

Abstract
Fusion peptides (FPs) in spike proteins are key players mediating early events in cell-to-cell fusion, vital for intercellular viral spread. A proline residue located at the central FP region has often been suggested to have a distinctive role in this fusion event. The spike glycoprotein from strain RSA59 (PP) of mouse hepatitis virus (MHV) contains two central, consecutive prolines in the FP. Here, we report that deletion of one of these proline residues, resulting in RSA59 (P), significantly affected neural cell syncytia formation and viral titers postinfection in vitro. Transcranial inoculation of C57Bl/6 mice with RSA59 (PP) or RSA59 (P) yielded similar degrees of necrotizing hepatitis and meningitis, but only RSA59 (PP) produced widespread encephalitis that extended deeply into the brain parenchyma. By day 6 postinfection, both virus variants were mostly cleared from the brain. Interestingly, inoculation with the RSA59 (P)–carrying MHV significantly reduced demyelination at the chronic stage. We also found that the presence of two consecutive prolines in FP promotes a more ordered, compact, and rigid structure in the spike protein. These effects on FP structure were due to proline's unique stereochemical properties intrinsic to its secondary amino acid structure, revealed by molecular dynamics and NMR experiments. We therefore propose that the differences in the severity of encephalitis and demyelination between RSA59 (PP) and RSA59 (P) arise from the presence or absence, respectively, of the two consecutive prolines in FP. Our studies define a structural determinant of MHV entry in the brain parenchyma important for altered neuropathogenesis.
Keywords: fusion protein, membrane fusion, plus-stranded RNA virus, neuroinflammation, neurodegeneration, central proline, fusion peptide, mouse hepatitis virus, spike glycoprotein, viral titer, syncytia formation, demyelination, neuropathology
Introduction
The fusion of the viral envelope with the host cell membrane can lead to the entry of the viral genome either through pore formation or the internalization of the virus into the host cell following endocytosis. This process has been widely studied using mutagenesis (1), structural biology (2), and fluorescence resonance transfer assays (3), coupled with in vitro studies, such as lipid systems involving synthetic peptides (4). However, little is known about the contribution of individual residues to this process and the downstream consequences, such as pathogenesis. Some consensus exists, such as the role of a cleavage site in fusion proteins in enhancing fusogenic activity, a hydrophobicity requirement of residues in the FP4 to facilitate membrane disruption, and a possible role of the heptad repeat during the fusion process and viral entry.
Viral FPs are typically short, apolar, and alanine/glycine-rich segments of the fusion protein, which are believed to participate in early events of the virus–host contact process (5). These polypeptides tend to show high amino acid residue conservation within a virus family but little similarity across families (1). The location of the FP varies in the primary structure of the fusion protein. It is often located adjacent to heptad repeats that preferentially interact with host lipid bilayers. Ideally, the FPs need to be membranotropic, yet in several cases they also include charged residues (2) and contain a proline residue at the center (6). The specific role of proline in FP has been experimentally investigated through mutation studies in several viruses, such as avian sarcoma/leucosis virus (7), Ebola virus (8), vesicular stomatitis virus (9), and hepatitis C virus (10). Interestingly, central proline-containing FPs are present across different classes of fusion proteins, although they are not conserved. This suggests that their role in the fusion process may be important but not always essential.
Although FP or fusion protein sequences are largely conserved within virus species and strains, small variations impart diversity and complexity to the fusion process that makes residue contributions difficult to elucidate. For example, a central proline in the FP of spike glycoprotein of the betacoronavirus genus appears to carry a variety of fusion mechanisms. In the JHM strain of MHV, fusion and entry can occur directly at the cell surface, after either receptor binding or endocytosis or both (11). Based on the strain of MHV and host cell type, the fusion mechanisms can differ and at times may appear distinct from each other, as observed in the case of MHV-A59 and MHV-2 (12, 13). Herein, MHV-A59 with an H716D mutation in the spike protein was found to be deficient in cell–cell fusion due to cleavage impairment (14). The alteration of the cleavage site sequence in MHV-A59 (corresponding to the MHV-2 cleavage sequence) also delayed cell–cell fusion, whereas the MHV-2 strain spike itself may not be cleaved at all for fusion. Interestingly, the MHV-A59 spike protein derived from mouse liver homogenates was not found to be cleaved at all, suggesting that cleavage is not essential for entry and spread in vivo (15). Even low pH, which can at times act as a trigger, does not show a consistent trend, as a cleavage-competent (cleavage site–engineered) MHV-2 spike protein has been shown to cause infection even at neutral pH (13). Two other mechanisms that are known to activate fusion through specific interactions with target cell receptors (like the most common CEACAM1 receptor) at neutral pH, or receptor priming at neutral pH followed by activation at a low pH, do not clarify the existing ambiguities regarding the precise combination of factors required to successfully initiate and drive each fusion process. Without knowledge of the exact combination of factors dominating a given fusion process, our understanding of this very important event remains incomplete, impairing our ability to fundamentally understand the origins of virus infection.
In 1990, Chambers et al. (16) proposed an internal FP (residues 929–944) as a candidate fusion domain based on its hydrophobicity and location adjacent to the heptad repeat domains in the demyelinating strain (MHV-A59). Previous studies in MHV-A59 FP have shown that mutagenesis/substitution of the methionine residue at position 936 with lysine (M936K) or leucine (M936L) of the 929–944 domains did not affect fusion. However, whereas substitution of the proline residue at position 938 with lysine (P938K) partially impaired fusion, replacing the same proline residue with a leucine residue did not have any effect on fusion (17). No studies are available to understand whether two consecutive proline residues play a role in demyelinating strain fusogenicity.
We were interested in identifying the distinct role of the central proline in the fusion peptide and whether its insertion/deletion could alter the kinetics of the fusion process and consequent neuropathogenesis. Thus, we engineered an internal FP mutant of MHV in which one proline has been deleted from RSA59, an isogenic spike protein recombinant strain of MHV (engineered from the parental strain MHV-A59), which encodes a spike glycoprotein that is expressed on the virion envelope. Spike protein mediates many biological properties of MHV, including receptor attachment, virus cell fusion during entry, cell-to-cell fusion during viral spread, and immune activation (18–20). The original RSA59 strain FP contains two consecutive prolines and enhanced GFP (EGFP). For the control study, we used RSMHV2, an isogenic recombinant strain of MHV-A59 (background genes are from the demyelinating strain MHV-A59) except for the spike protein. RSMHV2 encodes the spike protein from the nonfusogenic/nondemyelinating parental strain MHV-2. RSA59 and RSMHV2 differ in their ability to cause cell-to-cell fusion as well as their ability to cause demyelination (20–22). The RSMHV2 strain FP differs from RSA59 FP with only one central proline. Tables 1 and 2 depict the nomenclature of all of the parental recombinant strains and mutant strains. In vitro and in vivo studies were conducted to check the alteration of viral entry, spread, and neuropathogenesis of each strain. These were combined with computational and structural biology studies on the FP and spike protein fusion domain to understand the specific role of proline in altering the fusion phenotype. The studies reveal why and to what extent a central proline in FP of MHV can alter the fusion determinant and pathogenesis and provide generic insight into factors that play an important role in this process. The findings also reveal a suitable system to expand the study of cellular mechanisms of MHV spread and pathogenesis. Our study helps to understand how specific amino acid mutations in FP may result in alterations of virus infectivity and whether this insight can be used for therapeutic purposes.
Table 1.
Nomenclature of different parental strains, recombinant strains, and proline mutated strains of MHV
| MHV strains | Description |
|---|---|
| WT MHV-A59 (19, 44, 46) | Parental demyelinating strain of MHV which is fusion-positive |
| WT MHV2 (19, 32, 47) | Parental non-demyelinating strain of MHV which is fusion-negative |
| RSA59 (PP) (32) | EGFP-expressing isogenic spike protein recombinant strain of MHV-A59 (a demyelinating strain of MHV, expressing MHV-A59 spike gene in the MHV-A59 background). The FP has two central consecutive prolines |
| RSMHV2 (P) (13, 22) | EGFP-expressing isogenic spike protein recombinant strain of MHV2 (a non-demyelinating strain of MHV, expressing MHV-2 spike gene in the MHV-A59 background). The FP has only one central proline |
| RSA59 (P) | Proline-deleted FP mutant strain of RSA59 (PP). One proline (at position 939) has been deleted from EGFP-expressing isogenic spike protein recombinant strain of MHV-A59; RSA59 (PP). The FP has one central proline |
Table 2.
Nomenclature of spike protein constructs used for in silico studies
Results
In silico and structural biology
Overall structure of the MHV coronavirus fusion core
The MHV spike protein is a 180-kDa protein consisting of two subunits: S1 and S2. The S1 subunit contains the receptor-binding domain (RBD), and the S2 subunit contains an N-terminal FP, two heptad repeat domains (HR1 and HR2), a transmembrane domain (TM), and an intracellular cytoplasmic tail (IC) (Fig. 1a). Fig. 1b illustrates the entire amino acid sequence alignment of the MHV-A59 and MHV2 S2 domains highlighting FP and the cleavage signal site. Alignment of the full-length protein is shown in Fig. S1.
Figure 1.

Introduction to spike protein. Shown is a schematic of the domain structure of coronavirus spike, including receptor-binding domain (RBD) in the S1 subunit; the cleavage signal sequence (CSS) between S1 and S2; and putative FP, heptad repeat 1 (HR1), heptad repeat 2 (HR2), transmembrane domain (TM), and intracellular tail (IC) in the S2 subunit. a, the protein can be proteolytically cleaved at CSS into an S1 and S2 subunit, which are noncovalently linked. b, fusion domains of the MHV-A59 and MHV2 spike genes are aligned using ClustalW as described under “Materials and methods.” The cleavage site and FP are highlighted. FP with consecutive proline is more efficient in causing infection. RSA59 (PP) is more effective in its ability to cause cell-to-cell fusion compared with RSMHV2 (P) in N2a cells. A monolayer of N2a cells was infected with 1 MOI of virus from RSA59 (PP) and RSMHV2 (P) strain. The cells were incubated for 24 h at 37 °C with 5% CO2, fixed with 4% paraformaldehyde, and mounted in DAPI-containing mounting medium, and then epifluorescence microscopy was performed. Images were acquired with a Hamamatsu Orca-1 charge-coupled device camera and Image-Pro image analysis software. EGFP fluorescence (green) identifies virus-infected cells, and DAPI (blue fluorescence) stains the nucleus in the merged images. RSA59 (PP) strain infection induces giant syncytia formation (c) (multinucleated cells), but in RSMHV2 (P) strain infection (c), cell-to-cell fusion and giant syncytia formation are rare. The experiment was repeated five times.
MHV strains differ in fusogenic properties
RSA59 (PP) and RSMHV2 (P) both could infect Neuro2A (N2a), but they differed significantly in their cytopathic/fusogenic properties, as revealed by the size, rate, and degree of syncytial formation (Fig. 1c). Upon RSA59 (PP) infection, N2a cells started to fuse as early as 12 h (data not shown) and formed profuse syncytia, which commenced to increase with time until 24 h. At 24 h postinfection (p.i.), most of the giant syncytia started to dissolve, as all of the infected cells had lysed. In contrast, RSMHV2 (P)–infected individual cells rarely formed syncytia even after 24 h p.i. The experiments were repeated five times under the same conditions.
Sequence analysis of MHV spike proteins
In coronaviruses, the spike protein alone can traffic to the cell surface and is sufficient for the induction of cell-to-cell fusion (17, 23). Therefore, we questioned what differences therein could lead to altered fusion between RSA59 (PP) and RSMHV2 (P). The overall organization of the primary structure of spike proteins is shown in Fig. S1. Against the background of the 81% identical primary structure of S-MHV-A59 (PP) (spike protein of MHV-A59) and S-MHV-2 (P) (spike protein of MHV2), alterations were restricted to a 43-residue insertion in S-MHV-2 (P) between residues 496 and 538, another 3-residue deletion between residues 565 and 567 and between residues 901 and 902, and a single residue deletion at position 977. Interestingly, the 43-residue insertion was conserved in 23 other coronaviruses (including human coronavirus), and the degree of conservation varied, with cysteine being the most conserved residue, suggesting that it was more likely a deletion from S-MHV-A59 (PP) than an insertion in S-MHV-2 (P). There were no contrasting substitutions in the alignment, with only 11 substitutions from the fusion domain (all non-Gly and isolated mutations), with a deletion at amino acids 901 and 902 outside the heptad regions and unlikely to participate in any early fusion events (also verified from the structural model). Interestingly, the internal FPs for S-MHV-A59 (PP) (residues 929–952, TGATAAAMFPPWSAAAGVPFSLSV) and for S-MHV-2 (P) (residues 967–989, AGATVSAMFPWSAAAGVPFSLSV) were found to be 87.5% identical, excluding the proline deletion (Fig. 2a). The examination of the quaternary structure of the fusion domain of S-MHV-A59 (PP) obtained by cryo-EM (PDB code 3JCL) (24) showed an all α-helical structure for the fusion domain (Fig. 2b). The FP in the structure also had a segmented α-helical structure interspersed with two short loop regions (Fig. 2d). The comparative model for S-MHV-2 (P) showed similar features, although the orientations of the helices in the tertiary structures were such that they are not identically aligned (Fig. 2, c and e). Therefore, the structural role of the central proline appeared to be distinctive, with potential to distinguish the fusion activity of the two FPs; thus, it was investigated in more detail through molecular dynamics simulation and NMR studies.
Figure 2.

Trimeric model of the fusion domains S-MHV-A59 (PP), S-MHV-2 (P), energy landscapes, and their internal FPs. a, the sequence comparison of FP between S-MHV-A59 (PP) and S-MHV-2 (P) shows that S-MHV-A59 (PP) has two consecutive central prolines. A trimeric model of the fusion domain of S-MHV-A59 (PP) spanning residues 871–1116 from PDB structure 3JCL is shown in b, and the comparative model for the trimeric fusion domain was built from 3JCL for S-MHV-2 (P) spanning residue locations 911–1153 in c. The FP segment highlighted in b is also marked in a blue cartoon diagram, and proline is shown in a ball-and-stick model (d). The corresponding FP from S-MHV-2 (P) is shown in e. The snapshots shown are representatives of the largest structural clusters obtained from the MD simulation trajectories. Energy landscapes from simulation of trimeric S-MHV-A59 (P) and S-MHV-A59 (PP) show double potential wells. The population of the conformers in the trajectory can be assessed by the relative energy scale given in the key. It is also observed that the mutant S-MHV-A59 (P) has comparatively shallow potential wells (f).
Molecular dynamics (MD) of the spike protein fusion domain
Significant differences were observed between the energy landscapes obtained from equilibrium MD of the fusion domain in the spike proteins from S-MHV-2 and S-MHV-A59 (Fig. 2f). The insertion or deletion of a single proline in FP was responsible for this change in dynamics of the fusion domain from S-MHV-2 (P), S-MHV-A59 (P), and S-MHV-A59 (PP). The insertion of proline tended to contract the overall conformational state space. Because mutant S-MHV-A59 (P) showed larger deviations in the RMSD and radius of gyration compared with WT S-MHV-A59 (PP), we concluded that the deletion of proline relaxed the fusion domain. The studied fusion domains were sufficiently large (3 × 245 residues) to ensure that the results were comparable with the equilibrium MD of full-length spike proteins.
Proline-induced rigidity in FP
The dihedral fluctuation of the FP segment was suppressed whenever two consecutive prolines (PP) were present, in contrast to the presence of a single proline (P) in the chain (Fig. 3a). The mid-helical segment in FP (P) with a single proline had 20% or less helical occurrence in contrast to the FP in S-MHV-A59 (PP), where the equivalent segment had >90% occurrence of a helical conformation in the MD trajectory (Fig. 3b). Interestingly, the same segments showed an equally high helical occurrence (>90%) in methanol. The rigidity of the FP could be estimated from the hydrogen bond retention of proline in and around the FP (Table S1). For single-proline structures, the number of hydrogen bonds, as well as their occurrence, was low; in contrast, many bonds for diproline cases showed a >88% occurrence. These mainly corresponded to hydrogen bonding in the helix.
Figure 3.
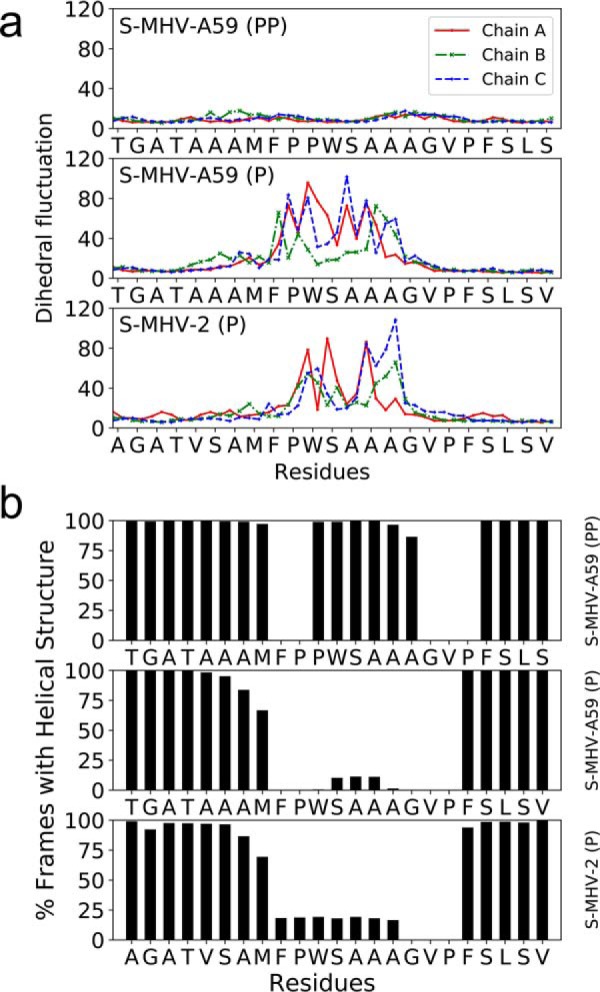
Dihedral fluctuations (in degrees) for S-MHV-A59 (PP), S-MHV-A59 (P), and S-MHV-2 (P) in water. Dihedral fluctuation is defined as the S.D. of the distribution of absolute difference from mean for a dihedral angle in the trajectory. The marker on the curve on either side of each tick mark for a residue label corresponds to ϕ and ψ backbone torsion angle for that residue (a). Statistics of helical structure detected by the program DSSP (48) for each residue in MD (fusion domain in water) trajectory around the FP (b).
Analysis of the NMR spectra of the FP fragment from S-MHV-A59 (PP)
The differential behavior of the FPs observed in the MD simulation arose from the basic stereochemistry of proline, limiting the allowed φ dihedral angles in the polypeptide backbone where it is located. To further investigate the same phenomenon, we studied a 16- and 24-residue segment of the FPs to understand their intrinsic behavior, with the caveat that these might not be reproducible when the FP was part of the full protein. The 1H-1H TOCSY NMR spectrum of the 16-residue FP showing a cross-peak between protons attached to Cα and N could be clearly attributed to two sets of conformers, one existing in cis and another in trans conformation (Fig. S2). The NOE cross-peak between Pro938 (P8) Hα and Pro939 (P9) Hδ confirmed that one of the peptides was in trans conformation (Fig. 4a and Fig. S3). The cis conformation was evidenced by a cross-peak between Pro938 (P8) Hα and Pro939 (P9) Hα (Fig. 4b). The presence of a cis-trans isomer in the polypeptide solution was also confirmed by the 1H-13C HSQC spectrum (Fig. S4). The cross-peak between Phe937 (F7) Hα and Pro938 (P8) Hδ confirmed the presence of the trans peptide conformation joining these two residues. A similar Hα-Hα cross-peak did not exist for Phe937 and Pro938, suggesting that only one cis peptide bond existed in the population. The peak volume estimates suggested approximately a 60:40 ratio for trans/cis relative to the population of isomers. The chemical shift dispersion (7.7–8.7 ppm) in the amide region (Fig. S3, blue/red labels) and secondary chemical shifts (Fig. S5) and 3JNHα couplings (Tables S2 and S3) suggested a flexible trans FP and FP in the restricted cis conformation. The 20 best structures calculated from the NMR distance constraints also showed the same in trans (Fig. 4, c and e) compared with cis (Fig. 4, d and f). The presence of a cis peptide induced a hairpin-like structure for the cis conformer with helical structures at both termini, in contrast to a largely irregular and extended structure for the trans conformer. It is interesting to note that the cis peptide, which induces a type VI turn, was not located at the center of the chain reversal section and that no hydrogen bonds stabilized the reversal of direction. In contrast, the 24-residue FP in water and D2O (10%) solvent suggested the presence of only the trans conformer in solution (Figs. S6–S9 and Table S4). The structure was mostly a random coil with a short helical segment on the C-terminal side of the proline residues. Notwithstanding, the polypeptide chain formed a closed loop–like structure that was far more compact than the 16-residue trans conformer in methanol (Fig. 4g). A short C-terminal segment downstream of the proline showed structural similarity between the 16- and 24-residue polypeptides.
Figure 4.

NMR-derived structure of FPs from S-MHV-A59 (PP) 1H-1H ROESY spectrum shows that the trans conformation can be identified by the dαδ NOE peak between P8 and P9 (red). The cross-peak between F7αH and P8δH indicates that P8 is in trans conformation (green) (a). The zoomed in picture of the 2D spectrum in b shows the dαα NOE between P8 and P9, which indicates the cis conformation (blue). Note that in both cases, the P8 exists in the trans conformation. The P8 and P9 in the polypeptide, corresponding to Pro938 and Pro939, respectively, in the FP, are shown using pink and orange colors for the 10 and 20 superposed structures determined for the respective trans and cis conformer from the NMR data (c and d). A corresponding ribbon diagram for one model is given in the bottom panel (e and f). g, a superposed structure comparing the 16-residue polypeptide (red) in methanol with the 24-residue polypeptide (blue) in water, both in trans conformation. h, root mean square fluctuation for the 16-residue FP fragment of S-MHV-A59 (PP) under combination of four MD simulation conditions (cis/trans) and (methanol/water). The FP shows greater fluctuation in water, even for the two prolines of interest, which are highlighted by a shaded background.
Proline dipeptide is more rigid under methanolic conditions
To further rationalize the observations, we used the 16-residue fusion fragment in both cis and trans form, as determined from the NMR spectra, and ran a 500-ns MD simulation in 100% methanol and water (Fig. 4h). The diproline segment was found to have the least fluctuation compared with all other segments of the FP. The magnitudes of fluctuations were greater in water than methanol. There was no isomerization event during the simulation. No notable difference in fluctuation behavior between cis and trans polypeptides in the individual solvent environments of methanol and water was observed. Thus, only proline and the solvent environment appear to be important for arresting the flexibility of a given FP structure.
In vitro
S-MHV-A59 (PP) (spike protein) traffics to the surface in in vitro transiently transfected HeLa cells
To detect the subcellular/surface localization of S-MHV-A59 (PP) (S-MHV-A59 (PP) YFP construct in pCAGGS), HeLa cells were transiently transfected and then examined by indirect immunofluorescence microscopy after 48 h of transient transfection, utilizing either YFP fluorescence or immunolabeling with anti-spike antibody.
Full-length S-MHV-A59 (PP) protein predominantly accumulated in the perinuclear region, but a large amount of the transiently expressed protein trafficked to the cell surface and induced cell-to-cell fusion to form a large syncytium (Fig. 5, a and c). To confirm the surface expression of spike protein and its ability to form syncytia, we also immunolabeled S-MHV-A59 (PP)–transfected cells with anti-MHV-A59 spike mAb. The fusion protein trafficked to the surface and was also retained in the perinuclear region (Fig. 5b). Immunolabeled cells also confirmed that the surface expression of spike protein can induce the formation of large syncytia (Fig. 5d). HeLa cells are devoid of the CEACAM1a spike receptor; thus, the syncytia induced by surface expression of the spike protein is a receptor-independent cell-to-cell fusion event. The spike protein that was expressed on the cell surface was quantified as described under “Materials and methods.” A significant percentage of spike protein was trafficked to the surface (Fig. 5e).
Figure 5.

S-MHV-A59 (PP) protein overexpression leads to its trafficking to the cell surface and causes syncytia formation. HeLa cells are transiently transfected with S-MHV-A59 (PP) (a–d). After 48 h of transfection, cells were either fixed with 95% ice-cold EtOH to visualize YFP (a and c) or immunolabeled with anti-spike antibody by a routine protocol as discussed under “Materials and methods” and observed (b and d). S-MHV-A59-(PP) can be seen surrounding the nucleus as well as on the cytoplasmic surface (a and b). Overexpression of spike to the surface leads to syncytia formation (c and d). The amount of spike protein surface expression was quantified as discussed under “Materials and methods.” ∼40% of total spike protein was expressed on the surface as compared with cytosolic fraction (e). Level of significance was determined by paired t test. n = 12; ****, p < 0.0001. Error bars, S.E.
Sequence comparison between RSA59 (PP) and RSA59 (P) spike gene
The mutant MHV strain RSA59 (P) with deleted proline was generated by a targeted RNA recombination technique, as described under “Materials and methods.” The entire spike gene of RSA59 (PP) and RSA59 (P) was sequenced and compared with the known MHV-A59 spike gene sequence (GenBankTM accession number 9629812) (14, 18, 25). Consensus sequencing was carried out by RT-PCR of viral mRNA amplified from RSA59 (PP)– and RSA59 (P)–infected L2 cells (14). The sequence analysis of the spike gene of RSA59 (P) was identical to the published sequence of the RSA59 (PP), excluding the deletion of one proline. Furthermore, the sequence analysis of the spike gene of RSA59 (PP) was completely identical to the published sequence of the MHV-A59 spike gene. No additional mutations were present in either RSA59 (PP) or RSA59 (P).
Differential fusogenic properties of RSA59 (PP) and RSA59 (P)
At 12 h p.i., RSA59 (PP) formed profuse discrete syncytia in N2a cells (Fig. 6a), but syncytia were rarely observed in RSA59 (P)–infected cultures (Fig. 6b). After 16 h of RSA59 (PP) infection, the syncytia grew bigger, and almost all of the N2a cells in the infected culture came in contact with each other (Fig. 6c); in contrast, RSA59 (P) cells were individually infected and started to fuse, but the numbers and sizes of syncytia were reduced compared with RSA59 (PP) (Fig. 6d). At 24 h p.i., larger syncytia started dissolving in RSA59 (PP)-infected culture, as almost all the infected cells lysed (Fig. 6e). However, in the case of RSA59 (P), the syncytia continued to grow in size and number with a large number of individually infected cells (Fig. 6f). The mean nuclei per syncytia were counted as described under “Materials and methods.” The number of nuclei involved in syncytia formation was significantly less in the case of RSA59 (P)–infected culture at 12 h and 16 h p.i. compared with RSA59 (PP) (Fig. 6g). At 24 h p.i, the syncytia could not be quantified, because most of the cells in RSA59 (PP)–infected cultures were dead.
Figure 6.

Comparison of syncytia formation in N2a cells infected by RSA59 (PP) and the proline-deleted mutant strain RSA59 (P). N2a cells were infected with RSA59 (PP) and RSA59 (P) at an MOI of 1. The green fluorescence is due to EGFP that is integrated into the RSA59 (PP) and RSA59 (P) genome. Images of DAPI (blue) and EGFP (green) were merged to construct the final images; a, c, and e are from RSA59 (PP)–infected N2a cells at 12, 16, and 24 h, respectively. b, d, and f show N2a cells infected by RSA59 (P) at 12, 16, and 24 h, respectively. Arrows, syncytia. The mean nuclei per syncytia were counted and plotted (g). A luciferase reporter assay was performed to compare the fusion efficiency of pMH54EGFP (PP) and pMH54EGFP (P). BHK-R cells were used as effector cells and HeLa cells as target cells. Effector cells were transfected with plasmids expressing the luciferase reporter gene. Target HeLa cells were transfected with plasmids expressing T7 RNA polymerase and spike plasmids. Luciferase activity was measured 36 h after co-culturing the effector and target cells. Relative luciferase units were determined using a luminometer (Berthold Detection Systems, Pforzheim, Germany). Experiments were repeated three times. Comparative fusion efficiency was measured in pMH54EGFP (PP)– and pMH54EGFP (P)–transfected cells after 36 h (h) (*, p < 0.05) and 48 h (i) (****, p < 0.0001). The plot shown here represents one of the experiments. The level of significance was calculated by unpaired t tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Error bars, S.E.
To quantify the fusion efficiency of spike proteins, a virus-free cell–cell fusion assay based on luciferase activity was performed as described previously (26, 27). As expected, the fusion power measured by the luciferase activity of pMH54EGFP (P) (spike plasmid with deleted proline)–expressing HeLa cells (target cells) with luciferase-expressing BHK-R (effector cells) was significantly reduced at 36 h (**, p < 0.01) and 48 h (****, p < 0.0001) post-co-culture compared with pMH54EGFP (PP) (spike plasmid with double proline)–transfected HeLa cells (Fig. 6, h and i).
Differential rate of fusion and replication kinetics of recombinant strains
The kinetics of viral fusion of recombinant viruses was studied by live cell imaging (Fig. 7a). A monolayer of N2a cells infected with RSA59 (PP) and RSA59 (P) at a multiplicity of infection (MOI) of 1 was observed for 350 min, after 8 h p.i. RSA59 (PP) established syncytia much faster than RSA59 (P).
Figure 7.

Time lapse imaging of RSA59 (PP) and and RSA59 (P) infection of N2a cells. a, still images of RSA59 (PP)– and RSA59 (P)–infected N2a cells from a time lapse video microscopy. The images were acquired 8 h p.i. and continued until 14 h p.i. The images were captured with a Hamamatsu Orca-1 charge-coupled device camera equipped with Image-Pro image analysis software. EGFP fluorescence (green) identifies the virus-infected cells. L2 cells were infected with RSA59 (PP) and RSA59 (P) at an MOI of 1 for differential growth kinetics (b). At time points 4, 8, 12, 20, and 24 h p.i., the infected cultures with medium were freeze-thawed three times and centrifuged, and viral titers were determined by a routine plaque assay. Viral titers at different time points were plotted in the GraphPad Prism version 6.01 software. The level of significance was calculated using unpaired t test. Experiments were repeated three times (n = 3); each dot represents mean of triplicates of one experiment. Furthermore, viral replication was studied by real-time PCR analysis of spike and nucleocapsid transcript at 0, 4, 8, and 12 h p.i. in L2 cells as mentioned under “Materials and methods.” Both the viruses were able to replicate efficiently, but S and N gene expression of RSA59 (P) was consistently less compared with RSA59 (PP) (c and d). The level of significance was measured using unpaired t test. *, p < 0.05; **,p < 0.01; ***, p < 0.001; ****, p < 0.0001.
The replication of RSA59 (P) was compared with RSA59 (PP) by analyzing the growth curves from 4 to 24 h p.i. A standard plaque assay was performed to calculate the titer from different time postinfected L2 cells, as mentioned under “Materials and methods.” RSA59 (P) showed a slower replication rate (Fig. 7b), as evidenced by a lower titer throughout the studied time points compared with RSA59 (PP). The two consecutive prolines in RSA59 (PP) consistently promoted a higher (∼10 times) titer compared with the proline deletion mutant RSA59 (P).
Furthermore, the replication of RSA59 (P) was compared with RSA59 (PP) by analyzing the relative transcript level of spike (S) and nucleocapsid (N) gene using real-time PCR at 0, 4, 8, and 12 h p.i. The RSA59 (P) was constantly able to replicate its S and N gene but relatively less compared with RSA59 (PP) (Fig. 7, c and d). The differential replication kinetics as well as fusion ability of spike between two viruses collectively contributed the difference in fusion efficiency of the two viruses.
In vivo
RSA59 (PP) and RSA59 (P) both induce necrotizing hepatitis at days 3 and 6 p.i. in liver tissues
Four-week-old C57Bl/6 male mice were infected intracranially with RSA59 (PP) and its mutated recombinant strain RSA59 (P). Day 3 and day 6 p.i. mice were sacrificed and studied for liver pathology. RSA59 (PP) and RSA59 (P) day 3 and 6 p.i. mice liver sections stained with hematoxylin and eosin (H&E) showed large foci of hepatitis and hepatic lesions throughout the liver section (Fig. 8, a, b, e, and f). For confirmation of hepatitis and the presence of inflammatory cells in the liver tissue, the sections were immunohistochemically labeled with anti-Iba1 (microglia/macrophage marker). Most of the inflammatory cells in both RSA59 (PP)– and RSA59 (P)–infected liver sections showed immunoreactivity for Iba1 (Fig. 8, c, d, g, and h).
Figure 8.
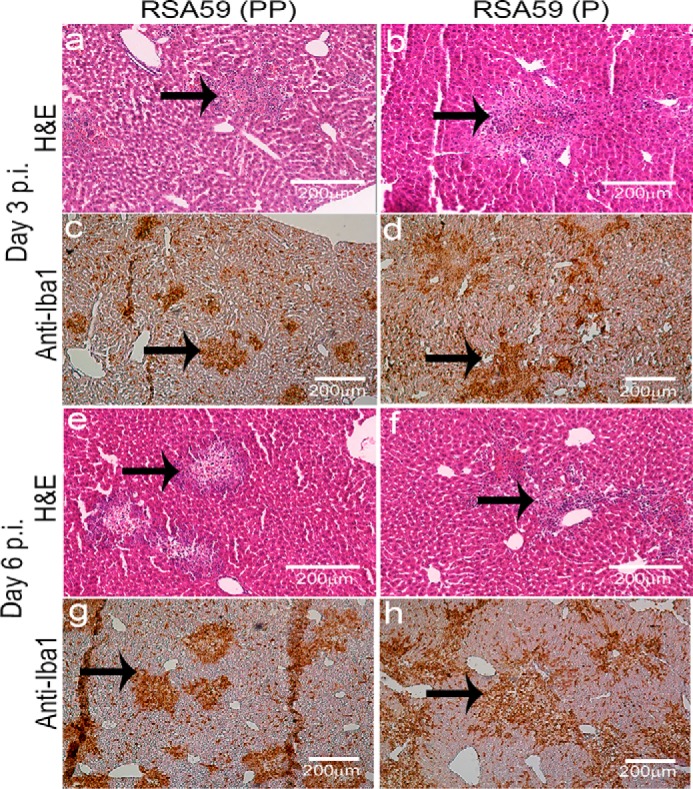
Liver pathology represents moderate to severe hepatitis following RSA59 (PP) and RSA59 (P). Liver sections of day 3 and 6 p.i. mice were stained with H&E. Similar liver pathology consisting of moderate to severe necrotizing and nonnecrotizing hepatitis was observed throughout the liver sections in both RSA59 (PP) (a and e) and RSA59 (P) (b and f) infection. Serial sections immunolabeled with anti-Iba1 antibody illustrate a similar degree of hepatic inflammation in RSA59 (PP) (c and d) and RSA59 (P) (g and h). Arrows, the presence of Iba1+ cells in hepatic lesions.
Differential neuropathological outcome in RSA59 (PP)– and RSA59 (P)–infected mice CNS
RSA59 (PP)–infected mice, similar to earlier studies (22), showed meningitis, encephalitis, and demyelination; in contrast, the mice infected with the proline mutant strain RSA59 (P) exhibited meningitis, with a reduced severity of encephalitis during the acute stage of infection and sequential myelin loss during the chronic stage of infection (day 30 p.i. as the peak of demyelination) in the CNS. This phenomenon was observed by viral antigen staining and immunohistoprofiling using serial sections of day 3 and 6 postinfected mouse brain tissues (Figs. 9–12) and day 30 spinal cord tissues (Fig. 14), as detailed below.
Figure 9.
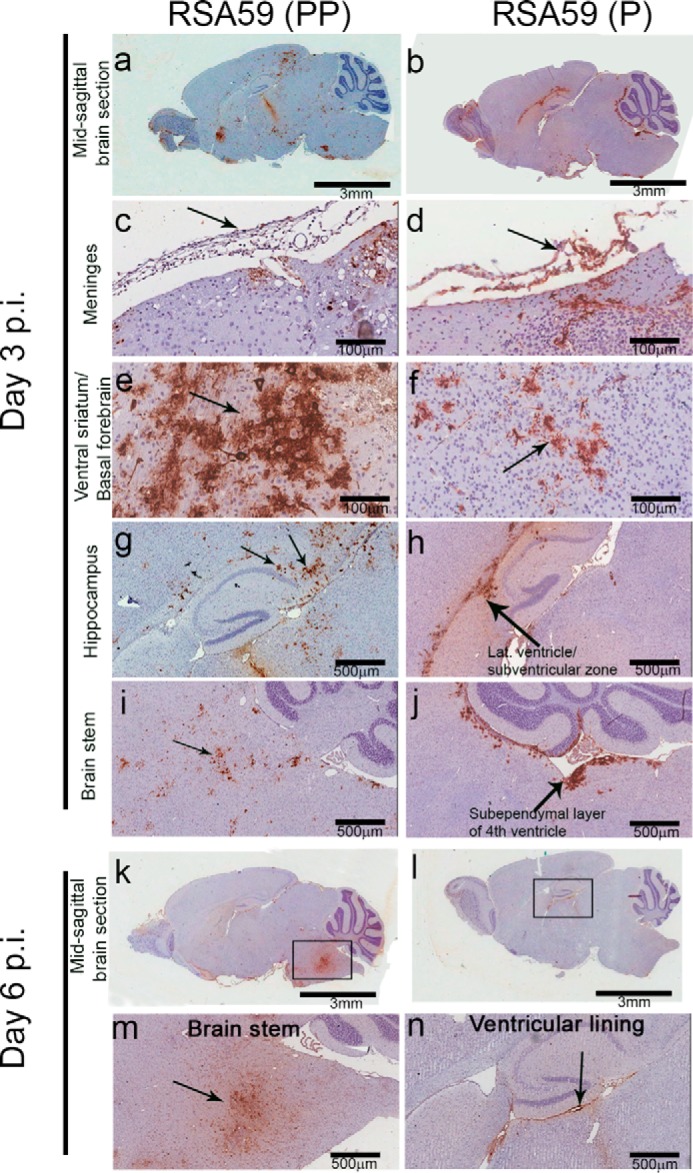
Differential distribution of viral antigen in different neuroanatomical regions from RSA59 (PP) and RSA59 (P) of day 3 and day 6 p.i. brains. Five-μm-thick mid-sagittal brain sections from RSA59 (PP)– and RSA59 (P)–infected mice were immunohistochemically stained with anti-N antibody (viral antigen). The scanned images of an entire sagittal section from RSA59 (PP) and RSA59 (P) day 3 p.i. are shown in a and b, respectively. In RSA59 (PP), viral antigen distribution was observed almost throughout the brain, including in the subpial layer of meninges (c), ventral striatum/basal forebrain (e), hippocampus (g), and brain stem (i). In contrast, in RSA59 (P)–infected brain sections, viral antigen distribution was limited to regions like the meninges (d), basal forebrain (f), lateral ventricle/subventricular zone (h), and also the subependymal layer of the fourth ventricle near the cerebellum (j). At day 6 p.i., RSA59 (PP)–infected mice showed distribution of viral antigen mainly in the posterior part of the brain stem (m) and deep cerebellar white matter with very little remnant of viral antigen in the ventricular lining (k). In comparison, RSA59 (P) viral antigen was observed to be restricted predominantly in the lateral ventricular lining (l and n). Arrows, virus antigen staining.
Figure 10.

Effect of proline residues on viral antigen distribution to different neuroanatomical regions of the brain. The distribution of viral antigen was quantified in different neuroanatomical regions of the brain based on scores as follows: 0, no infection; 1, very small foci of infection; 2, wide spread small foci of infection; 3, wide spread with large foci of infection. The mid-sagittal brain section was divided into several neuroanatomical regions (a). At day 3 p.i., RSA59 (PP) brain compared with RSA59 (P) showed significant difference in cerebral cortex, ventral striatum/basal forebrain, lateral ventricle/subventricular zone, hippocampus, thalamus, hypothalamus, midbrain, brain stem, and subependymal layer of the fourth ventricle (b–d). No significant difference was observed in meninges and cerebellum. Differences were also observed at day 6 p.i in different neuroanatomical regions, such as meninges, cerebral cortex, ventral striatum/basal forebrain, lateral ventricle/subventricular zone, thalamus, hypothalamus, midbrain, and brain stem (e–g) with no significant difference in hippocampus, cerebellum, and subependymal layer of the fourth ventricle (n = 7 mice/virus infection). The level of significance was determined by two-way ANOVA and Tukey's multiple-comparison test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Two-way ANOVA was performed to demonstrate the interaction between the type of infection (treatment) and amount of infection in different neuroanatomic regions of the brain. F(df, df) values for type of infection (treatment), amount of infection in different neuroanatomic regions, and their interaction at day 3 p.i. were F(2, 362) = 199.8 (****, p < 0.0001), F(10, 362) = 13.76 (****, p < 0.0001), and F(20, 362) = 16.35 (****, p < 0.0001), respectively, and at day 6 p.i. were F(2, 348) = 63.52 (****, p < 0.0001), F(10, 348) = 5.294 (****, p < 0.0001), and F(20, 348) = 7.220 (*, p < 0.0001), respectively. Error bars, S.E.
Figure 11.
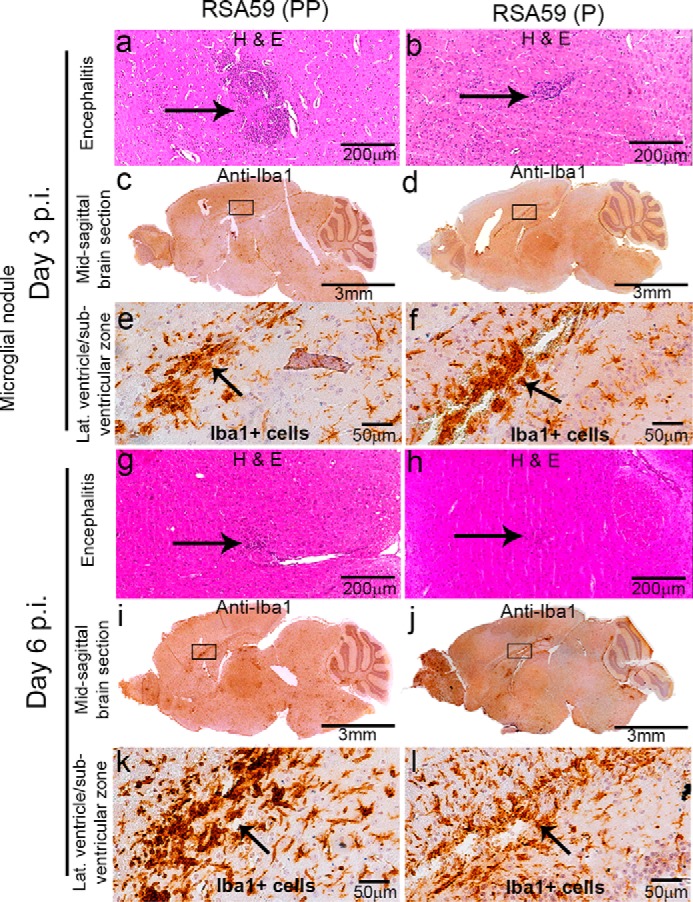
Neuroinflammation implied by Iba1+ cells in RSA59 (PP)– and RSA59 (P)–infected mice brain. Five-μm-thick mid-sagittal sections from RSA59 (PP)– and RSA59 (P)–infected mice at day 3 and 6 p.i. were stained with H&E and also immunohistochemically with anti-Iba1 (microglia/macrophage marker). H&E staining shows the observed encephalitis (in a different region of the brain). Arrows, inflamed region (microglia/macrophage with cigar-shaped nuclei) and lymphocytes in the vicinity of neurons in mice infected with RSA59 (PP) (a and g) and RSA59 (P) (b and h). At day 3 p.i., the presence of anti-Iba1+ staining indicates acute inflammation (encephalitis) in different neuroanatomic regions of RSA59 (PP)–infected brain (c; magnified image in e). Also, in RSA59 (P)–infected brain, Iba1+ cells were distributed throughout the brain parenchyma (d; magnified image in f). Representative high-magnification images show microglial/macrophage proliferation in the lateral ventricle/subventricular zone in RSA59 (PP)–infected (e) and RSA59 (P)–infected (f) brain. At day 6 p.i. in RSA59 (PP)–infected brain, Iba1+ cells were still present even when the viral antigen was cleared from the anterior part of the brain, like the basal forebrain and hippocampus, as well as in the posterior part, like the brain stem (i; magnified image in k). At day 6 p.i., in RSA59 (P)–infected brain, Iba1+ cells were widely distributed in the brain parenchyma with microglia/macrophage proliferation accumulated near the lateral ventricle/subventricular zone (j; magnified image in l). Arrows, accumulation of proliferated Iba1+ microglia/macrophage cells.
Figure 12.

Effect of proline residues on microglia/macrophage distribution to different neuroanatomical regions of the brain. The distribution as well as the level of inflammation was quantified as described under “Materials and methods.” RSA59 (PP) and RSA59 (P) infection showed significant neuroinflammation compared with control-infected mice. No significant difference was observed in distribution of microglia to different neuroanatomical regions of RSA59 (PP)–infected brain compared with RSA59 (P) at day 3 (a) and 6 p.i. (b) (n = 7 mice/virus infection). Only the lateral ventricular/subventricular zone showed significantly higher neuroinflammation in RSA59 (P)–infected compared with RSA59 (PP)–infected brain. Two-way ANOVA and Tukey's multiple-comparison test were performed to determine the statistical significance. Two-way ANOVA was performed to demonstrate the interaction between the type of infection (treatment) and amount of Iba1+ staining in different neuroanatomic regions of the brain. F(df, df) values for type of infection (treatment), amount of Iba1+ staining in different neuroanatomic regions, and their interaction at day 3 p.i. were F(2, 282) = 45.23 (****, p < 0.0001), F(8, 282) = 9.279 (****, p < 0.0001), and F(16, 282) = 2.850 (***, p < 0.001), respectively, and at day 6 p.i., the values were F(2, 262) = 61.12 (****, p < 0.0001), F(8, 262) = 5.185 (****, p < 0.0001), and F(16, 262) = 1.035 (not significant), respectively. Error bars, S.E.
Figure 14.
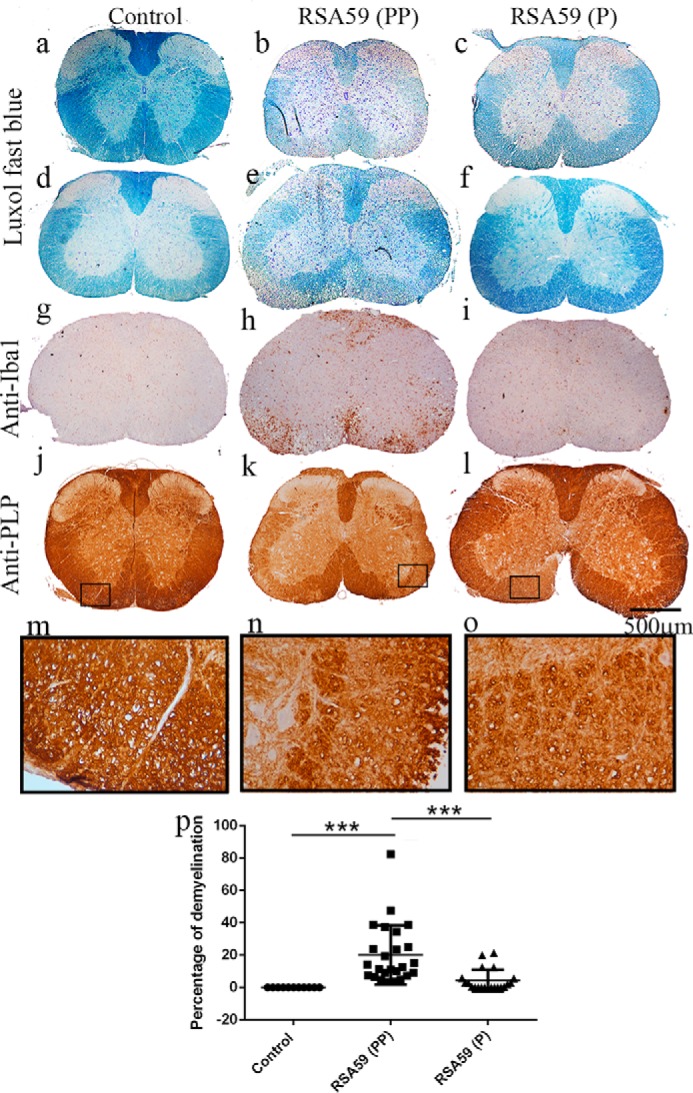
Differential intensity of demyelination observed between RSA59 (PP) and RSA59 (P) during the chronic stage of inflammation. Five-μm-thick spinal cord sections from day 30 p.i. RSA59 (PP) (b and e), RSA59 (P) (c and f), and control (a and d) mice were stained with LFB to detect myelin loss. No myelin loss was observed in mock-infected spinal cord sections (a and d). In contrast, extensive myelin loss was observed at multiple levels of spinal cord from RSA59 (PP)–infected mice (b and e), but in RSA59 (P)–infected mice, smaller patches of myelin loss were observed in very few sections of spinal cord (c and f). The corresponding serial sections were immunohistochemically labeled with anti-Iba1 (g–i) and anti-PLP antibody (j–l) to observe the presence of microglia/macrophage and morphological changes in myelin protein, respectively. RSA59 (PP)–infected sections showed the accumulation of Iba1+ cells (h) in the corresponding demyelination patches (e), whereas in RSA59 (P), very few scattered Iba1+ cells were evident (i), almost identical to mock infection (g). Anti-PLP staining also illustrated the loss of the myelin sheath in the white matter of RSA59 (PP)–infected spinal cord (k; high magnification image in n). No obvious loss of anti-PLP staining was observed in RSA59(P)–infected (l; magnified image in o) spinal cord sections. In the mock-infected spinal cord section, anti-PLP-stained myelin was intact (j; magnified image in m). Quantification of demyelination was plotted in a scatter diagram (p), and the level of significance was calculated by one-way ANOVA and Tukey's multiple-comparison test. n = 7; ***, p < 0.001. F(df, df) values for infection are F(2, 54) = 13.04 (****, p < 0.0001). Error bars, S.E.
Differential distribution of viral antigen in brain sections infected with RSA59 (PP) versus RSA59 (P)
Sagittal brain sections from RSA59 (PP)– and RSA59 (P)–infected mice at days 3 and 6 p.i. were tested for viral antigen distribution in different anatomic regions of the brain (Fig. 9, a, b, k, and l). Brain sections were labeled immunohistochemically with virus-specific anti-nucleocapsid (anti-N) antibody. Immunostaining data revealed that day 3 RSA59 (PP) p.i. brain section viral antigen was extensively distributed to different neuroanatomic regions, including meninges (Fig. 9c), the site of inoculation (near the lateral geniculate nucleus), ventral striatum/basal forebrain (Fig. 9e), hippocampus (Fig. 9g), and brainstem (Fig. 9i). In contrast, in day 3 RSA59 (P) p.i. brain tissues, the amount of viral antigen distribution was significantly reduced and restricted mainly to the meninges (Fig. 9d), the lateral ventricle/subventricular zone (Fig. 9h), the subependymal layer of the fourth ventricle (Fig. 9j), and, occasionally, the basal forebrain (Fig. 9f).
In day 6 p.i. mice, RSA59 (PP) viral antigen was mainly present in the posterior part of the brain (Fig. 9, k and m) and deep cerebellar white matter, with very little remnant in the ventricular lining. In comparison, at the same time p.i., RSA59 (P) viral antigen was observed to be restricted predominantly to the lateral ventricular lining (Fig. 9, l and n), and similar to day 3 p.i., the viral antigen level was significantly reduced compared with RSA59 (PP). Widespread dissemination of viral antigen in RSA59 (PP)–infected mouse brain sections compared with RSA59 (P) indicated that the deletion of one proline resulted in compromised viral antigen spreading. The titer of the obtained brain tissues confirmed the presence of virus in RSA59 (PP) at day 3 and 6 p.i. (105 to 108 pfu/mouse); in contrast, RSA59 (P) was present at day 3 p.i (105 pfu/mouse) and below the significance level of detection at day 6 p.i.
The viral antigen distribution of RSA59 (PP) and RSA59 (P) was quantified and compared in different neuroanatomic regions of the brain (Fig. 10a) as mentioned under “Materials and methods.” At day 3 p.i., RSA59 (PP) versus RSA59 (P) significantly differed in the cerebral cortex, ventral striatum/basal forebrain lateral ventricle/subventricular zone, hippocampus, thalamus, hypothalamus, midbrain, brain stem, and subependymal layer of the fourth ventricle. Conversely, no significant difference was observed in the meninges and cerebellum (Fig. 10, b–d). Similarly, significant differences were observed at day 6 p.i. in several neuroanatomic regions, such as the meninges, cerebral cortex, ventral striatum/basal forebrain, lateral ventricle/subventricular zone, thalamus, hypothalamus, midbrain, and brain stem (Fig. 10, e–g). No significant difference was observed in the hippocampus, cerebellum, and subependymal layer of the fourth ventricle. The mean difference and S.E. of the difference in different neuroanatomic regions are shown in Table 3 and plotted as a scatter diagram in Fig. 10 (b–g).
Table 3.
Mean difference and statistical significance as an effect of proline residues on viral antigen distribution in different neuroanatomical regions of RSA59 (PP)– and RSA59 (P)–infected mouse brain
*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; NS, no significance.
| Tukey's multiple-comparison test | Day 3 p.i. |
Day 6 p.i. |
||||
|---|---|---|---|---|---|---|
| Mean difference | S.E. | Significance | Mean difference | S.E. | Significance | |
| Meninges | ||||||
| Control versus RSA59 (PP) | −1.446 | 0.2316 | **** | −0.5385 | 0.2754 | NS |
| Control versus RSA59 (P) | −1.589 | 0.2316 | **** | −1.786 | 0.2716 | **** |
| RSA59 (PP) versus RSA59 (P) | −0.1429 | 0.1975 | NS | −1.247 | 0.236 | **** |
| Cerebral cortex | ||||||
| Control versus RSA59 (PP) | −1.357 | 0.2316 | **** | −1 | 0.2754 | *** |
| Control versus RSA59 (P) | −0.5714 | 0.2316 | * | −0.4286 | 0.2716 | NS |
| RSA59 (PP) versus RSA59 (P) | 0.7857 | 0.1975 | *** | 0.5714 | 0.236 | * |
| Ventricular striatum/basal forebrain | ||||||
| Control versus RSA59 (PP) | −1.786 | 0.2316 | **** | −1 | 0.2754 | *** |
| Control versus RSA59 (P) | −0.7857 | 0.2316 | ** | −0.2857 | 0.2716 | NS |
| RSA59 (PP) versus RSA59 (P) | 1 | 0.1975 | **** | 0.7143 | 0.236 | ** |
| Lateral ventricles/subventricular zone | ||||||
| Control versus RSA59 (PP) | −1.179 | 0.2316 | **** | −0.2885 | 0.2754 | NS |
| Control versus RSA59 (P) | −2.036 | 0.2316 | **** | −1.179 | 0.2716 | **** |
| RSA59 (PP) versus RSA59 (P) | −0.8571 | 0.1975 | **** | −0.8901 | 0.236 | *** |
| Hippocampus | ||||||
| Control versus RSA59 (PP) | −0.7143 | 0.2316 | ** | −0.3333 | 0.2797 | NS |
| Control versus RSA59 (P) | 0 | 0.2316 | NS | −0.1429 | 0.2716 | NS |
| RSA59 (PP) versus RSA59 (P) | 0.7143 | 0.1975 | *** | 0.1905 | 0.2411 | NS |
| Thalamus | ||||||
| Control versus RSA59 (PP) | −1.214 | 0.2316 | **** | −1.154 | 0.2754 | *** |
| Control versus RSA59 (P) | −0.07143 | 0.2316 | NS | −0.2143 | 0.2716 | ns |
| RSA59 (PP) versus RSA59 (P) | 1.143 | 0.1975 | **** | 0.9396 | 0.236 | *** |
| Hypothalamus | ||||||
| Control versus RSA59 (PP) | −1.429 | 0.2316 | **** | −0.8462 | 0.2754 | ** |
| Control versus RSA59 (P) | −0.2143 | 0.2316 | NS | 0 | 0.2716 | NS |
| RSA59 (PP) versus RSA59 (P) | 1.214 | 0.1975 | **** | 0.8462 | 0.236 | ** |
| Midbrain | ||||||
| Control versus RSA59 (PP) | −1.929 | 0.2316 | **** | −1.231 | 0.2754 | **** |
| Control versus RSA59 (P) | −0.1429 | 0.2316 | NS | −0.2143 | 0.2716 | NS |
| RSA59 (PP) versus RSA59 (P) | 1.786 | 0.1975 | **** | 1.016 | 0.236 | **** |
| Brain stem | ||||||
| Control versus RSA59 (PP) | −2.5 | 0.2316 | **** | −2.231 | 0.2754 | **** |
| Control versus RSA59 (P) | −0.3571 | 0.2316 | NS | −0.9286 | 0.2716 | ** |
| RSA59 (PP) versus RSA59 (P) | 2.143 | 0.1975 | **** | 1.302 | 0.236 | **** |
| Cerebellum | ||||||
| Control versus RSA59 (PP) | −0.8462 | 0.2348 | ** | −0.9231 | 0.2754 | ** |
| Control versus RSA59 (P) | −1.071 | 0.2316 | **** | −1 | 0.2797 | ** |
| RSA59 (PP) versus RSA59 (P) | −0.2253 | 0.2013 | NS | −0.07692 | 0.2453 | NS |
| Sub-ependymal layer of fourth ventricle | ||||||
| Control versus RSA59 (PP) | −0.9464 | 0.2316 | *** | −0.6923 | 0.2754 | * |
| Control versus RSA59 (P) | −2.161 | 0.2316 | **** | −1.077 | 0.2754 | *** |
| RSA59 (PP) versus RSA59 (P) | −1.214 | 0.1975 | **** | −0.3846 | 0.2404 | NS |
Distribution and intensification of microglia/macrophages in RSA59 (PP)– and RSA59 (P)–infected brain sections
Day 3 and day 6 p.i. sagittal brain sections from RSA59 (PP)– and RSA59 (P)–infected mice were stained with H&E. Histopathological studies during the acute phase (days 3 and 6 p.i.) of the infection revealed that both viruses produced encephalitis characterized by parenchymal lymphocytic infiltrates and microglial nodules (Fig. 11, a, b, g, and h) compared with control (mock-infected) (not shown). To further characterize the cells, the sagittal brain sections were immunohistochemically stained with anti-Iba1 (microglia/macrophage marker) antibodies. Anti-Iba1 immunostaining revealed that the microglia/macrophages were widely distributed throughout the brain section in both RSA59 (PP)–infected (Fig. 11, c, e, i, and k) and RSA59 (P)–infected (Fig. 11, d, f, j, and l) mice. Interestingly, in the RSA59 (PP)–infected brain, Iba1+ ramified microglia/macrophages largely followed the trajectory of the viral antigen distribution with occasional distribution to other regions where viral antigen is not present; in contrast, in RSA59 (P)–infected brain, Iba1+ ramified microglia/macrophages were scattered throughout the brain section irrespective of the presence or absence of viral antigen.
The distribution, as well as the level, of inflammation (Iba1+ cells) was quantified as mentioned under “Materials and methods.” No significant difference was observed in the distribution of microglia to different neuroanatomic regions of RSA59 (PP)– and RSA59 (P)–infected mice brain. Only the lateral ventricle/subventricular zone showed a significantly higher percentage of Iba1+ staining in RSA59 (P)–infected brain compared with RSA59 (PP). The mean difference and S.E. of the difference are shown in Table 4 and plotted in a scatter diagram in Fig. 12, a and b.
Table 4.
Mean difference and statistical significance as an effect of proline residues on microglia distribution in different neuroanatomical regions of infected mouse brain
*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; NS, no significance.
| Tukey's multiple-comparison test | Day 3 p.i. |
Day 6 p.i. |
||||
|---|---|---|---|---|---|---|
| Mean difference | S.E. | Significance | Mean difference | S.E. | Significance | |
| Cerebral cortex | ||||||
| Control versus RSA59 (PP) | −1.267 | 0.6780 | NS | −2.354 | 0.7639 | ** |
| Control versus RSA59 (P) | −1.940 | 0.6780 | * | −1.762 | 0.7530 | NS |
| RSA59 (PP) versus RSA59 (P) | −0.6733 | 0.5536 | NS | 0.5921 | 0.6430 | NS |
| Ventricular striatum/ basal forebrain | ||||||
| Control versus RSA59 (PP) | −1.420 | 0.6780 | NS | −3.671 | 0.7530 | **** |
| Control versus RSA59 (P) | −2.530 | 0.6766 | *** | −3.785 | 0.7530 | **** |
| RSA59 (PP) versus RSA59 (P) | −1.111 | 0.5641 | NS | −0.1141 | 0.6300 | NS |
| Lateral ventricles/subventricular zone | ||||||
| Control versus RSA59 (PP) | −3.278 | 0.7438 | **** | −5.269 | 0.8539 | **** |
| Control versus RSA59 (P) | −6.394 | 0.7533 | **** | −4.725 | 0.8417 | **** |
| RSA59 (PP) versus RSA59 (P) | −3.116 | 0.6189 | **** | 0.5441 | 0.7187 | NS |
| Hippocampus | ||||||
| Control versus RSA59 (PP) | −1.737 | 0.6780 | * | −2.890 | 0.7639 | *** |
| Control versus RSA59 (P) | −2.261 | 0.6780 | ** | −2.695 | 0.7530 | ** |
| RSA59 (PP) versus RSA59 (P) | −0.5240 | 0.5536 | NS | 0.1947 | 0.6430 | NS |
| Thalamus | ||||||
| Control versus RSA59 (PP) | −2.285 | 0.6780 | ** | −2.980 | 0.7530 | *** |
| Control versus RSA59 (P) | −2.141 | 0.6780 | ** | −2.723 | 0.7530 | ** |
| RSA59 (PP) versus RSA59 (P) | 0.1447 | 0.5536 | NS | 0.2570 | 0.6300 | NS |
| Hypothalamus | ||||||
| Control versus RSA59 (PP) | −0.6092 | 0.6780 | NS | −2.723 | 0.7530 | ** |
| Control versus RSA59 (P) | −1.739 | 0.6866 | * | −2.185 | 0.7530 | * |
| RSA59 (PP) versus RSA59 (P) | −1.130 | 0.5641 | NS | 0.5385 | 0.6300 | NS |
| Midbrain | ||||||
| Control versus RSA59 (PP) | −2.449 | 0.6780 | ** | −2.832 | 0.7639 | *** |
| Control versus RSA59 (P) | −2.003 | 0.6780 | ** | −2.150 | 0.7530 | * |
| RSA59 (PP) versus RSA59 (P) | 0.4465 | 0.5536 | NS | 0.6818 | 0.6430 | NS |
| Brain stem | ||||||
| Control versus RSA59 (PP) | −1.162 | 0.6780 | NS | −3.471 | 0.7530 | **** |
| Control versus RSA59 (P) | −1.486 | 0.6780 | NS | −3.777 | 0.7530 | **** |
| RSA59 (PP) versus RSA59 (P) | −0.3246 | 0.5536 | NS | 0.3054 | 0.6300 | NS |
| Cerebellum | ||||||
| Control versus RSA59 (PP) | −0.8965 | 0.6780 | NS | −1.182 | 0.9184 | NS |
| Control versus RSA59 (P) | −0.8741 | 0.7082 | NS | −1.028 | 0.9274 | NS |
| RSA59 (PP) versus RSA59 (P) | 0.02238 | 0.5901 | NS | 0.1547 | 0.6430 | NS |
The spread of RSA59 (P) virus antigen to the spinal cord is significantly reduced compared with RSA59 (PP)
Day 3 and 6 p.i. spinal cord cross-sections from control–, RSA59 (PP)–, and RSA59 (P)–infected mice were stained with viral antigen (Fig. 13, a–c and g–i). In RSA59 (PP)–infected mice, viral antigen (Fig. 13, b and h) was distributed both in gray matter and white matter cells, as observed in earlier studies (20, 22), but upon RSA59 (P) infection, very few cells were positive for viral antigen (Fig. 13, c and i) in the spinal cord sections, even at day 6 p.i., indicating an inability to spread and replicate in the spinal cord. Differential staining was quantified as described under “Materials and methods,” and it was quite evident that the reduction of viral antigen staining in RSA59 (P) was significantly different compared with RSA59 (PP) at day 6 p.i. (Table 5 and Fig. 13 (m and o)).
Figure 13.

Spread and distribution of viral antigen and microglia/macrophages in RSA59 (PP)– and RSA59 (P)–infected mice spinal cord. Control (mock-infected) and virus-infected spinal cord cross-sections at days 3 and 6 p.i. were stained with anti-N antibody (a–c and g–i) as well as anti-Iba1 antibody for microglia/macrophages (d–f and j–l). No obvious viral antigen staining was observed in mock-infected spinal cord sections (a and g). At day 3 p.i. in RSA59 (PP)–infected spinal cord, viral antigen distribution was observed both in gray and white matter (b), whereas very little viral antigen staining was apparent in RSA59 (P)–infected spinal cord (c). At day 6 p.i. in RSA59 (PP), the amount of viral antigen was significantly increased in the white matter as well as in the gray-white matter junction (h), but in RSA59 (P), viral antigen was almost cleared (i). Iba1+ cells were distributed in both RSA59 (PP)– and RSA59 (P)–infected spinal cord sections compared with control both at day 3 (d–f) and day 6 p.i. (j–l). Scale bar, 50 μm. The total viral antigen–positive areas (m and o) and Iba1-positive areas (n and p) were quantified as discussed under “Materials and methods.” The level of significance was determined by one-way ANOVA and Tukey's multiple-comparison test. n = 7; ***, p < 0.001. For anti-N staining, F(df, df) values at day 3 p.i. were F(2, 83) = 9.241 (****, p < 0.0001), and at day 6 p.i., they were F(2, 96) = 10.58 (****, p < 0.0001). For Iba1 staining, F(df,df) values at day 3 p.i. were F(2, 80) = 5.352 (*, p < 0.05), and at day 6 p.i., they were F(2, 98) = 7.530 (****, p < 0.0001). Error bars, S.E.
Table 5.
Mean difference and statistical significance as an effect of proline residues on viral antigen distribution in infected mouse spinal cord
**, p < 0.01; ***, p < 0.001; ****, p < 0.0001; NS, no significance.
| Tukey's multiple-comparison test | Day 3 p.i. |
Day 6 p.i. |
||||
|---|---|---|---|---|---|---|
| Mean difference | S.E. | Significance | Mean difference | S.E. | Significance | |
| Control versus RSA59 (PP) | −0.7895 | 0.1840 | *** | −2.475 | 0.6817 | ** |
| Control versus RSA59 (P) | −0.4307 | 0.1840 | NS | −0.08576 | 0.6657 | NS |
| RSA59 (PP) versus RSA59 (P) | 0.3588 | 0.1661 | NS | 2.390 | 0.5754 | *** |
Distribution and intensification of microglia/macrophages in RSA59 (PP)– and RSA59 (P)–infected spinal cord sections
Day 3 and 6 p.i. spinal cord cross-sections from control–, RSA59 (PP)–, and RSA59 (P)–infected mice were stained with anti-Iba1 (Fig. 13, d–f and j–l) and quantified based on the area of staining relative to the total area as described under “Materials and methods” (Fig. 13, n and p). The percentage area of activated microglia/macrophages in spinal cord sections of RSA59 (P) in day 3 and 6 p.i. mice was significantly increased compared with the control mice, but no difference was observed between RSA59 (PP) versus RSA59 (P) in day 3 and 6 p.i. mice. The mean difference and S.E. of the difference are shown in Table 6 and plotted as a scatter diagram (Fig. 13, n and p).
Table 6.
Mean difference and statistical significance as an effect of proline residues on microglia distribution in infected mouse spinal cord
**, p < 0.01; NS, no significance.
| Tukey's multiple-comparison test | Day 3 p.i. |
Day 6 p.i. |
||||
|---|---|---|---|---|---|---|
| Mean difference | S.E. | Significance | Mean Diff. | S.E. | Significance | |
| Control versus RSA59 (PP) | −0.3182 | 0.2126 | NS | −0.7573 | 0.2107 | ** |
| Control versus RSA59 (P) | −0.6799 | 0.2113 | ** | −0.7429 | 0.2189 | ** |
| RSA59 (PP) versus RSA59 (P) | −0.3617 | 0.1868 | NS | 0.01434 | 0.1857 | NS |
The intensity of myelin loss (demyelination) in RSA59 (PP)–infected mouse spinal cord is greater compared with RSA59 (P) infection during the chronic inflammation stage (day 30 p.i.)
Spinal cord sections from control–, RSA59 (PP)–, and RSA59 (P)–infected mice at day 30 p.i. were stained with Luxol Fast Blue (LFB) to detect demyelination. Data from LFB-stained cross-sections from the different levels of spinal cord showed that all of the mice (seven of seven mice) infected with RSA59 (PP) developed demyelination; in contrast, six of seven RSA59 (P)–infected mice developed demyelination. Even RSA59 (P)–infected mice showed demyelination (Fig. 14, c and f), but the degree and amount of demyelination were reduced compared with RSA59 (PP) (Fig. 14, b and e). The demyelination percentage was scored, and the area of the demyelinating plaques were quantified as discussed under “Materials and methods” (Table 7). The average ± S.E. demyelination score for RSA59 (PP) was 20.05 ± 4.650. RSA59 (P)–infected mice exhibited reduced demyelination at day 30 p.i. (Fig. 14p) with an average ± S.E. demyelination score of 4.267 ± 4.784. The average ± S.E. difference in demyelination scores of RSA59 (PP)– versus RSA59 (P)–infected mouse spinal cords at day 30 p.i. was found to be significant: 15.78 ± 3.804 (***, p < 0.001) (Fig. 14p).
Table 7.
Mean difference and statistical significance as an effect of proline residues on area with myelin loss
***, p < 0.001; NS, no significance.
| Tukey's multiple-comparison test | Day 30 p.i. |
||
|---|---|---|---|
| Mean difference | S.E. | Significance | |
| Control versus RSA59 (PP) | −20.05 | 4.65 | *** |
| Control versus RSA59 (P) | −4.267 | 4.784 | NS |
| RSA59 (PP) versus RSA59 (P) | 15.78 | 3.804 | *** |
Sections from the same mice were also immunostained with anti-Iba1 antibody to detect microglia/macrophages at day 30 p.i. Iba1+ cells were observed in the corresponding demyelinating areas of a given section (Fig. 14, g–i). The spinal cord cross-sections were also immunostained for anti-PLP (Fig. 14, j–l), which stains myelin. PLP staining confirmed that myelin sheath loss was comparatively higher in RSA59 (PP) than RSA59 (P) infection. The insets in Fig. 14n show the loss of PLP straining in RSA59 (PP); in contrast, no significant loss was observed in RSA59 (P) (Fig. 14o). Mock-infected spinal cord showed an intact myelin sheath (Fig. 14m).
Discussion
The fusion proteins of all of the enveloped viruses have a characteristic hydrophobic stretch of fusion peptide, which engages with target membrane to initiate fusion. In our study, we identified a minimal region of RSA59 spike protein responsible for the fusion property. This study was carried out by comparing the spike of RSMHV2, which is a nonfusogenic strain. In silico studies showed that the RSA59 fusion peptide has two consecutive prolines (at amino acids 938 and 939), whereas the RSMHV2 strain has a single proline residue (amino acid 976). A three-dimensional study of the spike proteins demonstrated that the observed differences in structure and function between the two strains could be due to the presence of the two consecutive prolines in the fusion peptide. The importance of the proline was further established through mutagenesis experiments, in which one proline deletion from the spike FP of RSA59 (PP) led to a significant loss of fusogenicity, entry, and syncytial formation in vitro. The deletion of one proline may destabilize the spike protein and result in impaired fusogenicity. Additionally, there could be slower or less transport of the S protein to the plasma membrane, which would correspondingly result in slower kinetics of cell fusion. In a previous study, it has been shown that a substitution mutant, P939L, showed similar delayed kinetics of cell fusion and virus growth (28). Earlier studies have shown that trafficking and retention of spike protein to different intracellular compartment depends upon the retention signals present in the cytoplasmic domain of the spike protein (29, 30). Both of our strains (RSA59 (PP) and RSA59 (P)) have same cytoplasmic sequence. Hence, it can be argued that both can traffic to surface equally. Further, we have observed that the deletion of one proline reduces the replication of RSA59 (P), so the differential fusion ability of the two viruses may be a cumulative result of defective trafficking, reduced replication, and conformational instability of spike to cause fusion.
Furthermore, in vivo studies using the proline mutant strains showed altered viral infectivity as a consequence of the impaired fusogenicity. Transcranial inoculation demonstrated differential patterns of neuropathogenicity during the acute stage of infection, although both strains exhibited a similar pattern and degree of necrotizing hepatitis and leptomeningitis. RSA59 (PP) infection showed a widespread viral antigen distribution in the brain parenchyma, whereas in RSA59 (P) infection, consecutive viral antigen spread was mainly restricted to the inoculation site and in meninges. At day 6 p.i., both RSA59 (PP) and RSA59 (P) were mostly cleared from the brain. Although Iba1+ cells were widely distributed throughout the brain parenchyma, accumulation of proliferated ones were near the lateral ventricular/subventricular region in the RSA59 (P)–infected brain, whereas, in cases of RSA59 (PP), proliferated Iba1+ cells were accumulated in the cerebral cortex, ventral striatum/basal forebrain, lateral ventricle/subventricular zone, hippocampus, hypothalamus, thalamus, midbrain, and brain stem following the trajectory of viral antigen distribution. Interestingly, there was a significant reduction in demyelination in RSA59 (P) strain–infected mouse spinal cord during the chronic stage of disease compared with the RSA59 (PP) strain. This is one of the first attempts to comprehensively address the role of two consecutive prolines in the FP of the spike protein toward neurotropic mouse hepatitis virus infectivity, fusogenicity, and neuropathogenicity. Our findings may have broader implications in understanding the mechanistic process of MHV-induced demyelination.
The question concerning the vital role of the two consecutive proline residues in altering the rate of viral infectivity, viral spread, and fusogenicity remains. Proline is not an amino but an imino acid, in which the side chain binds to the main-chain nitrogen atom to form a ring (and a secondary amino acid), thereby restricting the backbone φ torsion angle of the polypeptide chain. Thus, it is the most restricted of all of the naturally occurring amino acids, occupying the smallest area of the Ramachandran map (31). Its presence significantly reduces the intrinsic flexibility of the protein segment in which it is located. This point is strongly corroborated by our MD simulations, whereby the presence of a single proline can be seen to profoundly affect the fluctuation profile of a molecule by contracting its overall conformational space. These effects percolate globally and are not restricted to FP alone, and they can be seen based on differences in the energy landscapes of S-MHV-A59 (PP) and S-MHV-A59 (P). Focusing only on FP, the distinction is stark with diproline, showing almost no fluctuation compared with the single-proline FP. Part of the rigidity of the diproline-embedded FP is contributed by an increased number of hydrogen bonds in addition to proline's own conformational rigidity.
Directly demonstrating the role of proline by trapping the spike protein during a host-membrane rupture action is a challenging task. Indirectly examining its role through an assay using a synthetic membrane spike system under in vitro conditions offers an alternate avenue; however, given the complexity of real cell membranes, wherein receptor proteins mediate virus–host interactions, allowing for membrane penetration and cell entry, such studies also have great limitations. We attempted to balance the two scenarios through the use of in vivo histopathology studies in mouse brain tissue. The focus has been to estimate the difference in acute-stage viral spread in brain tissue, where host cell entry is the rate-limiting step controlling chronic-stage demyelination. The difference in spread can be unambiguously attributed to the presence or absence of the additional proline, which alone represents the difference between the engineered strains studied.
To clearly define the role of proline, we must study on a large scale the effects of neighboring amino acids on the entire S2 domain. A previous study has shown that a natural recombinant Penn 97-1 (a mosaic spike protein in which the S1 domain is from MHV-A59 and the S2 domain is from MHV-2) abolishes demyelination (19). This study can be revisited and a mutant strain constructed with RSMHV2 that carries two prolines at the center of its FP. It will be interesting to study the addition of proline with concomitant replacement of the cleavage sequence. Swapping the cleavage in combination with the proline mutation will help us to further delineate the role of two consecutive prolines in cell-to-cell fusion, as well as the subsequent demyelination, as a pathogenic outcome. As previous studies have demonstrated that the biological properties of viral persistence are essentially the failure of the immune system to clear the virus from CNS cells, it will also be interesting to study whether RSA59 (PP) and RSA59 (P) differ in their neural cell persistence and CNS-resident immune surveillance. It will also be exciting to examine the differential mechanism of RSA59 (PP) and RSA59 (P) in the transneuronal spread by which viruses may be synaptically linked to other neural cells, more specifically to oligodendrocytes. Taking into consideration previous findings, we are tempted to focus our future analyses on detailed glial cell tropism and to determine whether RSA59 (P) will spread less efficiently from gray matter neurons to white matter oligodendrocytes, successively damage oligodendrocytes, and, as a result, alter the pathology of demyelination.
In vitro spike protein trafficking studies in combination with real-time kinetics of viral spread and dissemination clearly demonstrated that the spike alone can initiate the fusion process irrespective of the presence of the murine CEACAM receptor protein and can be responsible for fusogenicity, which in turn may help in viral entry and subsequent neuropathogenesis to elicit encephalitis, myelitis during the acute stage of infection, and chronic-stage demyelination as a consequence of neuroimmune activation. In silico molecular modeling and NMR studies in combination with in vivo and in vitro experiments helped to dissect the possible mode of action of two prolines in the backbone of the spike fusion domain.
Overall, our observations of pathogenic differences due to proline deletion offer insights into viral spread based on fusion mechanisms, and these results are also pertinent to other virus–cell interactions. Interestingly, many fusion proteins do not have a central proline in their FP; however, the residues therein that improve FP rigidity (26) and cause the neighborhood structure to become more ordered within the membrane environment, adding kinetic efficiency to the fusion process. These insights are useful in understanding the mechanisms employed by the virus fusion apparatus and may help guide the development of therapeutic approaches to prevent intercellular viral spread and neuroinfection.
Materials and methods
Viral strains
The isogenic recombinant demyelinating strain RSA59 and nondemyelinating strain RSMHV2 engineered from the MHV-A59 and MHV-2 parental strains, respectively, were used as described previously (20, 22). All viral strains used in this study are tabulated in Table 1. Both strains express EGFPs (20, 22, 32). Parental recombinant viruses were propagated in murine 17Cl-1 cells. Plaque assays and purifications were carried out on murine L2 cells. Cells were maintained on plastic tissue culture flasks in Dulbecco's modified Eagle's medium (Gibco, Thermo Fisher Scientific) with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific), 1% penicillin (10,000 units/ml), 1% streptomycin (100 mg/ml) (HyClone, Thermo Fisher Scientific), 10 mm HEPES buffer solution (Invitrogen, Thermo Fisher Scientific), and 1.4% of 7.5% NaHCO3 in the presence of 5% CO2 at 37 °C.
Construction of MHV-A59 spike gene expression in the pYFPN1 vector
For transient transfection in HeLa cells, the full-length cDNA from pMH54 (a pseudo-defective interfering construct that contains all of the genes from the 3′ end of the MHV-A59 strain (19, 33) encoding the MHV-A59 spike) was subcloned into pYFPN1 (Addgene, Watertown, MA). The full-length cDNA of the MHV-A59 spike gene was amplified by PCR in a Robocycler (Stratagene, La Jolla, CA) using the Expand Long Template PCR system (Roche Diagnostics), starting with MHV-A59 spike gene cDNA as template. The resulting PCR products were digested with SacI and XmaI followed by ligation into the pYFP-N1 vector to generate an in-frame fusion protein. The ligated product was transformed into bacterial stocks, and DNA was isolated and purified using the Qiagen Midiprep kit (Qiagen, Germany) according to the manufacturer's instructions. Positive clones of S-MHV-A59 (PP)/pYFPN1 were screened by restriction digestion and sequenced to confirm the in-frame fusion of MHV-A59 (PP) spike protein in pYFPN1.
Transfection and immunofluorescence
For transient transfection in HeLa cells, the full-length cDNA encoding S-MHV-A59 (PP)/pYFPN1 was subcloned into the pCAGGS vector (Addgene). The full-length cDNA of S-MHV-A59 (PP)-YFP was amplified by PCR using MHV-A59-S/pYFP-N1 as a template. The PCR products were digested and ligated into the pCAGGS mammalian expression vector. The ligated product was transformed, and plasmid DNA was isolated and purified as described previously. Positive clones of S-MHV-A59 (PP)-YFP constructs in pCAGGS were screened and sequenced. HeLa cells were plated on 25-mm circular coverslips in 35-mm dishes 1 day prior to transfection and transfected with the generated S-MHV-A59 (PP)-YFP (S-MHV-A59 (PP) in Fig. 5) constructs using Fugene (Roche Diagnostics) at 1 μg/ml DNA using a Fugene/DNA ratio of 6:1 (1 μg) as described previously (29).
To evaluate YFP fluorescence after 48 h of transfection, the cells were washed with ice-cold PBS, fixed in ice-cold 95% EtOH for 20 min, washed three times with PBS, and mounted using Vectashield (Vector Laboratories, Burlingame, CA). To label surface S-MHV-A59 (PP)-YFP fusion proteins, transfected cells were washed with ice-cold PBS and incubated with rabbit anti-A59-S antibody for 10 min at 4 °C. The cells were then washed twice with ice-cold PBS and fixed in 4% paraformaldehyde in PBS for 10 min at room temperature. They were then washed three times with PBS, permeabilized with PBS + 0.5% Triton X-100, and blocked with PBS + 0.5% Triton X-100 + 2% heat-inactivated goat serum (PBS/GS). The cells were incubated with primary antibody diluted in PBS/GS for 1 h, washed, and labeled for 1 h with secondary antibody (Texas Red goat anti-rabbit) diluted in PBS/GS. The cells were then washed with PBS and mounted in Vectashield (Vector Laboratories) The cells were visualized by fluorescence microscopy using an Olympus IX-81 microscope system with a ×60 UPlanApo oil immersion objective with the iris diaphragm partially closed to limit the contribution of out-of-plane fluorescence and filter packs suitable for green (U-MWIBA BP460-490 DM505 BA515-550) and red (U-NMG BP530-550 DM570 BA590-800+) fluorescence. Images were acquired with a Hammatzu Orca-1 CCD camera and Image Pro image analysis software (Media Cybernetics, Silver Spring, MD).
To quantify the surface and cytosolic spike protein expression, Fiji (ImageJ 1.52g) software was used (34). The mean intensity/μm2 area of fluorescence was measured for the plasma membrane and for the cytoplasm, separately. The percentage of cytosolic versus surface fluorescence was plotted using GraphPad Prism version 6.01. The level of significance was calculated using paired Student's t test.
Pairwise global sequence alignment of spike protein
The spike protein details of the parental strains of RSA59 (PP) and RSMHV2 (P), namely MHV-A59 and MHV-2, respectively (MHV strains nomenclature shown in Table 2), are available under GenBankTM accession numbers 9629812 (14, 18, 25) and AF201929 (35, 36), respectively, and were obtained from the NCBI database (http://www.ncbi.nlm.nih.gov/). To compare the spike sequences, ClustalW software and the Gonnet PAM 250 matrix were used (37). The spike gene was also sequenced to confirm that the primary structure matched the parental strain (see “Viral spike gene sequencing”).
Structure modeling
The spike protein S-MHV-A59 (PP) has recently been solved using cryo-EM (24) (PDB code 3JCL). Residues 871–1116 of the trimeric fusion domain of 3JCL were used as template for comparative modeling of the fusion domains of S-MHV-A59 (P) and S-MHV-2 (P). Modeler software was used for comparative modeling (38).
MD simulation of the extended region around internal FPs
MD simulations were performed for an extended region of the quaternary structure covering the fusion domain, spanning residue locations 871–1116 of S-MHV-A59 (PP), 871–1115 of S-MHV-A59 (P), and 910–1154 of S-MHV-2 (P), respectively. We used periodic boundary conditions, the CHARMM force field, and SPC/E (SPC216) water for all MD simulations with GROMACS (version 5.0.5) software (39). The pH of the system was kept neutral as MHV spike proteins are known to be fusion-competent under such conditions (40). Each structure was centered in its respective cubic box, with a box dimension ensuring a distance of 1.0 nm between the edge of the molecule and the nearest box face. The proteins were solvated in water and energy-minimized, and then they were equilibrated for isothermal-isochoric and isothermal-isobaric states. The steepest descent energy minimization was performed to remove any overlapping contacts and to reduce the maximum force in the system to 1000 kJ/mol/nm. The solvated, steepest descent minimized structure was our starting and reference structure at tref = 0. Equilibration was performed using Berendsen temperature coupling with a total time of t = 2000 ps, temperature of 300 K, and isotropic pressure coupling with a total time of t = 2000 ps under a pressure of 1 atm. After minimization and equilibration, unconstrained MD simulation was carried out for 500 ns at 300 K and 1 atm pressure. The time step was 2 fs, and the frames were saved every 100 ps for all simulation steps mentioned above. The conformation analysis was performed using GROMACS utilities, MDTraj (41) and scripts written in house.
NMR data collection
Synthetic 24- and 16-residue polypeptides (TGATAAAMFPPWSAAAGVPFSLSV and ATAAAMFPPWSAAAGV) (USV Private Ltd., Mumbai, India) identical to the segment of the internal FP of S-MHV-A59 (PP) were obtained for NMR studies in water (H2O + D2O) and methanol (CD3OH), respectively. The sample was prepared at ∼1 mm concentration. A series of 1H-1D NMR experiments were recorded with 128 scans at different temperatures to calculate the temperature coefficients (hydrogen bond), and 2D NMR experiments were performed at 288/278 K for the water/methanol sample. The 2D NMR experiments, including 2D TOCSY, 2D ROESY, 2D 15N HSQC, and 2D 13C HSQC, were recorded for sequential assignments and structure calculation. BRUKER 800- and 700-MHz NMR spectrometers equipped with triple resonance cryogenically cooled probes were used for data collection; 32 scans and 64 scans with increments were used in the 2D TOCSY and 2D ROESY experiments, respectively.
NMR data analysis
NMR data for FP were processed and analyzed using BRUKER TOPSPIN version 3.2 software. The 2D TOCSY and 2D ROESY, 2D 1H-13C HSQC NMR experiments were used for residue-specific and sequential resonance assignments. A chemical shift difference plot was generated for 1Hα chemical shifts for secondary structure prediction. The structure calculation was performed using the Cyana program, and only the top 20 model structures were considered for analysis. One model each in cis and trans prolyl peptide form from the 16-residue FP was obtained for the 500-ns MD simulations in 100% methanol and water, as per the MD protocol described above.
Fusogenicity of the recombinant virus in N2a cells
N2a is a mouse neuroblastoma cell line (CCL-131) that was obtained from the American Type Culture Collection. N2a cells were seeded on glass coverslips. Monolayers of cells were infected with recombinant strains of the viruses at an MOI of 1. Infected cells were incubated at 37 °C in 5% CO2 for 12, 16, and 24 h. Cells were fixed in 4% paraformaldehyde (PFA) and mounted on glass slides using Vectashield with DAPI (Vector Laboratories). The slides were then observed under an epifluorescence microscope for EGFP excitation (Olympus IX-81 microscope system with a Hammatzu Orca-1 CCD) under a 470/40 blue band filter. Images were acquired with a Hamamatsu Orca-1 charge-coupled device camera and analyzed using Image-Pro image analysis software (Media Cybernetics, Silver Spring, MD). The mean nuclei per syncytia were quantified by counting the number of DAPI-stained nuclei inside the EGFP-expressing syncytia. A total of 15 random frames of ×40 images were selected from three different experiments for every time point per strain of virus. The level of significance was calculated using unpaired Student's t test.
Plasmid pMH54
pMH54EGFP was generated by inserting the spike gene from MHV-A59 and EGFP from the pEGFP-N1 vector, a synthetic defective interfering construct containing all 3′-end genes from MHV-A59 (19, 22, 32).
Generation of a new proline mutated strain of MHV
The oligonucleotide primers were designed with the desired mutation. Mutations were then inserted into pMH54EGFP using the QuikChange site-directed mutagenesis kit (Stratagene, now Agilent), after which plasmids were isolated and sequenced using primers 26486F (5′-AGACGGCAATGGACCTAGTG-3′) and 27019R (5′-TCTACAGCCTCAAGCCGAGT-3′). The plasmids were linearized by PacI restriction digestion. Synthetic RNA was synthesized from linearized plasmids using an in vitro transcription kit (Ambion, Austin, TX). Targeted RNA recombination was carried out between synthetic capped RNAs transcribed from pMH54EGFP containing the desired mutation, using a T7 polymerase transcription kit (Ambion), and fMHV was used as a recipient virus. Recombinant virus was generated as described previously (19, 22, 32). The resultant recombinant strains were identical to RSA59, except that one proline was deleted from FP. The recombinant strains were selected by replication in murine 17Cl-1 cells (18, 33). Candidate recombinants were plaque-purified three times, followed by amplification of the virus in 17Cl-1 cells as described below.
Virus-free cell-to-cell fusion assay
The cell-to-cell fusion assay was set up with minor modifications according to previously established methods (26, 27). BHK-R cells (42) designated as effector cells were transfected with plasmid pT7EMCLuc (gift from Vaibhav Tiwari, Midwestern University, Downers Grove, IL), which expresses the firefly luciferase gene under the control of the T7 promoter (27). HeLa cells in continuous culture, considered as target cells, were cotransfected with pCAGT7, which expresses T7 RNA polymerase with the chicken actin promoter and the CMV enhancer as well as with the spike-expressing plasmid pMH54EGFP (PP) (two prolines are present in FP). In parallel, HeLa cells were transfected with pCAGT7 and spike mutant plasmid with the deleted proline (pMH54EGFP (P)). At 36 h post-transfection, effector and target cells were trypsinized and co-cultured at a 1:1 ratio. The pT7EMCLuc-expressing effector cells and only T7 RNA polymerase-transfected target cells were used as the negative control (vector control). The luciferase reporter assay (Promega) was performed to estimate the luciferase activity at 36 and 48 h post-co-culture as described previously (27). Luciferase activity is considered a measurement of cell fusion ability and is directly correlated with the fusion property of the spike gene. Luciferase reporter activity was plotted using GraphPad Prism version 6.01 software. The level of significance between the two-proline–containing spike gene construct and the spike gene construct with a deleted proline was calculated using unpaired Student's t test.
Amplification and titer estimation of proline-mutated recombinant strain RSA59 (P) compared with parental RSA59 (PP)
Recombinant strains were amplified using a routine protocol (43, 44). A monolayer of 17Cl-1 cells in T75 flasks was infected with 3-fold plaque-purified (PP3) recombinant strains. The infected 17Cl-1 cells were incubated for 16 h at 37 °C in 5% CO2. Then the cells were maintained at −80 °C for 2 h. Infected cells were freeze (−80 °C)-thawed (room temperature) three times, and viral particles were collected in the supernatant by centrifugation at 500 rpm at 4 °C.
Plaque assay
The viral titer was estimated according to the following standard plaque assay protocol (43, 44). Briefly, L2 cells were seeded in 6-well plates. RSA59 (PP)– and RSA59 (P)–infected culture supernatants were serially diluted in 2% fetal bovine serum–containing medium. The L2 monolayer was then infected with 200 μl of each dilution. The plates were incubated at 37 °C for 1 h with intermittent rocking every 15 min. The cells were then overlaid with 1% agarose (1 volume of 2% dissolved agarose in double-distilled water with 1 volume of 2× L2 medium) and allowed to solidify. After 48 h of incubation, each well was overlaid with 1.4% agarose containing 0.04% neutral red, and the plaques were manually counted after 4–5 h of incubation. The titer was calculated as the pfu based on a mathematical calculation by fitting to the formula, titer = (number of plaques × dilution factor/ml). The logarithm base 10 value of the titer was plotted using GraphPad Prism version 6.01 as a scatter diagram. The level of significance was determined using unpaired Student's t test.
Viral spike gene sequencing
Aiming to sequence the spike genes from RSA59 (PP) and RSA59 (P), RT-PCR amplification was performed using the cytoplasmic RNA extracted from virus-infected L2 cells (MOI of either 1 or 2) harvested at 16 h postinfection. Complementary DNA was synthesized using oligonucleotide primers based on previously published MHV-A59 sequences (14). Primers were designed to amplify fragments of ∼600 bp. PCR products were purified with a QIAquick PCR purification kit (Qiagen, Chatsworth, CA) and sequenced by automated sequencing using the Taq dye terminator procedure according to the manufacturer's protocol (TaqDyeDeoxy Terminators cycle sequencing kit, Applied Biosystems). The primers used for sequencing were the same as those applied for amplification. Each fragment was sequenced in both directions, followed by analysis using the Sequencher version 5.4.6 program. Nucleotide sequences and predicted protein sequences were compared with the sequences available in NCBI GenBankTM using “BlastN and BlastP” searches and the FASTA program.
Growth kinetics of recombinant strains
A monolayer of L2 cells was infected with RSA59 (PP) or RSA59 (P) strains of MHV and incubated at 37 °C in 5% CO2. At different time points postinfection (4, 8, 12, 20, and 24 h), the cultures were transferred to −80 °C. The cells were freeze-thawed three times, and after a final round of thawing, dislodged cells with culture supernatant were centrifuged at 500 rpm for 15 min at 4 °C. Clear supernatants from infected culture plates were subjected to a routine plaque assay for titer estimation. The titer value was calculated according to routine procedures (43, 44) and plotted against the corresponding time postinfection. The level of significance was determined using unpaired Student's t test.
RNA isolation, reverse transcription, and quantitative PCR
RNA was extracted from 0.1 MOI, RSA59 (PP)– and RSA59 (P)–infected L2 cells (four culture dishes each at 0, 4, 8, and 12 h p.i.) using the TRIzol isolation protocol (Invitrogen, Thermo Fisher Scientific). Total RNA concentration was measured using a NanoDrop ND-100 spectrophotometer. Two μg of RNA was used to prepare cDNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative real-time PCR analysis was performed using a DyNAmo ColorFlash SYBR Green qPCR kit (Thermo Fisher Scientific) in a real-time PCR system (Applied Biosystems 7500) under the following conditions: initial denaturation at 95 °C for 7 min, 40 cycles of 95 °C for 10 s, 60 °C for 30 s, melting curve analysis at 60 °C for 30 s. Reactions were performed in quadruplicate. The following primer pairs were used: GAPDH, 5′-GCCCCTTCTGCCGATGC-3′ (forward) and 5′-CTTTCCAGAGGGGCCATCC-3′ (reverse), spike gene 5′-GCCAGTATACCATTTGTCTGTTACCT-3′ (forward) and 5′-CTACTACGTTTTTGTTTAG-3′ (reverse); and nucleocapsid (N) gene, 5′-AGGATAGAAGTCTGTTGGCTCA-3′ (forward) and 5′-GAAGTTAGCAAGGTCCTACG-3′ (reverse). Relative quantitation was achieved using the comparative threshold (ΔΔCt) method. mRNA expression levels of target genes (S and N gene) in infected samples were normalized with GAPDH and expressed as relative -fold change compared with expression level at 0 h. The level of significance was calculated using unpaired Student's t test
In vitro live imaging of the formation of syncytia
N2a cells were grown in 35-mm culture dishes. Almost 80% confluent monolayers of N2a cells were infected with an MOI of 1 of the viruses. Viral absorption was allowed for 1 h, with intermittent shaking every 15 min, after which the infected cells were washed and placed in fresh medium. Proline mutant recombinant virus spreading of RSA59 (PP) and RSA59 (P) and syncytial formation were monitored by time-lapse imaging using an Olympus IX-81 microscope system with a Hammatzu Orca-1 CCD. A stage top incubator UNIV2 D35 attached to a temperature and gas mixture controlling panel (incubation system of microscope, TOKAI HIT) was used to maintain the temperature at 37 °C and 5% CO2. Images were acquired using a ×40 objective with a Hamamatsu Orca-1 charge-coupled device camera and processed using ImageJ software. The virus-infected culture was monitored at 15-min intervals from 8 h until 14 h postinfection.
Inoculation of mice
Four-week-old, MHV free, C57Bl/6 (B6) male mice (In vivo Biosciences, Bangalore) were inoculated intracranially with RSA59 (PP) and RSA59 (P) at 20,000 pfu (50% LD50 dose of RSA59 (PP)) as described previously (22, 32). Seven mice (n = 7 in two experiments) were inoculated in each infection group for histopathological analysis at days 3, 6, and 30 p.i. Similarly, n = 3 mice/infection group were infected for estimation of the viral titer by routine plaque assay as discussed previously at days 3 and 6 p.i. Mice were monitored daily for mortality and signs of disease. Mock-infected (control) mice (n = 4/each day point) were inoculated similarly with uninfected PBS containing 0.75% BSA at a comparable dilution.
Ethics statement
The use of animals and all experimental procedures were reviewed and approved by the institutional animal care and use committee at the Indian Institute of Science Education and Research Kolkata. The animal protocols adhered to the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India.
Histopathological analyses
Mice were sacrificed at days 3, 6, and 30 p.i. and perfused transcardially with PBS followed by 4% PFA in PBS. Liver, brain, and spinal cord tissues were collected, postfixed in 4% PFA overnight, and embedded in paraffin. Brain and spinal cord tissues were sectioned at 5 μm. Brain sections were stained with H&E for evaluation of inflammation, whereas day 30 p.i. spinal cord tissue sections were stained with LFB to detect myelin damage. To confirm the expected virulence of the strains used, livers from the infected mice were embedded in paraffin, sectioned at 5 μm, and stained with H&E. All slides were coded and read in a blinded manner.
Immunohistochemical analyses
Serial sagittal sections of brain, spinal cord, and liver tissues were stained using the avidin-biotin-immunoperoxidase technique (Vector Laboratories) with 3,3′-diaminobenzidine as substrate and anti-Iba1 and anti-N antibody as primary antibodies (details in Table 8). Sequential spinal cord sections from day 30 p.i. mice were also immunolabeled with anti-PLP antibody.
Table 8.
List of antibodies and their dilutions used for immunohistochemical analysis
| Target cell/structure | Primary antibody | Dilution |
|---|---|---|
| Microglia/macrophage | Anti-Iba1 rabbit (microglia/macrophage marker, Wako Chemicals, catalog no. 019-19741) | 1:250 |
| Viral antigen | Mouse monoclonal anti-N, nucleocapsid protein of MHV-JHM, monoclonal clone 1-16-1, kindly provided by Julian Leibowitz (Texas A&M, College Station, TX) | 1:50 |
| Myelin sheath | Anti-PLP (proteolipid protein) rat IgG (gift from Judith B. Grinspan, Children's Hospital of Philadelphia) | 1:1 |
Quantification of histological slides
The extent of infection in different neuroanatomic regions of infected mice brain tissues was scored based on the amount of viral antigen present according to an arbitrary scale: 0, no apparent viral antigen staining; 1, very small foci of viral antigen-positive cells; 2, widespread but small foci of viral antigen-positive cells; 3, widespread large foci of viral antigen-positive cells. The brain has been divided into different neuroanatomic regions as shown under “Results” in Fig. 10a.
To quantify antiviral staining in spinal cord and microglial-based inflammation in different neuroanatomic regions of the brain and spinal cord, Fiji (ImageJ 1.52g) software was used (34). Image analysis was performed using the basic densitometric thresholding features of Fiji (ImageJ, NIH Image, and Scion Image). Image analysis was performed by first capturing the images at the highest magnification (×10) that allowed the entire section (i.e. scan area) to be visualized within a single image frame. The RGB image was color-deconvoluted into three different colors to separate the 3,3′-diaminobenzidine–specific staining. The background labeling was also subtracted from all images, and then the contrast was slightly enhanced to improve the resolution. The perimeter of each neuroanatomic region for the brain and spinal cord was digitally outlined, and the area was calculated in μm2. To ensure that all labeled cells were selected, a threshold value was defined for each image. The magnitude of viral staining and Iba1+ microglia/macrophage activation was defined as the percentage area of staining (ratio of target stained area to total selected area multiplied by 100). To ensure error-free data collection, the entire quantification procedure was performed by two investigators together and read in a blinded manner.
To determine the total white matter area and areas with myelin loss in day 30 RSA59 (PP) and RSA59 (P) p.i. mice, 4–5 LFB-stained spinal cord cross-sections from each mouse were randomly selected and analyzed using Fiji software (ImageJ 1.52g) (45). The total number of mice in each group was 7 (n = 7). The total perimeter of the white matter regions in each cross-section was outlined and calculated by summing the dorsal, ventral, and anterior white matter areas in each section. The total area of the demyelinated regions was also outlined and summed for each section separately. The percentage of spinal cord demyelination per section per mouse was obtained by dividing the total area of the demyelinating plaque over the total area of the calculated white matter and then multiplying by 100.
Statistical analysis
The infection and magnitude of inflammation within the different neuroanatomic regions of the brain are presented in Tables 3 and 4, respectively as the mean values ± S.E., and two-way ANOVA was used to calculate significance. In addition, multiple comparison was performed using Tukey's multiple-comparison test.
The level of significance for immunohistochemistry staining and LFB staining in spinal cord sections was calculated using one-way ANOVA and Tukey's multiple-comparison test. All data were plotted and analyzed using GraphPad Prism version 6.01 software. The level of significance and the means ± S.D. are presented in a scatter diagram. A p value < 0.05 (*) was considered statistically significant.
Author contributions
M. S. and A. K. data curation; M. S., D. P., and J. D. S. validation; M. S. and A. K. investigation; M. S., A. K., D. M., P. S., B. K., S. V., and S. R. methodology; M. S. and A. K. writing-original draft; D. M., D. P., and J. D. S. writing-review and editing; S. R., D. P., and J. D. S. supervision; L. C. K. visualization; J. D. S. conceptualization; J. D. S. funding acquisition; J. D. S. project administration.
Supplementary Material
Acknowledgments
We thank the Department of Biotechnology (DBT), New Delhi, for supporting the computational facilities and the Department of Science and Technology, New Delhi, for supporting the NMR facilities. We thank Ramachandra Dongre for helping with structure modeling from NMR data and IISER Kolkata for facilities. We also thank and acknowledge the IISER Kolkata Microscopy facility. We thank Dr. Kenneth S. Shindler and Dr. David Pleasure for critically reading and editing the manuscript. We thank Elsevier for editorial assistance.
This work was supported by the University Grant Commission, New Delhi, and DuPre fellowship, Multiple Sclerosis Society International Federation (MSIF), UK (to M. S.); fellowships from the Department of Biotechnology (DBT), New Delhi, and Council of Scientific and Industrial Research, New Delhi (to A. K. and D. M., respectively); and funding support for the experiments from the DBT, New Delhi (to J. D. S.). The authors declare that they have no conflicts of interest with the contents of this article. The funders had no role in the study design, data collection, and interpretation, or the decision to publish the work.
This article contains Tables S1–S4 and Figs. S1–S9.
- FP
- fusion peptide
- MHV
- mouse hepatitis virus
- p.i.
- postinfection
- EGFP
- enhanced GFP
- PDB
- Protein Data Bank
- MD
- molecular dynamics
- MOI
- multiplicity of infection
- H&E
- hematoxylin and eosin
- CNS
- central nervous system
- LFB
- Luxol Fast Blue
- GS
- goat serum
- 2D
- two-dimensional
- PFA
- paraformaldehyde
- DAPI
- 4′,6-diamidino-2-phenylindole
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ANOVA
- analysis of variance
- RMSD
- root mean square deviation
- TOCSY
- total correlation spectroscopy
- ROESY
- rotating-frame overhauser spectroscopy
- HSQC
- heteronuclear single quantum coherence spectroscopy.
References
- 1. Durell S. R., Martin I., Ruysschaert J. M., Shai Y., and Blumenthal R. (1997) What studies of fusion peptides tell us about viral envelope glycoprotein-mediated membrane fusion (review). Mol. Membr. Biol. 14, 97–112 10.3109/09687689709048170 [DOI] [PubMed] [Google Scholar]
- 2. Earp L. J., Delos S. E., Park H. E., and White J. M. (2005) The many mechanisms of viral membrane fusion proteins. Curr. Top Microbiol. Immunol. 285, 25–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sainz B. Jr., Rausch J. M., Gallaher W. R., Garry R. F., and Wimley W. C. (2005) Identification and characterization of the putative fusion peptide of the severe acute respiratory syndrome-associated coronavirus spike protein. J. Virol. 79, 7195–7206 10.1128/JVI.79.11.7195-7206.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pécheur E. I., Sainte-Marie J., Bienvenüe A., Hoekstra D. (1999) Peptides and membrane fusion: towards an understanding of the molecular mechanism of protein-induced fusion. J. Membr. Biol. 167, 1–17 10.1007/s002329900466 [DOI] [PubMed] [Google Scholar]
- 5. Martin I., and Ruysschaert J. M. (2000) Common properties of fusion peptides from diverse systems. Biosci. Rep. 20, 483–500 10.1023/A:1010454803579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. White J. M. (1990) Viral and cellular membrane fusion proteins. Annu. Rev. Physiol. 52, 675–697 10.1146/annurev.ph.52.030190.003331 [DOI] [PubMed] [Google Scholar]
- 7. Delos S. E., Gilbert J. M., and White J. M. (2000) The central proline of an internal viral fusion peptide serves two important roles. J. Virol. 74, 1686–1693 10.1128/JVI.74.4.1686-1693.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gómara M. J., Mora P., Mingarro I., and Nieva J. L. (2004) Roles of a conserved proline in the internal fusion peptide of Ebola glycoprotein. FEBS Lett. 569, 261–266 10.1016/j.febslet.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 9. Fredericksen B. L., and Whitt M. A. (1995) Vesicular stomatitis virus glycoprotein mutations that affect membrane fusion activity and abolish virus infectivity. J. Virol. 69, 1435–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drummer H. E., and Poumbourios P. (2004) Hepatitis C virus glycoprotein E2 contains a membrane-proximal heptad repeat sequence that is essential for E1E2 glycoprotein heterodimerization and viral entry. J. Biol. Chem. 279, 30066–30072 10.1074/jbc.M405098200 [DOI] [PubMed] [Google Scholar]
- 11. Nash T. C., and Buchmeier M. J. (1997) Entry of mouse hepatitis virus into cells by endosomal and nonendosomal pathways. Virology 233, 1–8 10.1006/viro.1997.8609 [DOI] [PubMed] [Google Scholar]
- 12. Eifart P., Ludwig K., Böttcher C., de Haan C. A., Rottier P. J., Korte T., and Herrmann A. (2007) Role of endocytosis and low pH in murine hepatitis virus strain A59 cell entry. J. Virol. 81, 10758–10768 10.1128/JVI.00725-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qiu Z., Hingley S. T., Simmons G., Yu C., Das Sarma J., Bates P., and Weiss S. R. (2006) Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 80, 5768–5776 10.1128/JVI.00442-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leparc-Goffart I., Hingley S. T., Chua M. M., Jiang X., Lavi E., and Weiss S. R. (1997) Altered pathogenesis of a mutant of the murine coronavirus MHV-A59 is associated with a Q159L amino acid substitution in the spike protein. Virology 239, 1–10 10.1006/viro.1997.8877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hingley S. T., Leparc-Goffart I., and Weiss S. R. (1998) The spike protein of murine coronavirus mouse hepatitis virus strain A59 is not cleaved in primary glial cells and primary hepatocytes. J. Virol. 72, 1606–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chambers P., Pringle C. R., and Easton A. J. (1990) Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J. Gen. Virol. 71, 3075–3080 10.1099/0022-1317-71-12-3075 [DOI] [PubMed] [Google Scholar]
- 17. Luo Z., and Weiss S. R. (1998) Roles in cell-to-cell fusion of two conserved hydrophobic regions in the murine coronavirus spike protein. Virology 244, 483–494 10.1006/viro.1998.9121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Phillips J. J., Chua M. M., Lavi E., and Weiss S. R. (1999) Pathogenesis of chimeric MHV4/MHV-A59 recombinant viruses: the murine coronavirus spike protein is a major determinant of neurovirulence. J. Virol. 73, 7752–7760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Das Sarma J., Fu L., Tsai J. C., Weiss S. R., and Lavi E. (2000) Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J. Virol. 74, 9206–9213 10.1128/JVI.74.19.9206-9213.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das Sarma J., Kenyon L. C., Hingley S. T., and Shindler K. S. (2009) Mechanisms of primary axonal damage in a viral model of multiple sclerosis. J. Neurosci. 29, 10272–10280 10.1523/JNEUROSCI.1975-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kenyon L. C., Biswas K., Shindler K. S., Nabar M., Stout M., Hingley S. T., Grinspan J. B., and Das Sarma J. (2015) Gliopathy of demyelinating and non-demyelinating strains of mouse hepatitis virus. Front. Cell. Neurosci. 9, 488 10.3389/fncel.2015.00488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Das Sarma J., Iacono K., Gard L., Marek R., Kenyon L. C., Koval M., and Weiss S. R. (2008) Demyelinating and nondemyelinating strains of mouse hepatitis virus differ in their neural cell tropism. J. Virol. 82, 5519–5526 10.1128/JVI.01488-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo Z. L., and Weiss S. R. (1998) Mutational analysis of fusion peptide-like regions in the mouse hepatitis virus strain A59 spike protein. Adv. Exp. Med. Biol. 440, 17–23 [DOI] [PubMed] [Google Scholar]
- 24. Walls A. C., Tortorici M. A., Bosch B. J., Frenz B., Rottier P. J. M., DiMaio F., Rey F. A., and Veesler D. (2016) Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531, 114–117 10.1038/nature16988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ziebuhr J., Snijder E. J., and Gorbalenya A. E. (2000) Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 81, 853–879 10.1099/0022-1317-81-4-853 [DOI] [PubMed] [Google Scholar]
- 26. Ou X., Zheng W., Shan Y., Mu Z., Dominguez S. R., Holmes K. V., and Qian Z. (2016) Identification of the fusion peptide-containing region in betacoronavirus spike glycoproteins. J. Virol. 90, 5586–5600 10.1128/JVI.00015-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tiwari V., Darmani N. A., Yue B. Y., and Shukla D. (2010) In vitro antiviral activity of neem (Azardirachta indica L.) bark extract against herpes simplex virus type-1 infection. Phytother. Res. 24, 1132–1140 10.1002/ptr.3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaufman G., Liu P., and Leibowitz J. L. (2014) Identification of novel functional regions within the spike glycoprotein of MHV-A59 based on a bioinformatics approach. Virus Res. 189, 177–188 10.1016/j.virusres.2014.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sadasivan J., Singh M., and Sarma J. D. (2017) Cytoplasmic tail of coronavirus spike protein has intracellular targeting signals. J. Biosci. 42, 231–244 10.1007/s12038-017-9676-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lontok E., Corse E., and Machamer C. E. (2004) Intracellular targeting signals contribute to localization of coronavirus spike proteins near the virus assembly site. J. Virol. 78, 5913–5922 10.1128/JVI.78.11.5913-5922.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chakrabarti P., and Pal D. (2001) The interrelationships of side-chain and main-chain conformations in proteins. Prog. Biophys. Mol. Biol. 76, 1–102 10.1016/S0079-6107(01)00005-0 [DOI] [PubMed] [Google Scholar]
- 32. Das Sarma J., Scheen E., Seo S. H., Koval M., and Weiss S. R. (2002) Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. J. Neurovirol. 8, 381–391 10.1080/13550280260422686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuo L., Godeke G. J., Raamsman M. J., Masters P. S., and Rottier P. J. (2000) Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J. Virol. 74, 1393–1406 10.1128/JVI.74.3.1393-1406.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Donnelly D. J., Gensel J. C., Ankeny D. P., van Rooijen N., and Popovich P. G. (2009) An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J. Neurosci. Methods 181, 36–44 10.1016/j.jneumeth.2009.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Das Sarma J., Fu L., Hingley S. T., Lai M. M., and Lavi E. (2001) Sequence analysis of the S gene of recombinant MHV-2/A59 coronaviruses reveals three candidate mutations associated with demyelination and hepatitis. J. Neurovirol. 7, 432–436 10.1080/135502801753170282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Das Sarma J., Fu L., Hingley S. T., and Lavi E. (2001) Mouse hepatitis virus type-2 infection in mice: an experimental model system of acute meningitis and hepatitis. Exp. Mol. Pathol. 71, 1–12 10.1006/exmp.2001.2378 [DOI] [PubMed] [Google Scholar]
- 37. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 38. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 39. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- 40. Kooi C., Cervin M., and Anderson R. (1991) Differentiation of acid-pH-dependent and -nondependent entry pathways for mouse hepatitis virus. Virology 180, 108–119 10.1016/0042-6822(91)90014-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McGibbon R. T., Beauchamp K. A., Harrigan M. P., Klein C., Swails J. M., Hernández C. X., Schwantes C. R., Wang L. P., Lane T. J., and Pande V. S. (2015) MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 10.1016/j.bpj.2015.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dveksler G. S., Pensiero M. N., Cardellichio C. B., Williams R. K., Jiang G. S., Holmes K. V., and Dieffenbach C. W. (1991) Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 65, 6881–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Compton S. R., Winograd D. F., and Gaertner D. J. (1995) Optimization of in vitro growth conditions for enterotropic murine coronavirus strains. J. Virol. Methods 52, 301–307 10.1016/0166-0934(94)00161-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lavi E., Gilden D. H., Wroblewska Z., Rorke L. B., and Weiss S. R. (1984) Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34, 597–603 10.1212/WNL.34.5.597 [DOI] [PubMed] [Google Scholar]
- 45. McGavern D. B., Murray P. D., and Rodriguez M. (1999) Quantitation of spinal cord demyelination, remyelination, atrophy, and axonal loss in a model of progressive neurologic injury. J. Neurosci. Res. 58, 492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lavi E., Fishman P. S., Highkin M. K., and Weiss S. R. (1988) Limbic encephalitis after inhalation of a murine coronavirus. Lab. Invest. 58, 31–36 [PubMed] [Google Scholar]
- 47. Keck J. G., Soe L. H., Makino S., Stohlman S. A., and Lai M. M. (1988) RNA recombination of murine coronaviruses: recombination between fusion-positive mouse hepatitis virus A59 and fusion-negative mouse hepatitis virus 2. J. Virol. 62, 1989–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.