagi Z, Brandner DD, Le P, McNeal DW, Gong X, Dou H, Fulton DJ, Beller A, Ngyuen T, Larson EB, Montine TJ, Keene CD, Back SA. Vasodilator dysfunction and oligodendrocyte dysmaturation in aging white matter. Ann Neurol 2018;83:142–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Muñoz Maniega S, Chappell FM, Valdés Hernández MC, Armitage PA, Makin SD, Heye AK, Thrippleton MJ, Sakka E, Shuler K, Dennis MS, Wardlaw JM. Integrity of normal-appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J Cereb Blood Flow Metab 2016;37:644–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoth KF, Tate DF, Poppas A, Forman DE, Gunstad J, Moser DJ, Paul RH, Jefferson AL, Haley AP, Cohen RA. Endothelial function and white matter hyperintensities in older adults with cardiovascular disease. Stroke. 2007;38:308–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nezu T, Hosomi N, Aoki S, Kubo S, Araki M, Mukai T, Takahashi T, Maruyama H, Higashi Y, Matsumoto M. Endothelial dysfunction is associated with the severity of cerebral small vessel disease. Hypertens Res 2015;38:291–297 [DOI] [PubMed] [Google Scholar]
- 92.Kearney-Schwartz A, Rossignol P, Bracard S, Felblinger J, Fay R, Boivin J-M, Lecompte T, Lacolley P, Benetos A, Zannad F. Vascular structure and function is correlated to cognitive performance and white matter hyperintensities in older hypertensive patients with subjective memory complaints. Stroke. 2009;40:1229–1236 [DOI] [PubMed] [Google Scholar]
- 93.Castro P, Azevedo E, Sorond F. Cerebral autoregulation in stroke. Curr Atheroscler Rep 2018;20:1–12 [DOI] [PubMed] [Google Scholar]
- 94.Paulson O, Strandgaard S, Edvinsson L. Cerebral autoregulation. Cerebrovasc Brain Metab Rev 1990;2:162–192 [PubMed] [Google Scholar]
- 95.Lassen N Cerebral blood flow and oxygen consumption in man. Physiol Rev 1959;39:183–238 [DOI] [PubMed] [Google Scholar]
- 96.Tan CO. Defining the characteristic relationship between arterial pressure and cerebral flow. J Appl Physiol (1985). 2012;113:1194–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Longden TA, Hill-Eubanks DC, Nelson MT. Ion channel networks in the control of cerebral blood flow. J Cereb Blood Flow Metab. 2016;36:492–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M, Gudermann T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J 2008;27:3092–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kroetsch JT, Levy AS, Zhang H, Aschar-Sobbi R, Lidington D, Offermanns S, Nedospasov SA, Backx PH, Heximer SP, Bolz SS. Constitutive smooth muscle tumour necrosis factor regulates microvascular myogenic responsiveness and systemic blood pressure. Nat Commun. 2017;8:14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol (Lond). 1998;508 (Pt 1):199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jarajapu YPR, Knot HJ. Relative contribution of rho kinase and protein kinase c to myogenic tone in rat cerebral arteries in hypertension. Am J Physiol Heart Circ Physiol 2005;289:H1917–1922 [DOI] [PubMed] [Google Scholar]
- 102.Kusano Y, Echeverry G, Miekisiak G, Kulik TB, Aronhime SN, Chen JF, Winn HR. Role of adenosine a2 receptors in regulation of cerebral blood flow during induced hypotension. J Cereb Blood Flow Metab 2010;30:808–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Strandgaard S Autoregulation of cerebral blood flow in hypertensive patients. The modifying influence of prolonged antihypertensive treatment on the tolerance to acute, drug-induced hypotension. Circulation. 1976;53:720–727 [DOI] [PubMed] [Google Scholar]
- 104.Strandgaard S, Jones J, MacKenzie E, Harper A. Upper limit of cerebral blood flow autoregulation in experimental renovascular hypertension in the baboon. Circ Res 1975;37:164–167 [DOI] [PubMed] [Google Scholar]
- 105.Birns J, Jarosz J, Markus HS, Kalra L. Cerebrovascular reactivity and dynamic autoregulation in ischaemic subcortical white matter disease. J Neurol Neurosurg Psychiatr 2009;80:1093–1098 [DOI] [PubMed] [Google Scholar]
- 106.Traon AP, Costes-Salon MC, Galinier M, Fourcade J, Larrue V. Dynamics of cerebral blood flow autoregulation in hypertensive patients. J Neurol Sci 2002;195:139–144 [DOI] [PubMed] [Google Scholar]
- 107.Lipsitz L, Gagnon M, Vyas M, Iloputaife I, Kiely D, Sorond F, Serrador J, Cheng D, Babikian V, Cupples L. Antihypertensive therapy increases cerebral blood flow and carotid distensibility in hypertensive elderly subjects. Hypertension. 2005;45:216–221 [DOI] [PubMed] [Google Scholar]
- 108.Zhang R, Witkowski S, Fu Q, Claassen JAHR, Levine BD. Cerebral hemodynamics after short- and long-term reduction in blood pressure in mild and moderate hypertension. Hypertension. 2007;49:1149–1155 [DOI] [PubMed] [Google Scholar]
- 109.Immink R, van den Born B, van Montfrans G, Koopmans R, Karemaker J, Van Lieshout J. Impaired cerebral autoregulation in patients with malignant hypertension. Circulation. 2004;110:2241–2245 [DOI] [PubMed] [Google Scholar]
- 110.Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Menard J, Zetterberg H, Wisniewski T, de Leon MJ. Clearance systems in the brain-implications for alzheimer disease. Nat Rev Neurol 2015;11:457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Morris AWJ, Sharp MM, Albargothy NJ, Fernandes R, Hawkes CA, Verma A, Weller RO, Carare RO. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol 2016;131:725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nedergaard M. Neuroscience. Garbage truck of the brain. Science. 2013;340:1529–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Absinta M, Ha S-K, Nair G, Sati P, Luciano NJ, Palisoc M, Louveau A, Zaghloul KA, Pittaluga S, Kipnis J, Reich DS. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by mri. Elife. 2017;6:780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the cns. Nat Immunol 2017;18:123–131 [DOI] [PubMed] [Google Scholar]
- 115.Faraco G, Park L, Anrather J, Iadecola C. Brain perivascular macrophages: Characterization and functional roles in health and disease. J Mol Med (Berl). 2017;95:1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Santisteban MM, Iadecola C. Hypertension, dietary salt, and cognitive impairment. J Cereb Blood Flow Metab 2018;in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol Regul Integr Comp Physiol 2003;284:R893–912 [DOI] [PubMed] [Google Scholar]
- 118.Kazama K, Wang G, Frys K, Anrather J, Iadecola C. Angiotensin-ii attenuates functional hyperemia in the mouse somatosensory cortex. Am J Physiol 2003;285:H1890–1899 [DOI] [PubMed] [Google Scholar]
- 119.Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, Iadecola C. Angiotensin-ii impairs neurovascular coupling in neocortex through nadph oxidase-derived radicals. Circ Res 2004;95:1019–1026 [DOI] [PubMed] [Google Scholar]
- 120.Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler Thromb Vasc Biol 2006;26:826–832 [DOI] [PubMed] [Google Scholar]
- 121.Capone C, Faraco G, Coleman C, Young CN, Pickel VM, Anrather J, Davisson RL, Iadecola C. Endothelin 1-dependent neurovascular dysfunction in chronic intermittent hypoxia. Hypertension. 2012;60:106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Faraco G, Park L, Zhou P, Luo W, Paul SM, Anrather J, Iadecola C. Hypertension enhances aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of app. J Cereb Blood Flow Metab 2016;36:241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang Y, Kimura-Ohba S, Thompson J, Rosenberg GA. Rodent models of vascular cognitive impairment. Transl Stroke Res 2016;7:407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Capone C, Faraco G, Peterson JR, Coleman C, Anrather J, Milner TA, Pickel VM, Davisson RL, Iadecola C. Central cardiovascular circuits contribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci 2012;32:4878–4886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Touyz RM, Briones AM. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens Res 2011;34:5–14 [DOI] [PubMed] [Google Scholar]
- 126.Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol 2007;27:303–309 [DOI] [PubMed] [Google Scholar]
- 127.Pacher P, Beckman J, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007;87:315–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Park L, Wang G, Moore J, Girouard H, Zhou P, Anrather J, Iadecola C. The key role of transient receptor potential melastatin-2 channels in amyloid-β-induced neurovascular dysfunction. Nature communications. 2014;5:5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ago T, Kuroda J, Kamouchi M, Sadoshima J, Kitazono T. Pathophysiological roles of nadph oxidase/nox family proteins in the vascular system. Review and perspective. Circ J 2011;75:1791–1800 [DOI] [PubMed] [Google Scholar]
- 130.Santhanam AV, d’Uscio LV, Smith LA, Katusic ZS. Uncoupling of enos causes superoxide anion production and impairs no signaling in the cerebral microvessels of hph-1 mice. J Neurochem 2012;122:1211–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pires PW, Girgla SS, McClain JL, Kaminski NE, van Rooijen N, Dorrance AM. Improvement in middle cerebral artery structure and endothelial function in stroke-prone spontaneously hypertensive rats after macrophage depletion. Microcirculation. 2013;20:650–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.van Veluw SJ, Shih AY, Smith EE, Chen C, Schneider JA, Wardlaw JM, Greenberg SM, Biessels GJ. Detection, risk factors, and functional consequences of cerebral microinfarcts. The Lancet Neurology. 2017;16:730–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Biesbroek JM, Weaver NA, Biessels GJ. Lesion location and cognitive impact of cerebral small vessel disease. Clin Sci 2017;131:715–728 [DOI] [PubMed] [Google Scholar]
- 134.Akoudad S, Wolters FJ, Viswanathan A, de Bruijn RF, van der Lugt A, Hofman A, Koudstaal PJ, Ikram MA, Vernooij MW. Association of cerebral microbleeds with cognitive decline and dementia. JAMA neurology. 2016;73:934–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Benjamin P, Trippier S, Lawrence AJ, Lambert C, Zeestraten E, Williams OA, Patel B, Morris RG, Barrick TR, Mackinnon AD, Markus HS. Lacunar infarcts, but not perivascular spaces, are predictors of cognitive decline in cerebral small-vessel disease. Stroke. 2018;49:586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Seo SW, Lee JM, Im K, Park J-S, Kim S-H, Kim ST, Ahn H-J, Chin J, Cheong H-K, Weiner MW, Na DL. Cortical thinning related to periventricular and deep white matter hyperintensities. Neurobiology of aging. 2012;33:1156–1167 [DOI] [PubMed] [Google Scholar]
- 137.Rosenberg GA. Extracellular matrix inflammation in vascular cognitive impairment and dementia. Clin Sci 2017;131:425–437 [DOI] [PubMed] [Google Scholar]
- 138.Guo S, Kim WJ, Lok J, Lee S-R, Besancon E, Luo B-H, Stins MF, Wang X, Dedhar S, Lo EH. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci USA. 2008;105:7582–7587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Habes M, Sotiras A, Erus G, Toledo JB, Janowitz D, Wolk DA, Shou H, Bryan NR, Doshi J, Volzke H, Schminke U, Hoffmann W, Resnick SM, Grabe HJ, Davatzikos C. White matter lesions: Spatial heterogeneity, links to risk factors, cognition, genetics, and atrophy. Neurology. 2018;91:e964–e975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Joutel A, Chabriat H. Pathogenesis of white matter changes in cerebral small vessel diseases: Beyond vessel-intrinsic mechanisms. Clinical Science. 2017;131:635–651 [DOI] [PubMed] [Google Scholar]
- 141.Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F, Wharton SB, Shaw PJ, O’Brien JT, Ince PG, Function MRCC, Ageing Neuropathology Study G. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke. 2006;37:1391–1398 [DOI] [PubMed] [Google Scholar]
- 142.Sam K, Crawley AP, Conklin J, Poublanc J, Sobczyk O, Mandell DM, Venkatraghavan L, Duffin J, Fisher JA, Black SE, Mikulis DJ. Development of white matter hyperintensity is preceded by reduced cerebrovascular reactivity. Ann Neurol 2016;80:277–285 [DOI] [PubMed] [Google Scholar]
- 143.van der Veen PH, Muller M, Vincken KL, Hendrikse J, Mali WPTM, van der Graaf Y, Geerlings MI, Group SS. Longitudinal relationship between cerebral small-vessel disease and cerebral blood flow: The second manifestations of arterial disease-magnetic resonance study. Stroke. 2015;46:1233–1238 [DOI] [PubMed] [Google Scholar]
- 144.Shi Y, Thrippleton MJ, Makin SD, Marshall I, Geerlings MI, de Craen AJ, van Buchem MA, Wardlaw JM. Cerebral blood flow in small vessel disease: A systematic review and meta-analysis. J Cereb Blood Flow Metab 2016;36:1653–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Farrall AJ, Wardlaw JM. Blood-brain barrier: Ageing and microvascular disease--systematic review and meta-analysis. Neurobiology of aging. 2009;30:337–352 [DOI] [PubMed] [Google Scholar]
- 146.Maclullich AM, Ferguson KJ, Reid LM, Deary IJ, Starr JM, Seckl JR, Bastin ME, Wardlaw JM. Higher systolic blood pressure is associated with increased water diffusivity in normal-appearing white matter. Stroke. 2009;40:3869–3871 [DOI] [PubMed] [Google Scholar]
- 147.Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L, Li H, Haring M, Hochgerner H, Romanov RA, Gyllborg D, Munoz Manchado A, La Manno G, Lonnerberg P, Floriddia EM, Rezayee F, Ernfors P, Arenas E, Hjerling-Leffler J, Harkany T, Richardson WD, Linnarsson S, Castelo-Branco G. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 2016;352:1326–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Arai K, Lo EH. An oligovascular niche: Cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci 2009;29:4351–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Back SA, Tuohy TMF, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, Trapp BD, Bebo BF, Rao MS, Sherman LS. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med 2005;11:966–972 [DOI] [PubMed] [Google Scholar]
- 150.Summers PM, Hartmann DA, Hui ES, Nie X, Deardorff RL, McKinnon ET, Helpern JA, Jensen JH, Shih AY. Functional deficits induced by cortical microinfarcts. Journal of Cerebral Blood Flow & Metabolism. 2017;37:3599–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Carnevale L, D’Angelosante V, Landolfi A, Grillea G, Selvetella G, Storto M, Lembo G, Carnevale D. Brain mri fiber-tracking reveals white matter alterations in hypertensive patients without damage at conventional neuroimaging. Cardiovasc Res 2018;361:878–811 [DOI] [PubMed] [Google Scholar]
- 152.Beauchet O, Celle S, Roche F, Bartha R, Montero-Odasso M, Allali G, Annweiler C. Blood pressure levels and brain volume reduction: A systematic review and meta-analysis. J Hypertens 2013;31:1502–1516 [DOI] [PubMed] [Google Scholar]
- 153.Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M. Cerebral arterial pulsation drives paravascular csf-interstitial fluid exchange in the murine brain. J Neurosci 2013;33:18190–18199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bedussi B, Almasian M, de Vos J, VanBavel E, Bakker EN. Paravascular spaces at the brain surface: Low resistance pathways for cerebrospinal fluid flow. Journal of Cerebral Blood Flow & Metabolism. 2017;38:719–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Azarpazhooh MR, Sposato LA, Hachinski V. Concomitant vascular and neurodegenerative pathologies double the risk of dementia. Alzheimers Dement 2018;14:148–156 [DOI] [PubMed] [Google Scholar]
- 156.Gottesman RF, Schneider ALC, Zhou Y, Coresh J, Green E, Gupta N, Knopman DS, Mintz A, Rahmim A, Sharrett AR, Wagenknecht LE, Wong DF, Mosley TH Jr.. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA. 2017;317:1443–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Lowe VJ, Graff-Radford J, Roberts RO, Mielke MM, Machulda MM, Petersen RC, Jack CR Jr. Age, vascular health, and alzheimer disease biomarkers in an elderly sample. Ann Neurol 2017;82:706–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Arvanitakis Z, Capuano AW, Lamar M, Shah RC, Barnes LL, Bennett DA, Schneider JA. Late-life blood pressure association with cerebrovascular and alzheimer disease pathology. Neurology. 2018;91:e517–e525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kester MI, van der Flier WM, Mandic G, Blankenstein MA, Scheltens P, Muller M. Joint effect of hypertension and apoe genotype on csf biomarkers for alzheimer’s disease. Journal of Alzheimer’s Disease. 2010;20:1083–1090 [DOI] [PubMed] [Google Scholar]
- 160.Korf ES, White LR, Scheltens P, Launer LJ. Midlife blood pressure and the risk of hippocampal atrophy: The honolulu asia aging study. Hypertension. 2004;44:29–34 [DOI] [PubMed] [Google Scholar]
- 161.Faraco G, Park L, Zhou P, Luo W, Paul SM, Anrather J, Iadecola C. Hypertension enhances abeta-induced neurovascular dysfunction, promotes beta-secretase activity, and leads to amyloidogenic processing of app. J Cereb Blood Flow Metab 2016;36:241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shih Y-H, Wu S-Y, Yu M, Huang S-H, Lee C-W, Jiang M-J, Lin P-Y, Yang T-T, Kuo Y-M. Hypertension accelerates alzheimer’s disease-related pathologies in pigs and 3xtg mice. Frontiers in aging neuroscience. 2018;10:623–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Carnevale D, Mascio G, D’Andrea I, Fardella V, Bell RD, Branchi I, Pallante F, Zlokovic B, Yan SS, Lembo G. Hypertension induces brain β-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension. 2012;60:188–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schreiber S, Drukarch B, Garz C, Niklass S, Stanaszek L, Kropf S, Bueche C, Held F, Vielhaber S, Attems J, Reymann KG, Heinze H-J, Carare RO, Wilhelmus MMM. Interplay between age, cerebral small vessel disease, parenchymal amyloid-β, and tau pathology: Longitudinal studies in hypertensive stroke-prone rats. J Alzheimers Dis 2014 [DOI] [PubMed] [Google Scholar]
- 165.Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti G. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of alzheimer disease. J. CLin. Invest 2007;117:3393–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Royea J, Zhang L, Tong XK, Hamel E. Angiotensin iv receptors mediate the cognitive and cerebrovascular benefits of losartan in a mouse model of alzheimer’s disease. J Neurosci 2017;37:5562–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.d’Uscio LV, He T, Katusic ZS. Expression and processing of amyloid precursor protein in vascular endothelium. Physiology (Bethesda). 2017;32:20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhu D, Shi J, Zhang Y, Wang B, Liu W, Chen Z, Tong Q. Central angiotensin II stimulation promotes beta amyloid production in sprague dawley rats. PLoS One. 2011;6:e16037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Austin SA, Katusic ZS. Loss of endothelial nitric oxide synthase promotes p25 generation and tau phosphorylation in a murine model of alzheimer’s disease. Circ Res 2016;119:1128–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tian M, Zhu D, Xie W, Shi J. Central angiotensin II-induced alzheimer-like tau phosphorylation in normal rat brains. FEBS Lett 2012;586:3737–3745 [DOI] [PubMed] [Google Scholar]
- 171.Tzourio C, Dufouil C, Ducimetiere P, Alperovitch A. Cognitive decline in individuals with high blood pressure: A longitudinal study in the elderly. Eva study group. Epidemiology of vascular aging. Neurology. 1999;53:1948–1952 [DOI] [PubMed] [Google Scholar]
- 172.Peila R, White LR, Masaki K, Petrovitch H, Launer LJ. Reducing the risk of dementia: Efficacy of long-term treatment of hypertension. Stroke. 2006;37:1165–1170 [DOI] [PubMed] [Google Scholar]
- 173.Haag MD, Hofman A, Koudstaal PJ, Breteler MM, Stricker BH. Duration of antihypertensive drug use and risk of dementia: A prospective cohort study. Neurology. 2009;72:1727–1734 [DOI] [PubMed] [Google Scholar]
- 174.Forette F, Seux ML, Staessen JA, Thijs L, Birkenhager WH, Babarskiene MR, Babeanu S, Bossini A, Gil-Extremera B, Girerd X, Laks T, Lilov E, Moisseyev V, Tuomilehto J, Vanhanen H, Webster J, Yodfat Y, Fagard R. Prevention of dementia in randomised double-blind placebo-controlled systolic hypertension in europe (syst-eur) trial. Lancet. 1998;352:1347–1351 [DOI] [PubMed] [Google Scholar]
- 175.Nih-funded researchers preview preliminary clinical trial results at aaic suggesting aggressive blood pressure control may lower risk of cognitive impairment. 2018
- 176.Zonneveld TP, Richard E, Vergouwen MD, Nederkoorn PJ, de Haan R, Roos YB, Kruyt ND. Blood pressure-lowering treatment for preventing recurrent stroke, major vascular events, and dementia in patients with a history of stroke or transient ischemic attack. Cochrane Database Systemic Reviews. 2018;7:CD007858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.McGuiness B, Todd S, Passmore P, Bullock R. The effects of blood pressure lowering on development of cognitive impairment and dementia in patients without apparent prior cerebrovascular disease. Cochrane Database Systemic Reviews. 2006;19:CD004034. [DOI] [PubMed] [Google Scholar]
- 178.Elias PK, Elias MF, D’Agostino RB, Cupples LA, Wilson PW, Silbershatz H, Wolf PA. Niddm and blood pressure as risk factors for poor cognitive performance. The framingham study. Diabetes Care. 1997;20:1388–1395 [DOI] [PubMed] [Google Scholar]
- 179.Wolf PA, Beiser A, Elias MF, Au R, Vasan RS, Seshadri S. Relation of obesity to cognitive function: Importance of central obesity and synergistic influence of concomitant hypertension. The framingham heart study. Curr Alzheimer Res 2007;4:111–116 [DOI] [PubMed] [Google Scholar]
- 180.Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, Tuomilehto J. Risk score for the prediction of dementia risk in 20 years among middle aged people: A longitudinal, population-based study. Lancet Neurol 2006;5:735–741 [DOI] [PubMed] [Google Scholar]
- 181.Virta JJ, Heikkila K, Perola M, Koskenvuo M, Raiha I, Rinne JO, Kaprio J. Midlife cardiovascular risk factors and late cognitive impairment. European Journal of Epidemiology. 2013;28:405–416 [DOI] [PubMed] [Google Scholar]
- 182.Gonzalez HM, Tarraf W, Harrison K, Windham BG, Tingle J, Alonso A, Griswold M, Heiss G, Knopman D, Mosley TH. Midlife cardiovascular health and 20-year cognitive decline: Atherosclerosis risk in communities study results. Alzheimers and Dementia. 2018;14:579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Qiu C, Winblad B, Fastbom J, Fratiglioni L. Combined effects of apoe genotype, blood pressure, and antihypertensive drug use on incident ad. Neurology. 2003;61:655–660 [DOI] [PubMed] [Google Scholar]
- 184.Andrews S, Das D, Anstey KJ, Easteal S. Interactive effect of apoe genotype and blood pressure and cognitive decline: The path through life study. Journal of Alzheimer’s Disease. 2015;44:1087–1098 [DOI] [PubMed] [Google Scholar]
- 185.Rodrigue KM, Rieck JR, Kennedy KM, Devous MD Sr., Diaz-Arrastia R, Park DC. Risk factors for beta-amyloid deposition in healthy aging: Vascular and genetic effects. JAMA neurology. 2013;70:600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rast P, Kennedy KM, Rodrigue KM, Robinson PRAW, Gross AL, McLaren DG, Grabowski T, Schaie KW, Willis SL. Apoee4 genotype and hypertension modify 8-year cortical thinning: Five occasion evidence from the seattle longitudinal study. Cerebral Cortex. 2018;28:1934–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gelber RP, Ross GW, Petrovitch H, Masaki KH, Launer LJ, White LR. Antihypertensive medication use and risk of cognitive impairment: The honolulu-asia aging study. Neurology. 2013;81:888–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tzourio C, Anderson C, Chapman N, Woodward M, Neal B, MacMahon S, Chalmers J, PROGRESS Collaborative Group. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Archives of Internal Medicine. 2003;163:1069–1075 [DOI] [PubMed] [Google Scholar]
- 189.Tully PJ, Peters R, Peres K, Anstey KJ, Tzourio C. Effect of ssri and calcium channel blockers on depression symptoms and cognitive function in elderly persons treated for hypertension: Three city cohort study. International Psychogeriatrics. 2018;30:1345–1354 [DOI] [PubMed] [Google Scholar]
- 190.Peters R, Collerton J, Granic A, Davies K, Kirkwood T, Jagger C. Antihypertensive drug use and the risk of cognitive decline in the very old: An observational study- the newcastle 85+ study. Journal of Hypertension. 2015;33:156–164 [DOI] [PubMed] [Google Scholar]
- 191.Maxwell CJ, Hogan DB, Ebly EM. Calcium-channel blockers and cognitive function in elderly people: Results from the canadian study of health and aging. CMAJ. 1999;161:501–506 [PMC free article] [PubMed] [Google Scholar]
- 192.Ahmed HA, Ishrat T, Pillai B, Fouda AY, Sayed MA, Eidahshan W, Waller WJ, Ergul A, Fagan SC. Ras modulation prevents progressive cognitive impairment after experimental stroke: A randomized, blinded preclinical trial. Journal of Neuroinflammation. 2018;15:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wharton W, Goldstein FC, Tansey MG, Brown AL, Tharwani SD, Verble DD, Cintron A, Kehoe PG. Rationale and design of the mechanistic potential of antihypertensives in preclinical alzheimer’s ( heart) trial. Journal of Alzheimer’s Disease. 2018;61:815–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wright JT Jr., Dunn JK, Cutler JA, Davis BR, Cushman WC, Ford CE, Haywood LJ, Leenen FH, Margolis KL, Papademetriou V, Probstfield JL, Whelton PK, Habib GB, Group ACR. Outcomes in hypertensive black and nonblack patients treated with chlorthalidone, amlodipine, and lisinopril. JAMA. 2005;293:1595–1608 [DOI] [PubMed] [Google Scholar]
- 195.Aaslid R, Lindegaard KF, Sorteberg W, Nornes H. Cerebral autoregulation dynamics in humans. Stroke. 1989;20:45–52 [DOI] [PubMed] [Google Scholar]