

Abstract
Cytochrome P450 (CYP) 3A4 is a major contributor to hepatic drug and xenobiotic metabolism in human adults. The related enzyme CYP3A5 is also expressed in adult liver and has broader age and tissue distributions. However, CYP3A5 expression is low in most Caucasians because of the prevalence of an allele that leads to an incorrectly spliced mRNA and premature termination of translation. When expressed, CYP3A5 expands metabolic capabilities and can augment CYP3A4-mediated drug metabolism, thereby reducing drug efficacy and potentially requiring dose adjustments. The extensive role of CYP3A4 in drug metabolism reflects in part the plasticity of the substrate-free enzyme to enlarge its active site and accommodate very large substrates. We have previously shown that the structure of the CYP3A5–ritonavir complex differs substantially from that of the CYP3A4–ritonavir complex. To better understand whether these differences are conserved in other CYP3A5 structures and how they relate to differential plasticity, we determined the X-ray crystallographic structure of the CYP3A5 substrate-free complex to 2.20 Å resolution. We observed that this structure exhibits a much larger active site than substrate-free CYP3A4 and displays an open substrate access channel. This reflected in part a lower trajectory of the helix F–F′ connector in CYP3A4 and more extensive π–CH interactions between phenylalanine residues forming the roof of the active-site cavity than in CYP3A5. Comparison with the CYP3A5–ritonavir complex confirmed conserved CYP3A5 structural features and indicated differences in plasticity between CYP3A4 and CYP3A5 that favor alternative ritonavir conformations.
Keywords: cytochrome P450, drug metabolism, protein structure, crystal structure, protein conformation, CYP3A4, CYP3A5, protein plasticity
Introduction
Subfamily 3A cytochromes P450 (CYP)2 contribute extensively to the metabolic clearance of orally administered drugs and other foreign compounds (1, 2) and can limit the efficacy of new drug candidates. Drug–drug interactions involving 3A enzymes can also lead to unwanted side effects during drug therapy. P450 3A4 is the most highly expressed of the 3A enzymes in adult human livers, whereas 3A7 predominates in the fetal and neonatal period (3, 4). P450 3A5 is expressed in adult human liver, as well as fetal liver, and exhibits a broader tissue distribution than 3A4 (5). However, 3A5 expression varies between individuals because of prevalent genetic differences that lead to nonproductive mRNA splicing and low or absent expression of the protein that varies in frequency between different ethnic groups (6). When abundant, 3A5 often augments drug clearance by 3A4 and can significantly reduce drug efficacy (7–10). Metabolic profiles differ between 3A4 and 3A5, leading to differential impacts on drug clearance (11) and to generation of alternative metabolites (12, 13). A recent study has identified a substrate for 3A5 that is poorly metabolized by 3A4 (14).
To better understand the structural basis for the metabolic differences exhibited by 3A4 and 3A5, an X-ray crystal structure of 3A5 complexed with ritonavir was determined previously and compared with the structure of the 3A4 ritonavir complex (15). Ritonavir exhibits very similar binding affinities for 3A4 and 3A5 but adopts different conformations in the two enzymes. The high binding affinity exhibited by both enzymes reflects, in part, the formation of coordinate covalent bond between the nitrogen of the thioazole group of ritonavir and the heme iron, as well as the relatively large size and hydrophobicity of ritonavir compared with most drugs, which is complemented by the hydrophobicity and size of the 3A4 and 3A5 active sites. Because the conformation of the 3A4 ritonavir complex, as well as the size and shape of the active site, differs significantly from the structure of substrate-free 3A4 (16), the conformational differences seen between the 3A4 and 3A5 ritonavir complex are likely to reflect, in part, regional differences in flexibility of the two proteins to accommodate ritonavir, as well as differences between the structures of substrate-free 3A4 and 3A5. The configurational flexibility exhibited by structures of 3A4 with different compounds is thought to contribute to the broad capacity of 3A4 to facilitate xenobiotic clearance, and even larger structural changes than seen for ritonavir (MW, 721) are exhibited by structures of 3A4 complexed with one molecule of erythromycin (MW, 734) or 2 molecules of ketoconazole (MW, 531) (17). This flexibility is also important for the opening and closing of the active site to allow substrate entry and substrate or product egress, as well as adaptations for binding different drugs such as midazolam (18) and the experimental drug 8-(4-methoxy-3-methylphenyl)-2,3,4,8-tetrahydroimidazo[1,5-a]pyrimidin-6-amine (19).
Determination of the structure of 3A5 in the absence of ritonavir or other substrates or inhibitors was undertaken in this study to understand how binding of ritonavir changes the structure of 3A5, as well as how the substrate-free structures of 3A5 and 3A4 differ, because these are reference states for configurational changes that occur when drugs bind to the substrate-free enzymes, as well as for free energy calculations for substrate binding. Conditions were identified for the generation of 3A5 crystals prepared in the absence of ritonavir or other substrates or inhibitors. Here we report that the apo 3A5 structure displays a much larger active-site cavity than that of apo 3A4 and exhibits an open substrate access channel, which closes when ritonavir binds in the cavity. These results also confirm fundamental structural differences between 3A5 and 3A4 that are likely to contribute to functional differences.
Results
Structure determination
The crystalized protein, 3A5C2dH, was engineered for crystallization and structure determination by removal of the N-terminal transmembrane helix and was used previously for determination of the structure of the 3A5–ritonavir complex (15). Molecular replacement provided initial phasing in the I222 space group with one chain in the asymmetric unit. The solution with a log likelihood of 1415 was obtained using the PDB code 1TQN structure of substrate-free 3A4 (20) as the search model, and it was used as the initial model. Solutions obtained using individual chains from the PDB code 5VEU structure of 3A5 ritonavir complex exhibited log likelihood scores that were roughly half of the value obtained using the 1TQN structure. Electron density maps indicated that this difference is likely to reflect the extensive changes elicited by the binding of ritonavir in 3A5 relative to the substrate-free 3A5. Additionally, these maps indicated that the structure of substrate-free 3A5 differed significantly from substrate-free 3A4, and extensive rebuilding was required for portions of the structure encompassing helices F through G and adjacent structural elements where the 3A5 apo structure differs from both of the 3A4 and 3A5 phasing models (Fig. 1, A and C).
Figure 1.

Conformational differences between apo 3A5 (yellow) and apo 3A4 (magenta; A and B), apo 3A5 and the 3A5 ritonavir complex (green; C and D), and apo 3A4 and the 3A4 ritonavir complex (cyan; E and F). Ritonavir is depicted as spheres. Heme is depicted as a stick model with an iron sphere. Heteroatom colors are nitrogen (blue), oxygen (red), iron (orange), and sulfur (yellow). Distances (dashed lines) are reported in angstroms.
Differences in the active-site architectures of substrate-free 3A5 and 3A4
The largest differences between the substrate-free structures of 3A5 and 3A4 are seen for the connector between helices F and F′, the helix B–B′ connector, and the turn in the C-terminal loop (Fig. 1A). The connectors between helices F and F′, as well as G′ and G in 3A4 (20, 21) and 3A5 (15), are longer than those typically seen in most mammalian P450s, where helices F and G extend across helix I to the opposite side of the substrate-binding site (22). As shown in Fig. 1 (A, C, and E), amino acid side chains on the two flexible connecters form the roof of the active site above the heme co-factor where the iron bound reactive intermediate is generated. There is a considerable difference between apo 3A4 and 3A5 in the topology of the polypeptide chain connecting the F and F′ helices (Fig. 1A). Because amino acid side chains in this region contribute to the size and shape of the active site, the surfaces of active-site cavities swept with a 1.4 Å probe were determined with open channels blocked at their narrowest constriction (Fig. 2, A versus C). The results indicated that the cavity volume for substrate-free 3A5 (1866 Å3) was much larger than that reported for apo 3A4 (1386 Å3) as estimated by the same method (20). The additional volume in apo 3A5 relative to apo 3A4 is greatest in the distal portion of the cavity, and an open substrate access channel is evident in apo 3A5 between Tyr-53, Glu-76, Phe-215, Leu-216, and Gln-479 (Fig. 3A). This access channel is sufficiently large to accommodate a molecule of testosterone as modeled in Fig. 3A, but it is closed in apo 3A4, in part by the lower trajectory of the F–F′ connector (Fig. 3B). A docking pose for testosterone in the 3A5 entrance channel is displayed to indicate the size of opening relative to a typical substrate, and the binding site represents a potential encounter complex to bind testosterone for passage into the active-site cavity. The entrance-bound testosterone is 16.7 Å from the heme iron with additional cavity volume to accommodate one or two additional molecules of testosterone. Studies of testosterone hydroxylation by 3A4 indicate that the binding of three testosterone molecules were required to trigger robust product formation (23), which may include one testosterone bound in the entrance channel.
Figure 2.

Active-site cavities are shown as semitransparent surfaces for apo 3A5 (A), 3A5 ritonavir complex (B), apo 3A4 (C), and 3A4 ritonavir complex (D) with adjacent amino acid side chains, the heme co-factor, and ritonavir are shown as stick models. Amino acid residues that differ between 3A4 and 3A5 are identified by bold labels. The viewpoint and scale are identical for each image.
Figure 3.
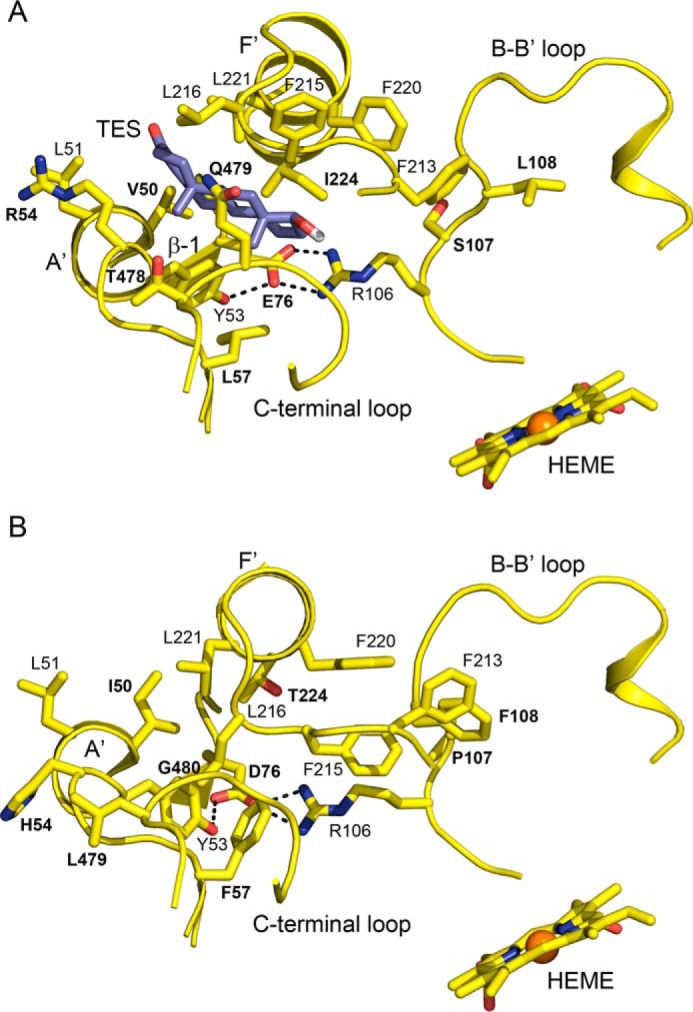
A view of the open entrance channel of the apo 3A5 active site with testosterone docked in the channel (A) and the closed entrance channel in apo 3A4 (B) that reflects the lower trajectory of F–F′ connector and interactions of Phe-107, Phe-215, and Phe-220 with Phe-213. The testosterone pose overlaps two glycerol molecules seen in the apo 3A5 structure (Fig. S1). The docking pose was identified by using Autodock Vina. The distance from testosterone (TES) to the heme iron is 16.7 Å. Amino acid residues that differ between 3A4 and 3A5 are identified by bold labels.
The smaller active-site cavity in substrate-free 3A4 structure reflects in part the π–CH interactions between several phenylalanine residues 213, 215, 220, and 241 in the helix FG region, Phe-304 on helix I, and Phe-108 on the helix B to B′ loop (Figs. 1B and 2C) to form an unusual roof of aromatic side chains above the active site (20, 21). Although 3A5 Phe-213 is positioned similarly for interactions with phenylalanines 241 and 304, additional interactions are not seen in 3A5 because of the different trajectories of the F–F′ and G′–G connectors. The longer length of 3A5 Phe-210 relative to 3A4 Leu-210 pushes the F–F′ connector upward and outward rather than lower and inward as seen for 3A4 (Fig. 1, A and B). This difference is also evident for the 3A4 and 3A5 ritonavir complexes (15). As a result, the distance between the Cα of residue 212 in the two structures is very large (5.5 Å) with 3A5 Lys-212 oriented outward into solvent and with 3A4 Arg-212 oriented downward into the active site (Fig. 1B) where it donates a hydrogen bond from an epsilon nitrogen to the carbonyl of Phe-304. Moreover, the 3A5 Phe-215 Cα is displaced upward by 6.2 Å relative to 3A4 and oriented outward contributing to the more open distal active site in 3A5. Additionally, 3A5 Leu-108 is too short to interact with Phe-213, and its Cα is displaced by 4.6 Å outward from that of 3A4 Phe-108 (Fig. 1B), reflecting a conformational difference for the portion of the helix B-B′ loop between Arg-105 and Pro-110 in 3A5 that turns outward relative to 3A4 (Fig. 1, A and B). This conformational difference reflects, in part, a twist of the polypeptide chain imparted by interaction of Arg-106 with Met-393 and Glu-76 in 3A5 (Fig. 1B) coupled with an opposite conformational effect of 3A4 Pro-107 (Ser-107 in 3A5) on the connector between Arg-106 and Pro-110.
The divergent trajectories of the helix F–F′ connector in the two structures also contribute to the differences seen for the conformation of the C-terminal loops of the two proteins (Fig. 1A). Overall, this comparison indicates that the greater distance between helix F–F′ and G′–G connectors in 3A5 reduces interactions of the phenylalanines and other hydrophobic residues in the distal active-site cavity that creates a larger (Fig. 2) and more open (Fig. 3) active-site cavity in 3A5 compared with 3A4.
Structural changes elicited by the binding of ritonavir to 3A5
Binding of ritonavir causes the central portion of 3A5 helix I to bow outward relative to the apo structure to accommodate the coordination of the thioazole moiety to the heme iron and the adjacent P1 phenyl group (Fig. 1C). Similar differences are also evident when the structures of the apo 3A4 and the 3A4 ritonavir complex are compared (Fig. 1E). Because this region is in the center of the structure, there are additional compensatory changes.
In addition, the P1 phenyl moiety of ritonavir engages with the phenylalanine cluster, displacing Phe-304 and triggering a reorganization of the phenylalanine cluster around the ritonavir P1 phenyl group in both the 3A5 and 3A4 complexes (Fig. 1, D and F). These changes relative to the apo protein are similar between the two enzymes, and the conformation adopted by ritonavir is similar near helix I in both proteins.
The conformations of ritonavir diverge between the two proteins with the P2 phenyl group adopting a π/π stacking interaction with the surface of the heme in 3A4 and a lower trajectory of the distal end of ritonavir under helix F′, which allows 3A4 phenylalanines 213, 215, and 220 to retain π–CH or π–π interactions (Fig. 1, E versus C). Helix A′ moves outward to accommodate the distal end of ritonavir in this conformation. The roof of the active site moves upward, which disrupts interactions between phenylalanines 108 and 213 that are offset by new interactions with ritonavir in 3A4.
The space under helix F′ is less available in 3A5 because of differences in the placement of the salt bridge network centered around 3A4 Asp-76 and 3A5 Glu-76 with Arg-106, Glu-374, Arg-372, and Tyr-53 (Fig. 2, B and D). Arg-106 of 3A5 is pushed inward by interactions with Met-393, which is larger than Val-393 in 3A4. This change in 3A5 is accommodated by the longer length of Glu-76 relative to Asp-76 in 3A4 to preserve the salt bridge (15). This network of salt bridges and hydrogen bonds is not significantly affected by ritonavir in 3A4, but Leu-108 moves inward to contact ritonavir while preserving the interaction of Arg-106 with Glu-76. The S107P difference between 3A5 and 3A4 is likely to contribute to the plasticity of Leu-108 and B–B′ connector of 3A5.
Comparison of the structure of the 3A5 ritonavir complex with the apo 3A5 structure indicates that engagement of the distal end of ritonavir with the flexible connector between helices F and F′ pushes the connector outward along with Phe-213 (Fig. 1D). Leu-108, Phe-215, Phe-220, and Phe-241 make adjustments to forge hydrophobic contacts with ritonavir (Figs. 1D and 2B). In 3A4, ritonavir pushes these portions of the helix F–G region upward without forcing the phenylalanine side chains out of the central cavity. In contrast, 3A5 Phe-215 and Phe-220 reorient into the cavity to engage with ritonavir. These changes reduce the size of the distal cavity and closes the substrate access channel seen in the apo 3A5 structure (Fig. 2, A versus B). Thus, the end points for ritonavir binding and inferred starting states (apo structures) for the binding of ritonavir to 3A4 and 3A5 differ substantially.
Discussion
Determination of the structure of 3A5 crystallized in the absence of a substrate or inhibitor was undertaken to better understand structural differences between 3A5 and 3A4 that underlie divergent functional properties and to better define how the binding of ritonavir to 3A5 alters the substrate-free 3A5 structure. Apo 3A5 exhibits a much larger active-site cavity compared with apo 3A4 and to a lesser extent that of the 3A5 ritonavir complex. The volumetric difference between apo 3A4 and apo 3A5 is associated with relatively large differences in the conformation of the helix F through helix F′ region, which is part of a larger loop-like structure that includes helices G′ and G and forms the roof over the active site. This region begins and ends where helices F and G interact with each other and the upper surface of helix I. The apex of the loop is the turn between helices F′ and G′, which resides outside of the helix B–B′ loop and is stabilized in that position by several polar interactions. The smaller volume of the apo 3A4 binding site reflects the lower trajectory of helix F and the inward sag of the connector between helices F and F′ that is stabilized by edge-to-face interactions between several phenylalanine residues that form the roof above the heme at a lower height than seen in 3A5. These interactions are less extensive in 3A5 in part because of the replacement of 3A4 Phe-108 with the shorter leucine residue that disrupts the chain of phenylalanine interactions that extends from the proximal to distal cavity in 3A4. Additional amino acid differences that alter the trajectory of the helix F to helix F′ region open the center of the loop-like structure of the F–G region in both the 3A5 apo and 3A5 ritonavir complexes (15) relative to the 3A4 apo and ritonavir complexes and prevent additional phenylalanine interactions in the distal cavity of apo 3A5.
In the structure of the 3A5 ritonavir complex, ritonavir binds in a conformation that exploits the more open distal cavity seen in the structure of apo 3A5. Additionally, the conformation of ritonavir seen in 3A4 that extends under helix F′ is blocked by the altered network of salt bridges and H-bonds in 3A5 (15). In contrast, the observed conformation of ritonavir in 3A4 is favored by the greater plasticity of the N-terminal region. The lower trajectory of ritonavir in 3A4 raises the roof of the cavity to a lesser extent because of the extensive phenylalanine interactions in the upper portion of the cavity. Yuki et al. (24) demonstrated that when explicit CH–π aromatic interactions were included in molecular dynamics simulations of the 3A4 carbamazepine complex, the stability of the active site increased, and the motion of carbamazepine in the cavity was more constrained. In contrast, the reduced volume of the active-site cavity in the 3A5–ritonavir complex when compared with apo 3A5 reflects the plasticity of the F–G region that adapts to the presence of ritonavir and changes to form favorable hydrophobic interactions with ritonavir that reduce the cavity size and allows ritonavir to extend into the center of the ring-like F–G region.
Family 3 P450s are known to catalyze the metabolic clearance for substrates as large as cyclosporine A (MW, 1203), as well as much smaller molecules such as testosterone (MW, 288), and the size of the apo 3A5 cavity is consistent with that of larger substrates such cyclosporine A. Interestingly, 3A5, but not 3A4, is prominently expressed in kidney (5), and greatly increased cyclosporine A metabolism is evident for microsomes derived from renal tissues where one or two functional 3A5 alleles are present (25). In this regard, the active-site cavity of substrate-free 3A5 is more similar in size to structures of 3A4 co-crystallized with erythromycin (MW, 734) or ketoconazole (MW, 531) characterized by Ekroos and Sjögren (17) that exhibit expanded distal active-site cavities. Erythromycin did not alter helix I but expanded the distal cavity (PDB code 2J0D) by raising the trajectory of the helix F–F′ connector upward and displacing Arg-212 out into solvent. The coordinate covalent binding of the ketoconazole imidazole moiety with the heme iron (PDB code 2V0M) led to outward distortion of helix I (Fig. 4) like that seen for ritonavir (Fig. 1F), and a dichlorophenyl group adjacent to imidazole moiety engaged with the phenylalanine cluster similar to that seen for P1 phenyl group of ritonavir. A second ketoconazole molecule was identified in the active-site cavity stacked above the heme-bound ketoconazole in an antiparallel orientation (Fig. 4). The second molecule greatly expanded the size of the cavity and opened the distal end similarly to that seen for apo 3A5. The 3A4 helix F–F′ connector was elevated to a greater extent than seen for ritonavir with Phe-215 oriented upward out of the cavity as seen for apo 3A5 (Fig. 3A). In the 3A4 erythromycin complex, Phe-215 was rotated inward to close the cavity, which is also seen in the 3A5 ritonavir complex relative to the apo 3A5 structure (Fig. 1D). In both cases, the distal ends of the erythromycin and ketoconazole reside under the helix F′, and the center of the helix F–G region remains closed in 3A4 because of the packing interactions between phenylalanines 108, 213, 220, and 241. In contrast, ritonavir resides in the more open ring formed by the 3A5 F–G region that reflects the larger distances between the F–F′ and G′–G connectors imparted by the F210L and L108F differences between 3A5 and 3A4, which contributes to diminished interactions between phenylalanine residues and the increased plasticity seen for the F–G region in 3A5. These differences are likely to underlie the differentiated functions of the two enzymes. Consistent with this notion, reciprocal substitutions of the divergent amino acids in 3A4 and 3A5, L210F and F108L, alter metabolite formation by 3A4 (26) and 3A5 (13, 14). As seen in Fig. 2B, the presence of ritonavir led to closure of the entrance channel seen in apo 3A5, but it remains unclear whether further reductions in the size of the 3A5 active site would occur when a significantly smaller substrates than ritonavir bind.
Figure 4.
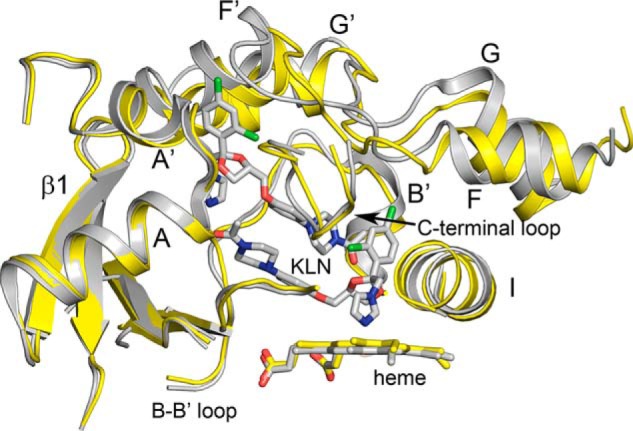
Superposition of 3A4 (gray) with two molecules of ketoconazole (KLN) bound in the active site (PDB code 2V0M) with apo 3A5 (yellow) using ALIGN with defaults in PyMOL. Portions of the proteins are shown as cartoons displaying secondary and tertiary structure. The imidazole nitrogen of the lower ketoconazole molecule is ligated to the heme iron, and together with the neighboring dichlorophenyl group, they displace a portion of helix I outward. A second molecule of ketoconazole stacks above the first in anti-parallel orientation with dichlorophenyl moiety residing in the entrance channel. The conformation of the helix F–G regions of the two structures are overlapped to greater extent than seen for structures of the 3A5 and 3A4 ritonavir complexes, and the 3A4 active site is greatly enlarged by the presence of the two ketoconazole molecules.
Experimental procedures
Protein expression and purification
P450 3A5 was modified for expression in Escherichia coli and to facilitate purification and crystallization by modification of the N terminus to remove the transmembrane helix, optimize codons for the truncated N terminus to increase expression, and substitution of a four-histidine tag for the five C-terminal amino acids that extend beyond the catalytic domain. Details of the construction of the expression plasmid pCW3A5C2, conditions for the expression of the protein in E. coli, and purification of the protein are documented in an earlier publication (15). The detergent CHAPS was omitted from the final wash and elution buffers for the carboxymethyl-agarose chromatography step, and the final size-exclusion chromatography step used previously was not required.
Crystallization conditions
The concentration of the protein solution was 450 μm as determined by the reduced CO-difference spectrum (27). The protein buffer was 0.1 m Na-HEPES (pH 7.4), 50 mm potassium acetate (pH 7.4), 20% glycerol (v/v), 0.5 mm EDTA, 10 mm β-mercaptoethanol, and 0.5 mm phenylmethylsulfonyl fluoride. The precipitant and well solution was 0.2 m sodium N-(2-acetamido)iminodiacetate, pH 6.5, containing 30% PEG 3350 (w/v). Crystallization was achieved by sitting-drop vapor diffusion against 0.5 ml of well buffer in a Cryschem plate (Hampton Research) at 24 °C. The sitting drop was formed by combining the protein solution, 10% (w/v) solution of the detergent Anapoe 20, and the precipitant in a 1:0.1:1 ratio (final drop size is 2.1 μl). The crystals were harvested and soaked in perfluoropolyether for 20 s prior to placing the crystal in liquid nitrogen for shipment and data collection.
Model building and refinement
The protein model was refined using data collected from a single crystal at 100 K on Beamline 12-2 at the Stanford Radiation Light Source to an apparent limiting resolution of 2.20 Å in the I222 space group. The X-ray diffraction reflections were integrated, merged, and scaled using XDS (28). Initial phases were obtained by molecular replacement using PDB code 1TQN structure of apo 3A4 as the search model and PHASER (29) as implemented in PHENIX (30). PHASER identified one chain in the asymmetric unit and identified the enantiomeric space group as I222 with a log likelihood 1415 using the 3A4 1TQN structure. COOT (31) was used for model building, and PHENIX (30) was used for model refinement. Statistics for data reduction, model refinement, and model quality are summarized in Table 1. φ–ψ angles for 96.9% of the residues are in the most favored range without any φ–ψ angle outliers or side-chain rotamer outliers. The fit to the reflections exhibits an R value of 0.209 and a free R value of 0.248 for 5% of reflections that were not included in the refinement of the model. The final, refined model was refined against X-ray diffraction data to limiting resolution of 2.20 Å. The model is complete for residues 26–496 with the exception of residues 260–269 and 280–287, respectively, in external loops between helices G to H and H to I that could not be modeled because of configurational heterogeneity. Additionally, 50 waters, 3 glycerols, and the heme co-factor are included in the model. Two of the glycerols are bound in the substrate access channel, and the third resides in the active site near the heme surface (Fig. S1).
Table 1.
Data collection and model refinement statistics
Statistics for the highest-resolution shell are shown in parentheses.
| Substrate free cytochrome P450 3A5 (CYP3A5), PDB code 6MJM | |
|---|---|
| Data collection | |
| Resolution range (Å) | 38.89–2.20 (2.26–2.20) |
| Space group | I 2 2 2 |
| Unit cells | |
| Lengths (Å) | 75.67, 100.69, 136.02 |
| Angles (°) | 90, 90, 90 |
| Wavelength (Å) | 0.97946 |
| Total reflections | 254,554 (18,651) |
| Unique reflections | 26,713 (1940) |
| Multiplicity | 9.7 (9.7) |
| Completeness (%) | 99.8 (98.9) |
| Mean I/σ(I) | 33.47 (3.87) |
| Wilson B-factor (Å3) | 55.60 |
| Rmerge | 0.032 (0.508) |
| Rmeas | 0.034 (0.536) |
| CC½ | 1.000 (0.924) |
| Model refinement | |
| Resolution range (Å) | 34.274–2.20 (2.279–2.20) |
| Reflections used in refinement | 26,710 (2482) |
| Reflections used for Rfree | 1353 (124) |
| Rwork | 0.2092 (0.2700) |
| Rfree | 0.2480 (0.3292) |
| Number of heavy atoms | 3740 |
| Protein | 3630 |
| Ligands | 61 |
| Solvent | 49 |
| Ramachandran favored (%) | 96.87 |
| Ramachandran outliers (%) | 0.00 |
| Average B-factor | 69.64 |
| Protein | 70.02 |
| Heme | 46.98 |
| Glycerols | 78.43 |
| Waters | 57.52 |
| TLS groups | 1 |
Graphic images were generated using PyMOL (32), and surfaces of substrate binding cavities were generated using VOIDOO (33) with a 1.4 Å probe. The atomic coordinates and structure factors (PDB code 6MJM) are deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University (New Brunswick, NJ).
Automated docking of testosterone in the Apo 3A5 structure
Automated docking studies employed VINA (34). AUTODOCK TOOLS (35) was used to define the search space and to generate PDBQT files that define rotatable bonds and charges from the coordinates for the apo 3A5 final model and for testosterone (Ligand Expo). OPEN BABEL (36) was used to extract PDB files for the docked ligand from the VINA output.
Author contributions
M.-H. H. and E. F. J. conceptualization; M.-H. H. and E. F. J. data curation; M.-H. H. and E. F. J. validation; M.-H. H. and E. F. J. investigation; M.-H. H. methodology; M.-H. H. and E. F. J. writing-review and editing; E. F. J. supervision; E. F. J. funding acquisition; E. F. J. writing-original draft; E. F. J. project administration.
Supplementary Material
Acknowledgment
We thank Üzen Savas for generation of the expression vector and critical reading of the manuscript.
This work was supported by National Institutes of Health Grant R01-GM031001 (to E. F. J.). The use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 6MJM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains Fig. S1.
- CYP or P450
- a generic term for a cytochrome P450 enzyme (individual P450s are identified using a number–letter–number designation based on amino acid sequence relatedness)
- PDB
- Protein Data Bank
- MW
- molecular weight.
References
- 1. Zanger U. M., Turpeinen M., Klein K., and Schwab M. (2008) Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem. 392, 1093–1108 10.1007/s00216-008-2291-6 [DOI] [PubMed] [Google Scholar]
- 2. Rendic S., and Guengerich F. P. (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 28, 38–42 10.1021/tx500444e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stevens J. C. (2006) New perspectives on the impact of cytochrome P450 3A expression for pediatric pharmacology. Drug Discov. Today 11, 440–445 10.1016/j.drudis.2006.03.002 [DOI] [PubMed] [Google Scholar]
- 4. Ince I., Knibbe C. A., Danhof M., and de Wildt S. N. (2013) Developmental changes in the expression and function of cytochrome P450 3A isoforms: evidence from in vitro and in vivo investigations. Clin. Pharmacokinet. 52, 333–345 10.1007/s40262-013-0041-1 [DOI] [PubMed] [Google Scholar]
- 5. Aoyama T., Yamano S., Waxman D. J., Lapenson D. P., Meyer U. A., Fischer V., Tyndale R., Inaba T., Kalow W., and Gelboin H. V. (1989) Cytochrome P-450 hPCN3, a novel cytochrome P-450 IIIA gene product that is differentially expressed in adult human liver. J. Biol. Chem. 264, 10388–10395 [PubMed] [Google Scholar]
- 6. Kuehl P., Zhang J., Lin Y., Lamba J., Assem M., Schuetz J., Watkins P. B., Daly A., Wrighton S. A., Hall S. D., Maurel P., Relling M., Brimer C., Yasuda K., Venkataramanan R., et al. (2001) Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 27, 383–391 10.1038/86882 [DOI] [PubMed] [Google Scholar]
- 7. Koudriakova T., Iatsimirskaia E., Utkin I., Gangl E., Vouros P., Storozhuk E., Orza D., Marinina J., and Gerber N. (1998) Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab. Dispos. 26, 552–561 [PubMed] [Google Scholar]
- 8. Williams J. A., Ring B. J., Cantrell V. E., Jones D. R., Eckstein J., Ruterbories K., Hamman M. A., Hall S. D., and Wrighton S. A. (2002) Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab. Dispos. 30, 883–891 10.1124/dmd.30.8.883 [DOI] [PubMed] [Google Scholar]
- 9. Yamazaki H., Nakamoto M., Shimizu M., Murayama N., and Niwa T. (2010) Potential impact of cytochrome P450 3A5 in human liver on drug interactions with triazoles. Br. J. Clin. Pharmacol. 69, 593–597 10.1111/j.1365-2125.2010.03656.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tseng E., Walsky R. L., Luzietti R. A. Jr., Harris J. J., Kosa R. E., Goosen T. C., Zientek M. A., and Obach R. S. (2014) Relative contributions of cytochrome CYP3A4 versus CYP3A5 for CYP3A-cleared drugs assessed in vitro using a CYP3A4-selective inactivator (CYP3cide). Drug Metab. Dispos. 42, 1163–1173 10.1124/dmd.114.057000 [DOI] [PubMed] [Google Scholar]
- 11. Niwa T., Murayama N., Emoto C., and Yamazaki H. (2008) Comparison of kinetic parameters for drug oxidation rates and substrate inhibition potential mediated by cytochrome P450 3A4 and 3A5. Curr. Drug Metab. 9, 20–33 10.2174/138920008783331121 [DOI] [PubMed] [Google Scholar]
- 12. Li X., Jeso V., Heyward S., Walker G. S., Sharma R., Micalizio G. C., and Cameron M. D. (2014) Characterization of T-5 N-oxide formation as the first highly selective measure of CYP3A5 activity. Drug Metab. Dispos. 42, 334–342 10.1124/dmd.113.054726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu Y., Hendrix C. W., and Bumpus N. N. (2012) Cytochrome P450 3A5 plays a prominent role in the oxidative metabolism of the anti-human immunodeficiency virus drug maraviroc. Drug Metab. Dispos. 40, 2221–2230 10.1124/dmd.112.048298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu J. J., Cao Y. F., Feng L., He Y. Q., Hong J. Y., Dou T. Y., Wang P., Hao D. C., Ge G. B., and Yang L. (2017) A naturally occurring isoform-specific probe for highly selective and sensitive detection of human cytochrome P450 3A5. J. Med. Chem. 60, 3804–3813 10.1021/acs.jmedchem.7b00001 [DOI] [PubMed] [Google Scholar]
- 15. Hsu M. H., Savas U., and Johnson E. F. (2018) The X-ray crystal structure of the human mono-oxygenase cytochrome P450 3A5–ritonavir complex reveals active site differences between P450s 3A4 and 3A5. Mol. Pharmacol. 93, 14–24 10.1124/mol.117.109744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sevrioukova I. F., and Poulos T. L. (2010) Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. U.S.A. 107, 18422–18427 10.1073/pnas.1010693107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ekroos M., and Sjögren T. (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 103, 13682–13687 10.1073/pnas.0603236103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sevrioukova I. F., and Poulos T. L. (2017) Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 114, 486–491 10.1073/pnas.1616198114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brändén G., Sjögren T., Schnecke V., and Xue Y. (2014) Structure-based ligand design to overcome CYP inhibition in drug discovery projects. Drug Discov. Today 19, 905–911 10.1016/j.drudis.2014.03.012 [DOI] [PubMed] [Google Scholar]
- 20. Yano J. K., Wester M. R., Schoch G. A., Griffin K. J., Stout C. D., and Johnson E. F. (2004) The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-Å resolution. J. Biol. Chem. 279, 38091–38094 10.1074/jbc.C400293200 [DOI] [PubMed] [Google Scholar]
- 21. Williams P. A., Cosme J., Vinkovic D. M., Ward A., Angove H. C., Day P. J., Vonrhein C., Tickle I. J., and Jhoti H. (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305, 683–686 10.1126/science.1099736 [DOI] [PubMed] [Google Scholar]
- 22. Johnson E. F., and Stout C. D. (2013) Structural diversity of eukaryotic membrane cytochrome p450s. J. Biol. Chem. 288, 17082–17090 10.1074/jbc.R113.452805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Denisov I. G., Baas B. J., Grinkova Y. V., and Sligar S. G. (2007) Cooperativity in P450 CYP3A4: linkages in substrate binding, spin state, uncoupling and product formation. J. Biol. Chem. 282, 7066–7076 10.1074/jbc.M609589200 [DOI] [PubMed] [Google Scholar]
- 24. Yuki H., Tanaka Y., Hata M., Ishikawa H., Neya S., and Hoshino T. (2007) Implementation of π–π interactions in molecular dynamics simulation. J. Comput. Chem. 28, 1091–1099 10.1002/jcc.20557 [DOI] [PubMed] [Google Scholar]
- 25. Dai Y., Iwanaga K., Lin Y. S., Hebert M. F., Davis C. L., Huang W., Kharasch E. D., and Thummel K. E. (2004) In vitro metabolism of cyclosporine A by human kidney CYP3A5. Biochem. Pharmacol. 68, 1889–1902 10.1016/j.bcp.2004.07.012 [DOI] [PubMed] [Google Scholar]
- 26. Wang H., Dick R., Yin H., Licad-Coles E., Kroetz D. L., Szklarz G., Harlow G., Halpert J. R., and Correia M. A. (1998) Structure-function relationships of human liver cytochromes P450 3A: aflatoxin B1 metabolism as a probe. Biochemistry 37, 12536–12545 10.1021/bi980895g [DOI] [PubMed] [Google Scholar]
- 27. Omura T., and Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes: II. Solubilization, purification, and properties. J. Biol. Chem. 239, 2379–2385 [PubMed] [Google Scholar]
- 28. Kabsch W. (2010) XDS. XDS. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeLano W. L. (2005) The case for open-source software in drug discovery. Drug Discov. Today 10, 213–217 10.1016/S1359-6446(04)03363-X [DOI] [PubMed] [Google Scholar]
- 33. Kleywegt G. J., and Jones T. A. (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 50, 178–185 10.1107/S0907444993011333 [DOI] [PubMed] [Google Scholar]
- 34. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Boyle N. M., Banck M., James C. A., Morley C., Vandermeersch T., and Hutchison G. R. (2011) Open Babel: an open chemical toolbox. J. Cheminform. 3, 33 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.