

Abstract
G protein–coupled receptor (GPCR) signaling is regulated by members of the protein kinase C (PKC) and GPCR kinase (GRK) families, although the relative contribution of each to GPCR function varies among specific GPCRs. The CXC motif receptor 4 (CXCR4) is a member of the GPCR superfamily that binds the CXC motif chemokine ligand 12 (CXCL12), initiating signaling that is subsequently terminated in part by internalization and lysosomal degradation of CXCR4. The purpose of this study is to define the relative contribution of PKC and GRK to CXCR4 signaling attenuation by studying their effects on CXCR4 lysosomal trafficking and degradation. Our results demonstrate that direct activation of PKC via the phorbol ester phorbol 12-myristate 13-acetate (PMA) mimics CXCL12-mediated desensitization, internalization, ubiquitination, and lysosomal trafficking of CXCR4. In agreement, heterologous activation of PKC by stimulating the chemokine receptor CXCR5 with its ligand, CXCL13, also mimics CXCL12-mediated desensitization, internalization, ubiquitination, and lysosomal degradation of CXCR4. Similar to CXCL12, PMA promotes PKC-dependent phosphorylation of serine residues within CXCR4 C-tail that are required for binding and ubiquitination by the E3 ubiquitin ligase AIP4 (atrophin-interacting protein 4). However, inhibition of PKC activity does not alter CXCL12-mediated ubiquitination and degradation of CXCR4, suggesting that other kinases are also required. Accordingly, siRNA-mediated depletion of GRK6 results in decreased degradation and ubiquitination of CXCR4. Overall, these results suggest that PKC and GRK6 contribute to unique aspects of CXCR4 phosphorylation and lysosomal degradation to ensure proper signal propagation and termination.
Keywords: G protein-coupled receptor (GPCR), CXC chemokine receptor type 4 (CXCR4), receptor desensitization, receptor internalization, receptor protein serine/threonine kinase, phorbol ester, receptor modification, chemokine, lysosomal trafficking, receptor down-regulation, heterotrimeric G protein, receptor endocytosis, receptor degradation, signal transduction, protein kinase C (PKC), cyclic AMP (cAMP)
Introduction
The signaling cascades elicited by the G protein–coupled receptor (GPCR)3 CXCR4 and its cognate ligand CXCL12 (SDF-1α) are involved in the normal development of the immune, cardiovascular, and central nervous systems (1–4) as well as playing an important role in the pathophysiology of several types of cancer (5). CXCR4 levels are up-regulated in at least 23 types of cancer and are correlated with a poor prognosis either by enhancing tumor growth or contributing to the tumor metastatic potential (6–10). In addition, WHIM syndrome, a rare immunodeficiency disorder characterized by warts, hypogammaglobulinemia, bacterial infections, and myelokathexis, is linked to truncations of CXCR4 C terminus (C-tail), which ultimately confer increased signaling capabilities to the receptor (11, 12). Despite its role in disease, the regulation of CXCR4 abundance or signaling remains poorly understood.
The magnitude and duration of CXCR4 signaling is under tight regulation at multiple steps of the signal transduction cascade, including at the level of the receptor itself (13). Receptor phosphorylation is regarded as a key step in mediating CXCR4 unresponsiveness to CXCL12, a process referred to as desensitization (13–16). CXCR4 phosphorylation is typically mediated by distinct serine/threonine kinases that belong to either the G protein–coupled receptor kinase (GRK) or protein kinase C (PKC) families (17). GRKs typically mediate receptor phosphorylation of agonist-bound receptor, leading to β-arrestin binding and subsequent G protein uncoupling, thereby terminating further receptor signaling (18). This is referred to as homologous desensitization (19). Second messenger–dependent protein kinases such as PKC also phosphorylate GPCRs, including CXCR4 (17), but they are able to do it in an agonist-independent manner; this is referred to as heterologous desensitization (18). Several GRKs, namely GRK6, GRK2, and GRK3, and several PKC isoforms have been implicated in phosphorylation of CXCR4 C-tail serine/threonine residues that contribute to CXCR4 desensitization (20–22).
Phosphorylation of CXCR4 is required for CXCL12-mediated receptor internalization and trafficking to lysosomes, a terminal degradative compartment (23, 24). Multiple phosphorylation sites on the C-tail have been implicated in CXCL12-mediated internalization of CXCR4 (23). CXCL12-induced phosphorylation of the C-tail is also required for targeting CXCR4 for lysosomal degradation (24). Site-specific phosphorylation of serine residues 324 and 325 (Ser-324/325) induced by CXCL12 promotes ubiquitination of nearby lysine residues, a prerequisite for lysosomal trafficking and receptor degradation (25). These residues are thought to be phosphorylated by PKC and GRK6, and whereas the contributions of PKC or GRK6 to CXCR4 internalization are known (17, 23, 26–30), their contributions to CXCR4 lysosomal trafficking remain unknown.
The purpose of this study is to define the relative contribution of PKC and GRK6 to CXCR4 lysosomal trafficking. Here we show that PMA, a phorbol ester that directly activates PKC (31), mimics many aspects of CXCL12 in promoting lysosomal trafficking of CXCR4. Similar to CXCL12, PMA promotes CXCR4 phosphorylation at Ser-324/325 and trafficking to a lysosomal degradative compartment. However, whereas PKC is dispensable for CXCL12-promoted ubiquitination and degradation, GRK6 is essential to target CXCR4 to a degradative compartment. Moreover, heterologous activation of PKC by CXCL13, the cognate ligand for the chemokine receptor CXCR5, promotes CXCR4 internalization and degradation. Accordingly, pretreatment with PMA or CXCL13 attenuates early CXCR4 heterotrimeric G protein Gαi signaling by CXCL12. Therefore, heterologous regulation of CXCR4 lysosomal trafficking by chemokines via PKC desensitizes CXCR4 signaling to its ligand CXCL12.
Results
PKC activation is sufficient to induce CXCR4 trafficking to lysosomes and degradation
Previous reports have provided evidence that direct activation of PKC by the phorbol ester PMA is sufficient to induce CXCR4 internalization in several cell types, although the fate of CXCR4 after internalization remains unclear (23, 28, 29). Here, we investigated whether direct PKC activation is sufficient to promote CXCR4 intracellular trafficking to terminal lysosomal compartments. In HEK293 cells stably expressing HA-tagged CXCR4, PMA induced dose-dependent (Fig. 1A) and time-dependent (Fig. 1B) degradation of HA-CXCR4. Based on these results, all subsequent experiments were performed at 3 h after the addition of 10 or 100 nm PMA. Next, we investigated whether PMA-mediated degradation was sensitive to endosomal acidification inhibitors such as chloroquine. In the absence of chloroquine, CXCL12 and PMA elicited 60–70% of HA-CXCR4 degradation after 3 h of CXCL12 stimulation (Fig. 1C). Pretreatment of HEK293 with chloroquine completely abrogated both CXCL12 and PMA-mediated HA-CXCR4 degradation (Fig. 1C), indicating that PMA, similar to CXCL12 (25), induces the delivery of CXCR4 to acidic compartments necessary for CXCR4 degradation.
Figure 1.

Characterization of PMA-mediated CXCR4 degradation. A and B, HEK293 cells stably expressing HA-CXCR4 were treated with increasing doses of PMA for 3 h (n = 6) (A) or for the indicated times with 10 nm PMA (n = 3) (B). Graphs represent the average amount of HA-CXCR4 degraded compared with its vehicle control at each dose (A) or time (B) ± S.D. Data were analyzed by one-way ANOVA, followed by Tukey's post hoc test. In A, the adjusted p values are for the following comparisons: 1 and 10 nm, * = 0.0185; 1 and 100 nm, ** = 0.0088; 1 and 1000 nm, *** = 0.0078. In B, the adjusted p values are for the following comparisons: 0.5 and 3 h, * = 0.0032; 0.5 and 6 h, ** = 0.0042. C, HEK293 cells stably expressing HA-CXCR4 were pretreated with 100 μm chloroquine (+CQ) or not (No CQ) for 30 min before treatment with 10 nm CXCL12, 10 nm PMA, or corresponding vehicle (n = 3) for 3 h. Representative immunoblots are shown. The positions of the molecular weight markers are indicated. Average percentage degradation of HA-CXCR4 was calculated as described under “Experimental procedures.” Error bars, S.D. In C, data were analyzed by two-way ANOVA, followed by Tukey's post hoc test. Adjusted p values are indicated.
To corroborate these results, we examined lysosomal trafficking of CXCR4 using multilabel fluorescence microscopy. HEK293 cells were transfected with CXCR4 tagged with YFP (CXCR4-YFP) and serum-starved for ∼3 h, followed by treatment with vehicle, CXCL12, or PMA for another 3 h. Following stimulation and fixation, LAMP1-positive lysosomes were immunostained for multilabel fluorescence microscopy and colocalization analysis. As shown in Fig. 2A, cells treated with PMA and CXCL12 displayed high levels of colocalization between CXCR4-YFP and LAMP1. The mean percentage of LAMP1-positive lysosomes colocalizing with CXCR4-YFP puncta was greater in cells treated with either PMA (∼22%) or CXCL12 (∼27%), compared with vehicle-treated cells (∼13%), with no significant difference between cells treated with PMA and CXCL12 (Fig. 2B). These data demonstrate that PMA, similar to CXCL12, elicits an intracellular itinerary that facilitates degradation of CXCR4 in terminal endocytic compartments.
Figure 2.
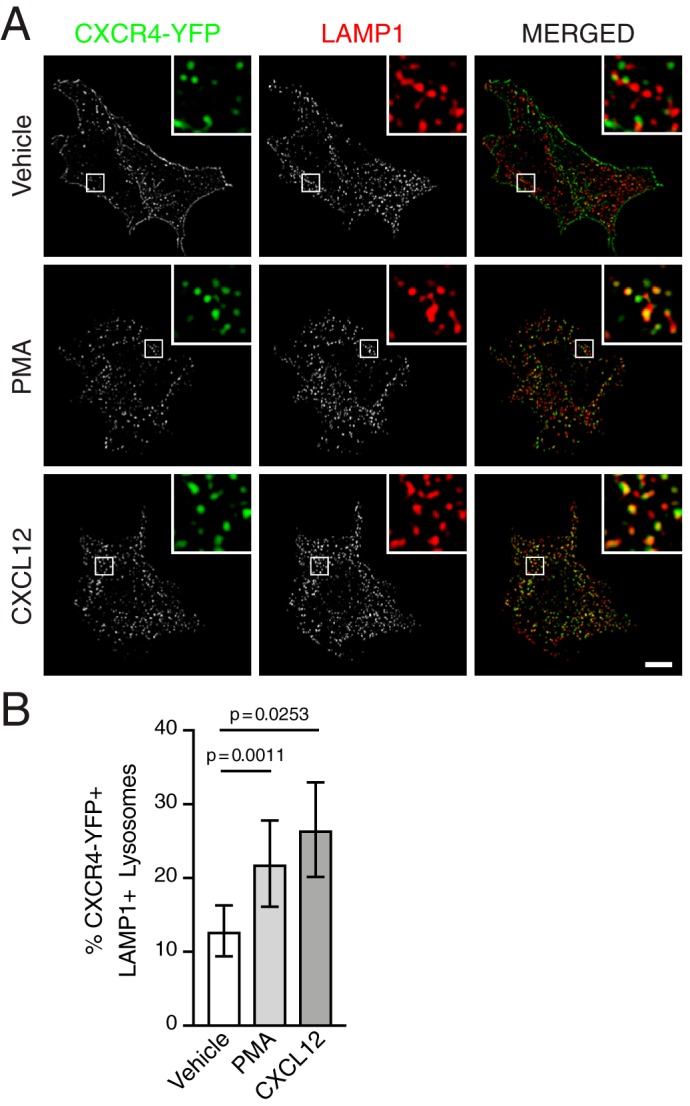
PMA and CXCL12 promote trafficking of CXCR4 to lysosomes. A and B, HEK293 cells transiently expressing HA-CXCR4-YFP were treated with either vehicle, 100 nm PMA, or 10 nm CXCL12 for 3 h and then coimmunostained with antibodies against LAMP1 for multilabel immunofluorescence microscopy and colocalization analysis. A, representative micrographs of cells treated with vehicle (top), PMA (middle), or CXCL12 (bottom) showing CXCR4-YFP+ puncta (left) and LAMP1+ lysosomes (center) in single-channel and merged images (right). Insets, boxed areas enlarged 5 times. Scale bar, 10 μm. B, quantification of the mean percentage of LAMP1+ lysosomes colocalized with CXCR4-YFP+ puncta in cells treated with vehicle, PMA, or CXCL12. The total number of LAMP1+ lysosomes and colocalization with CXCR4-YFP+ puncta were determined using 3D object-based analysis, as described under “Experimental procedures.” Bars, average from three independent experiments. Error bars, 95.00% CI. The following numbers of cells were used in the analysis: vehicle, n = 32 cells; PMA, n = 21 cells; and CXCL12, n = 31 cells. Data were analyzed using Welch's ANOVA followed by Tamhane's T2 multiple-comparison test. Adjusted p values are indicated.
Role of PKC in CXCR4 phosphorylation
The similarity between CXCL12 and PMA-mediated degradation raised the question of whether PKC is involved directly in CXCL12-mediated CXCR4 degradation. Previously, we have shown that CXCL12 stimulation drives CXCR4 degradation through transient phosphorylation of serine residues 324 and 325 (Ser-324/325) at the plasma membrane, using a custom antibody that specifically detects simultaneous phosphorylation of Ser-324 and Ser-325 (pSer324/325) by immunofluorescence microscopy (24). To determine whether PMA and CXCL12 elicit similar spatial patterns of Ser-324 and Ser-325 phosphorylation, HEK293 or HeLa cells transiently expressing CXCR4-YFP were serum-starved for 3.5 h and then stimulated with either CXCL12 or PMA for 30 min. The antibody against pSer324/325 showed robust staining at the cell periphery in both PMA- and CXCL12-treated cells compared with vehicle, suggesting simultaneous phosphorylation of Ser-324 and Ser-325 (Fig. 3A; quantified in Fig. 3B). To determine whether PKC is necessary for CXCL12-mediated phosphorylation of these residues, cells were treated with BisI, a selective PKC inhibitor (32), or vehicle before stimulation with CXCL12. Simultaneous phosphorylation of Ser-324 and Ser-325 was significantly inhibited by BisI compared with vehicle control (Fig. 4A; quantified in Fig. 4B). These results indicate that PKC is sufficient and necessary for simultaneous phosphorylation of Ser-324 and Ser-325 promoted by CXCL12.
Figure 3.
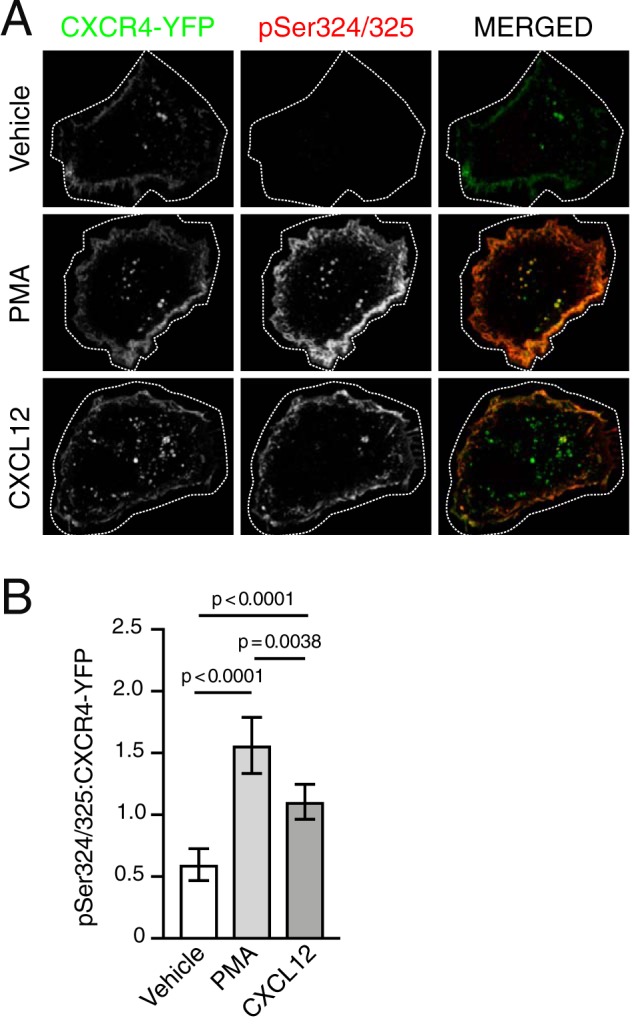
PKC is sufficient to mediate site-specific C-tail phosphorylation of CXCR4. A, representative micrographs of HEK293 cells transiently expressing HA-CXCR4-YFP and immunostained to detect simultaneous phosphorylation of serine residues 324 and 325 (pSer324/325). Cells were serum-starved for 3.5 h then stimulated with either vehicle, 10 nm PMA, or 10 nm CXCL12 for 30 min. Panels show HA-CXCR4-YFP (left) and pSer324/325 (center) as single-channel grayscale and pseudocolored, merged images (right) for the indicated treatment conditions. Images were processed in ImageJ, as described under “Experimental procedures.” For optimal viewing, HA-CXCR4-YFP and pSer324/325 signal intensities were adjusted with opacity screens in Photoshop. Screens for HA-CXCR-YFP were adjusted to offset between group variation in expression; screens for pSer324/325 were adjusted within each group to match adjustments in HA-CXCR4-YFP. Scale bar, 10 μm. B, quantification of pSer324/325:HA-CXCR4-YFP fluorescence intensities. Bars represent the average from three independent experiments. Error bars, 95.00% CI. For vehicle, n = 13 cells; for PMA, n = 15 cells; for CXCL12, n = 17 cells. Data are presented as mean pSer324/325:HA-CXCR4-YFP fluorescence intensity ratios. Data were analyzed using Welch's ANOVA followed by Tamhane's T2 multiple-comparison test. Adjusted p values are indicated.
Figure 4.
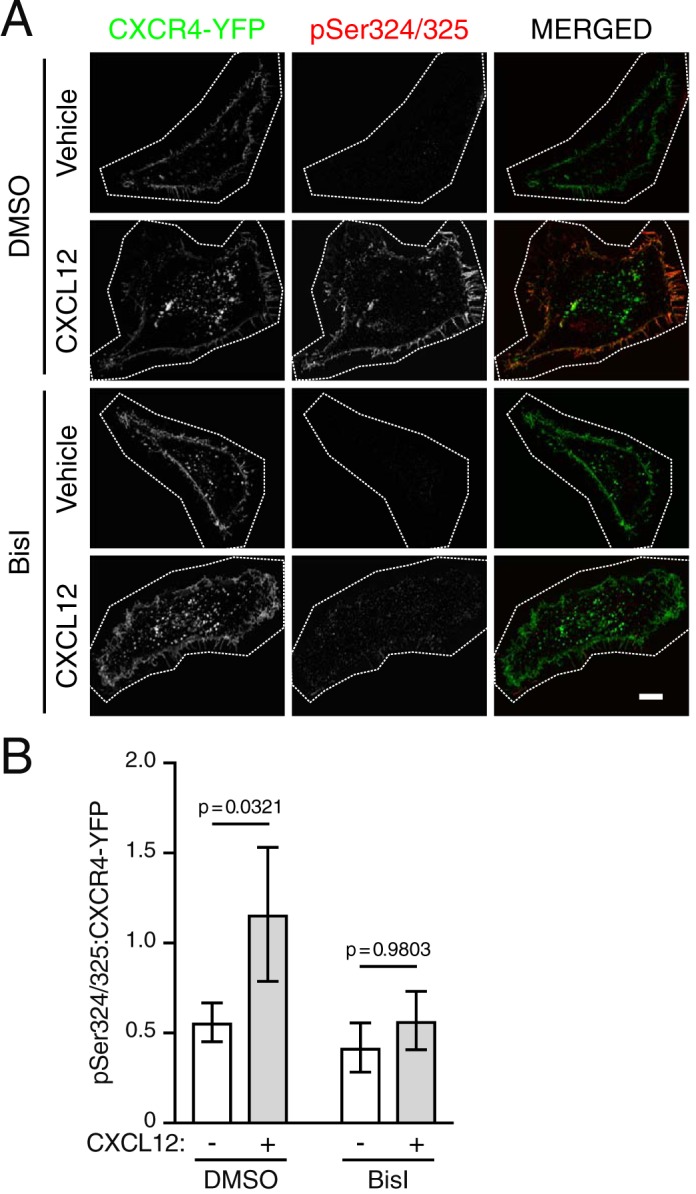
PKC is necessary for site-specific C-tail phosphorylation of CXCR4. A, representative micrographs of HEK293 cells transiently expressing HA-CXCR4-YFP and immunostained to detect simultaneous phosphorylation of serine residues 324 and 325 (pSer324/325). Cells were serum-starved for 3.5 h and then pretreated with either vehicle (DMSO) or BisI for 30 min and then stimulated with either vehicle or 10 nm CXCL12 for 30 min. Panels show HA-CXCR4-YFP (left) and pSer324/325 (center) as single-channel grayscale and pseudocolored, merged images (right) for the indicated treatment conditions. Images were processed in ImageJ, as described under “Experimental procedures.” For optimal viewing, HA-CXCR4-YFP and pSer324/325 signal intensities were adjusted with opacity screens in Photoshop. Screens for HA-CXCR-YFP were adjusted to offset between group variation in expression; screens for pSer324/325 were adjusted within each group to match adjustments in HA-CXCR4-YFP. Scale bar, 10 μm. B, quantification of pSer324/325:HA-CXCR4-YFP fluorescence intensities. Data are presented as mean pSer324/325:HA-CXCR4-YFP fluorescence intensity ratios. Bars, average from three independent experiments. Error bars, 95.00% CI. For DMSO:vehicle, n = 8 cells; for DMSO:CXCL12, n = 19 cells; for BisI:vehicle, n = 5 cells, for BisI:CXCL12, n = 14 cells. Data were analyzed using two-way ANOVA followed by Tukey's test for multiple comparisons. Adjusted p values are indicated.
Role of PKC in CXCR4 degradation and ubiquitination
To explore whether PKC is necessary for CXCR4 degradation, cells were pretreated with BisI, followed by treatment with CXCL12 or PMA for 3 h and immunoblotting. We performed these experiments in HEK293 cells transiently transfected with HA-CXCR4, as well as in HeLa cells, which endogenously express CXCR4 (33–36). Consistent with our previous data (33, 35, 37), CXCL12 promoted degradation of HA-CXCR4 in HEK293 cells (Fig. 5A; quantified in Fig. 5B) as well as endogenous CXCR4 in HeLa cells (Fig. 5C; quantified in Fig. 5D). Similarly, PMA promoted CXCR4 degradation in both HEK293 and HeLa cells; however, pretreatment with BisI did not impact CXCL12-promoted CXCR4 degradation in HEK293 or HeLa cells, whereas PMA-promoted degradation was completely inhibited by BisI pretreatment in both cell types (Fig. 5, A–D). These data indicate that PMA-mediated CXCR4 degradation is entirely due to activation of PKC, whereas PKC phosphorylation of CXCR4 induced by CXCL12 is dispensable to commit CXCR4 to the degradative pathway.
Figure 5.

PKC is sufficient, but not necessary, for CXCL12-mediated CXCR4 degradation. A–D, HEK293 cells transiently transfected with HA-CXCR4 (A) or HeLa cells (C) were pretreated with 10 μm BisI or DMSO equivalent for 30 min, before treatment with 10 nm each of CXCL12 or PMA for 3 h. A and C, representative immunoblots are shown. The positions of the molecular weight markers are indicated. B and D, degradation of HA-CXCR4 (n = 5–6) (B) and endogenous CXCR4 (4, 5) (D) was measured and calculated as described under “Experimental procedures.” Bars, average receptor degraded ± S.D. (error bars). Data were analyzed by two-way ANOVA followed by Tukey's post hoc test. Adjusted p values are indicated. E, HEK293 cells stably expressing HA-CXCR4 and transiently transfected with FLAG-ubiquitin were pretreated with 10 μm BisI or DMSO equivalent and stimulated for 20 min with 30 nm CXCL12 in the presence or absence of BisI. Detection of ubiquitinated CXCR4 was performed as described under “Experimental procedures.” Representative immunoblots are shown. F, CXCR4 ubiquitination was quantified by densitometric analysis. Ubiquitin levels were normalized to CXCR4 levels in immunoprecipitates, and CXCR4 ubiquitination is expressed as a fraction of control or the values from DMSO- and CXCL12-treated cells. Bars, average from three independent experiments ± S.D. Data were analyzed by two-way ANOVA, and there was a marginally significant effect between vehicle and CXCL12 (p = 0.0519).
Because phosphorylation of Ser-324/325 mediates AIP4 binding and ubiquitination (24), we next examined the role of PKC on the status of CXCR4 ubiquitination upon CXCL12 treatment in the presence of the PKC inhibitor BisI. Consistent with our previously published results (25, 33), CXCL12 elicits ubiquitination of CXCR4 (Fig. 5E; quantified in Fig. 5F). However, pretreatment with BisI had no effect on CXCR4 ubiquitination upon CXCL12 treatment (Fig. 5E; quantified in Fig. 5F). Because the antibody against pSer324/325 only recognizes dually phosphorylated CXCR4 (24), these results suggest that PKC activity may be responsible for the phosphorylation of only one of the residues in the Ser-324/325 doublet. To explore this further, we examined CXCR4 degradation of single serine mutants S324A and S325A transiently expressed in HEK293 cells in the presence of BisI. Degradation of these mutants was not impaired compared to the double-serine mutant S324A/S325A (Fig. 6A; quantified in Fig. 6B), as we have reported previously (24). Similarly, BisI did not impact degradation of the individual mutants to the level observed with the double S324A/S325A mutant. These data suggest that phosphorylation of either Ser-324 or Ser-325 is sufficient to support CXCR4 degradation by CXCL12, and although PKC may be sufficient, it also suggests that another kinase(s) is likely necessary for phosphorylation of these residues and CXCR4 degradation.
Figure 6.

Analysis of the role of Ser-324 and Ser-325 in PKC-mediated CXCR4 degradation. A, HEK293 cells transfected with WT and serine receptor mutants were pretreated with 10 μm BisI or vehicle (Veh; DMSO) for 30 min, before treatment with 10 nm CXCL12 for 3 h. Representative immunoblots are shown. The positions of the molecular weight markers are indicated. B, densitometric analysis of CXCR4 degradation. Bars, average receptor degraded from four independent experiments ± S.D. (error bars). Data were analyzed by two-way ANOVA, followed by Tukey's multiple-comparison test. The adjusted p values between the following comparisons are as follows. #, WT:vehicle versus S324/5A:vehicle = 0.0003; *, WT:BisI versus S324/5A:BisI = 0.0004; ##, WT:BisI versus S325A:BisI = 0.0175; **, S325A:vehicle versus S325A:BisI = 0.0523.
GRK6 contributes to CXCR4 ubiquitination and degradation
GRK6 has been previously implicated in phosphorylation of serine residues 324 and 325 induced by CXCL12 (17), suggesting that it may be involved in CXCR4 degradation. Thus, we addressed whether GRK6 participated in CXCL12-mediated degradation of CXCR4 by measuring degradation of endogenous CXCR4 following CXCL12 treatment in HeLa cells transfected with siRNA against GRK6. Compared with control siRNA, CXCR4 degradation was significantly attenuated by ∼60% in GRK6 siRNA-transfected cells (Fig. 7A; quantified in Fig. 7B). Pretreatment of BisI in the GRK6 siRNA background had no significant additional effect on CXCR4 degradation promoted by CXCL12 (data not shown). Thus, these data indicate that GRK6 has a distinctive role in targeting CXCR4 for degradation.
Figure 7.

Role of GRK6 in CXCR4 ubiquitination and degradation. A, HeLa cells transfected with control (Ctrl) or GRK6 siRNA were stimulated with vehicle or 10 nm CXCR4 for 3 h. Representative immunoblots are shown. The positions of the molecular weight markers are indicated. B, samples prepared in parallel from cells transfected with siCTRL or siGRK were analyzed by immunoblotting for GRK6 abundance. Representative immunoblots are shown. C, CXCR4 degradation was measured and calculated as described under “Experimental procedures.” Bars, average receptor degradation from six independent experiments ± S.D. (error bars) Data were analyzed by Student's t test. Adjusted p value is indicated. D, HEK293 cells stably expressing HA-CXCR4 were transfected with control (CTRL) or GRK6 siRNA and co-transfected 1 day later with FLAG-ubiquitin. The day of the experiment, cells were treated with 30 nm CXCL12 or vehicle for 20 min. Detection of ubiquitinated CXCR4 was performed as described under “Experimental procedures.” Representative immunoblots are shown. E, CXCR4 ubiquitination was quantified by densitometric analysis. Ubiquitin levels were normalized to CXCR4 levels in immunoprecipitates, and CXCR ubiquitination is expressed as a fraction of the siCTRL- and CXCL12-treated cells. Bars, average from three independent experiments ± S.D. Data were analyzed by two-way ANOVA, followed by Sidak's post hoc test. Adjusted p values are indicated. F, HeLa cells transfected as described in A were dissociated and aliquoted at 5 × 105 viable cells/tube. Each tube received the indicated concentrations of CXCL12 and was immediately incubated at 37 °C for 30 min. Thereafter, cells were stained with the mouse anti-CXCR4 12G5-PE antibody, as described under “Experimental procedures.” Data are presented as a percentage of the geometric mean fluorescent intensity of each sample relative to that of vehicle-treated cells. Data represent the average of 3–5 independent experiments ± S.D. The dashed line indicates the location of x axis segmentation. Data were analyzed by two-way ANOVA followed by Tukey's post hoc test. *, adjusted p value 0.0355.
Given the role of CXCR4 ubiquitination in CXCR4 degradation (24, 25), we investigated whether GRK6 contributes to receptor ubiquitination. CXCL12-induced ubiquitination of CXCR4 occurred normally in cells transfected with control siRNA, whereas GRK6 siRNA consistently decreased the fraction of ubiquitinated CXCR4 (Fig. 7D; quantified in Fig. 7E). Because impairment of internalization could also explain the impact on CXCR4 degradation and ubiquitination, we also assessed whether GRK6 siRNA impaired early steps of receptor endocytosis from the plasma membrane. For this purpose, HeLa cells were pretreated with increasing amounts of CXCL12 for 30 min at 37 °C, and the level of remaining endogenous CXCR4 was measured by staining with a PE-labeled anti-CXCR4 (12G5) and analyzed by flow cytometry. In control siRNA-transfected cells, CXCR4 showed a dose-dependent decrease in surface staining, consistent with receptor sequestration (Fig. 7E). Cells transfected with GRK6 siRNA also elicited a dose-dependent decrease in receptor levels upon CXCL12 treatment, but to a significantly lower extent (Fig. 7E). These data indicate that GRK6 contributes to CXCR4 endocytosis, but because CXCR4 internalization is not a rate-limiting step in its lysosomal trafficking (25), it is unlikely that this accounted for the defect observed at the level of degradation (Fig. 7B). In addition, because CXCR4 ubiquitination occurs at the level of the plasma membrane (25), the defect on internalization is unlikely to be responsible for decreased ubiquitination (Fig. 7D), further suggesting that GRK6 activity is required for proper ubiquitination of CXCR4. Taken together, our data suggest that GRK6 contributes to CXCR4 degradation by facilitating ubiquitination of CXCR4.
Heterologous regulation of CXCR4 lysosomal degradation by CXCR5
Next, we examined whether PKC activation elicited by signaling from other members of the chemokine receptor family could promote CXCR4 down-regulation. We focused on the chemokine receptor CXCR5 because it is endogenously expressed in HeLa cells and because its cognate ligand CXCL13 promotes signaling via a pertussis toxin–sensitive G protein and is likely to activate PKC in these cells (36). We first examined whether CXCL13 mediates CXCR4 internalization because it is a prerequisite for CXCR4 degradation (25). Stimulation with 100 nm CXCL13 promoted CXCR4 internalization, similar to levels that are observed with CXCL12 or PMA stimulation (Fig. 8A). Treatment with BisI almost completely blocked internalization by CXCL13 or PMA, but not CXCL12 (Fig. 8A). Additionally, CXCL13, similar to PMA, promoted rapid CXCR4 degradation (Fig. 8B; quantified in Fig. 8C), which was also almost completely blocked by BisI (Fig. 8D; quantified in Fig. 8E). Accordingly, PMA- or CXCL13-induced ubiquitination of CXCR4 was inhibited by BisI (Fig. 8F; quantified in Fig. 8G). Taken together, these data provide evidence that CXCR5 induces PKC-dependent heterologous internalization, ubiquitination, and degradation of CXCR4.
Figure 8.

Role of CXCR5 in cross-regulation of CXCR4. A, CXCR4 internalization was examined in HeLa cells stimulated with 10 nm CXCL12, 100 nm CXCL13, or 10 nm PMA for 5 min. Bars, average receptor internalized from five independent experiments ± S.D. B, CXCR4 degradation was examined in HeLa cells stimulated with 10 nm CXCL12 or 100 nm CXCL13 for the indicated times. Representative CXCR4 immunoblot is shown. Bottom, section of stain-free loading control used for densitometric analysis. The positions of the molecular weight markers are indicated. C, densitometric analysis of CXCR4 levels. Bars, average receptor degraded normalized to a stain-free loading control at each time point relative to vehicle. Error bars, S.D. D, HeLa cells were pretreated with DMSO 10 μm BisI or 10 μm BisV for 30 min, before treatment with 10 nm CXCL12, 100 nm CXCL13, or 10 nm PMA for 3 h. Whole-cell lysates were analyzed by immunoblotting for CXCR4. Bottom, GAPDH immunoblot shown as loading control. Positions of molecular weight markers are indicated. E, CXCR4 degradation was measured by densitometric analysis. Bars, average receptor degraded normalized to GAPDH with each ligand treatment compared with vehicle. Error bars, S.D. The data in A and E were analyzed by two-way ANOVA, followed by Tukey's multiple-comparison test. Adjusted p values are indicated. F, PKC is necessary for CXCL13-promoted ubiquitination of CXCR4. HEK293 cells transfected with FLAG-ubiquitin and HA-CXCR4 were treated with 10 nm PMA or 100 nm CXCL13 for 30 min in the presence or absence of 10 μm BisI. Detection of ubiquitinated CXCR4 was performed as described under “Experimental procedures.” Representative immunoblots are shown. G, CXCR4 ubiquitination was quantified by densitometric analysis. Ubiquitin levels were normalized to CXCR4 levels in immunoprecipitates, and CXCR4 ubiquitination is expressed as a fraction of control or the values from the DMSO and vehicle-treated cells. Bars, average from three independent experiments ± S.D. Data were analyzed by two-way ANOVA, followed by Tukey's post hoc test. The adjusted p value for vehicle:DMSO versus CXCL13:DMSO is indicated. *, adjusted p value 0.0467 for CXCL13:DMSO versus CXCL13:BisI.
CXCL13 affects agonist-induced CXCR4 signaling
We next examined whether heterologous degradation of CXCR4 was accompanied by a loss in receptor signaling. To address this, we pretreated HeLa cells with PMA or CXCL13 and then assessed CXCL12-elicited Gαi-dependent inhibition of adenylate cyclase (AC), which is typically quantified in the presence of the AC activator forskolin (FSK) (38). As expected, CXCL12 stimulation of HeLa cells for 10 min robustly inhibits FSK-induced cAMP production by ∼60% (Fig. 9A). Next, HeLa cells were pretreated with vehicle or multiple doses of PMA or CXCL13 for 30 min and subsequently challenged with 10 nm CXCL12 in the presence of FSK. In cells pretreated with vehicle, CXCL12 inhibited FSK-induced cAMP production (not shown), similar to levels observed without the pretreatment step (Fig. 9A). In contrast, pretreatment with PMA (Fig. 9B) or CXCL13 (Fig. 9C) dose-dependently attenuated CXCL12-mediated cAMP accumulation, indicating desensitization of CXCR4 signaling. Thus, the ability of PMA or CXCL13 to render CXCR4 insensitive to subsequent CXCL12 challenges correlates with their ability to commit CXCR4 to the degradative pathway, suggesting that heterologous regulation of CXCR4 down-regulation can impact signaling through its cognate ligand.
Figure 9.
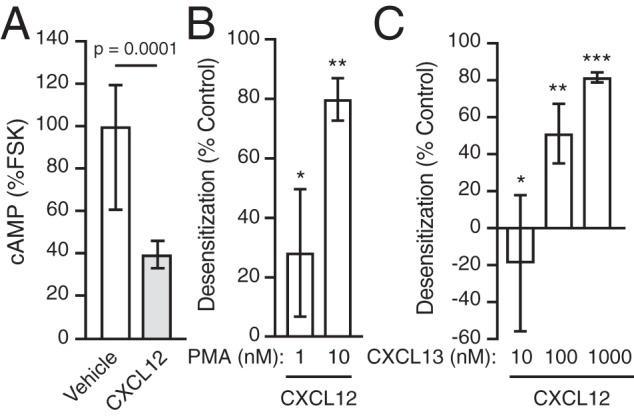
PMA and CXCL13 promote desensitization of CXCR4. A, HeLa cells were pulsed for 10 min with either vehicle or 10 nm CXCL12 in the presence of 1 μm FSK, after which cells were lysed and processed to determine the concentration of cAMP by an EIA assay, as described under “Experimental procedures.” Bars, average cAMP levels of eight samples from four independent experiments ± S.D. (error bars) normalized to cAMP levels of FSK in the absence of CXCL12. Data were analyzed by an unpaired t test. The adjusted p value is indicated. B, HeLa cells were pretreated with PMA (1 or 10 nm) or vehicle for 1 h at 37 °C. Subsequently, cells were pulsed for 10 min with 10 nm CXCL12 in the presence of 1 μm FSK. Cells were lysed, and cAMP levels were determined by EIA as in A. Bars, average desensitization of CXCL12-induced inhibition of cAMP levels from four independent experiments ± S.D. CXCL12-promoted inhibition of cAMP compared with vehicle in cells pretreated with 1 or 10 nm PMA was 72% ± 10 and 20% ± 7%, respectively. Data were analyzed by one-way ANOVA, followed by Tukey's post hoc test. The adjusted p values are as follows: *, vehicle versus 1 nm PMA = 0.0114; **, vehicle versus 10 nm PMA < 0.0001. C, cAMP levels were determined in HeLa cells transfected with pGlo22-cAMP sensor plasmid, as described under “Experimental procedures.” Cells were pretreated with either CXCL13 (10, 100, or 1000 nm) or vehicle for 30 min at 37 °C and then pulsed for 10 min with 10 nm CXCL12 at 25 °C. Luminescence recordings were made as described under “Experimental procedures.” Bars, average desensitization of CXCL12-induced inhibition of cAMP levels from three independent experiments ± S.D. CXCL12-promoted inhibition of cAMP in cells pretreated with 1, 10, or 1000 nm CXCL13 was 119 ± 31%, 49 ± 7%, and 18 ± 11%, respectively, compared with vehicle. Data were analyzed by one-way ANOVA, followed by Tukey's post hoc test. The adjusted p values compared with vehicle were as follows: *, for 1 nm CXCL13 = 0.5024; **, for 10 nm CXCL13 = 0.0135; ***, for 1000 nm CXCL13 = 0.0008.
Discussion
This study extends our knowledge of the role that kinases play in CXCR4 regulation. Here, we show that CXCR4 down-regulation can be heterologously regulated by PKC or via PKC-dependent CXCR5 signaling. The phorbol ester PMA, a potent activator of PKC, mimics several aspects of CXCL12-promoted endocytic trafficking of CXCR4, including internalization and lysosomal targeting, leading to CXCR4 degradation (Figs. 1 and 2). Similar to CXCL12 (24), PMA induces dual phosphorylation of C-tail Ser residues (Ser-324/325) in a PKC-dependent manner (Fig. 3). Although PKC is necessary and sufficient for Ser-324/325 phosphorylation, it is not necessary for promoting CXCL12-mediated CXCR4 ubiquitination and degradation (Fig. 5). This suggested that another kinase and/or phosphorylation of other C-tail sites were also able to support CXCL12-mediated degradation of CXCR4. Consistent with this hypothesis, depletion of GRK6 reduced CXCL12-mediated ubiquitination and degradation of CXCR4 (Fig. 7, A–E). Accordingly, GRK6 has been previously linked to CXCL12-mediated dual phosphorylation of Ser-324/325 within the C-tail of CXCR4 (17). We also provide evidence that heterologous regulation of CXCR4 can occur in a PKC-dependent mechanism through activation of CXCR5 by its cognate chemokine CXCL13, resulting in CXCR4 internalization, ubiquitination, and degradation (Fig. 8). Moreover, pretreatment of HeLa cells with either PMA or CXCL13 desensitizes early CXCR4 signaling, as evidenced by reduced cAMP inhibition when challenged acutely with CXCL12 (Fig. 9, B and C). Our data support the hypothesis that CXCR4 lysosomal trafficking is under tight heterologous regulation by PKC via activation of GPCRs, such as CXCR5, providing an additional layer of regulation that is likely required to fine-tune CXCR4 signaling.
PKC is responsible for phosphorylating multiple serine or threonine residues within the CXCR4 C-tail (27); however, phosphorylation of Ser-324/325 is particularly important because it is necessary for mediating CXCR4 lysosomal trafficking (24, 25). Dual phosphorylation of Ser-324/325 is required for recruitment of the E3 ubiquitin ligase AIP4 at or near the plasma membrane and subsequent ubiquitination of CXCR4, a prerequisite for its lysosomal trafficking and degradation (24). The precise PKC isoform responsible for phosphorylating Ser-324/325 cannot be discerned from our data because PMA activates (31) and BisI inhibits both conventional and novel PKC isoforms (39). However, a recent study suggests that a conventional PKC isoform may phosphorylate Ser-324/325 (40). Albeit sufficient, PKC is not necessary for CXCL12-stimulated ubiquitination and degradation of CXCR4 (Fig. 5). Inhibition of PKC with BisI completely blocked PMA-promoted degradation of CXCR4, but it did not impact its ubiquitination or degradation by CXCL12 even though BisI blocks phosphorylation of Ser-324/325 (Fig. 4). Because the antibody used to detect phosphorylation at Ser-324/325 specifically recognizes the dual phosphorylated sites, it is possible that phosphorylation of only one of these sites still occurs following stimulation with CXCL12 in the presence of BisI, albeit by a kinase other than PKC. We have previously shown that phosphomimetic mutants of CXCR4 at either Ser-324 or Ser-325 fully support AIP4 binding (24). Thus, phosphorylation of either Ser-324 or Ser-325 may be sufficient to support ubiquitination and degradation of CXCR4. Whether Ser-324 or Ser-325 is individually phosphorylated by CXCL12 in the presence of BisI remains unknown, but an MS-based approach has suggested that Ser-325 can be individually phosphorylated (17). Alternatively, CXCL12 may promote phosphorylation of other serine or threonine residues in the C-tail of CXCR4 in the presence of BisI, a number of which have been shown to be phosphorylated by CXCL12 stimulation (17, 40, 41). The CXCR4 C-tail has 18 Ser/Thr residues; however, the role of other residues in CXCR4 degradation in the context of PKC inhibition remains to be examined (17, 41).
In addition to PKC, CXCR4 is phosphorylated by several GRKs, including GRK6, GRK2, and GRK3 (17, 40). We focused on GRK6 in this study mainly because it has been linked to phosphorylation of Ser-324/325 (17, 21, 22). Accordingly, depletion of GRK6 by siRNA attenuated CXCR4 ubiquitination (Fig. 7D) and degradation (Fig. 7A), indicating that GRK6 is necessary for CXCR4 degradation. Similar to inhibiting PKC, it is possible that GRK siRNA impacts phosphorylation of either Ser-324 or Ser-325 individually, yet in contrast to inhibiting PKC, GRK6 siRNA reduces CXCR4 degradation by CXCL12 (Fig. 7A). One reason for this may be because in addition to Ser-324/325, GRK6 mediates phosphorylation of serine residues 330 and 339 following CXCL12 stimulation (17). Along with Ser-324/325, Ser-330 is part of a previously described degradation motif (25). Although we have determined that Ser-324/325 serve as a phospho-based interaction site for the E3 ubiquitin ligase AIP4 (24), the precise function of Ser-330 in CXCR4 lysosomal trafficking has yet to be defined. It has been previously shown that mutation of Ser-330 to alanine reduces CXCR4 degradation by CXCL12, but not its internalization (25), suggesting that phosphorylation of this residue is important for sorting CXCR4 into the degradative pathway at the level of the endosome. GRK6 also phosphorylates Ser-339 and adjacent Ser-338 (17), which are dually phosphorylated following stimulation with CXCL12 and also PMA (41). Mutation of these residues individually or simultaneously to alanine did not have an impact on CXCR4 degradation induced by PMA or CXCL12 (data not shown). Taken together, our data suggest that GRK6 may regulate CXCR4 lysosomal trafficking at the level of the plasma membrane and endosome via site-specific phosphorylation of CXCR4.
Although GRK6 may play an essential role in CXCR4 lysosomal degradation, it is possible that GRK2 and GRK3 may also have a role. GRK2 and GRK3 phosphorylate the C-tail of CXCR4 at sites that are not redundant to GRK6 (17, 40). There is evidence to suggest phosphorylation of Ser-324/325 is under hierarchical regulation (41) (i.e. CXCL12 promoted phosphorylation of distal C-tail serine residues Ser-346/347 may be a prerequisite for efficient phosphorylation of Ser-324/325 or Ser-338/339) (41). Ser-346/347 may be phosphorylated by GRK2 or GRK3 (40, 41), suggesting that these kinases may also be involved in CXCR4 ubiquitination and degradation. However, in a previous study examining C-tail truncation mutants without Ser-346/347, CXCR4 degradation by CXCL12 was not impacted (25). A reason for this may be because C-tail truncation mutants or Ser-346/347 point mutants only show reduced phosphorylation at Ser-324/325 (41), suggesting that sub-stoichiometric phosphorylation of these residues is not rate-limiting and will only modestly impact receptor turnover by ubiquitination and lysosomal degradation. This is important because mutations in the gene encoding CXCR4 lead to C-tail truncation mutants, which have been linked to the rare immunodeficiency disorder known as WHIM syndrome characterized by warts, hypogammaglobulinemia, infections, and myelokathexis (43). Most of these truncation receptor mutants have an intact degradation motif, which contains Ser-324/325 and Ser-330 (43). Although these mutant receptors are typically unable to efficiently terminate acute signaling (12), because the degradation motif is intact, they are likely able to undergo efficient lysosomal degradation. The implication is that heterologous degradation of CXCR4 may represent a bona fide strategy to control excessive CXCL12 signaling associated with WHIM syndrome.
We provide evidence that CXCR4 lysosomal trafficking is cross-regulated by the chemokine receptor CXCR5 (Figs. 8 and 9). The cognate ligand for CXCR5, CXCL13, robustly mimics PMA- and CXCL12-mediated internalization, ubiquitination, and degradation of CXCR4 (Fig. 8). BisI significantly attenuated CXCL13- and PMA-mediated regulation of CXCR4 internalization, suggesting that canonical Gαi-mediated signaling and PKC activation are responsible for these effects on CXCR4 trafficking. CXCL13 activation of PKC likely results in phosphorylation of Ser-324/325, the key serine residues that drive receptor ubiquitination and degradation (24, 25). A recent study has suggested that CXCR4 and CXCR5 cross-talk through receptor heterodimerization to possibly regulate their respective signaling (44). In contrast, our data are consistent with the classical heterologous second messenger–dependent mechanism of regulation, which may not require a heteromeric receptor complex. Heterologous regulation of CXCR4 trafficking by other chemokine receptors has been reported previously (45), which may represent a common manner of regulation in the chemokine receptor family (46). Agonist activation of CXCR1 mediates phosphorylation and internalization of CXCR4 in a PKC-dependent mechanism (45). In addition to heterologous regulation by GPCRs, CXCR4 is also heterologously regulated by non-GPCR signaling receptors. Activation of the antigen-specific B cell receptor (BCR) triggers CXCR4 internalization through a PKC-mediated mechanism (26). Whether CXCR1 or BCR activation also promotes CXCR4 lysosomal degradation remains to be examined.
Heterologous degradation of CXCR4 regulates its acute signaling. We provide evidence that PMA or CXCL13 leads to desensitization of acute CXCL12-mediated Gαi responses, namely inhibition of AC activity (Fig. 9). Because PMA or CXCL13 mediate CXCR4 internalization and sorting into the degradative pathway, it is likely that a loss in the full complement of receptor leads to a reduction in CXCR4 signaling. We propose that this is mediated in part by PKC phosphorylation of Ser-324/325, which promotes CXCR4 ubiquitination and degradation (24), suggesting a role for these residues in long-term down-regulation of signaling. However, desensitization of CXCR4 signaling is also mediated by β-arrestins (18). CXCL12-stimulated phosphorylation of the C-tail promotes β-arrestin recruitment to the receptor (17, 40) and rapid desensitization of CXCR4 signaling through G protein uncoupling (13). β-Arrestin-2 recruitment to CXCR4 upon CXCL12 stimulation may be mediated by GRK3, but also PKC (40); therefore, β-arrestins could account in part for the loss of CXCR4 signaling observed by PMA or CXCL13 pretreatment (Fig. 9). However, Ser-324/325 are not required for β-arrestin-1/2 recruitment to CXCR4 by CXCL12 (17). Previously, we have shown that Ser-324/325 are required for recruiting the E3 ubiquitin ligase AIP4 to CXCR4 (24), further suggesting that these resides have a role in long-term attenuation of signaling by controlling ubiquitination and lysosomal degradation of CXCR4. It is important to note that we have previously shown that β-arrestins do in fact mediate CXCR4 lysosomal degradation, but not CXCR4 ubiquitination, suggesting that they act at the level of the endosome and not the plasma membrane (47).
In summary, our study provides evidence that CXCR4 lysosomal degradation is subject to heterologous regulation via activation of CXCR5 and PKC. CXCR4 lysosomal degradation is subject to homologous regulation, which is at least in part mediated by GRK6. GRK6 or PKC mediates phosphorylation of Ser-324/325, which previously we have shown is essential for AIP recruitment, ubiquitination, and ultimately CXCR4 lysosomal degradation (24). Targeting CXCR4 heterologously via other GPCRs might represent a useful strategy to antagonize aberrant CXCR4 signaling found in some chronic diseases.
Experimental procedures
Cell lines, antibodies, and reagents
HEK293 (Microbix, Toronto, Canada) and HeLa cells (ATCC) were cultured in DMEM or EMEM supplemented with 10% FBS (Atlanta Biologicals). Plasmid DNA transfections were done using the Trans-IT transfection reagent from Mirrus (Madison, WI) according to the manufacturer's instructions as we have described previously (47) or polyethyleneimine (PEI; Polysciences Inc., catalogue no. 23966) (48). All siRNA transfections were done using Lipofectamine2000 (Invitrogen) according to the manufacturer's instructions, as we have described previously (36). Bisindolylmaleimide I (BisI; catalogue no. 0741) and V (BisV; catalogue no. 203303) were purchased from Tocris and Calbiochem, respectively. FSK was purchased from Biomol (Plymouth, PA). Isobutylmethylxanthine (IBMX), N-ethylmaleimide, poly-l-lysine, DMSO, and PMA were obtained from Sigma. The p-nitrophenyl phosphate and diethanolamine buffer (catalogue no. 9701861) was from Bio-Rad. For RNAi studies, the control and siGENOME SMARTpool targeting GRK6 was obtained from Dharmacon RNA Technologies (Lafayette, CO). Nonenzymatic cell dissociation solution, CellStripper, was from Mediatech (Manassas, VA). The mAb (2B11) and the phycoerythrin-labeled (12G5-PE) mAb against CXCR4 was obtained from BD. The mAb (5E11) against dual phosphorylated serine residues 324 and 325 was custom-made and described previously (24). The mouse monoclonal antibodies against actin and GAPDH were purchased from MP Biomedicals (Solon, OH) and Abcam, respectively. The rabbit anti-GRK6 antibody (C-20) was purchased from Santa Cruz Biotechnology. DNA constructs (HA-CXCR4, FLAG-ubiquitin) and antibodies (monoclonal and polyclonal anti-HA, anti-LAMP1, anti-FLAG, and anti-mouse IgG alkaline phosphatase–conjugated) have been described in detail previously (25, 37, 49).
Analysis of CXCR4 degradation
Degradation of CXCR4 was measured essentially as described previously (25). For studies requiring siRNA transfection, HEK293 or HeLa cells were transfected with 600 pmol/10-cm dish of control or GRK6 siRNA for a total of 72 h before performing degradation experiment. The day before experiments, cells were passaged into 6-well plates at 3.0 × 105 - 4.5 × 105 viable cells/well. Whenever using inhibitors, these were added in DMEM or EMEM for 30 min before agonist treatment unless otherwise noted. Subsequently, cells were incubated for 3 h with 10 nm CXCL12, 100 nm CXCL13, or 10 nm PMA using corresponding vehicle as control (water for CXCL12 or CXCL13 and ethanol for PMA) in the presence of 50 μg/ml cycloheximide and the inhibitors. Cells were lysed in 2× sample buffer (8% SDS, 10% glycerol, 0.7 m mercaptoethanol, 37.5 mm Tris-HCl, pH 6.5, and 0.003% bromphenol blue), and equal amounts were resolved by 10% SDS-PAGE, followed by immunoblotting. HA-CXCR4 was detected using a monoclonal anti-HA antibody, whereas endogenous CXCR4 was detected with an anti-CXCR4 (clone 2B11) antibody. Immunoblots were subsequently stripped and reprobed with anti-actin, or samples were run in parallel and immunoblotted for GAPDH. Bands were analyzed by densitometry and adjusted for loading using actin or GAPDH density as a correction factor.
Analysis of CXCR4 internalization by flow cytometry
HeLa cells transfected with control or GRK6 siRNA were dissociated using CellStripper according to the manufacturer's instructions. For all subsequent steps, cells were washed and collected in PBS containing 0.1% BSA unless otherwise noted. For each sample, 5 × 105 cells were incubated at 37 °C for 30 min with the indicated concentrations of CXCL12. After centrifugation in cold PBS, cells were fixed with a solution of 3.7% paraformaldehyde in PBS, followed by two washes with PBS. Cells were then stained with the monoclonal anti-CXCR4 (12G5-PE) for 1 h, followed by two PBS/BSA washes to remove unbound antibody. Fluorescent intensity was acquired using a FACSCanto II and analyzed using the FlowJo software package (TreeStar, Ashland, OR).
Analysis of CXCR4 internalization by whole-cell ELISA
Whole-cell ELISA was used to measure CXCR4 internalization, essentially as we have described previously (36, 48). HeLa cells grown on 10-cm dishes were transiently transfected with HA-CXCR4 (6 μg) using PEI. The next day, ∼2 × 105 cells were seeded onto poly-l-lysine–coated 24-well plates. The next day, cells were serum-starved for 1 h and pretreated with DMSO, 10 μm Bis I, or BisV for 30 min, followed by stimulation with 10 nm CXCL12, 100 nm CXCL13, or 10 nm PMA for 5 min. Cells were fixed with 3.7% paraformaldehyde in Tris-buffered saline (TBS) for 5–10 min at room temperature. Cells were washed three times with TBS and incubated for 45 min in TBS supplemented with 1% BSA to block nonspecific binding sites. Cells were incubated with an anti-HA mAb diluted to 1:1000 for 1 h at room temperature. Cells were washed and incubated with alkaline phosphatase–conjugated goat anti-mouse IgG diluted to 1:1000 in 1% BSA/TBS for 1 h at room temperature. After three rapid rinses with TBS, antibody binding was detected by adding 0.25 ml of the alkaline phosphatase substrate p-nitrophenyl phosphate diluted in diethanolamine buffer. Reactions were stopped by removing 0.1 ml and adding it to a well of a 96-well microtiter plate containing 0.1 ml of 0.4 m NaOH. The absorbance at 405 nm was read using a microtiter plate reader (FlexStation 3, Molecular Devices). The percentage of receptor internalization was calculated by subtracting the fraction of absorbance after agonist treatment and vehicle treatment (following background subtraction) from 1 and multiplying by 100.
Analysis of CXCR4 ubiquitination
Ubiquitination of CXCR4 was determined as we have described previously (49). Briefly, HEK293 cells stably expressing HA-CXCR4 were transfected with 3 μg of FLAG-tagged ubiquitin and 7 μg of empty vector (pCMV) per 10-cm dish using Transit-LT1 transfection reagent following the manufacturer's instructions. Additionally, HEK293 cells were transiently transfected with HA-CXCR4 (3 μg) and FLAG-ubiquitin (3 μg) using PEI transfection reagent. Twenty-four hours after transfection, cells were passed into four 6-cm dishes and grown for an additional 24 h. The day of the experiment, cells were washed two times with DMEM containing 20 mm HEPES and pretreated with vehicle/10 μm BisI for 30 min before treating with 30 nm CXCL12 for 20 min or 10 nm PMA or 100 nm CXCL13 for 30 min. Medium was aspirated, and cells were washed once with cold PBS followed by the addition of 1 ml of cold lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS) freshly supplemented with protease inhibitors (10 μg/ml each of aprotinin, pepstatin A, and leupeptin) and 20 nm N-ethylmaleimide. Subsequent HA-CXCR4 immunoprecipitation using 120–250 μg of lysate per sample and detection of ubiquitin-FLAG were performed as described previously (49).
Measurement of cAMP levels
HEK293 or HeLa cells grown on 10-cm dishes were transfected with plasmid DNA and control or GRK6 siRNA and split at 24–48 h later in 6-well plates (HeLa cells) or 24-well plates (HEK293 cells). At 48 h after transfection, cells were incubated with serum-free medium for 2–4 h, followed by a 30-min incubation at 37 °C with DMEM containing 1 mm IBMX and 20 mm HEPES (DMEM/IBMX). All subsequent incubations were performed at 37 °C in IBMX/DMEM. HEK293 cells were first treated with 1 μm FSK for 5 min and subsequently incubated with vehicle or increasing doses of CXCL12 for an additional 5 min. For HeLa cells, FSK and CXCL12 were added at the same time and incubated at 37 °C for 10 min. After treatment, agonists were removed by aspiration and one wash with cold PBS containing 1 mm IBMX. Thereafter, cells were lysed at ∼1 × 106 cells/ml using 0.1 n HCl, 0.5% Triton X-100 and incubated for 15 min at room temperature while rocking to allow for complete lysis. The concentration of cAMP in each sample was determined using a colorimetric cAMP EIA kit (Assay Designs) following the manufacturer's instructions. Samples and standards were run in duplicate and averaged. Concentration of cAMP was determined by extrapolation of sample average into the standard curve subjected to a four-parameter fit as recommended by the manufacturer. To calculate the effect of CXCL12 treatment on cAMP levels, FSK + CXCL12 values were divided by the cAMP levels of samples treated with FSK + vehicle multiplied by 100.
pGlo-based cAMP reporter assay
HeLa cells were transfected in a 10-cm dish with 2 μg of pGlo-22 sensor plasmid obtained from Mark Von Zastrow and Nikoleta Tsvetanova (University of California San Francisco) using PEI. Twenty-four hours later, cells were passed in triplicate into a white-walled 96-well plate (CELLSTAR) at a seeding density of 30,000 cells/well. The following day, cells were incubated for 1 h at 37 °C with phenol-free MEM (Gibco) containing 10% FBS (Atlanta Biologicals), 3.2 mm luciferin (GoldBio), and 50 μg/ml cycloheximide (Sigma). Cells were pretreated for 30 min with increasing concentrations of CXCL13 (0 nm, 10 nm, 100 nm, 1 μm) at 37 °C. Luminescent readings were taken at 25 °C, the optimal temperature for the pGlo sensor (50), at an integration of 1 s every minute for 20 min to obtain a baseline measurement using a Flex Station 3 plate reader (Molecular Devices). Then vehicle or 10 nm CXCL12 was added in the presence of 10 μm forskolin (Millipore-Sigma) and 500 μm IBMX (Biomol), and luminescence was measured for 10 min at 25 °C. Values were plotted for each condition, and the area under each curve was calculated to determine cAMP levels. Vehicle and CXCL12 values within the same pretreatment group were compared to determine percentage cAMP inhibition, with the resulting value being normalized to the control pretreatment group not receiving CXCL13.
Confocal immunofluorescence microscopy
For detection of phosphorylated CXCR4 serine residues 324 and 325, HEK293 or HeLa cells grown on 10-cm dishes were transfected with 5 μg of HA-CXCR4-YFP and passed 24 h later onto coverslips pretreated with polylysine, essentially as we have described previously (51). The next day, cells were washed once with DMEM plus 20 mm HEPES and serum-starved for 3–4 h in the same medium, followed by treatment with vehicle, 10 nm CXCL12, or 10 nm PMA for 15–30 min at 37 °C. For the PKC inhibitor experiment, cells were pretreated with 10 μm BisI or DMSO for 30 min at 37 °C. Following treatment, cells were fixed, permeabilized, and immunostained using a custom mouse mAb specific for dually phosphorylated serine residues 324 and 325 (pSer324/325) (Clone 5E11), as described previously (24, 52). Images were acquired using a Zeiss LSM 510 laser-scanning confocal microscope with a 63× W Apochromat oil-immersion objective. Image acquisition settings across parallel samples were identical. Image analysis was done with ImageJ software (National Institutes of Health). Individual cells were traced in each field of view to define regions of interest, and the mean fluorescence intensity for CXCR4-YFP and pSer324/325 was measured. For each region of interest, pSer324/325 intensity was divided by HA-CXCR4-YFP intensity to normalize CXCR4 phosphorylation to receptor expression.
For detection of LAMP1-positive lysosomes (LAMP1+) and colocalization analysis, HEK293 cells were grown in 6-well plates and transfected with 250 ng of HA-CXCR4-YFP. Cells were passaged onto PLL-coated coverslips and serum-starved as above, followed by treatment with vehicle, 10 nm CXCL12, or 10 nm PMA for 3 h. Following treatment, cells were fixed in ice-cold 4% paraformaldehyde, 0.1 m sodium phosphate buffer (pH 7.0) for 15 min and immunostained with an antibody against LAMP1, as we have described previously (37). Images were acquired as z-stacks at ×60 magnification (numerical aperture 1.4) using an Olympus Disk Spinning confocal microscope controlled by CellSens Dimensions software (Olympus). Acquisition settings for each channel were identical across parallel samples. z-Slices were spaced at 0.3 μm and acquired using the online deblur tool to remove out-of-focus light. The total number of LAMP1+ lysosomes and colocalization with CXCR4-YFP+ puncta were determined using 3D object-based analysis (42, 52, 53). Briefly, background was subtracted, and autothresholding was used to create binary images. HA-CXCR4-YFP+ and LAMP1+ puncta were segmented and quantified using the 3D Objects Counter Plugin (ImageJ) (52). Centroids from HA-CXCR4-YFP+ puncta and LAMP1+ objects from resultant labeled images were converted to binary masks and then multiplied to quantify the number of overlapping HA-CXCR4-YFP+ and LAMP1+ puncta.
Statistical analysis
Data are represented as the mean ± S.D. of at least three experiments or determinations. All statistical tests were done using GraphPad Prism version 7.0d for Mac OS X (GraphPad Software, La Jolla, CA). Student's t test was used to compare the difference between two groups, one-way analysis of variance (ANOVA) was used to compare the difference between three or more groups, and two-way ANOVA was used to compare the difference between different groups under different treatment conditions. ANOVA was followed by Tukey's or Bonferroni's post hoc test, as indicated in the figure legends. Colocalization studies were analyzed using Welch's ANOVA followed by Tamhane's T2 multiple-comparison test. A p value < 0.05 was considered significant. Specific values are provided in the figure panels or in the figure legends.
Author contributions
A. C., S. A. M., M. S. A., and M. R. R. data curation; A. C., S. A. M., M. S. A., M. R. R., and A. M. formal analysis; A. C., S. A. M., M. S. A., and M. R. R. investigation; A. C., S. A. M., M. S. A., and M. R. R. methodology; A. C. writing-original draft; A. C., S. A. M., M. S. A., M. R. R., and A. M. writing-review and editing; A. C. and A. M. conceptualization; A. M. supervision; A. M. funding acquisition; A. M. project administration.
Acknowledgments
We thank Dr. Nina Tsvetanova (Duke University) for generously providing reagents and advice to set up the pGLO sensor cAMP experiments. We thank Chad Koplinski (Protein Foundry, Milwaukee, WI) for advice and generosity with CXCL13 experiments.
This work was supported by National Institutes of Health Grant GM122889 (to A. M.). This work was also supported in part by institutional funds at Loyola University Chicago. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GPCR
- G protein–coupled receptor
- CXCR
- CXC motif chemokine receptor
- CXCL
- CXC motif chemokine ligand
- PKC
- protein kinase C
- GRK
- GPCR kinase
- AIP4
- atrophin-interacting protein 4
- PMA
- phorbol 12-myristate 13-acetate
- BisI and BisV
- bisindolylmaleimide I and V, respectively
- AC
- adenylate cyclase
- FSK
- forskolin
- DMEM
- Dulbecco's modified Eagle's medium
- EMEM
- Eagle's minimum essential medium
- PEI
- polyethyleneimine
- IBMX
- isobutylmethylxanthine
- TBS
- Tris-buffered saline
- 3D
- three-dimensional
- ANOVA
- analysis of variance
- CI
- confidence interval
- EIA
- enzyme immunoassay.
References
- 1. Nagasawa T., Hirota S., Tachibana K., Takakura N., Nishikawa S., Kitamura Y., Yoshida N., Kikutani H., and Kishimoto T. (1996) Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382, 635–638 10.1038/382635a0 [DOI] [PubMed] [Google Scholar]
- 2. Nagasawa T., Kikutani H., and Kishimoto T. (1994) Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc. Natl. Acad. Sci. U.S.A. 91, 2305–2309 10.1073/pnas.91.6.2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagasawa T., Nakajima T., Tachibana K., Iizasa H., Bleul C. C., Yoshie O., Matsushima K., Yoshida N., Springer T. A., and Kishimoto T. (1996) Molecular cloning and characterization of a murine pre-B-cell growth-stimulating factor/stromal cell-derived factor 1 receptor, a murine homolog of the human immunodeficiency virus 1 entry coreceptor fusin. Proc. Natl. Acad. Sci. U.S.A. 93, 14726–14729 10.1073/pnas.93.25.14726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tachibana K., Hirota S., Iizasa H., Yoshida H., Kawabata K., Kataoka Y., Kitamura Y., Matsushima K., Yoshida N., Nishikawa S., Kishimoto T., and Nagasawa T. (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393, 591–594 10.1038/31261 [DOI] [PubMed] [Google Scholar]
- 5. Chatterjee S., Behnam Azad B., and Nimmagadda S. (2014) The intricate role of CXCR4 in cancer. Adv. Cancer Res. 124, 31–82 10.1016/B978-0-12-411638-2.00002-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Woerner B. M., Warrington N. M., Kung A. L., Perry A., and Rubin J. B. (2005) Widespread CXCR4 activation in astrocytomas revealed by phospho-CXCR4-specific antibodies. Cancer Res. 65, 11392–11399 10.1158/0008-5472.CAN-05-0847 [DOI] [PubMed] [Google Scholar]
- 7. Schüller U., Koch A., Hartmann W., Garrè M. L., Goodyer C. G., Cama A., Sörensen N., Wiestler O. D., and Pietsch T. (2005) Subtype-specific expression and genetic alterations of the chemokinereceptor gene CXCR4 in medulloblastomas. Int. J. Cancer 117, 82–89 10.1002/ijc.21116 [DOI] [PubMed] [Google Scholar]
- 8. Li Y. M., Pan Y., Wei Y., Cheng X., Zhou B. P., Tan M., Zhou X., Xia W., Hortobagyi G. N., Yu D., and Hung M. C. (2004) Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 6, 459–469 10.1016/j.ccr.2004.09.027 [DOI] [PubMed] [Google Scholar]
- 9. Holm N. T., Byrnes K., Li B. D., Turnage R. H., Abreo F., Mathis J. M., and Chu Q. D. (2007) Elevated levels of chemokine receptor CXCR4 in HER-2 negative breast cancer specimens predict recurrence. J. Surg. Res. 141, 53–59 10.1016/j.jss.2007.03.015 [DOI] [PubMed] [Google Scholar]
- 10. Balkwill F. (2004) The significance of cancer cell expression of the chemokine receptor CXCR4. Semin. Cancer Biol. 14, 171–179 10.1016/j.semcancer.2003.10.003 [DOI] [PubMed] [Google Scholar]
- 11. Balabanian K., Lagane B., Pablos J. L., Laurent L., Planchenault T., Verola O., Lebbe C., Kerob D., Dupuy A., Hermine O., Nicolas J. F., Latger-Cannard V., Bensoussan D., Bordigoni P., Baleux F., et al. (2005) WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood 105, 2449–2457 10.1182/blood-2004-06-2289 [DOI] [PubMed] [Google Scholar]
- 12. Hernandez P. A., Gorlin R. J., Lukens J. N., Taniuchi S., Bohinjec J., Francois F., Klotman M. E., and Diaz G. A. (2003) Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 34, 70–74 10.1038/ng1149 [DOI] [PubMed] [Google Scholar]
- 13. Busillo J. M., and Benovic J. L. (2007) Regulation of CXCR4 signaling. Biochim. Biophys. Acta 1768, 952–963 10.1016/j.bbamem.2006.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCudden C. R., Hains M. D., Kimple R. J., Siderovski D. P., and Willard F. S. (2005) G-protein signaling: back to the future. Cell Mol. Life Sci. 62, 551–577 10.1007/s00018-004-4462-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goldsmith Z. G., and Dhanasekaran D. N. (2007) G protein regulation of MAPK networks. Oncogene 26, 3122–3142 10.1038/sj.onc.1210407 [DOI] [PubMed] [Google Scholar]
- 16. Gutkind J. S. (1998) The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J. Biol. Chem. 273, 1839–1842 10.1074/jbc.273.4.1839 [DOI] [PubMed] [Google Scholar]
- 17. Busillo J. M., Armando S., Sengupta R., Meucci O., Bouvier M., and Benovic J. L. (2010) Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J. Biol. Chem. 285, 7805–7817 10.1074/jbc.M109.091173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rajagopal S., and Shenoy S. K. (2018) GPCR desensitization: acute and prolonged phases. Cell. Signal. 41, 9–16 10.1016/j.cellsig.2017.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luttrell L. M., and Lefkowitz R. J. (2002) The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 [DOI] [PubMed] [Google Scholar]
- 20. Balabanian K., Levoye A., Klemm L., Lagane B., Hermine O., Harriague J., Baleux F., Arenzana-Seisdedos F., and Bachelerie F. (2008) Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. J. Clin. Invest. 118, 1074–1084 10.1172/JCI33187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fong A. M., Premont R. T., Richardson R. M., Yu Y. R., Lefkowitz R. J., and Patel D. D. (2002) Defective lymphocyte chemotaxis in β-arrestin2- and GRK6-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 99, 7478–7483 10.1073/pnas.112198299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vroon A., Heijnen C. J., Raatgever R., Touw I. P., Ploemacher R. E., Premont R. T., and Kavelaars A. (2004) GRK6 deficiency is associated with enhanced CXCR4-mediated neutrophil chemotaxis in vitro and impaired responsiveness to G-CSF in vivo. J. Leukocyte Biol. 75, 698–704 10.1189/jlb.0703320 [DOI] [PubMed] [Google Scholar]
- 23. Orsini M. J., Parent J. L., Mundell S. J., Marchese A., and Benovic J. L. (1999) Trafficking of the HIV coreceptor CXCR4: role of arrestins and identification of residues in the C-terminal tail that mediate receptor internalization. J. Biol. Chem. 274, 31076–31086 10.1074/jbc.274.43.31076 [DOI] [PubMed] [Google Scholar]
- 24. Bhandari D., Robia S. L., and Marchese A. (2009) The E3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol. Biol. Cell 20, 1324–1339 10.1091/mbc.e08-03-0308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marchese A., and Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 10.1074/jbc.C100527200 [DOI] [PubMed] [Google Scholar]
- 26. Guinamard R., Signoret N., Ishiai M., Marsh M., Kurosaki T., and Ravetch J. V., Masamichi I. (1999) B cell antigen receptor engagement inhibits stromal cell-derived factor (SDF)-1α chemotaxis and promotes protein kinase C (PKC)-induced internalization of CXCR4. J. Exp. Med. 189, 1461–1466 10.1084/jem.189.9.1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haribabu B., Richardson R. M., Fisher I., Sozzani S., Peiper S. C., Horuk R., Ali H., and Snyderman R. (1997) Regulation of human chemokine receptors CXCR4: role of phosphorylation in desensitization and internalization. J. Biol. Chem. 272, 28726–28731 10.1074/jbc.272.45.28726 [DOI] [PubMed] [Google Scholar]
- 28. Hezareh M., Moukil M. A., Szanto I., Pondarzewski M., Mouche S., Cherix N., Brown S. J., Carpentier J. L., and Foti M. (2004) Mechanisms of HIV receptor and co-receptor down-regulation by prostratin: role of conventional and novel PKC isoforms. Antivir. Chem. Chemother. 15, 207–222 10.1177/095632020401500404 [DOI] [PubMed] [Google Scholar]
- 29. Signoret N., Oldridge J., Pelchen-Matthews A., Klasse P. J., Tran T., Brass L. F., Rosenkilde M. M., Schwartz T. W., Holmes W., Dallas W., Luther M. A., Wells T. N., Hoxie J. A., and Marsh M. (1997) Phorbol esters and SDF-1 induce rapid endocytosis and down modulation of the chemokine receptor CXCR4. J. Cell Biol. 139, 651–664 10.1083/jcb.139.3.651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Amara A., Gall S. L., Schwartz O., Salamero J., Montes M., Loetscher P., Baggiolini M., Virelizier J. L., and Arenzana-Seisdedos F. (1997) HIV coreceptor downregulation as antiviral principle: SDF-1α-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J. Exp. Med. 186, 139–146 10.1084/jem.186.1.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Newton A. C. (2018) Protein kinase C: perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 53, 208–230 10.1080/10409238.2018.1442408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Toullec D., Pianetti P., Coste H., Bellevergue P., Grand-Perret T., Ajakane M., Baudet V., Boissin P., Boursier E., and Loriolle F. (1991) The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 266, 15771–15781 [PubMed] [Google Scholar]
- 33. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., and Benovic J. L. (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5, 709–722 10.1016/S1534-5807(03)00321-6 [DOI] [PubMed] [Google Scholar]
- 34. Alekhina O., and Marchese A. (2016) β-Arrestin1 and signal-transducing adaptor molecule 1 (STAM1) cooperate to promote focal adhesion kinase autophosphorylation and chemotaxis via the chemokine receptor CXCR4. J. Biol. Chem. 291, 26083–26097 10.1074/jbc.M116.757138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Verma R., and Marchese A. (2015) The endosomal sorting complex required for transport pathway mediates chemokine receptor CXCR4-promoted lysosomal degradation of the mammalian target of rapamycin antagonist DEPTOR. J. Biol. Chem. 290, 6810–6824 10.1074/jbc.M114.606699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. English E. J., Mahn S. A., and Marchese A. (2018) Endocytosis is required for C-X-C chemokine receptor type 4 (CXCR4)-mediated Akt activation and anti-apoptotic signaling. J. Biol. Chem. 293, 11470–11480 10.1074/jbc.RA118.001872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhandari D., Trejo J., Benovic J. L., and Marchese A. (2007) Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J. Biol. Chem. 282, 36971–36979 10.1074/jbc.M705085200 [DOI] [PubMed] [Google Scholar]
- 38. Horuk R. (1997) Adenylate cyclase assays to measure chemokine receptor function. Methods Enzymol. 288, 326–339 10.1016/S0076-6879(97)88022-6 [DOI] [PubMed] [Google Scholar]
- 39. Martiny-Baron G., Kazanietz M. G., Mischak H., Blumberg P. M., Kochs G., Hug H., Marmé D., and Schächtele C. (1993) Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J. Biol. Chem. 268, 9194–9197 [PubMed] [Google Scholar]
- 40. Luo J., Busillo J. M., Stumm R., and Benovic J. L. (2017) G protein-coupled receptor kinase 3 and protein kinase C phosphorylate the distal C-terminal tail of the chemokine receptor CXCR4 and mediate recruitment of β-arrestin. Mol. Pharmacol. 91, 554–566 10.1124/mol.116.106468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mueller W., Schütz D., Nagel F., Schulz S., and Stumm R. (2013) Hierarchical organization of multi-site phosphorylation at the CXCR4 C terminus. PLoS One 8, e64975 10.1371/journal.pone.0064975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wörz S., Sander P., Pfannmöller M., Rieker R. J., Joos S., Mechtersheimer G., Boukamp P., Lichter P., and Rohr K. (2010) 3D geometry-based quantification of colocalizations in multichannel 3D microscopy images of human soft tissue tumors. IEEE Trans. Med. Imaging 29, 1474–1484 10.1109/TMI.2010.2049857 [DOI] [PubMed] [Google Scholar]
- 43. Al Ustwani O., Kurzrock R., and Wetzler M. (2014) Genetics on a WHIM. Br. J. Haematol. 164, 15–23 10.1111/bjh.12574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. El-Haibi C. P., Sharma P., Singh R., Gupta P., Taub D. D., Singh S., and Lillard J. W. Jr. (2013) Differential G protein subunit expression by prostate cancer cells and their interaction with CXCR5. Mol. Cancer 12, 64 10.1186/1476-4598-12-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Richardson R. M., Tokunaga K., Marjoram R., Sata T., and Snyderman R. (2003) Interleukin-8-mediated heterologous receptor internalization provides resistance to HIV-1 infectivity: role of signal strength and receptor desensitization. J. Biol. Chem. 278, 15867–15873 10.1074/jbc.M211745200 [DOI] [PubMed] [Google Scholar]
- 46. Ali H., Richardson R. M., Haribabu B., and Snyderman R. (1999) Chemoattractant receptor cross-desensitization. J. Biol. Chem. 274, 6027–6030 10.1074/jbc.274.10.6027 [DOI] [PubMed] [Google Scholar]
- 47. Malik R., and Marchese A. (2010) Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 21, 2529–2541 10.1091/mbc.e10-02-0169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grimsey N., Lin H., and Trejo J. (2014) Endosomal signaling by protease-activated receptors. Methods Enzymol. 535, 389–401 10.1016/B978-0-12-397925-4.00022-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Caballero A., and Marchese A. (2011) Ubiquitination of GPCRs. Methods Mol. Biol. 746, 251–259 10.1007/978-1-61779-126-0_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Binkowski B. F., Fan F., and Wood K. V. (2011) Luminescent biosensors for real-time monitoring of intracellular cAMP. Methods Mol. Biol. 756, 263–271 10.1007/978-1-61779-160-4_14 [DOI] [PubMed] [Google Scholar]
- 51. Marchese A. (2016) Monitoring chemokine receptor trafficking by confocal immunofluoresence microscopy. Methods Enzymol. 570, 281–292 10.1016/bs.mie.2015.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bolte S., and Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 53. Obara B., Jabeen A., Fernandez N., and Laissue P. P. (2013) A novel method for quantified, superresolved, three-dimensional colocalisation of isotropic, fluorescent particles. Histochem. Cell Biol. 139, 391–402 10.1007/s00418-012-1068-3 [DOI] [PubMed] [Google Scholar]