

Abstract
Endoplasmic reticulum (ER) stress is thought to activate autophagy via unfolded protein response (UPR)-mediated transcriptional up-regulation of autophagy machinery components and modulation of microtubule-associated protein 1 light chain 3 (LC3). The upstream UPR constituents pancreatic EIF2-α kinase (PERK) and inositol-requiring enzyme 1 (IRE1) have been reported to mediate these effects, suggesting that UPR may stimulate autophagy via PERK and IRE1. However, how the UPR and its components affect autophagic activity has not been thoroughly examined. By analyzing the flux of LC3 through the autophagic pathway, as well as the sequestration and degradation of autophagic cargo, we here conclusively show that the classical ER stressor tunicamycin (TM) enhances autophagic activity in mammalian cells. PERK and its downstream factor, activating transcription factor 4 (ATF4), were crucial for this induction, but surprisingly, IRE1 constitutively suppressed autophagic activity. TM-induced autophagy required autophagy-related 13 (ATG13), Unc-51–like autophagy-activating kinases 1/2 (ULK1/ULK2), and GABA type A receptor–associated proteins (GABARAPs), but interestingly, LC3 proteins appeared to be redundant. Strikingly, ATF4 was activated independently of PERK in both LNCaP and HeLa cells, and our further examination revealed that ATF4 and PERK regulated autophagy through separate mechanisms. Specifically, whereas ATF4 controlled transcription and was essential for autophagosome formation, PERK acted in a transcription-independent manner and was required at a post-sequestration step in the autophagic pathway. In conclusion, our results indicate that TM-induced UPR activates functional autophagy, and whereas IRE1 is a negative regulator, PERK and ATF4 are required at distinct steps in the autophagic pathway.
Keywords: autophagy, endoplasmic reticulum stress (ER stress), unfolded protein response (UPR), protein degradation, signal transduction, activating transcription factor 4 (ATF4), autophagic sequestration, GABA type A receptor-associated protein (GABARAP), microtubule-associated protein 1 light chain 3 (LC3), pancreatic EIF2-α kinase (PERK), tunicamycin (TM), inositol-requiring enzyme 1 (IRE1), autophagic degradation
Introduction
Transmembrane and secretory proteins are translated in the endoplasmic reticulum (ER),5 where they are folded and undergo rigorous quality control to ensure cell function and protein homeostasis. When the folding capacity of the ER is exceeded (referred to as “ER stress”), the ER membrane-resident proteins PERK, IRE1, and ATF6 initiate the unfolded protein response (UPR) (1). PERK dimerizes and becomes activated via autophosphorylation, upon which it phosphorylates eIF2α. This in turn results in a general reduction in protein synthesis concurrent with increased translation of the transcription factor ATF4, which translocates to the cell nucleus to regulate gene expression. Furthermore, upon ER stress, IRE1 oligomerizes and acquires endoribonuclease activity, resulting in XBP1 mRNA splicing and production of the transcription factor–spliced XBP1 (XBP1s). Last, ER stress induces the translocation of ATF6 to the Golgi apparatus, where it is cleaved, releasing its cytoplasmic domain, which acts as a transcription factor. Together, the UPR-generated transcription factors initiate a transcriptional program aimed at increasing protein folding capacity and restoring protein homeostasis (1). Autophagy, the pathway for lysosomal degradation of intracellular material, has been proposed to play a key role in relieving cells of the burden of accumulating aberrant proteins. The first indication for this was provided by Klionsky and colleagues (2), who in 2006 demonstrated that ER stress can activate autophagy in yeast. Subsequently, various types of ER stress conditions were suggested to induce autophagy via the UPR also in mammalian cells, but as outlined below, further experimental evidence is needed to fully conclude how the UPR regulates autophagic activity.
Macroautophagy (herein referred to as autophagy) proceeds by the expansion of a condensed membrane cisterna (phagophore), which sequesters cellular material into a double-membrane or multimembrane vesicle (autophagosome) that subsequently fuses with lysosomes for degradation and recycling of the sequestered material (3). A set of evolutionarily conserved autophagy-related (ATG) proteins, first identified in yeast, orchestrate the generation and maturation of autophagosomes. Yeast Atg8 has been shown to play a central role in driving the autophagic pathway. In mammalian cells, seven different Atg8 orthologues, which segregate into the LC3 and GABARAP subfamilies, have been described. Of those, LC3B was the first mammalian Atg8 orthologue identified (4). During autophagy, the cytosolic form of LC3 (LC3-I) is converted to a lipidated, membrane-bound form (LC3-II) in a ubiquitin-like conjugation process that involves a number of ATGs. LC3-II localizes to phagophores, autophagosomes, and other membrane structures yet to be fully characterized (4–9). Local accumulation of LC3 on phagophores and autophagosomes may be detected as bright puncta by immunofluorescence microscopy, and LC3-II levels can be analyzed by Western blotting. After these discoveries, LC3 quickly became the most frequently used marker protein for studying mammalian autophagy, a trend that was further intensified after LC3 was reported to be a central player in autophagosome formation (10) and maturation (11, 12), as well as in cargo recruitment via interaction with autophagy receptors (13, 14). Consequently, studies on the relationship between ER stress and autophagy have strongly focused on deciphering signaling pathways and factors that modulate LC3. A number of different ER stressors have been reported to increase LC3-II levels and LC3 puncta in a manner that requires the action of PERK (15–19) and/or IRE1 (16, 20–24). Moreover, the PERK-eIF2α-ATF4 arm has been implicated in transcriptional up-regulation of numerous ATGs, including LC3B (25–28), ATG12 (15, 27, 28), ATG5 (25, 27), ULK1 (29), and BECN1, ATG16L1, ATG3, ATG7, ATG10, GABARAP, and GABARAPL1 (27), whereas the IRE1-XBP1s arm has been reported to up-regulate BECN1 (22) and ATG3 (30). Based on these observations, it has been generally inferred that UPR activates autophagy via a PERK/IRE1-driven transcriptional program. Additionally, IRE1 may promote JNK-mediated phosphorylation of BCL2 (21, 31), which in turn can increase the ability of Beclin-1 to enhance LC3 puncta formation (32).
Although valuable, these previously described effects of the UPR and its components on transcription of ATGs and lipidation of LC3 are not sufficient evidence by themselves to fully define how the UPR regulates functional autophagic activity, because (i) increased transcription and expression of components of the autophagic machinery may in some instances be a cellular attempt to compensate for reduced autophagic activity, and (ii) increases in cellular levels of lipidated LC3 may in some instances be the result of increased autophagy but in other cases the result of increased expression of LC3 and/or reduced LC3-II degradation caused by inhibition of autophagy at a late step in the pathway (33). To distinguish between those possibilities, one may assess the flux of LC3 through the autophagic pathway as well as analyze the sequestration and degradation of autophagic cargo (33). To date, the effect of the UPR on LC3 flux and autophagic cargo sequestration and degradation activity has not been thoroughly assessed.
Here, we employed various autophagy methods in combination with the classical ER stressor tunicamycin (TM; a glycosylation inhibitor) to investigate how the UPR and its components affect autophagic activity in mammalian cells. We find that TM enhances autophagic activity, as reflected by increased flux of LC3 through the pathway as well as increased sequestration and degradation of autophagic cargo. Moreover, our results reveal that TM-induced autophagy requires the action of the UPR components PERK and ATF4, whereas IRE1 plays an unexpected opposing role. Last, we demonstrate that PERK and ATF4 act at distinct steps in the autophagic pathway during TM-induced autophagy.
Results
Inhibition of N-linked glycosylation activates autophagy
To study how the UPR modulates autophagy, we treated LNCaP human prostate cancer cells with the classical ER stressor TM (2.5 μg/ml) and analyzed the flux of the autophagic membrane marker LC3 to lysosomes (33). The lipidated and membrane-attached form of LC3, LC3-II, is usually present on both the inner and outer membranes of the autophagosome, and the LC3-II that is present on the inner membrane is degraded after autophagosome–lysosome fusion (4, 33). Therefore, if TM would increase the flux of LC3-II to lysosomes, one would expect to observe an increase in the levels of LC3-II when LC3-II degradation is blocked by co-treatment with the lysosomal inhibitor bafilomycin A1 (Baf) (33). Indeed, LC3-II levels were significantly increased in LNCaP cells co-treated with TM (for 24 h) and Baf, compared with that observed in cells treated with TM or Baf alone (Fig. 1, A and B). Note that Baf was included only during the last 3 h of the 24-h treatment period, as recommended for this assay (33). Compared with treatment with Baf alone, co-treatment with TM plus Baf produced a ∼59% increase in LC3-II levels (Fig. 1B). A possible confounding factor in interpreting this assay is putative treatment effects on the expression of LC3, which may influence LC3-II levels (34), and as shown in Fig. 1A (and described below), TM did increase LC3 expression. To provide additional evidence, we generated an LNCaP cell line that expresses a tandem fluorescently tagged version of LC3, mTagRFP-mWasabi-LC3. This construct can be used to follow LC3 flux, because the green fluorescence of mWasabi is quenched in the acidic environment that arises upon fusion of autophagosomes with lysosomes, whereas the red fluorescence from mTagRFP is relatively resistant to acidic pH (33, 35). Thus, as illustrated in Fig. 1C, the fluorescent signal will change from yellow puncta when LC3 is attached to phagophores or autophagosomes (neutral pH) to red puncta when LC3 has reached the autolysosomes (acidic pH). The mTagRFP-mWasabi-LC3 construct is an improved version to follow autophagic membrane flux compared with earlier GFP-LC3–based constructs, because mWasabi is more acid-sensitive than GFP (35). As shown in Fig. 1D, treatment with TM (2.5 μg/ml) for 24 h led to a striking increase in the appearance of red puncta, similar to the phenotype observed upon a 3-h treatment with the mTOR inhibitor Torin1, which is a well-known autophagy inducer (33). In the presence of Baf, the puncta turned yellow (Fig. 1D), as expected from Baf-mediated neutralization of lysosomal acidity (36, 37). To quantify changes in mWasabi and mTagRFP fluorescence from a large number of cells, we subjected cells treated as in Fig. 1D to flow cytometry subsequent to a brief plasma membrane permeabilization with digitonin to wash out cytosolic mTagRFP-mWasabi-LC3 and thus selectively measure the fluorescent signals from membrane-attached mTagRFP-mWasabi-LC3. As shown in Fig. 1E, TM significantly increased the total fluorescence signal intensity from both mWasabi and mTagRFP, in a manner that was strikingly similar to that observed with Torin1. This indicates that TM increased both the number of autophagosomes and autolysosomes. On average, TM produced a ∼56% increase in mTagRFP fluorescence and a ∼66% increase in the sum of mWasabi and mTagRFP fluorescence. Importantly, compared with that observed in Baf-treated cells, the intensity from mTagRFP was substantially higher than that from mWasabi in both TM- and Torin1-treated cells, thus indicating active autophagic membrane flux under these conditions. The same was the case in DMSO control conditions (Fig. 1E), in agreement with the relatively high degree of basal autophagy in the LNCaP cell line (34, 38–41). Of note, there was some increase in the mTagRFP signal in Baf-treated cells (albeit far from the increase observed for the mWasabi signal), which likely reflects Baf-induced inhibition of mTagRFP degradation in autolysosomes. Compared with single treatment with Baf, TM, or Torin1 alone, co-treatment with TM + Baf or Torin1 + Baf led to a significant increase in both mWasabi and mTagRFP signals (Fig. 1E), further indicating that TM, like Torin1, increases the number of autophagic membrane structures destined for fusion with lysosomes.
Figure 1.

TM increases autophagic membrane flux in LNCaP cells. A, LNCaP cells were treated as indicated for 24 h, with Baf included the last 3 h only. Subsequently, protein extracts were prepared and subjected to immunoblotting for LC3 and α-tubulin as indicated. B, protein levels from three independent experiments as in A were quantified and normalized against α-tubulin (mean ± S.E. (error bars), n = 3). Red dots represent individual data points. Statistical significance was evaluated using repeated measures one-way ANOVA. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, illustration depicting the principle for the tandem fluorescent LC3 assay; mTagRFP-mWasabi-LC3 attached to phagophores and autophagosomes will be in an environment of neutral pH and therefore appear as yellow puncta (mix of green and red fluorescence). In contrast, mTagRFP-mWasabi-LC3 molecules that have reached the autolysosome will appear as red puncta because the green fluorescence from mWasabi, but not that from mTagRFP, is quenched in the acidic environment. D, LNCaP cells stably expressing mTagRFP-mWasabi-LC3 were treated for 24 h, as indicated, with Baf and Torin1 included the last 3 h only. Subsequently, cells were analyzed by live-cell fluorescence confocal microscopy. A representative image is shown for each treatment condition, and the white square insets in the top panels are shown in larger magnification in the bottom panels. Scale bar, 10 μm. E, LNCaP cells stably expressing mTagRFP-mWasabi-LC3 were treated for 24 h, as indicated, with Baf and Torin1 included the last 3 h only. Subsequently, cells were detached from the tissue culture plate and briefly treated with digitonin to permeabilize the plasma membrane and thereby deplete the cells of unconjugated, cytosolic mTagRFP-mWasabi-LC3 while preserving the conjugated, membrane-bound mTagRFP-mWasabi-LC3. Thereafter, the cells were analyzed for mWasabi and mTagRFP fluorescence intensity by flow cytometry. The results from three independent experiments are shown (mean ± S.E., n = 3). Black dots represent individual data points. Statistical significance was evaluated using repeated measures one-way ANOVA. **, p < 0.01; ***, p < 0.001.
LC3 flux is an indicator of autophagic flux. However, the flux of LC3 through the autophagic pathway does not always correlate with that of autophagic cargo (33, 41, 42), and autophagy does not always require LC3 (41, 43–45). Therefore, LC3-based assays should be accompanied by additional methods to monitor autophagy (33). The most direct approach to measure autophagic activity is to quantify the sequestration and degradation of autophagic cargo. To that end, we used two well-established and highly recommended (33) quantitative methods: the LDH sequestration assay (38–40, 46) and the long-lived protein degradation (LLPD) assay (47–49). During general autophagy, the ubiquitously expressed, soluble cytosolic enzyme lactate dehydrogenase (LDH) is nonselectively sequestered into autophagosomes along with cytosol and other autophagic cargo (46) (see illustration in Fig. 2A). In the presence of an inhibitor of autolysosomal degradation, such as Baf, the net autophagic sequestration rate can be accurately and quantitatively determined by a protocol that efficiently fractionates sequestered LDH from cytosolic LDH (38–40, 46). This method has been thoroughly validated through testing with a plethora of well-known chemical and genetic interferences with autophagosome formation in multiple cell types, demonstrating that it reliably monitors macroautophagic sequestration activity (34, 37–41, 46, 50, 51). To assess the effect of TM on LDH sequestration, LNCaP cells were treated with DMSO control or TM (2.5 μg/ml) for 21 h, followed by an additional 3-h treatment with DMSO (as a control) or Baf to block autolysosomal degradation of sequestered LDH. In DMSO control–pretreated cells, subsequent Baf treatment led to an LDH sequestration rate of ∼0.6%/h (Fig. 2A). This number is in good agreement with our earlier findings of the basal autophagic sequestration rate in this cell line (34, 38–41). When cells were pretreated with TM for 21 h before subsequent treatment with Baf, the sequestration rate was increased by ∼62% compared with that observed with Baf in DMSO-pretreated cells (Fig. 2A). The TM-induced increase was nearly half of that induced by Torin1, which, besides acute amino acid starvation, is the most efficient inducer of autophagic sequestration (33, 40, 41, 50). Treatment with TM thus induced a substantial increase in autophagic sequestration activity, estimated to be nearly half of that which can be maximally obtained.
Figure 2.

TM increases autophagic sequestration and degradation activity in LNCaP cells. A, LNCaP cells were treated for 24 h, as indicated, with Baf and Torin1 included the last 3 h only, followed by determination of LDH sequestration (mean ± S. E. (error bars), n = 4). Bottom, the LDH sequestration assay measures autophagic sequestration of cytosol (autophagosome formation), using endogenous LDH as a cargo probe and a crude fractionation protocol to separate cytosolic (nonsedimentable) from sedimentable LDH (38–40, 46). In the presence of the lysosomal inhibitor Baf, the degradation of the LDH that resides inside autophagosomes is blocked, allowing specific analysis of autophagic sequestration activity. B, LNCaP cells transfected with siCtrl or siULK1 + siULK2 were treated for 24 h, as indicated, with Baf included the last 6 h only. LLPD was measured at 18–24 h (mean ± S.E., n = 3). Bottom, the LLPD assay measures endogenous degradation of long-lived proteins and is a classical method for monitoring autophagic flux (33, 48, 49). The method is a true end point assay of the autophagic pathway (i.e. it monitors the product of protein degradation). The proportion of autophagic–lysosomal LLPD is determined by treatment with Baf as well as by RNAi-mediated silencing of key autophagy-related genes, such as ULKs (49). Red dots represent individual data points. Statistical significance was evaluated using repeated measures one-way ANOVA for A and repeated measures two-way ANOVA for B. *, p < 0.05; **, p < 0.01; ***, p < 0.001. N.S., not significant.
Next, we employed the LLPD assay (47–49) to investigate whether the increased autophagic sequestration observed with TM also results in increased autophagic degradation activity. During general autophagy, a substantial proportion of long-lived proteins are sequestered into autophagosomes and thereafter degraded in the autolysosomes. Following degradation, the resulting, free amino acids are released into the cytosol for cellular re-use (see illustration in Fig. 2B). In the LLPD assay, the amino acid degradation product (free, radioactive valine with our procedure) is specifically quantified (47–49). The LLPD assay is thus a true end point method for measuring autophagic cargo flux (33, 47–49). As shown in Fig. 2B, treatment of LNCaP cells with TM (2.5 μg/ml) significantly increased LLPD in the time period 18–24 h after the drug addition. Importantly, this was due to activation of the autophagic–lysosomal degradation pathway, because TM failed to significantly affect LLPD in the presence of Baf or in cells depleted of the autophagy-essential ATG proteins ULK1 and ULK2 (Fig. 2B and Fig. S1). As expected, and in agreement with earlier studies (34, 52), nearly 80% of the total amount of LLPD in DMSO control-treated cells in nutrient-rich medium was nonlysosomal (Baf-insensitive) and nonautophagic (insensitive to ULK1/ULK2 depletion). Due to this inherently high background in the LLPD assay (mostly due to proteasomal activity (34, 52)), the increases observed with autophagy stimuli appear small compared with the total levels. To obtain useful relative information, alterations in LLPD are therefore normally compared with the lysosomal-autophagic LLPD fraction (33, 47–49). When comparing the effect of TM on LLPD in the absence and presence of lysosomal (Baf) or autophagic inhibition (ULK1/ULK2 depletion or depletion of other ATGs, as shown below), we calculated that TM increases autophagic protein degradation activity by ∼65 ± 8% (mean ± S.E.), compared with basal levels in LNCaP cells. In all of these experiments, TM failed to produce a statistical significant increase in LLPD in the presence of lysosomal/autophagic inhibition. This indicates that the TM-induced increase in LLPD is predominantly due to TM increasing autophagy rather than other protein degradation processes. It has been reported that ER stress may increase proteasomal degradation as well as proteasome- and lysosome-independent ER-associated degradation (ERAD) pathways (53–55). However, TM did not increase chymotrypsin-like activity in LNCaP cells (Fig. S2A). Moreover, as shown in Fig. S2, B and C, TM-induced LLPD was unaffected by the proteasome inhibitor MG132 and the ERAD inhibitors kifunensine and eeyarestatin I at concentrations known to block proteasomal activity and ERAD (Fig. S2A) (56–58). There was an apparent slight decrease in the degree of TM-induced LLPD in the presence of MG132 (Fig. S2C), but this was not statistically significant (p = 0.2375, one-way analysis of variance (ANOVA)). Again, TM-induced LLPD was strongly diminished in the presence of Baf (Fig. S2C). Thus, we conclude that the effect of TM on LLPD is predominantly caused by an increase in autophagic–lysosomal protein degradation.
Taken together, our data strongly indicate that TM increases full autophagic flux in LNCaP cells, from productive autophagosome formation (cargo sequestration) to completion of the pathway (cargo degradation and release of degradation products from the autolysosomes). Of note, the observed TM-induced increases in LC3 flux, LDH sequestration, and autophagic LLPD were very similar (∼56–66% increase, depending on the assay), indicating a good correlation between autophagic membrane and cargo flux in TM-treated cells.
Maximal effects of TM on LLPD in LNCaP cells occurred in the concentration range 1.5–10 μg/ml after 18–24 h of treatment (Fig. S3A). Based on this, in subsequent experiments, we used 2.5 μg/ml TM and measured autophagy after 18–24 h. At this concentration, TM did not cause any cell death, even after 70 h of treatment (Fig. S3B). Moreover, unlike thapsigargin (TG; ER Ca2+ pump inhibitor) or A23187 (A23; Ca2+ ionophore), which deplete ER Ca2+ (Fig. S3C) and therefore block autophagosome formation (34), TM did not affect ER Ca2+ levels (Fig. S3C). Thus, the TM-induced autophagy we observed is not restricted by any negative effects that could have arisen from ER Ca2+ depletion.
Some reports have suggested that TM can reduce mTORC1 activity in cancer cells (59, 60). However, TM did not alter the levels of the mTORC1-phosphorylated forms of S6K (Thr-389) or ULK1 (Ser-757) (Fig. S3D), indicating that mTORC1 activity is not affected by TM in LNCaP cells.
TM causes aberrant protein folding by inhibiting the rate-limiting factor of N-linked glycosylation, GlcNAc-1-phosphate transferase (GPT) (61). If the effect of TM on autophagy was specifically caused by GPT inhibition, then depletion of GPT would be expected to also activate autophagy. Indeed, GPT-targeted RNAi induced a similar Baf-sensitive increase in LLPD as that observed with TM (Fig. 3 and Fig. S4A).
Figure 3.
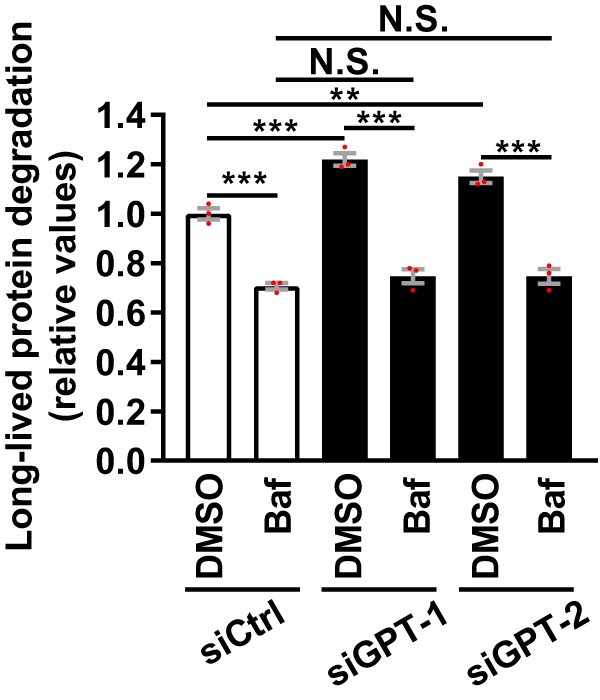
Silencing of GPT induces Baf-sensitive degradation of long-lived proteins. LNCaP cells were transfected with a nontargeting control siRNA (siCtrl) or two different GPT-targeting siRNAs. Subsequently, the cells were treated as indicated for 6 h, before measurement of LLPD (mean ± S.E. (error bars), n = 3). Red dots represent individual data points. Statistical significance was evaluated using repeated measures two-way ANOVA. **, p < 0.01; ***, p < 0.001. N.S., not significant.
The ability of TM to induce autophagy was not restricted to prostate cells or to cancer origin, because we observed significant, Baf-sensitive increases in LLPD also in HeLa cervical carcinoma cells and in nonmalignant RPE-1 retinal pigment epithelial cells (Fig. 4).
Figure 4.
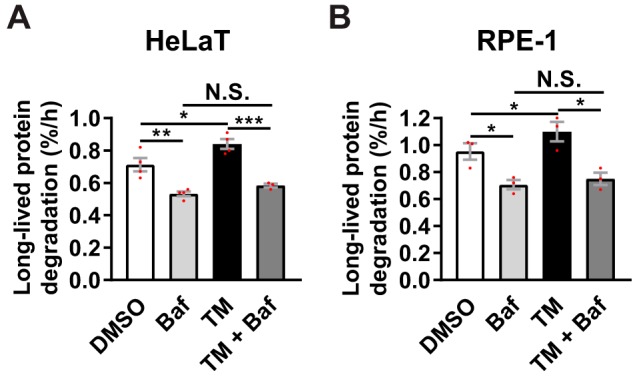
TM induces Baf-sensitive degradation of long-lived proteins in HeLa and RPE-1 cells. HeLaT (A) or RPE-1 (B) cells were treated for 24 h, as indicated, with Baf included the last 4 h only. LLPD was measured at 20–24 h (mean ± S.E. (error bars), n ≥ 3 for HeLaT, n = 3 for RPE-1). Red dots represent individual data points. Statistical significance was evaluated using regular two-way ANOVA in A (HeLaT) and repeated measures two-way ANOVA in B (RPE-1). *, p < 0.05; **, p < 0.01; ***, p < 0.001. N.S., not significant.
PERK and ATF4 are essential for, whereas IRE1 restricts, ER stress–induced autophagy
TM effectively activated the UPR in LNCaP cells, as demonstrated by increased levels of phosphorylated PERK and elevated protein levels of ATF4, CHOP, XBP1s, and BiP (Fig. 5A). TM-induced UPR could be discerned at 6 h, but not at 2 h, and was maintained for at least 18 h (Fig. 5A). Strikingly, TM-induced LLPD was completely abolished upon depletion of PERK (Fig. 5B and Fig. S4B). In contrast, and surprisingly, TM-induced LLPD was further increased upon IRE1 knockdown, whereas ATF6 silencing had no effect (Fig. 5B and Fig. S4B). Thus, whereas PERK is essential for TM-induced autophagy, IRE1 plays an opposing role. Knockdown of PERK under basal conditions did not alter LPPD (Fig. S4C), indicating that PERK is selectively required for autophagy during ER stress. Interestingly, however, autophagy was substantially enhanced by IRE1 depletion under basal conditions, as IRE1 knockdown increased LLPD in a manner that was abolished by Baf or ULK1/2 silencing (Fig. 5C). Moreover, IRE1 depletion significantly increased LDH sequestration in the presence of Baf (Fig. 5D). Thus, IRE1 restricts basal autophagy at a pre-sequestration step in the autophagic pathway, and this can largely explain the additional increase in LLPD observed upon knockdown of IRE1 in combination with TM (Fig. 5B). Increased LLPD upon IRE1 silencing was also observed in U2OS osteosarcoma cells (Fig. S4D), suggesting that IRE1 may limit basal autophagy in general and not only in LNCaP cells.
Figure 5.

PERK and ATF4 are essential, whereas IRE1 restricts ER stress–induced autophagy. A, LNCaP cells were treated as specified and immunoblotted for the indicated proteins. Upward mobility shift in the PERK band reflects PERK phosphorylation, as verified in the bottom panel; PERKi reverses the shift. One representative of two independent experiments is shown. The blots were spliced at the locations indicated by the dotted lines. B–G, LNCaP (B–F) or RPE-1 (G) were transfected with the indicated siRNAs and treated as specified for 24 h (B, E, and F), 6 h (C), 4 h (D), or 22 h (G). LLPD was measured at 18–24 h (B, E, and F), 0–6 h (C), or 18–22 h (G). LDH sequestration was determined after 4 h of DMSO or Baf treatment (D). Results are the mean ± S.E. (error bars), n ≥ 3 (B), n = 3 (C and G), n ≥ 3 (D), n ≥ 4 (E), n = 4 (F). Red dots represent individual data points. Statistical significance was evaluated using regular (B and E) or repeated measures (G) one-way ANOVA and regular (D) or repeated measures (C and F) two-way ANOVA. *, p < 0.05; **, p < 0.01; ***, p < 0.001. N.S., not significant.
Next, we assessed the role of ATF4. As shown in Fig. 5E, ATF4 depletion completely abolished TM-induced LLPD. Interestingly, LLPD levels in TM-treated, ATF4-depleted cells were even lower than basal levels (Fig. 5E). This indicates a requirement for ATF4 in basal autophagy as well as in ER stress–induced autophagy. Indeed, basal LLPD was significantly reduced by three of three ATF4-targeting siRNAs (Fig. 5F), and this was due to inhibition of autophagy, because ATF4 knockdown failed to reduce LLPD in ULK1/2-silenced cells (Fig. S4E). The reduction in basal LLPD observed upon ATF4 depletion was about half of that observed upon ULK1/2 depletion (Fig. 5, compare C and F). Thus, ATF4 is partially required for basal autophagy, which can partly explain the subbasal LLPD levels observed in TM-treated, ATF4-silenced cells. Interestingly, LLPD levels were always lower with the combination of TM and ATF4 depletion than with ATF4 depletion alone (Fig. 5F), indicating that TM induces degradation-inhibitory effects in the absence of ATF4. It remains to be examined whether this degradation-inhibitory effect is caused by IRE1-mediated repression of autophagy or by other mechanisms.
Finally, we examined whether PERK and ATF4 were essential for TM-induced autophagy also in a cell line other than LNCaP. Indeed, silencing of PERK or ATF4 completely abolished TM-induced LLPD in RPE-1 cells (Fig. 5G and Fig. S4F). In these cells, LLPD levels were below basal levels in both cases, which might suggest a partial requirement for PERK and ATF4 in basal autophagy in RPE-1 cells.
In summary, PERK and ATF4 are essential for TM-initiated ER stress–induced autophagy, whereas IRE1 acts as a brake. Moreover, ATF4 and IRE1 play opposing roles in autophagy under basal conditions.
RNA-Seq reveals candidate mediators of ATF4-dependent autophagy regulation, whereas PERK likely acts independently of transcription
Because both PERK and ATF4 were essential for TM-induced autophagy, and ATF4 is a transcription factor that normally acts downstream of PERK, we hypothesized that PERK mediates its effects via ATF4-dependent changes in gene transcription. In line with the notion of a transcriptional regulation of autophagy, TM increased autophagic activity in the time period of 18–24 h even if TM was washed out at 18 h (Fig. 6A). In contrast, and confirming that TM could be efficiently washed out, a 1-h treatment with TM was not sufficient to enhance autophagy at 18–24 h (Fig. 6A). Treatment with the potent PERK inhibitor GSK2606414 (“PERKi”) abolished TM-induced LLPD, indicating that PERK activity is essential for TM-induced autophagy (Fig. 6B). However, PERK activity was redundant during the last 6 h of TM treatment (Fig. 6B), whereas allowing PERK activity only for the first 6 h of TM treatment was insufficient for activation of LLPD in the 18–24-h time period (Fig. 6B). Together, our data indicate that TM causes PERK-dependent changes over the first 18 h of treatment that by themselves are sufficient for enhancing autophagy at 18–24 h. Of note, the level of LLPD under basal conditions (i.e. in the absence of TM) was unaffected by PERKi (Fig. 6B) or PERK knockdown (Fig. S4C), despite efficient reduction of basal p-eIF2α levels.
Figure 6.

ATF4 regulates gene expression independently of PERK in TM-treated LNCaP cells. A, LNCaP cells were treated with DMSO for 24 h or with TM, which was washed out and replaced with DMSO after 1 h (0–1 h) or 18 h (0–18 h) or kept throughout (0–24 h). LLPD was measured at 18–24 h (mean ± S.E. (error bars), n = 4). B, LNCaP cells were treated as indicated, with PERKi present in the specified time periods. LLPD was measured at 18–24 h (mean ± S.E., n ≥ 4). Immunoblotting validated PERK inhibition by PERKi. C, LNCaP cells were siRNA-transfected, followed by treatment for 18 h, as indicated, and subjected to RNA-Seq. The Venn diagram displays the numbers and overlap of differentially expressed genes (padj < 0.05, log2 FC ≥ 1) in TM-treated siCtrl cells versus DMSO siCtrl cells (brown) and in TM-treated siCtrl cells versus TM-treated cells suppressed/silenced for PERK (red) or ATF4 (green) (n = 4). Genes were considered to be PERK-regulated (red) when mRNA expression was significantly altered (padj < 0.05, log2 FC ≥ 1.0) by all PERK-interfering conditions (PERKi, siPERK-1, and siPERK-2). Genes were considered to be ATF4-regulated (green) when mRNA expression was significantly altered (padj < 0.05, log2 FC ≥ 1.0) by all ATF4-interfering conditions (siATF4-1 and siATF4-2). The analysis was performed in this manner to eliminate genes that were altered due to nonspecific/off-target effects from chemical and genetic interference with PERK and ATF4. D, heat map depictions of RNA-Seq data for the six ATG genes whose expression was up-regulated more than 2-fold by TM (padj < 0.05). Left, normalized log2 reads count (expression). Right, log2 FCs of TM + siCtrl versus DMSO + siCtrl, and TM + siATF4–1 or -2 versus TM + siCtrl (differential expression). Red dots represent individual data points. Statistical significance was evaluated using repeated measures (A) or regular (B) one-way ANOVA or DESeq2 version 1.10.1 Bioconductor package (C and D). ***, p < 0.001. N.S., not significant.
To probe for the putative PERK- and ATF4-regulated transcriptional program responsible for ER stress–induced autophagy, we performed global RNA-Seq of LNCaP cells treated with TM for 18 h in the absence or presence of interference with PERK (PERKi, or two different PERK-targeting siRNAs) or ATF4 (two different ATF4-targeting siRNAs). TM significantly altered the expression level of 2284 genes (padj < 0.05, log2 FC ≥ 1), and 431 of these alterations depended on ATF4 (Fig. 6C). Interestingly, ATF4 also controlled the expression of 123 genes that were not altered by TM treatment (Fig. 6C). Surprisingly, our analysis returned merely five genes whose expression was altered by interference with PERK (Fig. 6C). This indicates that PERK modulates autophagy independently of transcription in TM-treated LNCaP cells. Moreover, these results suggest that PERK and ATF4 are uncoupled in LNCaP cells and thus that they likely play distinct roles in ER stress–induced autophagy.
To gain insight into candidate mediators of ER stress–induced autophagy and to compare our results with previous reports that have indicated regulation of ATG gene expression by ER stress, we used the HUGO Gene Nomenclature Committee (HGNC) list of human ATGs to identify all autophagy-related genes whose expression was significantly altered by TM (padj < 0.05, any FC). This returned 18 genes, whose expression levels were up-regulated by TM, including MAP1LC3B, GABARAPL1, ATG16L1, GABARAP, ATG12, ATG5, ATG3, and BECN1 (Table S1), which have also previously been reported to be up-regulated by ER stress. The up-regulation of ATG16L1, GABARAP, ATG12, ATG5, ATG3, and BECN1 was, however, very modest (log2 FC ranging from 0.161 to 0.354), whereas the expression of MAP1LC3B and GABARAPL1 as well as that of four other genes (WIPI1, MAP1LC3B2, MAP1LC3A, ATG13) was increased more than 2-fold (log2 FC > 1) by TM (Table S1). Intriguingly, TM up-regulated the expression of all these six genes in an ATF4-dependent manner (Fig. 6D), suggesting that they may play an important role in ATF4-mediated autophagy regulation. Real-time RT-PCR analyses and Western blotting confirmed the up-regulation by TM at both the mRNA and protein levels (Fig. S5).
Next, we tested whether depleting any of these transcripts would diminish TM-induced autophagy. Because ATG8 isoforms may have overlapping functions in autophagy, a combined knockdown was performed for the LC3s and GABARAPL1. Depleting these mammalian ATG8s, or WIPI1 (Fig. S5, A and B) did not alter basal or TM-induced LLPD (Fig. 7, A and B). However, knockdown of ATG13 (Fig. S5C) strongly reduced both basal and TM-induced LLPD (Fig. 7B) as well as LDH sequestration (Fig. 7C). To test whether the observed induction of ATG13 by TM was necessary for TM-induced autophagy, we titrated the ATG13-targeting siRNA down to a concentration (0.025 nm) where basal autophagy was minimally affected (Fig. 7D, top) but where ATG13 mRNA and protein levels were reduced to near basal levels in the presence of TM (Fig. 7D, middle and bottom). Evidently, TM was still potently able to activate autophagy (Fig. 7D, top) even though ATG13 expression was no longer elevated by TM relative to basal levels (Fig. 7D, middle and bottom). When the siRNA concentration was gradually increased (from 0.025 to 0.2 nm), we observed a gradual decrease of basal LLPD that paralleled the decrease in LLPD observed in the presence of TM (Fig. 7D, top). Thus, although ATG13 is strongly required for both basal and TM-induced autophagy, the TM-mediated induction of ATG13 is not required for the ability of TM to activate autophagy.
Figure 7.

ER stress–induced autophagy requires ATG13 and GABARAPs, but not WIPI1 or LC3s. A and B, LNCaP cells were siRNA-transfected, followed by treatment for 24 h, as indicated. LLPD was measured at 18–24 h (mean ± S.E. (error bars), n = 3). C, LNCaP cells were siRNA-transfected, followed by treatment for 24 h, as indicated, with Baf included the last 3 h only. LDH sequestration was determined at 21–24 h (mean ± S.E., n ≥ 3). D, LNCaP cells were transfected with siCtrl (0.025 nm) or siATG13 at the specified concentrations and treated for 24 h, as indicated. In parallel, we determined LLPD at 18–24 h (top; mean ± S.D. of three replicates), p-ATG13 and ATG13 protein levels by immunoblotting (middle), and ATG13 mRNA levels by real-time RT-PCR (bottom; mean ± S.D. of three replicates). One representative of two independent experiments is shown. E, LNCaP cells were siRNA-transfected, followed by treatment for 24 h, as indicated. LLPD was measured at 18–24 h (mean ± S.E., n = 3). Red dots represent individual data points. Statistical significance was evaluated using repeated measures two-way ANOVA (A, B, and E) or regular one-way ANOVA (C). **, p < 0.01; ***, p < 0.001. N.S., not significant.
Basal and ER stress–induced autophagy is independent of LC3s but requires GABARAPs
Mammalian ATG8 proteins comprise the MAP1LC3 (LC3A, LC3B, and LC3C) and GABARAP (GABARAP, GABARAPL1, and GABARAPL2) families. LC3C expression is negligible in LNCaP cells (41), as confirmed by the LC3C RNA-Seq reads being below the cut-off value (data not shown). Thus, the finding that depletion of LC3A/B and GABARAPL1 did not alter LLPD (Fig. 7A and Fig. S5A) indicates that basal and TM-induced autophagy is independent of the LC3 family. To test whether GABARAPs are required and/or whether LC3s and GABARAPs may have overlapping functions, we simultaneously silenced all three GABARAP family members or both GABARAP and LC3 family members (Fig. S6), and measured the effects on LLPD. Strikingly, both basal and TM-induced autophagy were strongly reduced upon silencing of the GABARAP family, and no additional effect was observed upon simultaneous depletion of the LC3s (Fig. 7E). Again, knockdown of the LC3s alone did not affect autophagy (Fig. 7E).
ATF4 and PERK act at distinct steps in the autophagic pathway
The global mRNA expression analysis (Fig. 6C) strongly indicated that PERK and ATF4 are uncoupled in TM-treated LNCaP cells. In line with this, knockdown of PERK or inhibition of PERK activity did not reduce ATF4 protein levels in TM-treated cells (Fig. 8 (A and B) and Fig. S7A). Moreover, whereas the ∼4.4-fold increase in LC3-II levels observed in TM-treated cells was completely abolished upon ATF4 silencing (Fig. 8, A and B), inhibition of PERK activity had a minor or no effect (Fig. S7A), and depletion of PERK resulted in only a tendency of a moderate (∼30%) reduction in LC3-II levels (Fig. 8, A and B). Interestingly, TM induced an equivalent increase in the levels of LC3-I as in the levels of LC3-II, and LC3-I levels were modulated very similarly to LC3-II by both PERK and ATF4 (Fig. 8, A and B).
Figure 8.

ATF4 and PERK act at distinct steps in the autophagic pathway. A, LNCaP cells were siRNA-transfected, followed by treatment for 18 h, as specified, and immunoblotted for the indicated proteins. B, protein levels from three independent experiments as in A were quantified and normalized against α-tubulin (mean ± S.E. (error bars), n = 3). C, LNCaP cells were siRNA-transfected, followed by treatment for 24 h, as indicated, with Baf included the last 3 h only. LDH sequestration was determined at 21–24 h (mean ± S.E., n = 3). D, LNCaP cells were siRNA-transfected, followed by treatment for 24 h, as indicated. LLPD was measured at 18–24 h (mean ± S.E., n = 4). E, LNCaP cells were siRNA-transfected, and total LDH sequestration was determined after 24 h DMSO or TM treatment (mean ± S.E., n ≥ 3). F, major findings of our study were as follows. TM induces autophagy via ER stress in a manner that requires the action of ATF4 and PERK at distinct steps in the autophagic pathway. ATF4 likely acts via transcription and is essential for autophagosome formation, which occurs in an ULK1/2-, ATG13-, and GABARAPs-dependent manner. TM-induced ER stress stimulates LC3 transcription and elevates LC3-I and -II protein levels via ATF4, but the implications of this remain to be determined, as TM-induced autophagy did not appear to require the LC3 protein family. Of note, TM-induced autophagy is nevertheless associated with LC3 flux through the pathway (not depicted). PERK regulates autophagy after phagophore closure, in a manner that is independent of ATF4 and likely also independent of transcription. This novel function of PERK was revealed in LNCaP and HeLa cells, where TM up-regulated ATF4 protein levels independently of PERK. In other cell lines where PERK and ATF4 are coupled (e.g. RPE-1 and PC3), PERK may regulate autophagy via ATF4 as well as via the novel ATF4-independent function revealed in the current study. Red dots represent individual data points. Statistical significance in B–D was determined using repeated measures one-way ANOVA, and in E it was determined using regular two-way ANOVA. *, p < 0.05; **, p < 0.01; ***, p < 0.001. N.S., not significant.
Uncoupling of PERK and ATF4 was not restricted to LNCaP cells, as depletion of PERK also failed to reduce TM-mediated up-regulation of ATF4 protein levels in HeLa cells (Fig. S7B). However, PERK and ATF4 were partly coupled in RPE-1 cells (Fig. S7B). LNCaP cells are defective in the phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase PTEN (phosphatase and tensin homolog) and therefore have a constitutive high level of phosphatidylinositol 3,4,5-trisphosphate–mediated signaling (62, 63). To test whether this could be important for PERK-ATF4 uncoupling, we assessed PERK-ATF4 coupling in the PTEN-null cell line PC3 (62). Strikingly, TM-induced up-regulation of ATF4 protein levels was strongly dependent on PERK in PC3 cells (Fig. S7B), demonstrating that PTEN status is not relevant for PERK-ATF4 uncoupling. Furthermore, this indicates that the degree of linearity of the PERK-ATF4 axis can vary considerably from cell line to cell line.
Next, we compared the effects of PERK and ATF4 silencing on autophagic sequestration activity. Strikingly, knockdown of ATF4 completely abolished TM-induced LDH sequestration in LNCaP cells, whereas silencing of PERK had no effect (Fig. 8C). The same was found in HeLa cells (Fig. S7C). This confirms the absolute requirement for ATF4 in TM-initiated ER stress–induced autophagy. Additionally, it demonstrates that ATF4, but not PERK, acts at a step prior to phagophore closure. To further assess whether the reversal of TM-induced LLPD by PERK silencing (Fig. 5B) was due to abrogation of autophagy, we blocked autophagic LLPD by depleting ULK1/2 or ATG13 and tested whether PERK silencing would still reduce LLPD. As shown in Fig. 8D, PERK silencing completely abolished TM-induced LLPD but did not reduce LLPD levels in TM-treated cells depleted of ULK1/2 or ATG13. This clearly demonstrates that PERK indeed is required for TM-induced autophagic protein degradation, whereas it is dispensable for autophagic sequestration, indicating an obligatory role for PERK at a step after autophagosome formation during ER stress. To further test this conclusion, we performed the LDH sequestration assay in the absence of Baf (i.e. in the absence of deliberate inhibition of LDH degradation). If PERK is required at a step after autophagosome formation during TM-induced autophagy, one would expect an accumulation of sequestered LDH in TM-treated cells depleted of PERK. Indeed, we observed that TM induced a significant accumulation of sequestered LDH in PERK-silenced LNCaP cells (Fig. 8E). In contrast, there was no increase in sequestered LDH in TM-treated cells depleted of ATF4 (Fig. 8E). Moreover, PERK depletion had no effect in the absence of TM, confirming our finding from the LLPD assay that interference with PERK has no effect on autophagy in nonstressed cells (Fig. 6B and Fig. S4C). We did not observe any significant accumulation of sequestered LDH in control-transfected cells treated with TM in the absence of Baf (Fig. 8E). This was expected; because the half-life of autophagosomes is very short (∼10 min) (64, 65), the steady-state number of autophagosomes is very low in conditions of uninhibited autophagic–lysosomal flux. Confirming these findings in LNCaP cells, the combination of TM treatment and transfection with PERK-targeting siRNA also led to a significant accumulation of sequestered LDH in HeLa cells in the absence of Baf, whereas this did not occur upon transfection with control or ATF4-targeting siRNAs (Fig. S7D). In conclusion, our data strongly indicate that during TM-initiated ER stress–induced autophagy, PERK and ATF4 have distinct yet essential functions at different steps in the autophagic pathway.
Discussion
By employing functional autophagy assays, we have for the first time firmly demonstrated that ER stress and the resultant UPR can activate autophagic cargo sequestration and degradation activity in mammalian cells. Moreover, we for the first time dissect the roles played by the various UPR components in the regulation of autophagic activity. Our investigations conclusively show that TM-initiated ER stress induces autophagy in both malignant and nonmalignant human cell lines. PERK and ATF4 are absolutely essential for this effect, whereas IRE1, in contrast, acts as a negative regulator of autophagy. Remarkably, we found that PERK could act completely independently of ATF4 in two different cell lines (LNCaP and HeLa). By using assays that differentiate between autophagic sequestration (the result of autophagosome formation) and autophagic degradation (the result of autophagosome–lysosome fusion), we reveal that ATF4 is specifically required for autophagosome formation, whereas PERK acts at a post-sequestration step in the autophagic pathway (Fig. 8F).
Our data indicate that TM-initiated ER stress–induced autophagy requires the GABARAP protein family, whereas the LC3 proteins appear to be redundant. The same was the case for basal autophagy. These findings add to an emerging broader picture pointing to GABARAP rather than LC3 proteins as being the crucial Atg8-orthologous players in mammalian autophagic processes. Thus, GABARAPs, but not LC3s, are required for starvation-induced bulk autophagy (41, 43), Parkin-dependent mitophagy (44, 45), basal aggrephagy (45), basal autophagy (this study), and ER stress–induced autophagy (this study). The realization that LC3 may be dispensable for many, if not all, types of autophagy has important implications, because most studies during the last ∼15 years have used LC3 modulation (analyses of LC3 expression and lipidation) as the major or only readout for monitoring autophagy. Previous studies on the relationship between ER stress and autophagy are no exceptions. They have primarily focused on dissecting how ER stress and the UPR modulate LC3 expression and lipidation, assuming that it would reflect how ER stress modulates autophagy. However, because our data indicate that the LC3s are redundant for TM-initiated ER stress–induced autophagy, the general effect of ER stress on autophagic activity is unlikely to be mediated via modulation of LC3. It is interesting to note that TM-initiated ER stress did induce flux of LC3 to acidic environments, indicating increased transport of LC3-containing membranes to lysosomes. Thus, even though TM-induced autophagy did not seem to require LC3s, the autophagic membranes engaged in ER stress–induced autophagy are likely LC3-positive. It is highly probable that LC3s play a role in selective autophagy, but it remains to be determined whether, and under which conditions, the LC3s play a nonredundant role or whether the GABARAPs always play the dominant role. Because LC3s can have numerous nonautophagic functions (5–9), it is also conceivable that the ER stress–induced increase in LC3 expression is part of a cellular stress response that is unrelated to autophagy. Further studies are required to elucidate the functional implications of ER stress–induced elevation of LC3 expression and LC3-II. Of note, our results indicate that the major effects of ER stress on LC3 are mediated by ATF4, in good agreement with its previously described role in controlling LC3B transcription (25–28). Besides LC3B, our data additionally indicate that ER stress elevates the transcription of LC3A and LC3B2 via ATF4.
It remains to be determined whether ER stress stimulates the LC3 lipidation process per se or whether the stress-induced increase in LC3-II levels merely is the result of increased transcription of LC3 mRNA and thus more production of LC3-I. The latter alternative appears to be the case in cells treated with the ER stress–inducing calcium modulators A23 and TG, because we have shown that they increase the levels of LC3B mRNA, LC3-I, and LC3-II in a highly correlative manner (34). We later found these increases to be dependent on ATF4.6 TM also increased LC3B mRNA, LC3-I, and LC3-II levels to very similar degrees (5.8-, 4.8-, and 4.4-fold, respectively) and in an ATF4-dependent manner. Thus, our data obtained with A23 and TG (34), as well as TM (this study), are in line with the notion that ER stress increases LC3-II levels through increased transcription and production of LC3-I rather than through stimulation of the lipidation machinery.
Besides LC3B, LC3B2, and LC3A, our RNA-Seq analysis indicated that TM-induced ER stress alters the expression of hundreds of other genes via ATF4. Because ATF4 is a transcription factor and TM-induced autophagy was completely abolished by ATF4 knockdown, it is very likely that TM stimulates autophagy via ATF4-mediated transcriptional alterations. Besides LC3s, TM enhanced the expression of several other ATGs, and among them, WIPI1, GABARAPL1, and ATG13 were up-regulated more than 2-fold in an ATF4-dependent manner. However, knockdown of WIPI1 or GABARAPL1 had no effect on autophagy, and therefore their increased expression is unlikely to be mediating the effect of TM. Moreover, although we found ATG13 to be strongly required for both basal and TM-induced autophagy, our fine RNAi titration clearly showed that the TM-mediated induction of ATG13 was dispensable for the ability of TM to activate autophagy. In summary, our data suggest that no single ATF4-mediated ATG gene expression change is responsible for the effects of TM on autophagy. Rather, TM-induced autophagy may depend on the combined action of many ATF4-regulated genes, which may include ATGs as well as non-ATGs. It will be a challenging future task to decipher the exact ATF4-mediated gene transcript changes that are critical for ER stress–induced autophagy.
Intriguingly, our results indicate that IRE1 limits autophagy under both basal and TM-induced ER stress conditions. This finding seemingly contradicts previous publications that have proposed IRE1 and XBP1 as mediators of ER stress–induced autophagy (16, 20–24, 30). However, those studies assessed the autophagy-regulatory effects of IRE1 (16, 20, 21, 23, 24) or XBP1 (22, 30) solely by LC3-based assays or analyses of the expression of ATGs, and although those approaches may provide indications about effects on autophagy, they are insufficient by themselves to draw firm conclusions about effects on autophagic activity (i.e. the sequestration and degradation of autophagic cargo) (33). In line with our results, one study suggested that IRE1 (but not XBP1) negatively regulated autophagic degradation of an aggregation-prone mutant Huntington protein (66). However, it was not directly tested whether the increased levels of the mutant Huntington protein aggregates that were observed upon ectopic expression of IRE1 were caused by inhibition of autophagy or, vice versa, whether the decrease in aggregate levels observed upon knockdown or knockout of IRE1 was caused by increased autophagic activity. Another study showed that knockdown of IRE1 or XBP1 in a motoneuron cell line increased the clearance of aggregated mutant superoxide dismutase 1 protein (67). Pharmacologic and genetic interference demonstrated that the increased clearance of these aggregates in XBP1 knockdown cells was caused by autophagy. However, it was not tested whether this was also the case in IRE1 knockdown cells. We have yet to identify what is mediating the increase in autophagy that we observed upon IRE1 knockdown. The indications so far suggest that the limiting effect of IRE1 on autophagy is predominantly mediated independently of IRE1 activity and thus independently of XBP1. First, our preliminary data showed that pharmacologic inhibition of IRE1 activity had little effect on basal and TM-induced LLPD (data not shown). Second, although IRE1 shows a low level of basal activity in LNCaP cells (34), TM strongly activated IRE1, as witnessed by the massive increase in XBP1s protein levels upon TM treatment. Thus, if the inhibitory effect of IRE1 on autophagy was mediated by IRE1 activity, one would expect a much larger increase in autophagy upon IRE1 knockdown under TM-induced ER stress conditions than under basal conditions. However, we observed that IRE1 depletion increased autophagic protein degradation to nearly the same degree under basal conditions as under TM-induced ER stress conditions. We speculate that the IRE1 protein may serve a structural role or interact with other molecules in a manner that limits autophagy. An activity-independent role of an upstream UPR sensor like IRE1 is not without precedence, because PERK has been described to serve several structural roles in the cell independently of its kinase activity (68–70). Although the effects of IRE1 on autophagy may be mediated independently of its enzymatic activity, the herein described effects of PERK on autophagy are most likely not, because PERKi abolished TM-induced autophagy as efficiently as knockdown of PERK.
A very significant novel finding in our study is the revelation, as we observed in both LNCaP and HeLa cells, that ER stress–inducing conditions can activate ATF4 independently of PERK. The failure of PERK to mediate up-regulation of ATF4 protein levels in LNCaP cells was not due to a failure of PERK to transmit its signal to eIF2α, because interference with PERK efficiently reduced p-eIF2α levels in TM-treated cells and nevertheless did not reduce ATF4 protein levels. This also indicates that other eIF2α kinases of the integrated stress response do not compensate for loss of PERK activity and, therefore, that the integrated stress response is not responsible for the maintenance of ATF4 protein levels upon interference with PERK in TM-treated cells. Mammalian ER stress signaling pathways were initially assessed in knockout MEFs, where ER stressors completely failed to elevate ATF4 levels in PERK−/− MEFs (71). A more recent study has, however, indicated that the relationship between PERK and ATF4 is not that strict in all cell types, because PERK depletion only slightly reduced ATF4 protein levels and did not affect the expression of the ATF4 target gene ATF3 in ER-stressed SH-SY5Y neuroblastoma cells, despite efficient down-modulation of p-eIF2α by PERK knockdown (72). We observed even more extreme differences among the cell lines we tested; the TM-induced up-regulation of ATF4 protein levels was completely dependent on PERK in PC3 cells but only partially dependent on PERK in RPE-1 cells, whereas ATF4 was up-regulated independently of PERK in TM-treated LNCaP and HeLa cells. It has been noted that PERK transduces signals also via transcriptional mediators other than ATF4, because PERK alters the expression of many more genes than those altered by ATF4 in TM-treated MEFs (73) as well as in hepatocytes from TM-treated mice (74). Here, we demonstrate that PERK-initiated signal transduction is completely different in LNCaP cells. Not only did interference with PERK fail to affect TM-induced elevation of ATF4 protein expression, but additionally, our RNA-Seq experiments showed that whereas ATF4 altered the expression of hundreds of genes, interference with PERK had negligible effects on gene transcription. This is in sharp contrast to what would have been expected if PERK transduced signals to activate downstream transcription factor(s). Thus, the cellular effects of PERK are mediated independently of ATF4 and most likely also independently of transcriptional changes in TM-treated LNCaP cells. This revelation enabled us to uncover a completely novel role of PERK in autophagy regulation. Thus, we could pinpoint that PERK acts at a post-sequestration step in the autophagic pathway, in a manner that is essential for TM-induced autophagy and that is not mediated through ATF4 or transcriptional changes. Our kinetic experiments with PERKi indicated that PERK activity is required during the first 18 h of TM treatment for TM to enhance autophagy, but interestingly, in the 18–24-h time period, PERK activity was not needed for autophagy to proceed. Taken together, we propose that a PERK-mediated signaling pathway, which remains to be identified, leads to post-translational modifications of proteins that act at a post-sequestration step in the autophagic pathway. Interestingly, it was recently shown that PERK can activate MKK4 and thereby p38 MAPK and LAMP2A on lysosomes, leading to enhanced chaperone-mediated autophagy (CMA), a distinct form of autophagy that involves direct import of cytosolic proteins into lysosomes (75). However, this mechanism likely does not explain the reduced autophagic protein degradation that we observed in PERK-depleted cells, because in that case, PERK silencing would have reduced long-lived protein degradation also in ULK1/2- and ATG13-depleted cells (as ULK1/2 and ATG13 are dispensable for CMA). Nevertheless, our study and the above-mentioned (75) collectively reveal a previously unrecognized and central role of PERK in regulating macroautophagic and CMA-mediated degradation of cellular proteins in an ATF4- and transcription-independent manner. Preclinical and clinical evidence indicate a broad potential for the use of pharmacological modulators of PERK activity in the treatment of a variety of pathologies, including cancer, diabetes, and neurodegenerative diseases (76). Thus, the novel function of PERK in autophagy regulation that we describe here will be highly relevant to study further and to take into account when assessing clinical treatment modalities that aim to modulate PERK activity.
Although we in the current study have analyzed effects of ER stress with general autophagy methods (and not methods that specifically detect selective autophagy), and we have demonstrated that TM-initiated ER stress–induced autophagy includes the sequestration of nonselective cargo, we consider it very likely that ER stress also induces selective autophagy. In fact, nonselective autophagy may very often reflect “bystander autophagy” that occurs during selective autophagy (77). The ER stressor DTT has been reported to induce selective autophagy of the ER (“ER-phagy”) in yeast (78, 79) as well as in mammalian cells (80), the latter requiring a novel ER-phagy–specific receptor, CCPG1 (80). Moreover, ER-phagy was elegantly shown to occur upon recovery from treatment with the ER stressor cyclopiazonic acid, in a manner that required the translocon component Sec62, acting as an autophagy receptor (81). It remains to be determined whether TM-induced ER stress activates ER-phagy. Importantly, it also remains to be determined whether ER-phagy under ER stress conditions is mediated by the UPR or not. ER-phagy occurred in the absence of UPR upon overexpression of Sec62 in mammalian cells (81) and independently of the UPR upon overexpression of a single integral-membrane protein in yeast, even though the overexpression induced UPR (82). Moreover, ER-phagy takes place at a constitutive level in mammalian cells (83) and yeast (82), as well as upon starvation or treatment with mTOR inhibitors in mammalian cells (80, 83, 84) and yeast (85–87). Several lines of evidence indicate that ER-phagy is important for resolving ER stress (79, 81, 85, 88). However, evidence is lacking as to whether it is the UPR itself that is activating ER-phagy. The GABARAP-interacting ER-phagy receptor CCPG1 was shown to be induced at the transcriptional level by ER stressors (80), but it was not determined whether this was mediated by the UPR. Interestingly, our RNA-Seq data show that CCPG1 was strongly induced by TM in LNCaP cells in an ATF4-dependent manner (Tables S3, S7, and S8). Also the expression of another recently described ER-phagy receptor, RTN3 (84), was up-regulated by TM in an ATF4-dependent manner (Tables S3, S7, and S8). This suggests that the UPR may indeed stimulate ER-phagy, in a manner that involves ATF4-mediated transcriptional up-regulation of ER-phagy receptors. It will be important to follow up these and additional aspects of ER stress–induced autophagy in future studies.
In the current study, we used the classical ER stressor TM to study how the UPR affects autophagy. The effect of TM on autophagy was phenocopied by knockdown of the specific target of TM, GPT. Moreover, TM induced a full-blown UPR, and the UPR components PERK and ATF4 were essential for TM-induced autophagy. Thus, the effects observed with TM in the current study are highly likely to be specifically mediated via GPT inhibition, which leads to accumulation of unfolded proteins in the ER (ER stress) and induction of the UPR. Although TM-induced autophagy clearly required the UPR, the role of the UPR in regulating the autophagic process under other ER stress–inducing conditions remains to be determined. In particular, it will be important to understand how other factors in the cell affect the ability of the UPR to induce autophagy (e.g. in the context of cells or tissues treated with therapeutic drugs that induce ER stress). We have previously shown that although the ER stressors thapsigargin (an ER Ca2+ pump inhibitor from which promising tumor-targeting pro-drugs have been formulated (89)) and A23187 (a calcium ionophore) induce the UPR and increase LC3-II levels in LNCaP and U20S cancer cells, they both block general autophagy due to depletion of intraluminal ER Ca2+ and in a manner that is not regulated by the UPR (34). The inhibitory effects of ER Ca2+ depletion are thus dominant over the autophagy-stimulatory effects of the UPR. Recently, we have proceeded to examine the effects of 2-deoxyglucose (2-DG) on autophagy. 2-DG blocks glycolysis and may have potential in anti-cancer therapy (90). 2-DG is structurally similar to mannose and therefore interferes with oligosaccharide synthesis, leading to aberrant N-linked glycosylation (91), ER stress, and UPR induction (92). Based on monitoring increases in LC3-II levels and LC3 puncta, 2-DG has been suggested to activate autophagy via ER stress (93). We have found that even though 2-DG activated the UPR in LNCaP cells (94) and enhanced cellular LC3-II levels, it did not increase autophagic protein degradation (data not shown). The lack of autophagy induction was not due to induction of cell death by 2-DG. Rather, we believe the reason for the lack of UPR-induced autophagy in 2-DG–treated cells may be the decrease in cellular ATP levels that 2-DG causes via inhibition of glycolysis (93), as general autophagy is an energy-requiring process (95) that is positively regulated by glucose-derived ATP production (96). These examples illustrate how the effect of UPR on autophagy can be regulated by other cellular factors or processes. In future studies, it will be important to broadly address how autophagy is affected by different ER stress–inducing conditions and to determine how various cellular factors, processes, and signaling pathways can either restrict or amplify UPR-induced autophagy.
In summary, our study has revealed a number of novel and important relationships between ER stress/UPR and autophagy in mammalian cells. (i) TM-induced ER stress activates autophagy in a manner that does not appear to require LC3. This suggests that the implications of ER stress–induced modulation of LC3 should be re-examined. ER stress appears to affect LC3 expression predominantly via ATF4-dependent stimulation of LC3 transcription. (ii) ER stress induces a canonical form of macroautophagy, which depends on ULK1/2, ATG13, and GABARAP proteins. (iii) PERK and ATF4 are essential for TM-initiated ER stress–induced autophagy. In contrast, IRE1 acts as a negative regulator of autophagy under both ER stress–inducing and basal conditions and in a manner that likely does not require IRE1 activation. (iv) ATF4 has functional activity also under basal conditions, which does not appear to result from UPR, and which is partially required for basal autophagy in LNCaP cells. (v) ATF4 and PERK play distinct roles in TM-initiated ER stress–induced autophagy, with ATF4 being essential for autophagosome formation and PERK being essential at a post-sequestration step in the autophagic pathway. These ER stress–induced events drive the autophagic process all the way to completion (i.e. to the final stage of degradation and release of the sequestered autophagic cargo).
Experimental procedures
Cell culture
LNCaP (CRL-1740) and PC3 (CRL-1435) cells were obtained from ATCC. LNCaP cells were used at passage numbers below 30. RPE-1 and HeLaT cells were kind gifts from Harald Stenmark (Oslo, Norway) and Jan Parys (Leuven, Belgium), respectively. All cell lines were cultured in RPMI 1640 (Gibco 21875) supplemented with 10% fetal bovine serum (FBS; Sigma F7524, batch BCBT0730) (“complete medium”) and 5% CO2 at 37 °C.
Chemicals and cell treatments
Tunicamycin (Sigma, T7765) was dissolved in DMSO to a 5 mg/ml stock solution and, unless otherwise stated, used at a final concentration of 2.5 μg/ml in LNCaP and RPE-1 and 1 μg/ml in HeLaT and PC3 cells. Bafilomycin A1 (Enzo, BML-CM110) was dissolved in DMSO to a 0.2 mm stock solution and used at a final concentration of 50 nm when used for 4–6 h and 100 nm when used for 3 h. Torin1 (Tocris, 4247) was dissolved in DMSO to a 0.5 mm stock solution and used at a final concentration of 50 nm. GSK2606414 (“PERKi”), a kind gift from Dr. Jeffrey M. Axten (GlaxoSmithKline), was dissolved in DMSO to a 10 mm stock solution and used at a final concentration of 100 nm. MG132 (Calbiochem, 474790) was dissolved in DMSO to 5 mm and used at 5 μm. Kifunensine (Toronto Research Chemicals Inc., K450000) was dissolved in H2O to 1 mg/ml and used at 10 μg/ml. Eeyarestatin I (Merck-Millipore, 324521) was dissolved in DMSO to 8 mm and used at 8 μm. A23187 and thapsigargin (Sigma, C7522 and T9033, respectively) were dissolved in DMSO to 10 and 1 mm stock solutions and used at the indicated final concentrations.
Cell death measurements
Cell death was determined as described previously (97, 98). Briefly, LNCaP cells were seeded in poly-d-lysine–coated 96-well plates 2 days prior to treatment, when cells received complete medium supplemented with thapsigargin or tunicamycin in combination with 2.5 μg/ml propidium iodide, and were subjected to live-cell fluorescence imaging using an IncuCyte ZOOM instrument (Essen Bioscience). Multiple pictures were taken every 3 h, and cell death was analyzed as the ratio of red fluorescence confluence to total cell confluence (based on phase-contrast images), as determined by the IncuCyte software algorithms.
Measurement of endoplasmic reticulum Ca2+ levels
To monitor ER calcium changes directly, we used the genetically encoded Ca2+ indicator G-CEPIA1er (99). The plasmid was introduced to LNCaP cells using the GenJetTM In Vitro DNA Tranfection Reagent (Signagen) according to the manufacturer's protocol, followed by live-cell imaging using the IncuCyte ZOOM to analyze the intensity of the green fluorescence signal. Relative ER Ca2+ content was plotted as a function of total green intensity, starting directly after the addition of the indicated treatments (tunicamycin or thapsigargin). At the end of the experiment, the calcium ionophore A23187 was added to determine the level of green fluorescence in cells with fully depleted ER Ca2+. All values were normalized to the values measured after the addition of A23187.
LDH sequestration assay
LDH sequestration was determined as described previously (38, 39), with slight modifications. All experiments were performed in complete medium, and DMSO or TM were present throughout the treatment period (0–24 h), whereas Torin1 or Baf was included only during the last 3 h (21–24 h). Cells were harvested in trypsin-EDTA (0.25%) and collected in complete medium. After centrifugation at 300 × g for 5 min at 4 °C, the supernatant was aspirated, and cells were resuspended in 400 μl of isotonic sucrose (10%). To selectively disrupt the plasma membrane, cells were subjected to an electric pulse (2000 V and 1.2 microfarads in a 1 × 1 × 5-cm electrode chamber) with our homemade apparatus and subsequently mixed with 400 μl of phosphate-buffered sucrose (100 mm sodium monophosphate, 2 mm DTT, 2 mm EDTA, and 1.75% sucrose, pH 7.5) to a total volume of 800 μl. 140 μl of cell disruptate was removed for total LDH measurements (“LDHTotal”) and was stored overnight at −80 °C. 600 μl of cell disruptate was resuspended in 900 μl of resuspension buffer (50 mm sodium monophosphate, 1 mm EDTA, 1 mm DTT) supplemented with 0.5% BSA and 0.01% Tween 20. Cell corpses, containing autophagic vacuoles, were sedimented by centrifugation at 18,000 × g for 45 min at 4 °C. Supernatant was aspirated, and the pellet (“LDHSediment”) was stored overnight at −80 °C. The following day, both LDHTotal and LHDSediment were diluted in resuspension buffer supplemented with Triton X-405 (Sigma) to a final concentration of 1%. After a short spin (18,000 × g for 10 min at 4 °C), the enzymatic activity of LDH in LDHTotal and LHDSediment was measured as described previously (39) using homemade reagents, mixing 4 volumes of 65 mm imidazole (pH 7.5), 0.75 mm pyruvate with one volume of 65 mm imidazol (pH 7.5), 1.8 mm NADH. LDH sequestration activity was calculated as percentage of sedimentable LDH in experimentally treated cells minus percentage of sedimentable LDH in untreated cells (background), divided by the incubation time with Baf. For a detailed description of the protocol, see Ref. 39.
LLPD assay
LLPD was determined as described previously (49). Cells were seeded in 24-well plates in 0.5 ml of RPMI 1640, 10% FBS (complete medium) containing 0.1 μCi/ml 14C-labeled l-valine (Vitrax, VC 308) and incubated for 2 days. Unincorporated radioactivity was removed by washing each well with 0.5 ml of complete medium containing 10 mm nonradioactive (“cold”) l-valine (Sigma, V0513) (CML). Subsequently, cells were chased for 18 h (or 1 h in the GPT knockdown experiments) in the absence or presence of treatments in 0.5 ml of CML. Short-lived proteins were washed out with 0.5 ml of CML, followed by incubation in 0.25 ml of CML containing various treatments for another 4–6 h. For harvesting, the plates were cooled down on ice for 2 min. Each well then received 50 μl of ice-cold PBS, 2% BSA (Sigma) and subsequently 200 μl of ice-cold 25% TCA (Sigma), followed by overnight shaking at 4 °C. The following day, the solution from each well was transferred to an Eppendorf tube and centrifuged at 5000 × g for 10 min at 4 °C. The supernatant (the TCA-soluble fraction) was transferred to a scintillation tube and mixed with 4 ml of Opti-Fluor (PerkinElmer Life Sciences) by rigorous vortexing. The TCA-insoluble fraction that remained in the tubes (pellet) and in the wells each received 250 μl of 0.2 m KOH to dissolve the precipitated protein. Tubes were rotated, and the plates were agitated (on a shaker) for 1 h at room temperature. Dissolved proteins from the tubes, and the plate were merged and transferred to a scintillation tube and mixed with 4 ml of Opti-Fluor by rigorous vortexing. Radioactivity was determined in the TCA-soluble (containing 14C-labeled l-valine from degraded protein) and -insoluble fractions by liquid scintillation counting. The degradation rate for long-lived proteins was calculated as the percentage of radioactivity in the TCA-soluble fraction relative to the total radioactivity in the TCA-soluble and nonsoluble fractions, divided by the post-chasing incubation time.
Chymotrypsin-like activity
Cells were seeded in 96-well plates and allowed to settle for 1 day before adding the treatments. Proteasome Glo Reagent (Promega) was prepared and administered according to the manufacturer's protocol, and after 15 min, luminescence was measured with a plate reader (Infinite F200 Pro, Tecan). The amount of luminescence is directly proportional to chymotrypsin-like activity.
Generation of mTagRFP-mWasabi-LC3–expressing LNCaP cell line
LNCaP cells stably expressing the tandem fluorescent LC3 reporter mTagRFP-mWasabi-LC3 were generated by lentiviral transduction. The plasmid encoding mTagRFP-mWasabi-LC3 was a kind gift from Prof. Jian Lin (Peking University, China) (35), and the probe sequence was subcloned into a Gateway ENTRY vector by standard molecular biology techniques. From this vector, a lentiviral transfer vector was generated by recombination with pCDH-EF1a-GW-IRES-BSD (a gateway-enabled derivative of pCDH-EF1a-MCS-IRES-Puro (Systems Biosciences). Lentivirus particles were packaged using a third-generation packaging system (Addgene plasmid numbers 12251, 12253, and 12259) as described previously (100, 101). Cells were then transduced with low virus titers, and stable cell pools were generated by selection with blasticidin (3 μg/ml).
Live-cell confocal fluorescence microscopy
LNCaP cells stably expressing mTagRFP-mWasabi-LC3 were seeded in 8-well Lab-Tek chamber slides (VWR 734-2062) at a density of 4 × 104 cells/well 2 days prior to experiments. The cells were treated with TM (2.5 μg/ml, 24 h) or Torin 1 (50 nm, 3 h) in the absence or presence of Baf (100 nm) for the final 3 h. Subsequently, the cells were immediately imaged using a Zeiss LSM780 laser-scanning confocal microscope (Carl Zeiss MicroImaging LLC) equipped with an arrgon laser multiline (458/488/514 nm) and a DPSS-561 10 laser (561 nm). The objective used was a Zeiss plan-apochromat ×63/1.40 oil DIC M27. Images were acquired using ZEN 2010 software (Carl Zeiss MicroImaging) and processed by ImageJ (National Institutes of Health).
Flow cytometry
LNCaP cells stably expressing mTagRFP-mWasabi-LC3 were seeded in 12-well plates at a density of 3 × 105 cells/well 2 days prior to experiments. The cells were treated with TM (2.5 μg/ml, 24 h) or Torin 1 (50 nm, 3 h) in the absence or presence of Baf (100 nm) for the final 3 h. Cells were detached by Accutase (Sigma A6964) and washed in PBS plus 0.5% FBS. To selectively measure membrane-conjugated mTagRFP-mWasabi-LC3, cells were depleted for unconjugated, cytosolic mTagRFP-mWasabi-LC3 by gentle plasma membrane permeabilization with digitonin (30 μg/ml, 3 min, room temperature), followed by one wash with PBS plus 0.5% FBS. Signal intensities of mTagRFP and mWasabi were acquired (from 10,000 events for each condition in each experiment) by an LSR II flow cytometer (BD Biosciences) using the 561-nm (45-milliwatt) and 488-nm (100-milliwatt) laser lines and the FACS Diva software (BD Biosciences). The data were processed by FlowJo software.
Quantitative real-time RT-PCR
RNA isolation, cDNA synthesis, and RT-PCR were carried out as described previously (34), with slight modifications. RNA was isolated by ReliaPrep RNA Miniprep Systems (Promega) according to the manufacturer's protocol and reverse-transcribed to cDNA using SuperScript VILO Master Mix (Applied Biosystems). In a 96-well plate, 9 ng of cDNA was mixed with 0.5 μl of TaqMan probe (Applied Biosystems) and 5 μl of TaqMan Fast Advanced Master Mix (Applied Biosystems). PCR amplification was performed in duplicate or triplicate series using the ABI 7900HT FAST Sequence Detection System (Applied Biosystems). The cycling conditions were 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. Transcript levels relative to those in DMSO control samples were determined using the comparative Ct method (102) and normalization to the geometric mean Ct value of GAPDH and TBP (103). The following TaqMan gene expression assays (Applied Biosystems) were used: ATF4 (Hs00909568_g1), ATF6 (Hs00232586_m1), ATG13 (Hs00207186_m1), GPT (Hs00609750_g1), GABARAP (Hs00925899_g1), GABARAPL1 (Hs00740588_mH), GABARAPL2 (Hs00371854_m1), GAPDH (Hs99999905_m1), IRE1 (Hs00176385_m1), MAP1LC3A1 (Hs00261291_m1), MAP1LC3A2 (Hs00738808_m1), MAP1LC3B (Hs00797944_s1), PERK (Hs00984003_m1), TBP (Hs99999910_m1), ULK1 (Hs01124636_m1), ULK2 (Hs00979043_m1), and WIPI1 (Hs00924447_m1).
RNA-Seq and analysis
LNCaP cells were seeded in 12-well plates; reverse-transfected with siCtrl, siPERK-1, siPERK-2, siATF4–1, or siATF4–2 for 2 days; and subjected to various treatments for 18 h. Total RNA was isolated using QIAshredder (Qiagen) in combination with an RNeasy Mini Kit (Qiagen), with on-column DNase digestion according to the manufacturer's protocol. RNA integrity was assessed using an RNA 6000 Nano Kit (Agilent Technologies) on a Bioanalyzer (Agilent Technologies). All 28 samples submitted for sequencing (four independent experiments with seven conditions per experiment) had RNA integrity ≥ 9.7. Raw reads were aligned to the human genome (hg19), and splice junctions were mapped using TopHat version 2.1.0 (104) with default parameters. Gene-based read counts were obtained using featureCount version 1.5.0-p1 (105), and DESeq2 version 1.10.1 The Bioconductor package (106) was used for normalization and statistical evaluation with negative binomial model between conditions on the count data. We obtained an alignment rate in the range of 88.5–93.0% (Table S2). All treatment groups showed excellent biclustering across the four independent experiments (Fig. S8). The differential gene expression analyses performed in Fig. 6 (C and D) and Table S1 are based on Tables S3–S8.
SDS-PAGE and immunoblot analysis
Preparation of whole-cell lysates, SDS-PAGE, and immunoblot analysis was performed as described previously (34). Primary antibodies against the following proteins were used: α-tubulin (Abcam ab7291), ATF4 (Cell Signaling Technology (CST), 11815), ATG13 (CST, 13468), p-ATG13 (Rockland, 600-401-C49), BiP (CST, 3177), CHOP (CST, 2895), p-eIF2α (CST, 3398), GABARAP (MBL International Corp., PM037), GABARAPL1 (Abcam, ab86497), GABARAPL2 (MBL International, PM038), IRE1 (CST, 3294), LC3 (CST, 2775), PERK (CST, 3192), p-S6K (CST, 9205), ULK1 (CST, 8054), p-ULK1 (CST, 6888), WIPI1 antiserum (a kind gift from Dr. Tassula Proikas-Cezanne, Eberhard Karls University Tuebingen), and XBP1s (BioLegend, 647502, clone 143F). Secondary, horseradish peroxidase–conjugated goat anti-rabbit (Dako) and rabbit anti-mouse (Dako) antibodies were used at 1:5000 dilutions.
siRNA transfection
siRNA transfections were carried out as described previously (34), with slight modifications. Cells were reverse-transfected using Opti-MEM Reduced Serum Medium (Gibco) and Lipofectamine RNAiMax (Invitrogen) with a final siRNA concentration of 10 nm per siRNA, unless otherwise stated. Experiments were initiated 2 days after transfection, except for GPT knockdown, which was initiated 3 days after transfection. The following siRNAs were used (all Silencer® Select siRNAs from Ambion): Silencer® Select Negative Control #1 (4390843), siATF4-1 (s1704), siATF4-2 (s1702), siATF4–3 (1703), siATF6-1 (s223544), siATF6-2 (s223545), siATG13 (s18879), siGPT-1 (s4242), siGPT-2 (s4243), siPERK-1 (s18101), siPERK-2 (s18103), siIRE1-1 (s200430), siIRE1–2 (s200431), siGABARAP (s22362), siGABARAPL1 (s24333), siGABARAPL2 (s22387), siMAP1LC3A (s39157), siMAP1LC3B (s224886), siULK1 (s15964), siULK2 (s18706), and siWIPI1 (s30082).
Statistical analyses
For all experiments where tests of significance were performed, approximate normal distribution of the data was assumed. “t test” denotes Student's t test and was carried out to compare the means of two groups. Paired and unpaired sample t tests were employed for paired and unpaired samples, respectively. When more than two means were compared, ANOVA was performed. One-way ANOVA with Tukey post hoc test was employed when samples were grouped by one factor. Two-way ANOVA with Holm–Sidak post hoc test was employed when samples were grouped by two factors. Repeated measures ANOVA and regular ANOVA were employed for paired and unpaired samples, respectively. All statistical tests were performed using GraphPad Prism. The number of independent experiments performed is denoted by n. S.E. denotes the variation between values from at least three independent experiments (n ≥ 3), whereas S.D. denotes the variation between values of biological or technical replicates within one representative experiment. The statistical analyses of the RNA-Seq data are described under “RNA-Seq and analysis.”
Data access
The RNA-Seq data are available under GEO accession number GSE108212. The raw differential gene expression data used to perform the analyses in Fig. 6 (C and D) are included in Tables S3–S8.
Author contributions
M. L. and N. E. conceptualization; M. L., M. L. T., P. S., A. H., A. B., J. S., and N. E. data curation; M. L., M. L. T., P. S., A. H., A. B., J. S., and N. E. formal analysis; M. L., M. L. T., P. S., and N. E. investigation; M. L., M. L. T., P. S., A. B., and N. E. methodology; M. L. and N. E. writing-original draft; M. L., M. L. T., P. S., A. H., A. B., J. S., and N. E. writing-review and editing; J. S. and N. E. supervision; N. E. funding acquisition; N. E. project administration.
Supplementary Material
Acknowledgments
We acknowledge Georgios Magklaras and his computing team at NCMM for IT support; Alphonso Urbanucci for discussions on the experimental set-up for the RNA-Seq experiments; and Frank Sætre, Ulrikke Dahl Brink, and Linn F. Kymre for technical support.
This work was supported by grants from the Research Council of Norway (Project 230686) and the University of Oslo, the Nansen Foundation, the Anders Jahre Foundation, and the Legacy in memory of Henrik Homan (to N. E.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S8 and Table S1–S8.
The RNA-Seq data are available under GEO accession number GSE108212.
N. Engedal, unpublished results.
- ER
- endoplasmic reticulum
- ANOVA
- analysis of variance
- ATG
- autophagy-related
- A23
- A23187
- Baf
- bafilomycin A1
- CMA
- chaperone-mediated autophagy
- LDH
- lactate dehydrogenase
- LLPD
- long-lived protein degradation
- MEF
- mouse embryonic fibroblast
- PERKi
- PERK inhibitor (GSK2606414)
- TG
- thapsigargin
- TM
- tunicamycin
- UPR
- unfolded protein response
- XBP1s
- spliced XBP1
- p-
- phosphorylated
- ERAD
- ER-associated degradation
- GPT
- GlcNAc-1-phosphate transferase
- FC
- -fold change
- PTEN
- phosphatase and tensin homolog
- 2-DG
- 2-deoxyglucose
- FBS
- fetal bovine serum
- CST
- cell signaling technology
- padj
- adjusted p value.
References
- 1. Hetz C., and Papa F. R. (2018) The unfolded protein response and cell fate control. Mol. Cell. 69, 169–181 10.1016/j.molcel.2017.06.017 [DOI] [PubMed] [Google Scholar]
- 2. Yorimitsu T., Nair U., Yang Z., and Klionsky D. J. (2006) Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281, 30299–30304 10.1074/jbc.M607007200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bento C. F., Renna M., Ghislat G., Puri C., Ashkenazi A., Vicinanza M., Menzies F. M., and Rubinsztein D. C. (2016) Mammalian autophagy: how does it work? Annu. Rev. Biochem. 85, 685–713 10.1146/annurev-biochem-060815-014556 [DOI] [PubMed] [Google Scholar]
- 4. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., and Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bestebroer J., V'kovski P., Mauthe M., and Reggiori F. (2013) Hidden behind autophagy: the unconventional roles of ATG proteins. Traffic 14, 1029–1041 10.1111/tra.12091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cadwell K., and Debnath J. (2018) Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol. 217, 813–822 10.1083/jcb.201706157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanson H. H., Kang S., Fernández-Monreal M., Oung T., Yildirim M., Lee R., Suyama K., Hazan R. B., and Phillips G. R. (2010) LC3-dependent intracellular membrane tubules induced by γ-protocadherins A3 and B2: a role for intraluminal interactions. J. Biol. Chem. 285, 20982–20992 10.1074/jbc.M109.092031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramkumar A., Murthy D., Raja D. A., Singh A., Krishnan A., Khanna S., Vats A., Thukral L., Sharma P., Sivasubbu S., Rani R., Natarajan V. T., and Gokhale R. S. (2017) Classical autophagy proteins LC3B and ATG4B facilitate melanosome movement on cytoskeletal tracks. Autophagy 13, 1331–1347 10.1080/15548627.2017.1327509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Subramani S., and Malhotra V. (2013) Non-autophagic roles of autophagy-related proteins. EMBO Rep. 14, 143–151 10.1038/embor.2012.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weidberg H., Shvets E., Shpilka T., Shimron F., Shinder V., and Elazar Z. (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 29, 1792–1802 10.1038/emboj.2010.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pankiv S., Alemu E. A., Brech A., Bruun J. A., Lamark T., Overvatn A., Bjørkøy G., and Johansen T. (2010) FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 188, 253–269 10.1083/jcb.200907015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Itoh T., Kanno E., Uemura T., Waguri S., and Fukuda M. (2011) OATL1, a novel autophagosome-resident Rab33B-GAP, regulates autophagosomal maturation. J. Cell Biol. 192, 839–853 10.1083/jcb.201008107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., and Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- 14. Rogov V., Dötsch V., Johansen T., and Kirkin V. (2014) Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 53, 167–178 10.1016/j.molcel.2013.12.014 [DOI] [PubMed] [Google Scholar]
- 15. Kouroku Y., Fujita E., Tanida I., Ueno T., Isoai A., Kumagai H., Ogawa S., Kaufman R. J., Kominami E., and Momoi T. (2007) ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 14, 230–239 10.1038/sj.cdd.4401984 [DOI] [PubMed] [Google Scholar]
- 16. Sakaki K., Wu J., and Kaufman R. J. (2008) Protein kinase Cθ is required for autophagy in response to stress in the endoplasmic reticulum. J. Biol. Chem. 283, 15370–15380 10.1074/jbc.M710209200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim K. W., Moretti L., Mitchell L. R., Jung D. K., and Lu B. (2010) Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2α in caspase-3/7-deficient cells. Oncogene 29, 3241–3251 10.1038/onc.2010.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiu W., Zhang J., Dekker M. J., Wang H., Huang J., Brumell J. H., and Adeli K. (2011) Hepatic autophagy mediates endoplasmic reticulum stress-induced degradation of misfolded apolipoprotein B. Hepatology 53, 1515–1525 10.1002/hep.24269 [DOI] [PubMed] [Google Scholar]
- 19. Hart L. S., Cunningham J. T., Datta T., Dey S., Tameire F., Lehman S. L., Qiu B., Zhang H., Cerniglia G., Bi M., Li Y., Gao Y., Liu H., Li C., Maity A., et al. (2012) ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 122, 4621–4634 10.1172/JCI62973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ogata M., Hino S., Saito A., Morikawa K., Kondo S., Kanemoto S., Murakami T., Taniguchi M., Tanii I., Yoshinaga K., Shiosaka S., Hammarback J. A., Urano F., and Imaizumi K. (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231 10.1128/MCB.01453-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castillo K., Rojas-Rivera D., Lisbona F., Caballero B., Nassif M., Court F. A., Schuck S., Ibar C., Walter P., Sierralta J., Glavic A., and Hetz C. (2011) BAX inhibitor-1 regulates autophagy by controlling the IRE1α branch of the unfolded protein response. EMBO J. 30, 4465–4478 10.1038/emboj.2011.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Margariti A., Li H., Chen T., Martin D., Vizcay-Barrena G., Alam S., Karamariti E., Xiao Q., Zampetaki A., Zhang Z., Wang W., Jiang Z., Gao C., Ma B., Chen Y. G., et al. (2013) XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J. Biol. Chem. 288, 859–872 10.1074/jbc.M112.412783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaufman D. R., Papillon J., Larose L., Iwawaki T., and Cybulsky A. V. (2017) Deletion of inositol-requiring enzyme-1α in podocytes disrupts glomerular capillary integrity and autophagy. Mol. Biol. Cell 28, 1636–1651 10.1091/mbc.e16-12-0828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shimodaira Y., Takahashi S., Kinouchi Y., Endo K., Shiga H., Kakuta Y., Kuroha M., and Shimosegawa T. (2014) Modulation of endoplasmic reticulum (ER) stress-induced autophagy by C/EBP homologous protein (CHOP) and inositol-requiring enzyme 1α (IRE1α) in human colon cancer cells. Biochem. Biophys. Res. Commun. 445, 524–533 10.1016/j.bbrc.2014.02.054 [DOI] [PubMed] [Google Scholar]
- 25. Rouschop K. M., van den Beucken T., Dubois L., Niessen H., Bussink J., Savelkouls K., Keulers T., Mujcic H., Landuyt W., Voncken J. W., Lambin P., van der Kogel A. J., Koritzinsky M., and Wouters B. G. (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Invest. 120, 127–141 10.1172/JCI40027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rzymski T., Milani M., Pike L., Buffa F., Mellor H. R., Winchester L., Pires I., Hammond E., Ragoussis I., and Harris A. L. (2010) Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 29, 4424–4435 10.1038/onc.2010.191 [DOI] [PubMed] [Google Scholar]
- 27. B'chir W., Maurin A. C., Carraro V., Averous J., Jousse C., Muranishi Y., Parry L., Stepien G., Fafournoux P., and Bruhat A. (2013) The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699 10.1093/nar/gkt563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang J., Kang R., Huang H., Xi X., Wang B., Wang J., and Zhao Z. (2014) Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 10, 766–784 10.4161/auto.27954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pike L. R., Singleton D. C., Buffa F., Abramczyk O., Phadwal K., Li J. L., Simon A. K., Murray J. T., and Harris A. L. (2013) Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J. 449, 389–400 10.1042/BJ20120972 [DOI] [PubMed] [Google Scholar]
- 30. Sharma M., Bhattacharyya S., Sharma K. B., Chauhan S., Asthana S., Abdin M. Z., Vrati S., and Kalia M. (2017) Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 98, 1027–1039 10.1099/jgv.0.000792 [DOI] [PubMed] [Google Scholar]
- 31. García-Navas R., Munder M., and Mollinedo F. (2012) Depletion of l-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy 8, 1557–1576 10.4161/auto.21315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pattingre S., Bauvy C., Carpentier S., Levade T., Levine B., and Codogno P. (2009) Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 284, 2719–2728 10.1074/jbc.M805920200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klionsky D. J., Abdelmohsen K., Abe A., Abedin M. J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C. M., Adams P. D., Adeli K., Adhihetty P. J., Adler S. G., Agam G., Agarwal R., Aghi M. K., et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Engedal N., Torgersen M. L., Guldvik I. J., Barfeld S. J., Bakula D., Sætre F., Hagen L. K., Patterson J. B., Proikas-Cezanne T., Seglen P. O., Simonsen A., and Mills I. G. (2013) Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 9, 1475–1490 10.4161/auto.25900 [DOI] [PubMed] [Google Scholar]
- 35. Zhou C., Zhong W., Zhou J., Sheng F., Fang Z., Wei Y., Chen Y., Deng X., Xia B., and Lin J. (2012) Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 8, 1215–1226 10.4161/auto.20284 [DOI] [PubMed] [Google Scholar]
- 36. Bowman E. J., Siebers A., and Altendorf K. (1988) Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. U.S.A. 85, 7972–7976 10.1073/pnas.85.21.7972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K. J., Coppes R. P., Engedal N., Mari M., and Reggiori F. (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455 10.1080/15548627.2018.1474314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luhr M., Szalai P., and Engedal N. (2018) The lactate dehydrogenase sequestration assay: a simple and reliable method to determine bulk autophagic sequestration activity in mammalian cells. J. Vis. Exp. 137, e57971 10.3791/57971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luhr M., Szalai P., Sætre F., Gerner L., Seglen P. O., and Engedal N. (2017) A simple cargo sequestration assay for quantitative measurement of nonselective autophagy in cultured cells. Methods Enzymol. 587, 351–364 10.1016/bs.mie.2016.09.064 [DOI] [PubMed] [Google Scholar]
- 40. Seglen P. O., Luhr M., Mills I. G., Sætre F., Szalai P., and Engedal N. (2015) Macroautophagic cargo sequestration assays. Methods 75, 25–36 10.1016/j.ymeth.2014.12.021 [DOI] [PubMed] [Google Scholar]
- 41. Szalai P., Hagen L. K., Sætre F., Luhr M., Sponheim M., Øverbye A., Mills I. G., Seglen P. O., and Engedal N. (2015) Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp. Cell Res. 333, 21–38 10.1016/j.yexcr.2015.02.003 [DOI] [PubMed] [Google Scholar]
- 42. Saetre F., Hagen L. K., Engedal N., and Seglen P. O. (2015) Novel steps in the autophagic-lysosomal pathway. FEBS J. 282, 2202–2214 10.1111/febs.13268 [DOI] [PubMed] [Google Scholar]
- 43. Engedal N., and Seglen P. O. (2016) Autophagy of cytoplasmic bulk cargo does not require LC3. Autophagy 12, 439–441 10.1080/15548627.2015.1076606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nguyen T. N., Padman B. S., Usher J., Oorschot V., Ramm G., and Lazarou M. (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215, 857–874 10.1083/jcb.201607039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vaites L. P., Paulo J. A., Huttlin E. L., and Harper J. W. (2018) Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/MON1 regulator C18orf8/RMC1 in macro and selective autophagic flux. Mol. Cell. Biol. 38, e00392–17 10.1128/MCB.00392-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kopitz J., Kisen G. O., Gordon P. B., Bohley P., and Seglen P. O. (1990) Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol. 111, 941–953 10.1083/jcb.111.3.941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bauvy C., Meijer A. J., and Codogno P. (2009) Assaying of autophagic protein degradation. Methods Enzymol. 452, 47–61 10.1016/S0076-6879(08)03604-5 [DOI] [PubMed] [Google Scholar]
- 48. Klionsky D. J., Cuervo A. M., and Seglen P. O. (2007) Methods for monitoring autophagy from yeast to human. Autophagy 3, 181–206 10.4161/auto.3678 [DOI] [PubMed] [Google Scholar]
- 49. Luhr M., Sætre F., and Engedal N. (2018) The long-lived protein degradation assay: an efficient method for quantitative determination of the autophagic flux of endogenous proteins in adherent cell lines. Bio-Protocol 8, e2836 10.21769/BioProtoc.2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kjos I., Borg Distefano M., Sætre F., Repnik U., Holland P., Jones A. T., Engedal N., Simonsen A., Bakke O., and Progida C. (2017) Rab7b modulates autophagic flux by interacting with Atg4B. EMBO Rep. 18, 1727–1739 10.15252/embr.201744069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pattingre S., Petiot A., and Codogno P. (2004) Analyses of Gα-interacting protein and activator of G-protein-signaling-3 functions in macroautophagy. Methods Enzymol. 390, 17–31 10.1016/S0076-6879(04)90002-X [DOI] [PubMed] [Google Scholar]
- 52. Fuertes G., Martín De Llano J. J., Villarroya A., Rivett A. J., and Knecht E. (2003) Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem. J. 375, 75–86 10.1042/bj20030282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Donoso G., Herzog V., and Schmitz A. (2005) Misfolded BiP is degraded by a proteasome-independent endoplasmic-reticulum-associated degradation pathway. Biochem. J. 387, 897–903 10.1042/BJ20041312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mancini R., Aebi M., and Helenius A. (2003) Multiple endoplasmic reticulum-associated pathways degrade mutant yeast carboxypeptidase Y in mammalian cells. J. Biol. Chem. 278, 46895–46905 10.1074/jbc.M302979200 [DOI] [PubMed] [Google Scholar]
- 55. Shenkman M., Tolchinsky S., and Lederkremer G. Z. (2007) ER stress induces alternative nonproteasomal degradation of ER proteins but not of cytosolic ones. Cell Stress Chaperones 12, 373–383 10.1379/CSC-281.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Elbein A. D., Tropea J. E., Mitchell M., and Kaushal G. P. (1990) Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J. Biol. Chem. 265, 15599–15605 [PubMed] [Google Scholar]
- 57. Tsubuki S., Saito Y., Tomioka M., Ito H., and Kawashima S. (1996) Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J. Biochem. 119, 572–576 10.1093/oxfordjournals.jbchem.a021280 [DOI] [PubMed] [Google Scholar]
- 58. Fiebiger E., Hirsch C., Vyas J. M., Gordon E., Ploegh H. L., and Tortorella D. (2004) Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol. Biol. Cell 15, 1635–1646 10.1091/mbc.e03-07-0506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jin H. O., Seo S. K., Woo S. H., Kim E. S., Lee H. C., Yoo D. H., An S., Choe T. B., Lee S. J., Hong S. I., Rhee C. H., Kim J. I., and Park I. C. (2009) Activating transcription factor 4 and CCAAT/enhancer-binding protein-β negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic. Biol. Med. 46, 1158–1167 10.1016/j.freeradbiomed.2009.01.015 [DOI] [PubMed] [Google Scholar]
- 60. Qin L., Wang Z., Tao L., and Wang Y. (2010) ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 6, 239–247 10.4161/auto.6.2.11062 [DOI] [PubMed] [Google Scholar]
- 61. Heifetz A., Keenan R. W., and Elbein A. D. (1979) Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry 18, 2186–2192 10.1021/bi00578a008 [DOI] [PubMed] [Google Scholar]
- 62. Vlietstra R. J., van Alewijk D. C., Hermans K. G., van Steenbrugge G. J., and Trapman J. (1998) Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 58, 2720–2723 [PubMed] [Google Scholar]
- 63. Carson J. P., Kulik G., and Weber M. J. (1999) Antiapoptotic signaling in LNCaP prostate cancer cells: a survival signaling pathway independent of phosphatidylinositol 3′-kinase and Akt/protein kinase B. Cancer Res. 59, 1449–1453 [PubMed] [Google Scholar]
- 64. Pfeifer U. (1978) Inhibition by insulin of the formation of autophagic vacuoles in rat liver: a morphometric approach to the kinetics of intracellular degradation by autophagy. J. Cell Biol. 78, 152–167 10.1083/jcb.78.1.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schworer C. M., Shiffer K. A., and Mortimore G. E. (1981) Quantitative relationship between autophagy and proteolysis during graded amino acid deprivation in perfused rat liver. J. Biol. Chem. 256, 7652–7658 [PubMed] [Google Scholar]
- 66. Lee H., Noh J. Y., Oh Y., Kim Y., Chang J. W., Chung C. W., Lee S. T., Kim M., Ryu H., and Jung Y. K. (2012) IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 21, 101–114 10.1093/hmg/ddr445 [DOI] [PubMed] [Google Scholar]
- 67. Hetz C., Thielen P., Matus S., Nassif M., Court F., Kiffin R., Martinez G., Cuervo A. M., Brown R. H., and Glimcher L. H. (2009) XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306 10.1101/gad.1830709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Muñoz J. P., Ivanova S., Sánchez-Wandelmer J., Martínez-Cristóbal P., Noguera E., Sancho A., Díaz-Ramos A., Hernández-Alvarez M. I., Sebastián D., Mauvezin C., Palacín M., and Zorzano A. (2013) Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361 10.1038/emboj.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. van Vliet A. R., Giordano F., Gerlo S., Segura I., Van Eygen S., Molenberghs G., Rocha S., Houcine A., Derua R., Verfaillie T., Vangindertael J., De Keersmaecker H., Waelkens E., Tavernier J., Hofkens J., Annaert W., Carmeliet P., Samali A., Mizuno H., and Agostinis P. (2017) The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol. Cell 65, 885–899.e6 10.1016/j.molcel.2017.01.020 [DOI] [PubMed] [Google Scholar]
- 70. Verfaillie T., Rubio N., Garg A. D., Bultynck G., Rizzuto R., Decuypere J. P., Piette J., Linehan C., Gupta S., Samali A., and Agostinis P. (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891 10.1038/cdd.2012.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., and Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 72. Armstrong J. L., Flockhart R., Veal G. J., Lovat P. E., and Redfern C. P. (2010) Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J. Biol. Chem. 285, 6091–6100 10.1074/jbc.M109.014092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D. F., Bell J. C., Hettmann T., Leiden J. M., and Ron D. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 10.1016/S1097-2765(03)00105-9 [DOI] [PubMed] [Google Scholar]
- 74. Fusakio M. E., Willy J. A., Wang Y., Mirek E. T., Al Baghdadi R. J., Adams C. M., Anthony T. G., and Wek R. C. (2016) Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 27, 1536–1551 10.1091/mbc.E16-01-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li W., Zhu J., Dou J., She H., Tao K., Xu H., Yang Q., and Mao Z. (2017) Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 8, 1763 10.1038/s41467-017-01609-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Axten J. M. (2017) Protein kinase R(PKR)-like endoplasmic reticulum kinase (PERK) inhibitors: a patent review (2010–2015). Expert Opin. Ther. Pat. 27, 37–48 10.1080/13543776.2017.1238072 [DOI] [PubMed] [Google Scholar]
- 77. An H., and Harper J. W. (2018) Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 20, 135–143 10.1038/s41556-017-0007-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bernales S., McDonald K. L., and Walter P. (2006) Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4, e423 10.1371/journal.pbio.0040423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schuck S., Gallagher C. M., and Walter P. (2014) ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J. Cell Sci. 127, 4078–4088 10.1242/jcs.154716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Smith M. D., Harley M. E., Kemp A. J., Wills J., Lee M., Arends M., von Kriegsheim A., Behrends C., and Wilkinson S. (2018) CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev. Cell 44, 217–232.e11 10.1016/j.devcel.2017.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fumagalli F., Noack J., Bergmann T. J., Cebollero E., Pisoni G. B., Fasana E., Fregno I., Galli C., Loi M., Soldà T., D'Antuono R., Raimondi A., Jung M., Melnyk A., Schorr S., et al. (2016) Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 18, 1173–1184 10.1038/ncb3423 [DOI] [PubMed] [Google Scholar]
- 82. Lipatova Z., and Segev N. (2015) A role for macro-ER-phagy in ER quality control. PLoS Genet. 11, e1005390 10.1371/journal.pgen.1005390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Khaminets A., Heinrich T., Mari M., Grumati P., Huebner A. K., Akutsu M., Liebmann L., Stolz A., Nietzsche S., Koch N., Mauthe M., Katona I., Qualmann B., Weis J., Reggiori F., et al. (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358 10.1038/nature14498 [DOI] [PubMed] [Google Scholar]
- 84. Grumati P., Morozzi G., Hölper S., Mari M., Harwardt M. I., Yan R., Müller S., Reggiori F., Heilemann M., and Dikic I. (2017) Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 6, e25555 10.7554/eLife.25555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lipatova Z., Shah A. H., Kim J. J., Mulholland J. W., and Segev N. (2013) Regulation of ER-phagy by a Ypt/Rab GTPase module. Mol. Biol. Cell 24, 3133–3144 10.1091/mbc.e13-05-0269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mochida K., Oikawa Y., Kimura Y., Kirisako H., Hirano H., Ohsumi Y., and Nakatogawa H. (2015) Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522, 359–362 10.1038/nature14506 [DOI] [PubMed] [Google Scholar]
- 87. Chen S., Cui Y., Parashar S., Novick P. J., and Ferro-Novick S. (2018) ER-phagy requires Lnp1, a protein that stabilizes rearrangements of the ER network. Proc. Natl. Acad. Sci. U.S.A. 115, E6237–E6244 10.1073/pnas.1805032115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Moretti J., Roy S., Bozec D., Martinez J., Chapman J. R., Ueberheide B., Lamming D. W., Chen Z. J., Horng T., Yeretssian G., Green D. R., and Blander J. M. (2017) STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell 171, 809–823.e13 10.1016/j.cell.2017.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Doan N. T., Paulsen E. S., Sehgal P., Møller J. V., Nissen P., Denmeade S. R., Isaacs J. T., Dionne C. A., and Christensen S. B. (2015) Targeting thapsigargin towards tumors. Steroids 97, 2–7 10.1016/j.steroids.2014.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xi H., Kurtoglu M., and Lampidis T. J. (2014) The wonders of 2-deoxy-d-glucose. IUBMB Life 66, 110–121 10.1002/iub.1251 [DOI] [PubMed] [Google Scholar]
- 91. Datema R., and Schwarz R. T. (1978) Formation of 2-deoxyglucose-containing lipid-linked oligosaccharides: interference with glycosylation of glycoproteins. Eur. J. Biochem. 90, 505–516 10.1111/j.1432-1033.1978.tb12630.x [DOI] [PubMed] [Google Scholar]
- 92. Kurtoglu M., Gao N., Shang J., Maher J. C., Lehrman M. A., Wangpaichitr M., Savaraj N., Lane A. N., and Lampidis T. J. (2007) Under normoxia, 2-deoxy-d-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol. Cancer Ther. 6, 3049–3058 10.1158/1535-7163.MCT-07-0310 [DOI] [PubMed] [Google Scholar]
- 93. Xi H., Kurtoglu M., Liu H., Wangpaichitr M., You M., Liu X., Savaraj N., and Lampidis T. J. (2011) 2-Deoxy-d-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 67, 899–910 10.1007/s00280-010-1391-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Itkonen H. M., Engedal N., Babaie E., Luhr M., Guldvik I. J., Minner S., Hohloch J., Tsourlakis M. C., Schlomm T., and Mills I. G. (2015) UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene 34, 3744–3750 10.1038/onc.2014.307 [DOI] [PubMed] [Google Scholar]
- 95. Plomp P. J., Wolvetang E. J., Groen A. K., Meijer A. J., Gordon P. B., and Seglen P. O. (1987) Energy dependence of autophagic protein degradation in isolated rat hepatocytes. Eur. J. Biochem. 164, 197–203 10.1111/j.1432-1033.1987.tb11011.x [DOI] [PubMed] [Google Scholar]
- 96. Moruno-Manchón J. F., Pérez-Jiménez E., and Knecht E. (2013) Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem. J. 449, 497–506 10.1042/BJ20121122 [DOI] [PubMed] [Google Scholar]
- 97. Sehgal P., Szalai P., Olesen C., Praetorius H. A., Nissen P., Christensen S. B., Engedal N., and Møller J. V. (2017) Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 292, 19656–19673 10.1074/jbc.M117.796920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Szalai P., and Engedal N. (2018) An image-based assay for high-throughput analysis of cell proliferation and cell death of adherent cells. Bio-Protocol 8, e2835 10.21769/BioProtoc.2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Suzuki J., Kanemaru K., Ishii K., Ohkura M., Okubo Y., and Iino M. (2014) Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 5, 4153 10.1038/ncomms5153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dull T., Zufferey R., Kelly M., Mandel R. J., Nguyen M., Trono D., and Naldini L. (1998) A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K., and Kaufman P. D. (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529 10.1371/journal.pone.0006529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 103. Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., and Speleman F. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., Pimentel H., Salzberg S. L., Rinn J. L., and Pachter L. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liao Y., Smyth G. K., and Shi W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 106. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.