

Abstract
Malaria parasites repair DNA double-strand breaks (DSBs) primarily through homologous recombination (HR). Here, because the unrepaired DSBs lead to the death of the unicellular parasite Plasmodium falciparum, we investigated its recombinase, PfRad51, as a potential drug target. Undertaking an in silico screening approach, we identified a compound, B02, that docks to the predicted tertiary structure of PfRad51 with high affinity. B02 inhibited a drug-sensitive P. falciparum strain (3D7) and multidrug-resistant parasite (Dd2) in culture, with IC50 values of 8 and 3 μm, respectively. We found that B02 is more potent against these P. falciparum strains than against mammalian cell lines. Our findings also revealed that the antimalarial activity of B02 synergizes with those of two first-line malaria drugs, artemisinin (ART) and chloroquine (CQ), lowering the IC50 values of ART and CQ by 15- and 8-fold, respectively. Our results also provide mechanistic insights into the anti-parasitic activity of B02, indicating that it blocks the ATPase and strand-exchange activities of PfRad51 and abrogates the formation of PfRad51 foci on damaged DNA at chromosomal sites, probably by blocking homomeric interactions of PfRad51 proteins. The B02-mediated PfRad51 disruption led to the accumulation of unrepaired parasitic DNA and rendered parasites more sensitive to DNA-damaging agents, including ART. Our findings provide a rationale for targeting the Plasmodium DSB repair pathway in combination with ART. We propose that identification of a specific inhibitor of HR in Plasmodium may enable investigations of HR's role in Plasmodium biology, including generation of antigenic diversity.
Keywords: plasmodium, DNA repair, homologous recombination, drug screening, malaria, artemisinin, B02, comet assay, PfRad51, small-molecule inhibitor
Introduction
Malaria continues to be one of the biggest public health problems in this era. Up to 303 million new malaria cases are reported each year with almost a half-million of them resulting in death (1). Although artemisinin (ART)3-based combination therapies have helped reduce the World's malaria burden, the parasite's ability to develop resistance has overcome our efforts to curb this disease. Thus, there is an urgent need to come up with new anti-malaria drugs. To that end, one of the approaches could be the identification of novel drug targets. Here, we propose that targeting the DNA double-strand break (DSB) repair pathway of the malaria parasites could be a viable approach.
DSBs are inevitable in the life cycle of Plasmodium falciparum. Their sources could be endogenous, exogenous, or even physiologic. Whatever the source of DSBs, each DSB needs to be mended to ensure viability of a unicellular organism (2). In Plasmodium, DSBs are repaired primarily by the HR pathway. Parasites lacking functional HR machinery fail to repair DSBs and succumb to death (3), suggesting that the HR pathway cannot be compensated by any other DSB repair mechanisms in these parasites. Mice infected with parasites with defective HR live longer with significantly lower parasite burden (3).
In eukaryotes, Rad51 protein plays a central role during HR. Rad51 coats the ssDNA and forms nucleoprotein filament during strand invasion (4). Its ATP hydrolysis activity is required for product release during the synapsis step (5). The two Walker motifs (A and B) are responsible for its ATP-binding and hydrolysis activities (6). The ortholog of Rad51 from P. falciparum has been identified (7). PfRad51 shows ssDNA-dependent ATP hydrolysis activity and strand-exchange activity (8). A mutation in the Walker-A motif (PfRad51K143R) not only abrogates its ATPase activity but also exhorts a dominant-negative effect by inhibiting the ATPase activity of WT PfRad51 proteins (3). Yeast two-hybrid data indicate that PfRad51 interacts with itself (3), suggesting that PfRad51 also forms multimeric complexes like other eukaryotic Rad51 proteins. Using yeast as a surrogate model, it has been demonstrated that PfRad51 can repair DSBs, perform gene conversions, and facilitate gene targeting at the correct genomic locus (3).
A large-scale screening of the NCBI chemical library using a high-throughput fluorescence resonance energy transfer (FRET) assay, for identification of inhibitors of human Rad51 (hRad51)–mediated DNA strand exchange, has identified 17 compounds (9). Few of them were very specific to hRad51 as they did not inhibit the bacterial RecA, which is the structural and functional ortholog of Rad51 (9). Separate studies have identified additional inhibitors of hRad51 (10, 11), and some of them were further chemically modified to increase their specificity (12). Here, we report the in silico screening of small molecules for docking onto the predicted three-dimensional structure of PfRad51. One of the top-scoring ligands (B02) inhibits the ATPase activity of purified recombinant PfRad51 protein and its self-interaction. We further provide mechanistic insights for the anti-parasitic activity of B02 that it abrogates the formation of PfRad51 foci on the damaged DNA at chromosomal sites, probably by blocking the self-interaction of the PfRad51 proteins. This leads to the failure to repair damaged parasitic DNA and renders the parasites more sensitive toward DNA-damaging agents. B02 inhibits parasite growth more selectively than that of mammalian cell lines. We found out that the anti-parasitic activity of B02 can be potentiated by anti-malarial drugs chloroquine (CQ) and ART. Furthermore, we report that the anti-parasitic activity of B02 synergizes with ART and CQ but not with pyrimethamine (PY).
Results
Homology modeling of PfRad51 and in silico screening of small-molecule inhibitors
Because the X-ray crystal structure of PfRad51 is unavailable, we wished to develop an in silico 3D structure of PfRad51 by homology modeling. To achieve this, we have used the available crystal structure of the yeast Rad51 (ScRad51) as a template. The modeled structure of PfRad51 revealed the presence of all the characteristic features of the ScRad51 structure, namely the Walker motifs, the L1 site, the L2 site, and the dimerization surfaces 1 and 2 (Fig. 1, A and B). This is in corroboration with the sequence identity observed between PfRad51 and ScRad51 (Fig. S1). To predict the chemical compounds that might have anti-PfRad51 activity, we have prepared a virtual ligand library of small molecules that have previously established anti-Rad51/RecA activity (9–11). Seventeen ligands were chosen (Fig. S2) that inhibit the activity of eukaryotic Rad51 or Escherichia coli RecA proteins. With each of the 17 ligands, we performed blindfold docking, by setting the entire protein as the search space. This allowed us to identify the most preferred sites for binding, namely the Walker site and the dimerization site. The binding affinities of each ligand as predicted by different models are listed in Table 1. The docking scores from each of the models were totaled for each docking site to represent a measure of total affinity of ligand binding. A cutoff value of −30 kcal/mol was set because the binding affinity of ATP, which is the natural ligand of PfRad51, was found to be near this value. At the Walker site five ligands showed maximum affinity. These ligands are as follows: A04, A08, A07, A05, and B02. Similarly, at the dimerization site eight ligands showed maximum affinity. These ligands are as follows: A04, A08, A07, A05, B02, A10, A03, and RI-2. Interestingly, two well-characterized inhibitors of human Rad51, RI-1 and RI-2, exhibited weak docking at the Walker site of PfRad51. The binding affinity of RI-1 to the dimerization site of PfRad51 was also found to be below the cutoff value. Such differential binding affinity of RI-1 can be explained by the fact that the sequence of the ATP-binding site of PfRad51 and hRad51 are similar but not identical (Fig. S1) (13). Overall, A04, A08, A07, A05, and B02 showed high affinity toward docking at both the sites. Of these five ligands, B02 is commercially available, and hence it was used for the experimental validation of our bioinformatics prediction. The docking of B02 on PfRad51 is depicted in Fig. 1, C and D. The docking of the other top-scoring ligands (A04, A07, and A08) at the Walker A site is shown in Fig. S3A. The docking of the three top-scoring ligands (A04, A08, and A10) at the dimerization face is shown in Fig. S3B.
Figure 1.
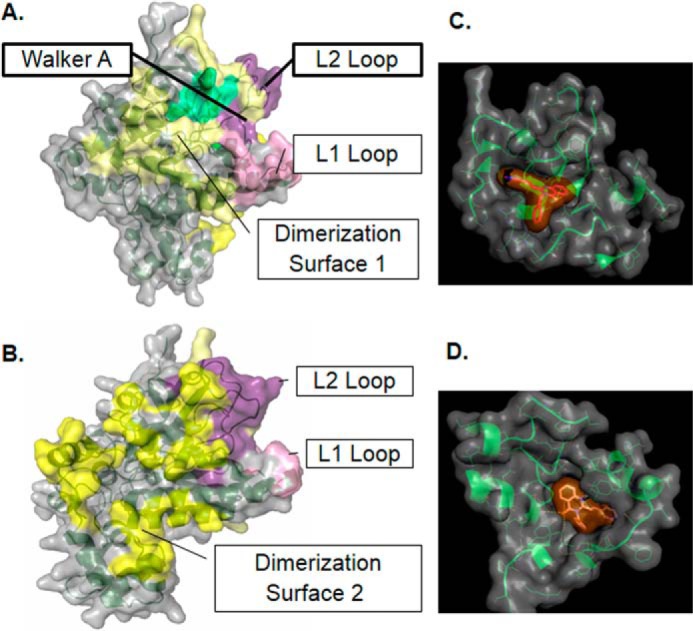
Docking of B02 onto the predicted tertiary structure of PfRad51. Front view (A) and rear view (B) of the homology model of PfRad51. C, docked conformation of B02 at the Walker site. D, docked conformation of B02 at the dimerization site.
Table 1.
Binding affinities of docked ligands (in −kcal/mol)
A = 3LDA, B = 3LDA_MIN, C = 1SZP, and D = ITASSER_AUTO.

B02 binds to PfRad51 and inhibits its ATP hydrolysis and three-strand exchange activities
To study the effect of B02 on PfRad51's activity, we measured the inhibition of ssDNA-dependent ATPase activity and strand-exchange activity of PfRad51 in the presence of B02. Our docking study has predicted that B02 binds at the Walker A motif of Rad51, a site for ATP binding (Fig. S3A). To test this prediction, we have done binding assays using a fluorescence quenching experiment. PfRad51 has a single tryptophan residue (Trp-170) that, when excited at 295 nm, emits fluorescence with an emission wavelength maximum at 332 nm (Fig. S3C). In the presence of increasing concentrations of B02, a dose-dependent quenching of the intrinsic fluorescence was observed (Fig. S3C). From the Stern-Volmer plot, the association constant (Ka) value was calculated as 0.8 μm−1 (Fig. 2A). As ATP is the natural substrate that binds to the Rad51 protein, we have also calculated the Ka value of ATP binding, which was determined as 0.22 μm−1. Thus, the association of B02 with PfRad51 is greater than the association of ATP and PfRad51. We have used radicicol as a negative control in our experiment as it did not show any quenching of the intrinsic fluorescence of the PfRad51 protein.
Figure 2.

B02 binds to PfRad51 and inhibits its ATP hydrolysis activity. A, quenching of intrinsic fluorescence of Trp-170 of PfRad51 upon B02 binding. Stern-Volmer plots showing the ratio of intrinsic fluorescence (F0) and quenched fluorescence (F1) at different concentrations of ligand binding are shown. The excitation was at 295 nm, and the emission was recorded at 332 nm. Data are the mean ± S.D. from three experiments. B, ssDNA-dependent ATPase activity of PfRad51 in the presence of various concentrations of drugs (as indicated on the x axis) as indicated. C, at 200 μm ATP concentration and 60 μm ssDNA, the activity of 1 μm PfRad51 is inhibited by B02 with an IC50 value of 8.48 μm. Mean and standard errors from three independent experiments are plotted. D, three-strand exchange activity of PfRad51 is inhibited by B02 with an IC50 value of 7.96 μm. Mean and standard errors from three independent experiments are plotted. LD, linear dsDNA (substrate); NC, nicked circular DNA (product).
Because ATP binding is a prerequisite for the ATP hydrolysis activity of Rad51, we hypothesized that B02 might inhibit the ATPase activity of PfRad51. We performed the ATPase assay with purified PfRad51 proteins in the presence of varying concentrations (0.01–40 μm) of B02, and we measured the percent activity by taking the activity of PfRad51 in the absence of any inhibitor as 100%. We observed a sharp decline in the ATPase activity of the protein with an increasing concentration of B02 (Fig. 2A). To ascertain whether such an inhibitory effect is specific to B02, we included two more chemical compounds, namely RI-1 and radicicol, and we investigated their effect on the ATPase activity of PfRad51. RI-1 has previously been found to inhibit the strand-exchange activity of hRad51 (10). However, radicicol has been reported to bind to the Bergerat fold and inhibit the ATPase activity of Hsp90 and TopoVIB proteins (14). In our experiments, RI-1 exhibited moderate inhibition to the ATPase activity of PfRad51 (Fig. 2A). This finding is consistent with our docking study, which predicted a weaker binding affinity of RI-1 with PfRad51. However, radicicol did not show any inhibitory effect to the ATPase activity of PfRad51 (Fig. 2B). This corroborates well with the prediction that radicicol does not bind to PfRad51 (data not shown). Thus, radicicol has acted as a negative control in our experiment. To obtain the IC50 value of B02 inhibition on the ATPase activity of PfRad51, we have plotted the percent inhibition of the ATPase activity in the presence of various concentrations of B02. The IC50 of PfRad51 inhibition by B02 was determined to be 8.48 μm (Fig. 2C). We further investigated the effect of B02 on the three-strand exchange activity of PfRad51 using circular single-stranded ϕX174 DNA and linear double-stranded ϕX174 DNA as substrates. As observed earlier (8), PfRad51 generated the intermediate joint molecules at the end of 30 min and the product nicked circular DNA at the end of 90 min (Fig. S3D). However, in the presence of B02, a dramatic inhibition of the product formation was observed in the presence of 20 μm B02 (Fig. S3E). The IC50 of such inhibition was determined to be 7.96 μm (Fig. 2D). In a previous study, the IC50 of B02 against hRad51-mediated strand-exchange reaction was found to be 27.4 μm (9). Thus, it seems that B02 has a higher specificity toward PfRad51.
B02 inhibits the repair of damaged Plasmodium DNA
Because HR is the primary DSB repair mechanism of Plasmodium, we hypothesized that B02 could block the repair of damaged Plasmodium DNA, thereby impairing parasite growth. To this end, we performed an MMS sensitivity assay. MMS creates numerous single-stranded and dsDNA breaks (DSB) in the Plasmodium genome (15). Each of the DSBs need to be repaired to ensure parasite survival. Thus, a decrease in parasite survival upon MMS treatment reflects a lack of DSB repair. We observed that parasites that are not exposed to B02 do not show MMS hypersensitivity, suggesting that these parasites are able to repair MMS-induced DSBs. This result is consistent with previous reports (7, 15, 16). Parasites, if treated with lower concentrations of B02 (10 nm), also do not show any significant hypersensitivity to MMS. However, treatment of parasites with higher concentrations of B02 (100 nm to 10 μm) results in significant hyper-sensitivity toward MMS (Fig. 3A). Such effects cannot be scored at a much higher B02 concentration (20 or 50 μm) due to excessive toxicity.
Figure 3.

B02 inhibits the repair of damaged Plasmodium DNA. A, MMS sensitivity of parasites treated with B02 as determined by return–to–growth assay. Growth of parasites that are neither treated with B02 nor with MMS represents 100% survival. Error bars indicate S.D. (n = 3 experiments); asterisks indicate values significantly different from the control, as follows: **, p < 0.01; NS, not significant. B, comet assay visualization of the persistence of damaged DNA upon MMS treatment in the presence or absence of B02 (8 μm). Control represents undamaged DNA. Shortening of the comet length indicates the repair of damaged DNA during recovery time. C, quantitative measurement of comet tail length at different time points ranging from pre-DNA damage to post-DNA damage. The average tail length of comets at each time point (n >50) was calculated. The mean of the averages from six independent experiments is plotted. Asterisks indicate values significantly different from the control, as follows: ***, p < 0.001; **, p < 0.01; NS, not significant.
To visualize and quantify the persistence of damaged parasitic DNA in the presence of B02, we performed the comet assay. Treatment with MMS creates DSBs, and the extent of damaged DNA can be quantified as a function of the tail length in a comet assay. Under normal circumstances, P. falciparum DNA repair machinery takes between 10 and 20 h to repair DSBs, and as a result, the tail length decreases (15). We treated P. falciparum in vitro culture with 0.05% MMS for 6 h to generate damaged DNA. Because such treatment was not lethal for the parasites (Fig. S4), the parasites were allowed to recover for 40 h in the presence or absence of B02 (10 μm). The extent of damaged DNA was measured at different time intervals (10, 20, and 40 h post-MMS treatment). Comets with long tails were observed upon MMS treatment. Such comets were absent in the control samples. In the absence of B02, the tail length of the comets was found to decrease with time. An initial rapid recovery was observed (at the end of 10 h) followed by a slower recovery. At the end of 40 h, the tail length of the comets was comparable with the untreated control samples (Fig. 3B, upper panel). In the presence of B02, an initial rapid recovery (10 h) was observed, albeit to a lesser extent. Interestingly, no further recovery was observed even until the last time point (40 h) (Fig. 3B, lower panel). Our data indicate an inefficient repair of damaged parasitic DNA in the presence of B02. Fig. 3C illustrates the graphical representation of the mean value of tail length (displacement of the tail's center of mass relative to the center of the head) with a recovery period in the presence and absence of B02. Taken together, our results suggest that B02 inhibits the repair of MMS-induced DSB repair in the parasite genome by inhibiting the activity of PfRad51.
B02 inhibits the formation of PfRad51 foci upon DNA damage
To gain further mechanistic insights into the mode of action of B02, we investigated whether PfRad51 foci formation is affected in the presence of B02. The cytoplasmic Rad51 proteins enter the nucleus upon DNA damage and oligomerize to form a nucleoprotein filament on ssDNA, which is generated due to the nucleolytic processing of a DSB. Such oligomerization of Rad51 onto the ssDNA represents the accumulation of numerous Rad51 proteins at the broken junctions that appear as foci under a fluorescent microscope (17). These foci are known as Rad51 foci and are crucial for the repair of DSB by the HR mechanism. It should be noted that other DSB repair proteins are also recruited at these DNA damaged sites. Formation of such a DNA damage-induced foci has been reported in P. falciparum as well (18). We performed an indirect immunofluorescence assay (IFA) to detect nuclear foci formation of PfRad51. When in vitro cultures of P. falciparum were treated with 0.05% MMS, distinct nuclear foci of PfRad51 were observed (Fig. 4A). No such nuclear focus was observed for the control (untreated) parasites. In the presence of B02, a marked reduction in the number of foci formation was observed upon MMS treatment. Quantification of the data revealed that there is almost an 80% reduction in the number of foci in the presence of B02 (Fig. 4B). As a control, we treated parasites with B02 alone, and as expected, PfRad51 foci were not observed.
Figure 4.

B02 inhibits the formation of PfRad51 foci upon DNA damage. A, IFA displays PfRad51 foci (FITC) upon treatment with the DNA-damaging agent MMS (3rd row). Such foci are not visible in the control cells (untreated with MMS) neither in the presence (2nd row) nor in the absence of 8 μm B02 (1st row). MMS-induced PfRad51 foci formation are inhibited in the presence of 8 μm B02 (4th row). DAPI staining indicates the location of the parasite nucleus. B, quantitative analysis of PfRad51 foci formation. Percent of focus formation is defined as the number of infected RBC having PfRad51 foci (FITC-stained) of 100 infected RBC (DAPI-stained). The mean and the standard deviations from three independent experiments are plotted. Two-tailed t test was performed to obtain the statistical significance. The p value is indicated at the top. C, self-interaction of PfRad51 is inhibited in the presence of 8 μm B02. pGADC1 and PGBDUC1 are the parent plasmids encoding the GAL4 activation domain and DNA-binding domain, respectively. DNA fragments corresponding to the full-length WT PfRAD51 ORF were fused to the GAL4 activation domain in pGADC1 and fused to the DNA-binding domain in pGBDUC1. Two-hybrid interactions were tested with yeast strain PJ694A, which bears the ADE2 gene as one of the reporters. Yeast cells harboring both plasmids were patched on control plates (SC–Ura–Leu) as well as experimental plates (SC–Ura–Leu–Ade) to test for protein–protein interactions in the absence or in the presence of 8 μm B02 (as indicated at the bottom).
Homodimerization of Rad51 is a prerequisite for Rad51 foci formation. The dimerization of Rad51 takes place in a front–to–rear fashion, where the ATP-binding site (the front side) of one monomer lies in close proximity to the dimerization face (the rear side) of the other monomer, also the adenine ring of ATP mediates dimer interactions (19). Because our docking studies also predicted that B02 might inhibit PfRad51 dimerization by blocking the dimerization site, as it competes for the ATP-binding site, we wanted to explore whether PfRad51 could form homodimers in the presence of B02. To this end, we have employed yeast two-hybrid analysis (Y2H). Earlier, it was observed that PfRad51 was able to interact with itself, and such self-interaction could be detected by Y2H (3). Similar Y2H experiments were performed with both bait and prey vectors having PfRAD51, and a positive interaction was observed as robust growth on triple dropout plates (Sc–Leu–Ura–Ade). Lack of growth on triple dropout plates indicated the lack of interaction. We found out that PfRad51 is able to interact with itself (Fig. 4C) in the absence of B02. However, in the presence of B02 no such interaction was observed. To ascertain that the lack of growth on the triple dropout plates containing B02 is due to the lack of interaction between the bait–protein and the prey–protein (in this case both are PfRad51) and not due to any toxic effect of B02, yeast cells harboring the bait–plasmid and the prey–plasmid were grown in double-dropout plates (Sc–Leu–Ura) in the presence of B02. Robust growth was observed. These results demonstrate that B02 inhibits the homodimerization of PfRad51 proteins. This could be one of the reasons behind the dramatic reduction in the formation of PfRad51 foci in the presence of B02.
B02 inhibits the intra-erythrocytic developmental cycle (IDC) of P. falciparum
To investigate the effects of B02 on the growth of P. falciparum, we monitored the intra-erythrocytic development of the in vitro culture of P. falciparum 3D7 in the presence of different concentrations of B02. Previous studies have demonstrated that malarial parasites lacking a functional HR mechanism exhibit lower parasitemia during long-term propagation in vivo. This finding is suggestive of a failure of the parasite to repair naturally occurring DSBs in its genome, which results in the elimination of such parasites from the parasite population (3). We reasoned that Plasmodium recombinase inhibitor B02 should also have a similar effect on the parasite's survival. To test this hypothesis, we treated synchronous trophozoite–stage parasites with different concentrations of B02 and monitored their growth after 48 h by counting the parasitemia from thin smears stained with Giemsa. B02 treatment resulted in a dose-dependent inhibition of parasite growth (Fig. 5A). The dose-response curve yielded an IC50 value of 8.25 μm (Fig. 5B). To validate this finding, we employed a second method of parasite counting, namely the SYBR Green method. In this method, the fluorescent dye SYBR Green I intercalates between the base stacks of parasite DNA, hence the measured fluorescent intensity is directly proportional to the number of parasites. First, we plotted the relative fluorescence intensity versus the parasitemia to obtain a standard curve. The correlation coefficient was found to be very high (r2 = 0.997). We observed a similar dose-dependent inhibition of the parasite growth. The IC50 value was determined to be 7.45 μm (Fig. 5C). To investigate whether B02 could inhibit the growth of other strains of P. falciparum, we tested the effect of B02 on the in vitro culture of one of the drug-resistant strains of P. falciparum, namely Dd2. We found out that B02 inhibits the IDC of Dd2 as well (IC50 = 3.73 μm) (Fig. 5D). We also investigated the toxic effects of B02 in two mammalian cell lines: mouse cell line MEF and human cell line HepG2, and we observed that B02 has a killing effect against these cell lines with an IC50 value of 18.2 and 11.93 μm, respectively (Fig. S5). Thus, the selectivity of B02 action toward P. falciparum is found to be much higher when compared with the mammalian cell lines (Table 2). The selectivity indexes for different parasite strains are found to vary between 2 and 5.
Figure 5.

B02 inhibits intra-erythrocytic development of P. falciparum. A, synchronous trophozoite stage 3D7 cultures were grown for 48 h in the presence of various concentrations of B02 (as marked on the x axis). After 48 h, percent of parasitemia of different cultures was counted. The mean and standard deviations from three parallel experiments are plotted. B, inhibition of parasite growth at various concentrations of B02 is plotted to obtain the IC50 value. Growth of P. falciparum 3D7 strain was monitored by the Giemsa staining method. Parasite growth in the absence of B02 is considered as zero inhibition. Mean and standard deviations from three independent experiments are plotted. C and D, inhibition of growth of P. falciparum 3D7 and Dd2 as measured by SYBR Green I method. C, inset, depicts the standard curve of parasitemia versus SYBR Green I fluorescence intensity.
Table 2.
IC50 of B02 in different P. falciparum strains as compared with mammalian cell lines
| Strain/cell line | IC50 (mean ± S.D.) | Selectivity index |
|---|---|---|
| μm | ||
| 3D7 | 7.45 ± 0.89 | 2.44 |
| Dd2 | 3.73 ± 0.35 | 4.88 |
| HepG2 | 11.93 ± 0.23 | 1.52 |
| MEF | 18.2 ± 0.7 | 1 |
Inhibitory effect of B02 on IDC of P. falciparum can be potentiated by DHA and CQ
To further investigate the anti-parasitic effects of B02, we used three well-established anti-malarial drugs, namely ART, CQ, and PY. Because ART causes DSBs in the Plasmodium genome (20), we predicted that the inhibitory effect of B02 could be potentiated in the presence of ART. We have used DHA as an ART derivative throughout our experiments. We observed that in the presence of 60 nm DHA (the IC50 concentration against 3D7 strain), the action of B02 is potentiated by 2.86-fold (Table 3). Similar potentiation (2.85-fold) was observed in Dd2 strain in the presence of 65 nm DHA (the IC50 concentration against Dd2 strain). A more dramatic potentiation was observed in the presence of CQ. In the presence of 26 nm CQ in 3D7 strain and 72 nm CQ in Dd2 strain, 9- and 5.2-fold potentiation of B02 was observed. Interestingly, the presence of PY did not result in any potentiation of B02 in either strain. This observation underscores the specificity of DHA and CQ in the potentiation of B02 action.
Table 3.
IC50 of B02 in combination with other anti-malarial drugs
| Strain | Combination of drugs | IC50 (mean ± S.D.) | Potentiation factor |
|---|---|---|---|
| μm | |||
| 3D7 | B02 (alone) | 7.45 ± 0.89 | 1 |
| B02 (DHA)a | 2.6 ± 0.2 | 2.86 | |
| B02 (CQ)b | 0.82 ± 0.04 | 9 | |
| B02 (PY)c | 7.59 ± 0.38 | 0.98 | |
| Dd2 | B02 (alone) | 3.73 ± 0.35 | 1 |
| B02 (DHA)d | 1.31 ± 0.05 | 2.85 | |
| B02 (CQ)e | 0.72 ± 0.07 | 5.2 | |
| B02 (PY)f | 3.85 ± 0.4 | 0.97 |
a IC50 concentration of DHA in 3D7 strain was used.
b IC50 concentration of CQ in 3D7 strain was used.
c IC50 concentration of PY in 3D7 strain was used.
d IC50 concentration of DHA in Dd2 strain was used.
e IC50 concentration of CQ in Dd2 strain was used.
f IC50 concentration of PY in Dd2 strain was used.
We wanted to investigate whether the potentiation of B02 activity by DHA could alter the selectivity of the drug toward the parasite strains. We have treated the mammalian cell lines (MEF and HepG2) with varying concentrations of B02 in the presence of 60 nm DHA and determined the IC50 value of B02 in such combination assays. We observed that B02 has greater selectivity for the parasite strains when combined with DHA than when administered alone. For the 3D7 strain, the selectivity of B02 in combination with DHA was found to be 3.12 (Table 4) as opposed to 2.44 when B02 was administered alone. For the multidrug-resistant strain Dd2, the selectivity index was found to be 6.19 in combination therapy (Table 4) versus 4.88 for B02 alone.
Table 4.
IC50 of B02 in combination with DHA in different P. falciparum strains as compared with mammalian cell lines
| Strain/cell line | IC50 (mean ± S.D.) | Selectivity index |
|---|---|---|
| μm | ||
| 3D7 | 2.6 ± 0.2 | 3.12 |
| Dd2 | 1.31 ± 0.05 | 6.19 |
| HepG2 | 7.6 ± 0.43 | 1.06 |
| MEF | 8.12 ± 0.6 | 1 |
B02 lowers the IC50 value of DHA and CQ in synergistic ways
We wanted to investigate the interactions between DHA and B02. Because DHA causes DSBs in the Plasmodium genome, and HR is the main mechanism to repair such breaks in malarial parasites, we predicted that inhibition of HR mechanism by B02 would render the parasites more sensitive toward DHA action. We have determined the IC50 of DHA in our experimental setup. To this end, we have treated synchronous trophozoite stage parasites with different concentrations of DHA for 48 h followed by parasite quantification by the SYBR Green method. We observed dos-dependent inhibition of parasite growth, and the IC50 value was determined to be 59.7 nm. When similar experiments were performed in the presence of IC50 concentrations of B02 (8 μm), a dramatic decrease of 15.34-fold in the IC50 value was observed (3.89 nm) (Table 5). Similarly, B02 also potentiated the anti-malarial activities of CQ by 8.33-fold (Table 5). We sought to investigate whether such interactions of DHA or CQ with B02 are synergistic or additive in nature. For that we performed a fixed-ratio drug combination assay. The mean FIC values derived from the dose-response curve for each combination are compiled in Table 6. The sum of FICs is plotted in isobolograms (Fig. 6). The isobologram demonstrates that the profound effect of B02 on the anti-malarial action of DHA is synergistic (Fig. 6, A and B). Similarly, the interaction between B02 and CQ is also found to be synergistic (Fig. 6, C and D).
Table 5.
IC50 of DHA and CQ in combination with B02
| Strain | Combination of drugs | IC50 (mean ± S.D.) | Potentiation factor |
|---|---|---|---|
| nm | |||
| 3D7 | DHA (alone) | 59.7 ± 3.27 | 1 |
| DHA (B02)a | 3.89 ± 2.04 | 15.34 | |
| CQ (alone) | 26.4 ± 1.98 | 1 | |
| CQ (B02)a | 3.17 ± 0.47 | 8.33 | |
| Dd2 | DHA (alone) | 64.8 ± 3.56 | 1 |
| DHA (B02)b | 14.1 ± 3.06 | 4.6 | |
| CQ (alone) | 63.5 ± 2.33 | 1 | |
| CQ (B02)b | 9.8 ± 0.71 | 6.48 |
a IC50 concentration of B02 in 3D7 strain was used.
b IC50 concentration of B02 in Dd2 strain was used.
Table 6.
Mean FIC values of drug combinations
| Strain | DHA: B02 | FICDHA | FICB02 | ΣFIC | CQ: B02 | FICCQ | FICB02 | ΣFIC |
|---|---|---|---|---|---|---|---|---|
| 3D7 | 5:0 | 1 | 0 | 1 | 5:0 | 1 | 0 | 1 |
| 4:1 | 0.41 | 0.12 | 0.53 | 4:1 | 0.57 | 0.14 | 0.71 | |
| 3:2 | 0.35 | 0.27 | 0.62 | 3:2 | 0.39 | 0.27 | 0.66 | |
| 2:3 | 0.25 | 0.50 | 0.75 | 2:3 | 0.21 | 0.34 | 0.55 | |
| 1:4 | 0.13 | 0.67 | 0.80 | 1:4 | 0.14 | 0.63 | 0.77 | |
| 0:5 | 0 | 1 | 1 | 0:5 | 0 | 1 | 1 | |
| Dd2 | 5:0 | 1 | 0 | 1 | 5:0 | 1 | 0 | 1 |
| 4:1 | 0.74 | 0.10 | 0.84 | 4:1 | 0.84 | 0.08 | 0.92 | |
| 3:2 | 0.63 | 0.20 | 0.83 | 3:2 | 0.65 | 0.18 | 0.83 | |
| 2:3 | 0.46 | 0.32 | 0.78 | 2:3 | 0.54 | 0.24 | 0.78 | |
| 1:4 | 0.34 | 0.49 | 0.83 | 1:4 | 0.38 | 0.51 | 0.89 | |
| 0:5 | 0 | 1 | 1 | 0:5 | 0 | 1 | 1 |
Figure 6.

Synergy of DHA and CQ with recombinase inhibitor B02. A and B, isobologram of DHA–B02 combination in 3D7 and Dd2 strains, respectively. C and D, isobologram of CQ–B02 combination in 3D7 and Dd2 strains, respectively. Fixed-ratio drug combination assays were performed. FIC, fraction inhibitory concentration. Each point represents the mean IC50 of drug combination from three independent experiments. The solid line is plotted between the IC50 values of each drug when used alone to emphasize the concave nature of the isobolograms.
Discussion
The data presented in this article provide compelling evidence that B02 inhibits DSB repair in P. falciparum. First, it inhibits the ATPase activity of PfRad51, which is important for the product release during HR. Second, by binding at the dimerization site, B02 blocks the dimerization of PfRad51, which is the first step for the formation of the higher-order nucleoprotein filament. Finally, B02 inhibits the formation of the PfRad51 foci at the damaged DNA ends, leading to the persistence of an unrepaired broken DNA. Our findings provide the first “proof of the concept” that parasitic HR pathways can be inhibited to attenuate parasitic growth, and such attenuation works in a synergistic way with two of the very potent malaria drugs: artemisinin and chloroquine. Our data also demonstrate that the native B02 on its own has a higher selectivity toward PfRad51 compared with hRad51 and is also more potent for inhibiting the growth of malaria parasite compared with the mammalian host. Lead compound optimization might lead to an even better selectivity against PfRad51 compared with hRad51. It could also be possible that certain chemical modifications of B02 might have anti-malarial action at lower nanomolar concentrations. Additionally, it is not necessary to inhibit PfRad51 per se; other key proteins of Plasmodium HR pathway that are less conserved in humans could serve as better targets. For example, the DNA repair nucleases (such as PfalMre11, PfExo1, etc.), or the RecQ helicases (PfBLM and PfWRN) have minimal sequence conservation with their human counterparts. Future experiments might explore the suitability of these DNA repair proteins as potential anti-malarial targets.
B02 and ART potentiate each other's action. This can be explained by the fact that ART creates numerous DSBs in the Plasmodium genome (20), and such DSBs can only be repaired by the HR pathways in this parasite. B02 is a potent inhibitor of the Plasmodium HR pathway, and it blocks the repair of ART-induced DSBs and thus potentiates the action of ART. B02 and CQ also potentiate each other's action. Currently, we do not have any mechanistic explanations to this. However, two plausible explanations could be as follows. First, CQ inhibits heme polymerization in malarial parasites, thereby generating free radicals that can potentially create DSBs in the parasitic genome. This way CQ may potentiate the action of B02. Although this is a plausible explanation, currently it is not established whether CQ treatment leads to creation of DSBs in parasite genome or not. Future experiments may shed important light into this possibility. A second possibility is that CQ is a well-established inhibitor of the human DNA repair kinase, Chk1. If CQ also inhibits the Plasmodium DNA repair kinase, it might lead to an unrepaired DSB, thereby potentiating the action of B02. As of now, the DNA repair kinases remain unidentified in Plasmodium. So, it is not known whether CQ also inhibits the Plasmodium DNA damage-response signaling pathway or not. PY, however, inhibits Plasmodium dihydrofolate reductase and thereby blocks purine and pyrimidine biosynthesis. Thus it is unlikely to create DSBs in a parasite genome. Probably for that reason we did not observe any synergism between PY and B02.
Our results demonstrated that B02 synergizes ART action. However, the asymmetric concave nature of the isobologram is very interesting, and this can be explained in the following way. By definition, when the combined effect of two drugs is greater than the additive effect of the two, it is known as synergism. In such a case, both drugs must have their individual effects. In contrast, if one of the drugs does not have any effect on its own, but becomes a potent drug only in the presence of the second drug, it is called potentiation (21). As discussed earlier, Plasmodium parasites encounter numerous DSBs that are generated spontaneously during their normal course of propagation, and such breaks are usually repaired by the HR. Parasites lacking a functional HR mechanism fail to mend such DSBs thereby succumbing to death (22). Similarly, if selected parasites are treated with B02, then such parasites cannot repair DSBs, and they also meet the same fate. Moreover, ARTs have multiple targets: DNA, proteins, membrane lipids, and possibly several other uncharacterized cellular targets. As mentioned earlier, activated ARTs create DSBs, and in most cases such DSBs are repaired by the efficient HR pathway of the parasite (unless the number of DSBs is too overwhelming for the parasite to repair). Thus, it is unlikely that ART-mediated parasite deaths are primarily due to the generation of DSBs. It is probably due to other cellular damages caused by ART. As our model suggests (Fig. 7) in the case of B02 and ART interactions, the DSBs created by the ARTs remain unrepaired as the PfRad51-mediated HR mechanism is inhibited by B02, and such unrepaired DSBs lead to parasite death. Thus, B02 potentiates the action of ART as far as the unrepaired DSB-mediated parasite killing is concerned. Therefore, in the absence of B02, ARTs kill parasites by damaging other cellular targets, and in the presence of B02, ARTs kill parasites by damaging the DNA as well. Thus, of the many types of cellular damages that ARTs create, only one of them (namely DSB) can be synergized by B02, explaining the asymmetric nature of the concave isobologram.
Figure 7.

Model of parasite killing by the combination of ART and B02. ART is activated by Fe2+ source within the parasite to generate activated ART*, which creates DNA double-strand breaks and several other cellular damages (protein alkylations, membrane lipid peroxidation, etc.). DSBs are repaired by PfRad51-mediated HR pathway leading to survival of parasites. In the presence of B02, the parasitic HR mechanism is blocked resulting in unrepaired DSBs that eventually lead to parasite death. Similarly, the other kinds of cellular damages if remaining unrepaired also lead to parasite death. However, this second process of parasite killing is independent of B02 action.
Whole-genome sequencing of ART-resistant mutant strains reveals that there are sequence polymorphisms in 38 Plasmodium genes between ART-sensitive strains and ART-resistant strains. Interestingly, no such polymorphism has been found in genes that are predicted to be involved in the HR pathway (23, 24). Thus, it is reasonable to propose that the HR pathways of ART-sensitive parasites and ART-resistant parasites are not likely to be different. In other words, B02 might synergize with ART's activity and lower the IC50 value of ART even in the ART-resistant parasite strains. This hypothesis can be experimentally tested in the future. We have noticed that the multidrug-resistant strain Dd2 is more sensitive to B02 than that of the sensitive strain 3D7. Currently, we do not have any mechanistic insights into this finding. However, one likely explanation could be that the strain Dd2 carries mutations in 12 DNA repair genes (25); thus, it may have a less efficient DNA repair mechanism.
In addition to serving as a potential anti-malarial chemical compound, B02 can also be used as a chemical knockout strategy to explore the role of PfRad51 in Plasmodium biology. For example, it will now be possible to investigate whether or not PfRad51 is involved in the diversification of var gene sequences by carrying out recombination between closely related, yet nonidentical var genes. Such studies will throw important lights into our understanding of antigenic variation, a potent immune evasion strategy of the parasite.
Experimental procedures
Materials
We have used dihydroartemisinin (DHA) as an artemisinin derivative (ART). DHA, B02, RI-1, and radicicol were purchased from Sigma. DHA, B02, and RI-1 were dissolved in DMSO. Radicicol was dissolved in ethanol.
Parasite culture
P. falciparum 3D7 or Dd2 cultures were maintained in RPMI 1640 medium supplemented with 1% albumax and 0.005% hypoxanthine in human erythrocytes with 5% hematocrit at 37 °C as described earlier (2).
ATP hydrolysis assay and three-strand exchange assay for PfRad51
Recombinant PfRad51 protein was expressed and purified as described earlier (3). We measured the ATPase activity of PfRad51 in the presence of BO2, RI-1, and radicicol by using EnzChek phosphate assay kit (Molecular Probe) as described earlier (26). Briefly, the enzyme (2 μm PfRad51 protein) was incubated with a fixed concentration of the substrate (200 μm ATP) and ϕX174 ssDNA (60 μm) in the presence or absence of varying concentrations of individual inhibitors, and the formation of the product (Pi) was measured. The following concentrations of the inhibitors were used in our assay: B02 (3, 6, 13, 15, 18, 20, 24, 27, 35, 40, 50, 100, 200, and 300 μm); RI-1 (5, 10, 20, 30, 40, 50, 60, 80, 100, 200, and 300 μm); radicicol (0.1, 10, 100, and 300 μm). ATPase activity of PfRad51 determined in the absence of any inhibitor was taken as 100%, and accordingly, the percent inhibition values were calculated for various concentrations of the inhibitors. For determination of the IC50 value, the ATPase activity of PfRad51 1 μm enzyme was incubated with various concentrations of B02 (ranging from 0.1 to 1000 μm). The data were plotted in a semi-log graph to determine the IC50 value with the help of the GraphPad Prism software. Three-strand exchange assays were performed as described earlier (8). Briefly, ϕX174 ssDNA (5 μm) and linear double-stranded ϕX174 DNA (15 μm) were incubated with 1 μm PfRad51 in the presence of B02 (10, 20, 50, and 100 μm) or DMSO. Formation of nicked circular dsDNA product at 90 min was measured from the band intensity using ImageJ software. The product formation in the absence of B02 was taken as 100%, and accordingly, the percent inhibition values were calculated for various B02 concentrations. The date obtained from three independent experiments was plotted to determine the IC50 value.
Fluorescence quenching assay for ligand binding to PfRad51
To study the selective binding of B02 with PfRad51 protein, an intrinsic fluorescence spectra of the protein was recorded using the Jasco FP-8500 spectrofluorometer. Briefly, 1 μm PfRad51 protein in phosphate buffer was taken in a quartz cuvette with a 1-cm path length, and the excitation wavelength of 295 nm was used. The emission spectra of PfRad51 protein was recorded in both the absence and presence of B02. The maximum fluorescence emission at 332 nm wavelength was recorded for PfRad51 (F0) and in the presence of varying B02 concentrations (0.05 to 0.4 μm) (F1). A blank run (0.1 m phosphate buffer with vehicle) was subtracted from each spectral reading. The ratio of fluorescence intensities (F1/F0) was calculated at every drug concentration. A Stern-Volmer plot was plotted taking the mean value from three independent experiments, and the association constant (Ka), which is the slope of the graph, was determined.
Inhibition of P. falciparum in vitro culture by B02
Highly synchronous P. falciparum cultures of the trophozoite stage were treated with various concentrations of B02 (1 nm to 100 μm) for 48 h. The percent of parasitemia was measured by two independent methods, Giemsa staining method and SYBR Green-I–based assay. For the Giemsa staining method, thin smear slides were prepared, fixed with methanol, and stained with Giemsa solution and for counting the number of parasitized erythrocytes in random, adjacent microscopic fields equivalent to about 12,000 erythrocytes under oil immersion. The slides were counted by two independent scientists. The SYBR Green-I–based parasite quantification was performed as described previously (27). Briefly, 100 μl of in vitro culture was taken in 96-well plates containing an equal volume of lysis buffer having a SYBR Green reagent (20 mm Tris-HCl (pH 7.5), 5 mm EDTA, 0.008% saponin, and 0.08% Triton X-100) and was incubated at room temperature in the dark for 1 h. Fluorescence intensities were measured with the multimode reader (Spectramax m2e) at excitation and emission wavelengths of 485 and 530 nm, respectively. Each assay was repeated three times for reproducibility. Corrected fluorescent readings were obtained by subtracting the background readings of the culture without parasites. The 50% inhibitory concentration (IC50) of B02 was determined by plotting the concentration of the drug versus the percent inhibition on a semi-log graph by using GraphPad Prism.
The MEF cell line and human liver cell line (HepG2) were cultured in a culture medium of Dulbecco's modified Eagle's medium (Gibco), supplemented with 10% fetal bovine serum (Gibco), 0.5 mg/ml gentamicin (Sigma), and incubated at 37 °C in a humidified atmosphere of 5% CO2. Following treatments with various concentrations of B02 for the stipulated times, the cells were collected by trypsinization and enumerated the number of viable and dead cells by trypan blue (Sigma) dye exclusion method using a hemocytometer under microscope.
Interaction of B02 with DHA or CQ by fixed-ratio isobologram method
To determine the interactions between different drug combinations, synchronous P. falciparum cultures of the trophozoite stage were used. For the determination of the IC50 of DHA, parasite cultures were incubated in the presence of various concentrations of DHA (0.1 nm to 1 μm) for 48 h followed by parasite quantification by SYBR Green-I based method. The IC50 value was taken from a semi-log plot of percent inhibition versus concentrations of DHA using GraphPad Prism. Similarly, the IC50 of CQ was also determined. Fixed ratio of drug combination was performed as mentioned (28, 29). Briefly, in combination assay DHA/CQ and B02 were combined in four fixed ratios (4:1, 3:2, 2:3, and 1:4). Approximately 8-fold IC50 concentrations of each compound were taken as 100% so that the IC50 of the individual compound falls in between the third and fourth of a 2-fold serial dilution. The effect of each of the drug combinations (along with their 2-fold serial dilutions) on the intra-erythrocytic development of the parasite was assayed in triplicate in a flat-bottom 96-well plate. Each experimental well contained a total volume of 200 μl of medium with or without drug and 1% parasitemia with 5% hematocrit. After seeding the wells, plates were incubated at 37 °C for a 48-h asexual cycle, followed by estimation of a parasite count by the SYBR Green I–based method. The IC50 value for each combination of drugs was identified by plotting the semi-log graph by using GraphPad Prism software. The FIC for each drug was determined by using the equation FIC = IC50 of drug in mixture/IC50 of drug alone. The interaction between B02 and DHA was identified by using the FIC values of both drugs using Equation 1.
The isobologram was prepared by using GraphPad Prism software. ∑FIC <1 represents synergism; ∑FIC ≥1 and <2 represent additive interaction; and ∑FIC ≥ 2 represents antagonism. Similar equation was used for CQ and B02 combination.
Inhibition of the repair of damaged DNA in the presence B02
For determining the IC50 of B02 upon treatment with DNA-damaging agent MMS, the cultures were divided into two equal parts. One part was treated with 0.005% MMS for 6 h followed by washing off MMS and returned to growth in complete medium containing various concentrations of B02 (as mentioned earlier) for one generation (48 h). The second aliquot of the culture was maintained in the presence of B02 without prior treatment with MMS. The culture without MMS and B02 treatment acted as the positive control.
Comet assay
The extent and recovery of DNA damage were assessed by alkaline comet assay. Briefly, synchronous trophozoite parasites were treated with 0.05% MMS for 6 h. Higher concentration of MMS was used in this assay to create numerous DSBs in the parasite genome so that they can be easily visualized on the gel. For measuring the viability of the parasites due to the MMS treatment, an aliquot of the culture was returned to growth in fresh culture medium without MMS, and the parasitemia were determined after 48 h as described elsewhere (7). For performing comet assay, after the treatment the washed parasites were returned to growth for 40 h in the presence or absence of B02 (8 μm). Parasites were harvested at different time points (at 0 h and after 10, 20, and 40 h). Alkaline comet assays were performed with saponin-lysed parasites as described earlier (20). The comets were visualized using a laser scanning confocal Microscope (Carl Zeiss). From each sample the extent of DNA damage and tail migration (horizontal distance from the end of the head to the end of the tail) was calculated using Comet Assay IV software. Quantification of data were done by using GraphPad Prism software.
IFA
IFA of parasites were performed according to the protocol described elsewhere (18). Briefly, parasite cultures pre-treated with B02 (8 μm) for 12 h were divided into two parts. One part was treated with 0.05% MMS for 6 h, and the other part remained untreated. Similarly, parasite cultures that were not pre-treated with B02 were also treated with MMS. As a control, parasites that were neither treated with B02 nor with MMS were also analyzed. Cold methanol-fixed slides were treated with anti-PfRad51 antibody (1:100 dilution) followed by incubation with anti-rabbit IgG (FITC-conjugated) (1:200 dilution). Parasite nuclei were stained with DAPI (10 μg/ml) and visualized using a laser-scanning confocal microscope (Carl Zeiss). Parasite samples not treated with MMS acted as the “no DNA damage control.” Parasite samples treated with B02 but not with MMS were also used as a control to demonstrate that B02 treatment per se does not induce DNA damage.
Yeast two-hybrid analysis
Yeast two-hybrid analysis was performed as described earlier (3). Briefly, the strain NRY13 (3) was grown in Sc–Leu–Ura medium with or without B02 (8 μm) up to mid-log phase. To ensure the presence of the bait and the prey plasmid, cells were patched on an Sc–Leu–Ura plate with or without B02. To score for protein–protein interaction, cells were also patched on an Sc–Leu–Ura–Ade plate with or without B02 and allowed to grow for 72 h.
Bioinformatics analysis
Homology model of PfRad51 protein was generated by using offline tool MODELLER (30), as well as the most widely used on-line server I-TASSER (31). Homology models of Rad51 were developed from the X-ray crystal structures of yeast Rad51 (PDB (32) codes 1SZP (33), 3LDA (34), 1XU4 (35), 1PZN (36), and Rad A (PDB code 2ZUC (37). Best models were selected based on the C-Scores in I-TASSER and DOPE Score in MODELLER. However, for further analysis only I-TASSER models were selected because the large gaps in the template X-ray structures of Rad51 could not be modeled efficiently in MODELLER. Models were viewed in PyMOL (38). Models were analyzed using MolProbity (39). To rectify finer issues, a short partially constrained molecular dynamics simulation–based energy minimization was run using the YASARA Minimization Server (http://www.yasara.org/minimizationserver.htm)4 (40). This analysis allowed us to obtain the final relaxed structures which are ready for docking studies.
Most of the ligands were obtained from the ZINC Database (41), as ready to dock 3D models with different bond types at different pH environments. Ligands, A04 and B01, were obtained as 2D models from NCBI PubChem and converted to 3D structures on the PRODRG server (42).
The docking search grid box was defined to encompass residues in and around the ATPase domain of the Rad51 protein models to obtain the most effective binding sites. For docking with Autodock Vina (43), we converted the obtained ligands from mol2 files into respective PDBQT files using MGLTools suite (44). For rigid receptor docking, we converted the protein models into single PDBQT files. For docking flexible residues, we also converted the protein models into a flexible-residue PDBQT file. (Residues set as flexible were the residues of the Walker domain that are well-conserved across different organisms and species.) Because the architecture of AutoDock Vina is such that it allows for variability of results from run to run, we ran the docking calculations a total of three times for both rigid and flexible docking to obtain a more concordant set of results.
In DOCK 6.6 (45), ligand models were fed directly as mol2 files. The protein models, however, needed to be processed. First, a receptor surface is created after removal of all hydrogen atoms from the model. It is then used to identify ligand-binding cavities by generating spheres that fit onto the surface and then by clustering the intersecting spheres. Cluster 3 in the case of PfRad51 was selected for docking as it is present in the ATPase domain of the protein model. The PfRad51 cluster 3 had to be manually trimmed as it encompassed more than the ATPase domain. A box of sufficient size (5 Å in PfRad51) encompassing the cluster was then defined as a docking grid and was pre-calculated to minimize calculations while docking. Thereafter, docking calculations were run, and the top docked poses for each ligand were generated. The generated poses were then rescored using the AMBER score (46) (GBSA score) function of DOCK 6. This was used to perform short molecular dynamics simulations of the ligand as well as the proximal residues of the protein model to obtain an induced fit of the ligand to the protein (47). The docking protocol was verified by docking ATP, the native ligand of the ATPase domain of PfRad51 protein. We observed binding modes similar to that present in the X-ray structures of yeast Rad51 in the top five docked poses.
Author contributions
P. V., D. D., N. S., S. B., and M. K. B. investigation; S. B. and M. K. B. supervision; M. K. B. conceptualization; M. K. B. resources; M. K. B. formal analysis; M. K. B. funding acquisition; M. K. B. writing-original draft.
Supplementary Material
Acknowledgments
Support from Department of Science and Technology-FIST and University Grant Commission-SAP to the Department of Biochemistry is acknowledged. Computational work incorporated in the paper was carried out at the Department of Biotechnology and funded the Bioinformatics Infrastructure Facility, University of Hyderabad. We thank Mrinnanda Bhattacharya for line editing of the manuscript.
This work was supported by grants from DST-SERB, Government of India Grant DST-SERB/2015/000999 (to M. K. B.), and UPE-II (University of Hyderabad) (to M. K. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- ART
- artemisinin
- DSB
- DNA double-strand break
- HR
- homologous recombination
- CQ
- chloroquine
- PY
- pyrimethamine
- PDB
- Protein Data Bank
- FIC
- fractional inhibitory concentration
- ssDNA
- single-strand DNA
- MMS
- methyl methanesulfonate
- IFA
- immunofluorescence assay
- DAPI
- 4,6-diamidino-2-phenylindole
- RBC
- red blood cell
- MEF
- mouse embryonic fibroblast
- IDC
- intra-erythrocytic developmental cycle
- Y2H
- yeast two-hybrid
- DHA
- dihydroartemisinin.
References
- 1. World Health Organization. (2015) World Malaria Report 2015. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2. Frankenberg-Schwager M., and Frankenberg D. (1990) DNA double-strand breaks: their repair and relationship to cell killing in yeast. Int. J. Radiat. Biol. 58, 569–575 10.1080/09553009014551931 [DOI] [PubMed] [Google Scholar]
- 3. Roy N., Bhattacharyya S., Chakrabarty S., Laskar S., Babu S. M., and Bhattacharyya M. K. (2014) Dominant negative mutant of Plasmodium Rad51 causes reduced parasite burden in host by abrogating DNA double-strand break repair. Mol. Microbiol. 94, 353–366 10.1111/mmi.12762 [DOI] [PubMed] [Google Scholar]
- 4. Stark J. M., Hu P., Pierce A. J., Moynahan M. E., Ellis N., and Jasin M. (2002) ATP hydrolysis by mammalian RAD51 has a key role during homology-directed DNA repair. J. Biol. Chem. 277, 20185–20194 10.1074/jbc.M112132200 [DOI] [PubMed] [Google Scholar]
- 5. Chi P., Van Komen S., Sehorn M. G., Sigurdsson S., and Sung P. (2006) Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair 5, 381–391 10.1016/j.dnarep.2005.11.005 [DOI] [PubMed] [Google Scholar]
- 6. Symington L. (2002) Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 66, 630–670 10.1128/MMBR.66.4.630-670.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bhattacharyya M. K., and Kumar N. (2003) Identification and molecular characterisation of DNA damaging agent induced expression of Plasmodium falciparum recombination protein PfRad51. Int. J. Parasitol. 33, 1385–1392 10.1016/S0020-7519(03)00212-1 [DOI] [PubMed] [Google Scholar]
- 8. Bhattacharyya M. K., Bhattacharyya nee Deb S., Jayabalasingham B., and Kumar N. (2005) Characterization of kinetics of DNA strand-exchange and ATP hydrolysis activities of recombinant PfRad51, a Plasmodium falciparum recombinase. Mol. Biochem. Parasitol. 139, 33–39 10.1016/j.molbiopara.2004.09.007 [DOI] [PubMed] [Google Scholar]
- 9. Huang F., Motlekar N. A., Burgwin C. M., Napper A. D., Diamond S. L., and Mazin A. V. (2011) Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 6, 628–635 10.1021/cb100428c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Budke B., Logan H. L., Kalin J. H., Zelivianskaia A. S., Cameron McGuire W., Miller L. L., Stark J. M., Kozikowski A. P., Bishop D. K., and Connell P. P. (2012) RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 40, 7347–7357 10.1093/nar/gks353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Normand A., Rivière E., and Renodon-Cornière A. (2014) Identification and characterization of human Rad51 inhibitors by screening of an existing drug library. Biochem. Pharmacol. 91, 293–300 10.1016/j.bcp.2014.07.033 [DOI] [PubMed] [Google Scholar]
- 12. Budke B., Kalin J. H., Pawlowski M., Zelivianskaia A. S., Wu M., Kozikowski A. P., and Connell P. P. (2013) An optimized Rad51 inhibitor that disrupts homologous recombination without requiring Michael acceptor reactivity. J. Med. Chem. 56, 254–263 10.1021/jm301565b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bhattacharyya M. K., Norris D. E., and Kumar N. (2004) Molecular players of homologous recombination in protozoan parasites: implications for generating antigenic variation. Infect. Genet. Evol. 4, 91–98 10.1016/j.meegid.2004.01.008 [DOI] [PubMed] [Google Scholar]
- 14. Chalapareddy S., Chakrabarty S., Bhattacharyya M. K., and Bhattacharyya S. (2016) radicicol-mediated inhibition of topoisomerase VIB-VIA activity of the human malaria parasite Plasmodium falciparum. mSphere 1, e00025–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gopalakrishnan A. M., and Kumar N. (2013) Opposing roles for two molecular forms of replication protein A in Rad51-Rad54-mediated DNA recombination in Plasmodium falciparum. MBio 4, e00252–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Badugu S. B., Nabi S. A., Vaidyam P., Laskar S., Bhattacharyya S., and Bhattacharyya M. K. (2015) Identification of Plasmodium falciparum DNA repair protein Mre11 with an evolutionarily conserved nuclease function. PLoS ONE 10, e0125358 10.1371/journal.pone.0125358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haaf T., Golub E. I., Reddy G., Radding C. M., and Ward D. C. (1995) Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. U.S.A. 92, 2298–2302 10.1073/pnas.92.6.2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitra P., Banu K., Deshmukh A. S., Subbarao N., and Dhar S. K. (2015) Functional dissection of proliferating-cell nuclear antigens (1 and 2) in human malarial parasite Plasmodium falciparum: possible involvement in DNA replication and DNA damage response. Biochem. J. 470, 115–129 10.1042/BJ20150452 [DOI] [PubMed] [Google Scholar]
- 19. Kokabu Y., and Ikeguchi M. (2013) Molecular modeling and molecular dynamics simulations of recombinase Rad51. Biophys. J. 104, 1556–1565 10.1016/j.bpj.2013.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gopalakrishnan A. M., and Kumar N. (2015) Antimalarial action of artesunate involves DNA damage mediated by reactive oxygen species. Antimicrob. Agents Chemother. 59, 317–325 10.1128/AAC.03663-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Breitinger H.-G., and Acree B. (eds) (2012) Drug Synergy–Mechanisms and Methods of Analysis, Toxicity and Drug Testing. InTech. 10.5772/30922 [DOI] [Google Scholar]
- 22. Klonis N., Creek D. J., and Tilley L. (2013) Iron and heme metabolism in Plasmodium falciparum and the mechanism of action of artemisinins. Curr. Opin. Microbiol. 16, 722–727 10.1016/j.mib.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 23. Ariey F., Witkowski B., Amaratunga C., Beghain J., Langlois A. C., Khim N., Kim S., Duru V., Bouchier C., Ma L., Lim P., Leang R., Duong S., Sreng S., Suon S., et al. (2014) A molecular marker of artemisinin resistant Plasmodium falciparum malaria. Nature 505, 50–55 10.1038/nature12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miotto O., Amato R., Ashley E. A., MacInnis B., Almagro-Garcia J., Amaratunga C., Lim P., Mead D., Oyola S. O., Dhorda M., Imwong M., Woodrow C., Manske M., Stalker J., Drury E., et al. (2015) Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 47, 226–234 10.1038/ng.3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gupta D. K., Patra A. T., Zhu L., Gupta A. P., and Bozdech Z. (2016) DNA damage regulation and its role in drug-related phenotypes in the malaria parasites. Sci. Rep. 6, 23603 10.1038/srep23603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Achanta S. S., Varunan S. M., Bhattacharyya S., and Bhattacharyya M. K. (2012) Characterization of Rad51 from apicomplexan parasite Toxoplasma gondii: an implication for inefficient gene targeting. PLoS ONE 7, e41925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chalapareddy S., Bhattacharyya M. K., Mishra S., and Bhattacharyya S. (2014) radicicol confers mid-schizont arrest by inhibiting mitochondrial replication in Plasmodium falciparum. Antimicrob. Agents Chemother. 58, 4341–4352 10.1128/AAC.02519-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fivelman Q. L., Adagu I. S., and Warhurst D. C. (2004) Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob. Agents Chemother. 48, 4097–4102 10.1128/AAC.48.11.4097-4102.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tallarida R. J. (2006) An overview of drug combination analysis with isobolograms. J. Pharmacol. Exp. Ther. 319, 1–7 10.1124/jpet.106.104117 [DOI] [PubMed] [Google Scholar]
- 30. Webb B., and Sali A. (2014) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 47, 5.6.1–32 10.1002/0471250953.bi0506s47 [DOI] [PubMed] [Google Scholar]
- 31. Yang J., Yan R., Roy A., Xu D., Poisson J., and Zhang Y. (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Conway A. B., Lynch T. W., Zhang Y., Fortin G. S., Fung C. W., Symington L. S., and Rice P. A. (2004) Crystal structure of a Rad51 filament. Nat. Struct. Mol. Biol. 11, 791–796 10.1038/nsmb795 [DOI] [PubMed] [Google Scholar]
- 34. Chen J., Villanueva N., Rould M. A., and Morrical S. W. (2010) Insights into the mechanism of Rad51 recombinase from the structure and properties of a filament interface mutant. Nucleic Acids Res. 38, 4889–4906 10.1093/nar/gkq209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu Y., Qian X., He Y., Moya I. A., and Luo Y. (2005) Crystal structure of an ATPase-active form of Rad51 homolog from Methanococcus voltae: insights into potassium dependence. J. Biol. Chem. 280, 722–728 10.1074/jbc.M411093200 [DOI] [PubMed] [Google Scholar]
- 36. Shin D. S., Pellegrini L., Daniels D. S., Yelent B., Craig L., Bates D., Yu D. S., Shivji M. K., Hitomi C., Arvai A. S., Volkmann N., Tsuruta H., Blundell T. L., Venkitaraman A. R., and Tainer J. A. (2003) Full-length archaeal Rad51 structure and mutants: mechanisms for RAD51 assembly and control by BRCA2. EMBO J. 22, 4566–4576 10.1093/emboj/cdg429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang Y. W., Ko T. P., Lee C. D., Chang Y. C., Lin K. A., Chang C. S., Wang A. H., and Wang T. F. (2009) Three new structures of left-handed RADA helical filaments: structural flexibility of N-terminal domain is critical for recombinase activity. PLoS ONE 4, e4890 10.1371/journal.pone.0004890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schrödinger, LLC (2015) The PyMOL Molecular Graphics System, Version 1.7.1.0, Schrödinger, LLC, New York [Google Scholar]
- 39. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., and Karplus K. (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77, Suppl. 9, 114–122 10.1002/prot.22570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Irwin J. J., Sterling T., Mysinger M. M., Bolstad E. S., and Coleman R. G. (2012) ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768 10.1021/ci3001277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schüttelkopf A. W., and van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 10.1107/S0907444904011679 [DOI] [PubMed] [Google Scholar]
- 43. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lang P. T., Brozell S. R., Mukherjee S., Pettersen E. F., Meng E. C., Thomas V., Rizzo R. C., Case D. A., James T. L., and Kuntz I. D. (2009) Dock 6: combining techniques to model RNA–small molecule complexes. RNA 15, 1219–1230 10.1261/rna.1563609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A. (2004) Development and testing of a general Amber force field. J. Comput. Chem. 25, 1157–1174 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- 47. Graves A. P., Shivakumar D. M., Boyce S. E., Jacobson M. P., Case D. A., and Shoichet B. K. (2008) Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J. Mol. Biol. 377, 914–934 10.1016/j.jmb.2008.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.