

Abstract
Heme oxygenase (HO) catalyzes heme degradation, a process crucial for regulating cellular levels of this vital, but cytotoxic, cofactor. Two HO isoforms, HO1 and HO2, exhibit similar catalytic mechanisms and efficiencies. They also share catalytic core structures, including the heme-binding site. Outside their catalytic cores are two regions unique to HO2: a 20-amino acid–long N-terminal extension and a C-terminal domain containing two heme regulatory motifs (HRMs) that bind heme independently of the core. Both HO isoforms contain a C-terminal hydrophobic membrane anchor; however, their sequences diverge. Here, using hydrogen–deuterium exchange MS, size-exclusion chromatography, and sedimentation velocity, we investigated how these divergent regions impact the dynamics and structure of the apo and heme-bound forms of HO1 and HO2. Our results reveal that heme binding to the catalytic cores of HO1 and HO2 causes similar dynamic and structural changes in regions (proximal, distal, and A6 helices) within and linked to the heme pocket. We observed that full-length HO2 is more dynamic than truncated forms lacking the membrane-anchoring region, despite sharing the same steady-state activity and heme-binding properties. In contrast, the membrane anchor of HO1 did not influence its dynamics. Furthermore, although residues within the HRM domain facilitated HO2 dimerization, neither the HRM region nor the N-terminal extension appeared to affect HO2 dynamics. In summary, our results highlight significant dynamic and structural differences between HO2 and HO1 and indicate that their dissimilar C-terminal regions play a major role in controlling the structural dynamics of these two proteins.
Keywords: heme oxygenase, enzyme catalysis, enzyme mechanism, protein dynamics, membrane protein, hydrogen exchange mass spectrometry, enzyme kinetics, conformational change, heme homeostasis, membrane anchor
Introduction
Heme oxygenase catalyzes the degradation of heme to biliverdin, carbon monoxide, and iron in a reaction that requires oxygen as well as seven electrons supplied from NADPH via cytochrome P450 reductase (CPR).4 The reaction is catalyzed by both isoforms of human heme oxygenase, heme oxygenase-1 (HO1) and heme oxygenase-2 (HO2), with similar catalytic efficiency (1) and proceeds in the catalytic core region of both of the enzymes. The highly α helical core, which has 55% sequence identity and a high degree of structural homology between HO1 and HO2, is the site of heme binding. More specifically, within the core is a pocket created by two α helices, the proximal and distal helices (2, 3) (see Fig. 1). The proximal helix includes a conserved histidine residue (His25 in human HO1 and His45 in human HO2) that coordinates the iron of the heme molecule. The distal helix contains conserved glycine residues that impart a high degree of flexibility to this helix, allowing it to interact with the bound heme.
Figure 1.

The structures and sequences of HO1 and HO2. Helices A1–A7 are labeled and indicated by color on the structure of the catalytic core of heme-bound HO1 (Protein Data Bank (PDB) code 1N3U) and the catalytic core of heme-bound HO2 (PDB code 2RGZ). Helices A1–A7, which cover the same indicated regions of both HO2 and HO1, are also indicated using the same color scheme above the linear sequence comparison of HO2 and HO1. An asterisk (*) indicates identical residues, a colon (:) indicates highly similar residues, and a period (.) indicates weakly similar residues. Gray boxes indicate small helical regions that are not numbered. The heme-ligating histidine residues of each protein (His25 in HO1 and His45 in HO2) are in the brown box. The disulfide bond between Cys265 and Cys282 is also indicated. All the constructs studied here that contain the HRMs in the C-terminal tail are oxidized and form a disulfide bond, incapable of binding heme, unless otherwise noted. Constructs of varying amino acid lengths were studied here. Constructs have been truncated on the N-terminal end and/or the C-terminal end as indicated by the blue arrows. A previous alignment differs in the region between HO2 residues 263 and 289 and matches Lys274 (HO2) with Arg254 (HO1) (3).
Despite the noted similarities between HO1 and HO2 in the catalytic cores, regions of the proteins outside the core differ in sequence. One region of interest that differs greatly between HO1 and HO2 is at the N terminus. HO1 is a 288-amino acid protein, whereas HO2 is composed of 316 amino acids, with the difference in the number of amino acids primarily due to a 20-amino acid extension at the N terminus of HO2. The X-ray crystal structures of C-terminally truncated HO2 lacked electron density for the first 28 amino acids (3). Furthermore, the N-terminal extension of HO2 has been shown to exist in an unfolded, dynamic random coil by NMR relaxation experiments (4). However, the functional significance of this region in HO2 remains unclear.
The regions in HO1 and HO2 immediately C-terminal to the catalytic core domain are not present in the X-ray crystal structures (residues 224–288 are missing in HO1, and residues 249–316 are missing in HO2), share little sequence similarity, and are related to different functional aspects of the proteins. The region between the core and membrane anchor of HO1 includes a proposed protein degradation signal sequence (PEST domain) (5) that is important for the signal peptide peptidase–mediated cleavage and subsequent translocation of HO1 into the cytosol and nucleus under hypoxic conditions (6–8). The proposed signal peptide peptidase cleavage site at Ser275-Phe276 is in the membrane anchor of HO1 (8). The short sequence is not conserved between HO1 and HO2, consistent with the observation that HO2 is not translocated to the nucleus under hypoxic conditions (9). Rather, the corresponding region in HO2, referred to as the C-terminal tail, includes two heme regulatory motifs (HRMs), centered at Cys265-Pro266 and Cys282-Pro283. Cys265 and Cys282 form a disulfide bond (10, 11) (Fig. 1). Upon reduction of the disulfide bond, Cys265 and a nearby histidine residue (His256) form an independent binding site for heme. Cys282 also coordinates the iron of an additional molecule of heme albeit with lower affinity than either the other HRM (Cys265/His256) or the core (His45) (12). The functional significance of heme binding to the HRMs of HO2 is unclear, although heme binding to HRMs of several other proteins has been associated with protein degradation (13). It is noteworthy that the sequence of HO1 contains no cysteines.
The membrane anchor regions are known not only to associate HO1 and HO2 with the endoplasmic reticulum (ER) membrane but also to promote self-association to form dimers and higher-order oligomers. A previous study showed that full-length HO1 formed dimers/oligomers while associated with the ER membrane (14). Furthermore, Trp270 is a residue within the membrane anchor region predicted to be at the interface between HO1 monomers, and mutation of this residue to Asn weakened oligomerization and increased susceptibility to cleavage (14). Gel-filtration studies later confirmed that full-length HO1 does form higher-order oligomers in vitro when purified from Sf9 cells with an N-terminal streptavidin tag, but HO1 does not oligomerize to the extent that HO2 does (9). The significance of the membrane anchor to the function and structure of HO1 and HO2 may thus be greater than previously assumed.
Biophysical studies of HO1 and HO2 have primarily utilized C-terminally truncated, soluble forms of the proteins to link structure and function. Minimal information is available regarding how the N- and C-terminal regions, where HO1 and HO2 sequences differ the most, affect the structures, dynamics, and heme-binding properties of the proteins. Therefore, with particular attention paid to the N- and C-terminal regions of HO1 and HO2, we probed full-length and truncated forms of HO1 and HO2 by hydrogen–deuterium exchange MS (HDX-MS), size-exclusion chromatography (SEC), and sedimentation velocity (SV). We demonstrate that, whereas the N-terminal region of HO2 appears to not affect dynamics of the catalytic core region, the C-terminal region alters both its dynamics and structure without affecting its steady-state activity, heme-binding affinity, or spectral features of the bound heme. Furthermore, the differences in dynamics and structure between the truncated and full-length forms of HO2 are unaffected by heme occupancy at the HRM when heme is bound to the core. In contrast, the C-terminal residues of HO1 appear to have very little influence on the structure and dynamics of the HO1 catalytic core. Thus, the C-terminal regions of these two proteins play a major role in conferring the differences in their structure and dynamics.
Results
Truncation of the C-terminal regions of HO2 and HO1 has no effect on steady-state activity or heme-binding properties
Full-length HO2 (HO2(1–316)) was purified as described previously for full-length HO1 (HO1(1–288)) (15). Native HO2(1–316) was purified to homogeneity without needing to introduce substitution, unlike HO1(1–288) in which an R254K stabilizes the protein from degradation (15). Accordingly, the HO1 and HO2 sequences (Fig. 1) do not align well around Arg254 of HO1, although an alternative alignment (3) matches this residue with Lys274 of HO2. SEC analysis of HO2(1–316) demonstrates that the full-length protein, like R254K HO1(1–288) (16), exists as a high-molecular-weight complex that elutes in/near the void volume (700,000 Da) (Fig. 2). The elution profile of HO2(1–316), in contrast to HO1 (16), lacks any significant peaks that correspond to truncated products (16).
Figure 2.

Characterization of full-length HO2. A, comparison of the spectra of heme-bound (with disulfide bond between Cys265 and Cys282) forms of HO2(1–288) (dashed line) and HO2(1–316) (solid line). Spectra are of 3 μm protein in 50 mm Tris (pH 8.0), 50 mm KCl buffer at 20 °C. B, HO2(1–316) analysis by size-exclusion chromatography. 10 μm heme-bound protein (with disulfide bond intact) was run at 1 ml min−1 in 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl and 0.1 mm EDTA using a Shodex KW-803 column on a Shimadzu HPLC with an LC-10AT pump system. Protein eluted in/near the void volume (700,000 Da) of the column. C, heme oxygenase activity with varying concentrations of truncated CPR was monitored. Activity is reported as nmol of bilirubin formed/min/mg of HO. HO2(1–316) activity is shown in squares, R254K HO1(1–288) activity is shown in circles, and the solid lines represent the fits of the data to determine Km and Vmax (listed in Table 1). mAU, milliabsorbance units. Error bars represent S.D.
Our results demonstrated that the C-terminal membrane-spanning region has little effect on heme binding, steady-state activity, or disulfide bond formation. The spectra of the heme-bound forms of HO2(1–316) and HO2(1–288) were very similar (Fig. 2). Heme off-rates, as measured by apo-H64Y/V68F-myoglobin (“green heme”) assay (17), a reflection of heme-binding affinity (12, 18–20), were very similar (Fig. 2 and Table 1). Steady-state kinetic analysis of HO2(1–316) reveals a Km for truncated CPR and Vmax values for conversion of heme to bilirubin very similar to those reported previously for HO2(1–288) (Table 1) (21). Lastly, like HO2(1–288) (10–12), HO2(1–316) forms a disulfide bond between Cys265 and Cys282, based on a 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) assay in the presence of guanidine hydrochloride, which reveals only one free thiol, presumably Cys127 (Table 1).
Table 1.
Comparison of heme-binding properties, activity, and thiol oxidation state for HO2(1–288), HO2(1–316), HO1(1–265), and R254K HO1(1–288)
| Protein construct | Heme bindinga: koff (s−1) | Steady-state activityb: Km for trCPR (μm) and Vmax (nmol min−1 mg−1) | Free thiols per proteinc |
|---|---|---|---|
| HO2 | |||
| HO2(1–288) | (1.76 ± 0.03) × 10−3 (32%) | 0.7 ± 0.1d | 0.93 |
| (5.82 ± 0.06) × 10−4 | 296.9 ± 4.6d | ||
| HO2(1–316) | (1.00 ± 0.02) × 10−2 (12%) | 0.6 ± 0.2 | 0.89 |
| (5.754 ± 0.009) × 10−4 | 230 ± 30 | ||
| HO1 | |||
| HO1(1–265) | (7.2 ± 0.01) × 10−3 (22%) | 0.5 ± 0.1d | N.A.e |
| (2.72 ± 0.02) × 10−4 | 370 ± 12d | ||
| HO1(1–288), R254K | (3.1 ± 0.1) × 10−2 (6%) | 1.0 ± 0.3 | N.A.e |
| (3.29 ± 0.002) × 10−4 | 320 ± 30 |
a Data were obtained by monitoring the change in absorbance at 600 nm in competition assay between apo-H64Y/V68F-myoglobin (“green heme”) and the heme-bound form of the constructs indicated. Data were fit using a double-exponential equation, hence two rates for each reaction. The percentage of the total absorbance change of the fast phase is indicated in parentheses.
b Heme oxygenase activity with varying concentrations of truncated CPR (trCPR) was monitored as shown in Fig. 2C. Activity is reported as nmol of bilirubin formed/min/mg of HO.
c Free thiols were measured using a DTNB assay in the presence of 6 m guanidine hydrochloride.
d Values as reported in Ref. 21.
e Not applicable. HO1 contains no cysteines.
Likewise, full-length R254K HO1(1–288) has similar heme off-rates and steady-state kinetic profiles (Table 1) as truncated HO1(1–265). It has been reported previously that the heme-bound spectra of both constructs are similar (15).
Furthermore, full-length HO1 and HO2 constructs display steady-state activities similar to each other when assayed with truncated CPR (Table 1). Heme-off rates were only slightly slower for the HO2 constructs, which could indicate a slightly greater affinity for heme than the HO1 constructs. However, the difference is only about 2-fold, so a more detailed kinetic analysis would be needed to confirm any difference in affinity.
Heme binding to the catalytic core of HO1 primarily affects local dynamics within the heme-binding pocket
In both HO1 and HO2, the binding site for heme in the catalytic core region is mostly formed by two helices: the proximal helix (A1), which includes the conserved histidine residue (His25 in human HO1 and His45 in human HO2) that coordinates the iron of the heme molecule, and the flexible, distal helix (A5) (Fig. 1). The flexibility of the distal helix was noted in the X-ray crystal structure (2) of a C-terminally truncated, soluble form of HO1 with heme bound in the catalytic core. Two molecules of HO1 were observed in the asymmetric unit, with one molecule in a “closed” conformation in which the distal helix is more tightly packed against the bound heme in the closed conformation than in the other, “open” conformation (2). The closed conformation has a more ordered solvent structure, which is involved in a hydrogen-bonded network with the catalytically important residue, Asp140, of the distal helix, than that of the open conformation (23). Furthermore, the apo form of HO1 was crystallized in the open conformation (23). This conformational flexibility likely serves to facilitate substrate (heme) binding and product (biliverdin) release in the open conformation and reaction specificity in the closed conformation. HDX-MS was used to probe the dynamics of HO1 in the heme-bound and apo forms of both C-terminally truncated HO1 that spans residues 1–265 (HO1(1–265)) and full-length HO1 (HO1(1–288)).
Decreased deuterium exchange was observed in helices containing residues important for heme binding (Fig. 3A). The proximal helix in the heme-bound forms of both HO1(1–265) and HO1(1–288) exhibited significantly less HDX at all time points (except 4 h) relative to their respective apo forms. The largest differences in exchange between the apo and heme-bound states occurred at the early (10-s and 1-min) time points where the protection from exchange due to the addition of heme was between 4.0 and 5.0 Da (relative values; see “Experimental procedures”). Protection from exchange in the HO1 distal helix was also observed, with a decrease in uptake of 2.0–3.0 Da in the heme-bound state. Protection from exchange upon the addition of heme was also observed around residue 180 for both HO1(1–265) and HO1(1–288) at later time points. This region corresponds to α-helix A6 in HO1 and includes Lys179 and Arg183, two residues that assist in orienting heme in the binding pocket to direct specific hydroxylation at the α-meso-carbon through interactions with the propionates of the heme (2, 23). These residues, together with Lys18 and Lys22 on the proximal helix, which also experiences protection from exchange, are proposed to influence the conformation of HO1. This group of four residues promotes formation of the open conformation in the absence of heme due to electrostatic repulsion among these basic residues and encourages establishment of the closed state in the presence of heme through charged interactions with the propionates (2, 23).
Figure 3.

A and B, differences in deuterium incorporation in the heme-bound versus apo forms of HO1 and HO2. The differences in deuterium level are shown for HO1 constructs 1–265 and 1–288 at labeling times 10 s, 1 min, 10 min, 1 h, and 4 h, left to right of each graph (A), and HO2 constructs 1–248, 1–288, and 1–316 at labeling times 5 s, 10 s, 30 s, 1 min, 10 min, 1 h, and 4 h, left to right in each graph (B). In each panel, the peptic peptides where deuterium was measured are arranged N- to C-terminal, top to bottom, with approximate sequence numbering on the left side. Deuterium differences between heme-bound and apo forms were calculated using the equation Dheme − Dapo for each comparison and then colored according to the scale shown. A guide to the regions of each protein is shown at the far right (colored as in Fig. 1). The additional N-terminal amino acids of HO2 and the disulfide bond between Cys265 and Cys282 are in gray as these features are unique to HO2. All data used to create this figure can be found in Excel File S1. C, mapping the data from the 1-min differences in A and B onto the structures for HO1 (PDB code 1N3U) and HO2 (PDB code 2RGZ). Heme is shown in stick form in green in each model.
Unexpected, however, was the protection from exchange within residues 65–80, again at later time points (10 min to 4 h). This region corresponds to the end of α-helix A2 and the region between A2 and A3, which includes a small α-helical region composed of residues 75–77. Residues 65–80 appear to be on the surface of the protein, and the region may be flexible given its slightly higher temperature factor relative to the regions around it (2). Furthermore, as observed in the X-ray crystal structures of HO1, this region exhibits some root mean square deviation of backbone atoms between apo- and heme-bound HO1 (23). In addition, residues 65–80 are near the distal helix, suggesting that coordination of heme stabilizes interaction between these two regions, leading to the observed protection.
Overall, the areas of protection observed upon heme binding in both HO1(1–265) and HO1(1–288) are consistent with the idea that heme binding promotes the closed conformation. Furthermore, the overall pattern of protection is highly similar between the truncated and full-length HO1 constructs, suggesting that the hydrophobic residues in the C-terminal membrane anchor region do not influence the dynamics associated with the binding of heme (Fig. 3).
Effects of heme binding to the catalytic core of HO2 varies with the length of the C terminus
As discussed in detail above, the binding site for heme in the catalytic core region is mostly formed by the proximal (A1) and the flexible, distal (A5) helices. Given the high sequence identity and high degree of structural homology between HO1 and HO2, we expected that they would exhibit similar dynamic features. Furthermore, we predicted that, as with HO1, the C-terminal membrane anchor region of HO2 would not markedly affect the dynamics of heme binding. HDX-MS was used to probe the dynamics of HO2 in both the apo form and in forms with heme bound in the core at His45. The apo and heme-bound forms contained a disulfide bond between Cys265 and Cys282. We investigated three constructs: a C-terminally truncated HO2 that includes the N-terminal region and the catalytic core, spanning residues 1–248 (HO2(1–248)); a C-terminally truncated HO2, spanning residues 1–288 (HO2(1–288)), that includes the N-terminal region, the catalytic core, and the HRM region; and full-length HO2 (HO2(1–316)) (Fig. 3B). In all three HO2 constructs, the pattern of protection was as seen with HO1 (Fig. 3A): protection from exchange in helices A1 (proximal), A2, A5 (distal), and A6. Beyond these helices, however, there were several regions of HO2 that experienced protection from exchange upon heme binding that are specific for HO2 (i.e. not observed in HO1).
One of these specifically protected regions, observed in all three HO2 constructs, is helix A3, just after residue 100 (Fig. 3B). Helix A3 is quite distant from the heme-binding site and is separated from the distal helix by a small helical region composed of residues 185–187, a region of low sequence homology between HO2 and HO1. Any stabilization of the distal helix upon heme binding would likely need to be communicated through HO2 residues 185–187, or nearby residues, to helix A3. Unfortunately, these residues were not covered by any peptic peptides where deuterium levels could be measured (Figs. 3B and S1), so a firm conclusion cannot be drawn.
Weak protection in the heme-bound form of HO2 was observed at longer time points in helix A7 of all three HO2 constructs. Helix A7 includes Phe234, which along with Val54, Phe53, and Phe57, create a hydrophobic region within the heme-binding site that faces the α-meso edge of the heme, which is selectively targeted in the HO reaction (3). Protection around residue 265 was observed in HO2(1–288); this area is not part of the HO2(1–248) construct and, although the region is present in HO2(1–316), no information is available regarding its dynamics due to lack of peptides in this region. Cys265 (part of an HRM) can either form a disulfide bond with Cys282 (also part of an HRM) or ligate heme (12). The disulfide bond, however, is present in both apo and heme-bound forms of HO2(1–288) used in these experiments. Thus, protection would seem to be a function of heme binding to the core of the protein by an unknown mechanism.
Finally, specific protection around residue 290 was observed with HO2(1–316). Although coverage within the membrane anchor region of HO2 was poor, protection was observed for those residues that did have coverage. In contrast, protection was not observed for any residues in the membrane anchor of HO1 despite its better peptide coverage than for HO2 (Fig. 3A). The membrane anchor plays a key role in oligomerization of HO1 and HO2 (9, 14); therefore, the protection of residues in the membrane anchor of HO2, but not HO1, could possibly relate to the previous observation that HO2 oligomerizes to a greater extent than HO1 in vitro (9). Overall, these results show several differences in dynamics between HO1 and HO2 that were unexpected based on catalytic core tertiary structure and sequence analysis. However, it should be noted that heme-bound HO2 did not crystalize in the closed conformation observed with heme-bound HO1 (3), suggesting that the core of HO2, even in the heme-bound state, could be more dynamic than HO1.
The most surprising result for the HO2 HDX-MS experiments was the pattern of protection observed with full-length HO2 versus the C-terminally truncated proteins, HO2(1–288) and HO2(1–248) (Fig. 3B). Significantly more peptides exhibited protection in apo- versus heme-bound HO2(1–316), but the protection, particularly in the proximal helix, is mild. Nearly every region of the catalytic core of HO2(1–316) experienced some level of mild protection from exchange in the heme-bound state as compared with the apo form. Perhaps the most distinct region of protection observed only with HO2(1–316) was in helix A4. Interestingly, the HO2 sequence in this region has low homology with HO1 and includes Cys127-Pro128, which has been referred to as HRM3 of HO2 but has not been shown to bind heme. Helix A4 is at the dimer interface between the two molecules of HO2 in the X-ray crystal structure (3). Helix A7, also at the dimer interface, exhibited increased protection from exchange in HO2(1–316) relative to the truncated forms of HO2 upon heme binding. Contributions from heme binding at His45 cannot be separated from the contributions from formation of dimers or higher-ordered species in this particular experiment. In sum, the data illustrate that there is a substantial effect of the C-terminal membrane anchor on the dynamics of HO2 upon heme binding.
Heme binding to the HRMs does not affect HO2 dynamics when heme is bound to the core
The above experiments investigated the effect of heme binding to the core of HO1 and HO2 on protein dynamics, when the HRMs of HO2 are linked by a disulfide bond. Reduction of the disulfide bond introduces two additional heme-binding sites in HO2 (12) (HO1 lacks cysteine). We used HDX-MS to investigate whether heme binding to these sites affects HO2 dynamics. Deuterium incorporation was monitored for the two HO2 constructs spanning the HRM region (HO2(1–288) and HO2(1–316)) in two forms: one with heme bound at the core and the HRMs in a disulfide bond and the other with heme bound in both, at the core and the HRMs. No significant difference in deuterium exchange was observed between the two states in either the truncated or the full-length protein (Fig. S2). Thus, heme binding to the HRMs does not affect HO2 dynamics when the core contains heme.
Unlike HO1, full-length HO2 is more dynamic than the C-terminally truncated forms
To more fully investigate the dynamics of HO2 that are independent of the effects of heme binding, deuterium incorporation was monitored for the apo (Fig. 4) and heme-bound forms (with disulfide bond when HRMs present) of three HO2 constructs, HO2(1–248), HO2(1–288), and HO2(1–316) (Fig. S3). Comparing the apo and heme-bound forms of HO2(1–248) and HO2(1–288) (Figs. 4 and S3), only minor changes in deuterium uptake were observed upon the addition of the two HRMs to the C terminus. In contrast, the 28 amino acids from 289–316, which includes the C-terminal membrane anchor, had a large impact on the dynamics of HO2. The full-length construct, HO2(1–316), incorporated more deuterium than either of the shorter constructs, HO2(1–248) and HO2(1–288). The increase in deuterium incorporation is seen throughout many regions of HO2 at all time points, suggesting that HO2(1–316) in both the apo and heme-bound forms is significantly more dynamic than the C-terminally truncated forms of HO2.
Figure 4.

HDX-MS comparisons of the apo states of HO2 constructs with various lengths of C-terminal residues. A, the impact of residues 249–288 on the apo state of HO2. Deuterium differences were calculated from Dapo HO2(1–288) − Dapo HO2(1–248) and colored according to the scale shown. B, the impact of residues 249–316 on the apo state of HO2. Deuterium differences were calculated from Dapo HO2(1–316) − Dapo HO2(1–248) and colored according to the scale shown. C, the impact of residues 289–316 on the apo state of HO2. Deuterium differences were calculated from Dapo HO2(1–316) − Dapo HO2(1–288) and colored according to the scale shown. D, representative deuterium incorporation graphs for regions where differences were observed in A–C. All data used to create this figure can be found in Excel File S1.
However, in contrast, the C-terminal membrane anchor does not seem to impact the overall conformation or dynamics of apo-HO1 (Fig. 5). The only changes in dynamics experienced upon the addition of the last 23 C-terminal residues in HO1 was at a single time point (10 min) in the region of residues 65–80, which incorporated just slightly more deuterium in the full-length construct compared with the truncated form.
Figure 5.
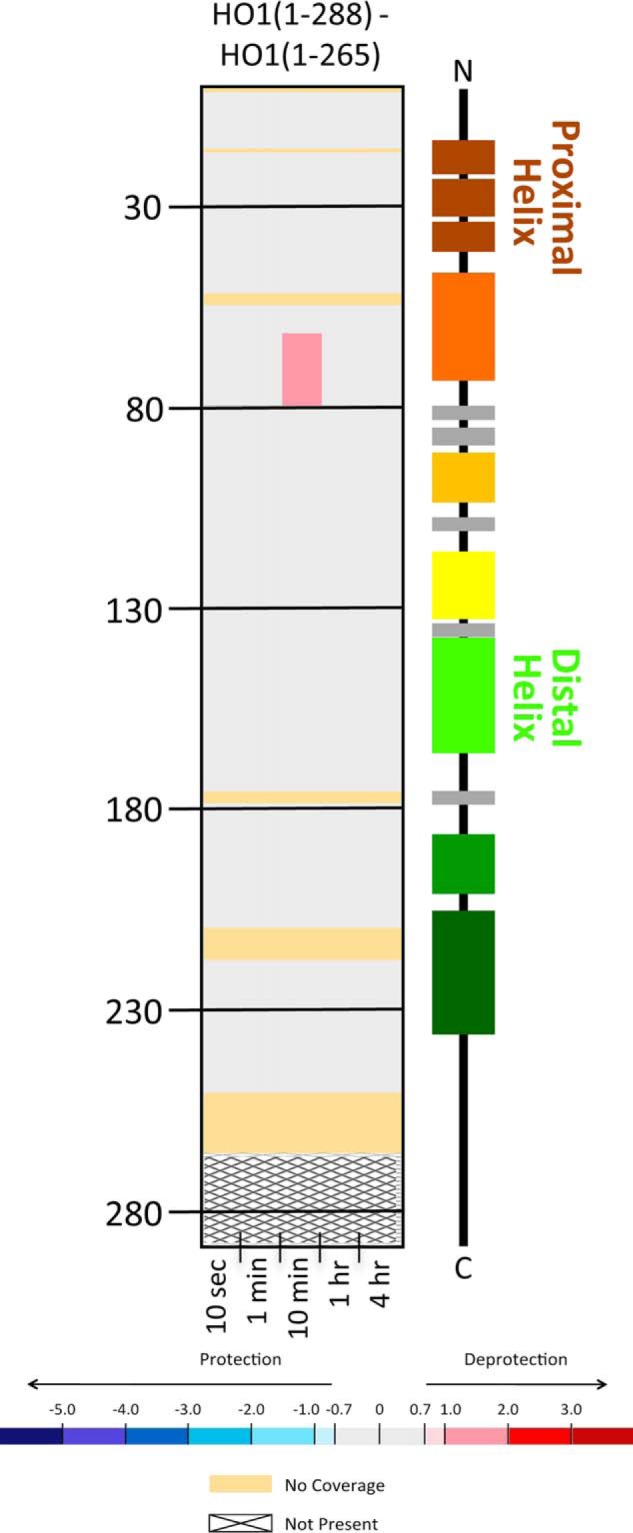
HDX-MS comparisons of the apo states of HO1 constructs with two lengths of C terminus. Deuterium differences were calculated from Dapo HO1(1–288) − Dapo HO2(1–265) and colored according to the scale shown. All data used to create this figure can be found in Excel File S1.
HO2(1–288), but not HO1(1–265), forms dimers in solution
To probe what role quaternary structure may play in the observed differences between HO2 and HO1, we probed the oligomeric state of the proteins by SEC and SV. The results indicate that regions of HO1 or HO2 outside the membrane anchor contribute to their oligomeric state (Figs. 6 and 7). Because the full-length forms of both HO1 and HO2 oligomerize and elute in the void volume of size-exclusion columns in the absence of membranes (Fig. 2) (9, 16), the C-terminally truncated forms were used in this study and were purified by nickel-nitrilotriacetic acid (Ni-NTA)–agarose affinity chromatography and incubated with heme prior to SEC. HO1(1–265) (with 30.3-kDa actual mass) eluted as a single peak corresponding to 46.1 kDa, which is presumably a monomer (Fig. 5), showing little propensity to form higher-ordered species. In contrast, the HO2(1–288) (33.0 kDa) elution profile included at least three peaks corresponding to 50.3 (52%), 74.4 (13%), and 95.9 kDa (35%). SV experiments with HO2(1–288) showed two species with a major peak at an apparent sedimentation coefficient of 2.6S (molecular mass ∼40 kDa) and a minor peak at 4.2S (molecular mass ∼79 kDa) (Fig. 7). The major and minor peaks were assigned as the monomer and dimer, respectively. The major peak in the SEC profile, corresponding to 50.3 kDa, was thus assigned as monomer. Other minor poorly resolved peaks are observed in the SEC elution profile, suggesting that HO2 undergoes reversible association (24, 25). The propensity of HO2 to form higher-ordered species was independent of heme binding, as the fraction of HO2(1–288) monomer in various states (apo, heme-bound at the core, and heme-bound at the core and HRMs) were similar (Fig. S4).
Figure 6.

HO1 and HO2 analysis by size-exclusion chromatography. 25 μm heme-bound protein was loaded onto a Superdex 75 Increase 10/300 GL column at 0.8 ml min−1 in 50 mm Tris buffer (pH 8.0) containing 50 mm KCl. Representative traces are shown in A with peaks indicated by the asterisk (*) being the monomer peak. The area of the monomer peak as a percentage of the total area of all peaks was quantified and plotted in B and C.
Figure 7.
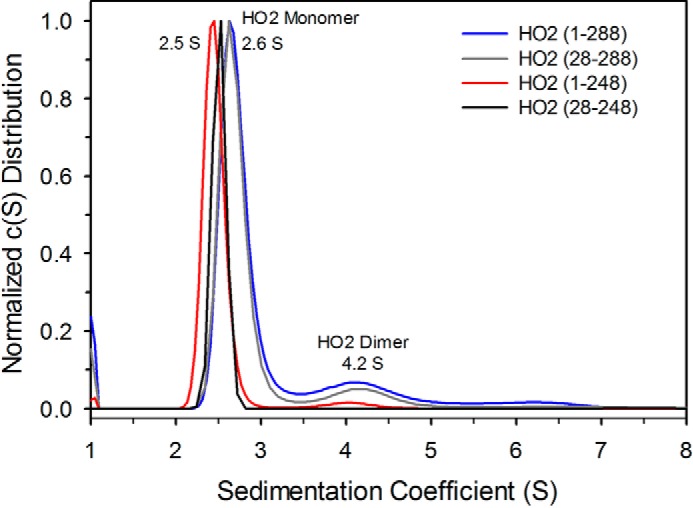
Sedimentation velocity analysis of HO2 proteins. Sedimentation coefficient distributions for HO2(1–288) (blue trace), HO2(28–288) (gray trace), HO2(1–248) (red trace), and HO2(28–248) (black trace) are shown. All the proteins show a major peak corresponding to a monomer (2.5S and 2.6S), whereas a minor peak (4.2S), assigned as a dimer, is observed only for HO2(1–288) and HO2(28–288).
The N-terminal region has little to no influence on the structure and dynamics of HO2
Because of the differential ability of HO2(1–288) and HO1(1–265) to dimerize, we probed the potential role of the N-terminal region, an extension found only on HO2, in dimerization. Based on NMR relaxation experiments, the N-terminal extension of HO2 exists in an unfolded, dynamic random coil (4). Successive, short N-terminal deletions of HO2(1–288) revealed a small increase in the fraction of protein in the monomeric state as the N-terminal region was shortened (Fig. 6B). SV also showed a slight decrease in the dimer peak for HO2(28–288) relative to HO2(1–288) (Fig. 7). However, HDX-MS experiments indicated that the N-terminal residues do not influence the dynamics of HO2 (Fig. S5). No difference in deuterium incorporation was observed upon comparing HO2(1–248) with HO2(28–248), HO2(1–288) with HO2(28–288), or HO2(1–316) with HO2(28–316). Furthermore, peptides in the region of residues 1–27 are close to their highest level of deuterium incorporation at the earliest time points (Fig. S5), consistent with the region being completely unstructured, which is in agreement with the prior NMR relaxation experiments (4). In apo-HO2(1–316), the N-terminal region did incorporate just slightly less deuterium than either apo-HO2(1–288) or apo-HO2(1–248) (Fig. 4), but the effect was very modest and likely involved only one or two backbone amide hydrogens.
The HRM-containing region influences the structure and dynamics of HO2
Besides the N-terminal extension, the other major structural difference between HO2 and HO1 is that HO2 contains two redox-active HRMs, which lie between the catalytic core and the membrane anchor. The HDX-MS experiments (above; Figs. 3 and 4) did not highlight any effect of this region on HO2 dynamics or structure. Therefore, SEC and SV experiments were performed to probe this region for a potential role in dimerization. Successive, short C-terminal deletions of HO2(1–288) revealed that there was not a significant change in the fraction of protein in the monomeric state as the C-terminal region was shortened; then there was an abrupt change when the protein was truncated to HO2(1–248) (Fig. 6C). HO2(1–248) and HO2 (28–248) do not exhibit a dimer peak in SV, further indicating a role for the C-terminal region in dimerization of HO2 (Fig. 7).
HDX-MS experiments were then designed to investigate the behavior of the HO2 HRM-containing region in the absence of the core region. A construct spanning residues 213–288 (HO2(213–288)), which includes helix A7 and the two C-terminal HRMs, has been characterized previously (12). As with HO2(1–288), the two HRMs of HO2(213–288) form a disulfide bond that, when reduced, presents two additional heme-binding sites. Furthermore, as demonstrated above for HO2(1–288) (Fig. S2), the disulfide bond–containing and the reduced, heme-bound forms of HO2(213–288) take up roughly the same levels of deuterium (Fig. S6). Therefore, we proceeded with our analysis using the disulfide bond–containing proteins.
When exchange data of apo-HO2(1–288) was compared with that of apo-HO2(213–288), the only region with a lower level of deuterium incorporation in the HO2(1–288) construct was peptide 213–240 (Fig. 8), which is part of helix A7 in the catalytic core. The HDX-MS data indicate that these residues have less deuterium uptake, perhaps from defined structure, only when they are part of HO2(1–288) containing the HO2 catalytic core and were lacking structure when expressed alone in HO2(213–288) (Fig. 8A and Excel File S1). Deuterium uptake (Excel File S1) for both constructs (1–288 and 213–288) shows that deuterium levels reach a high plateau at even the earliest time points for residues 213–288, and the profile lacks the characteristic gradual gain in deuterium incorporation observed in regions with α-helical or β-sheet structure. Because the amount of deuterium is so high, it is likely that these residues are highly dynamic, which is in agreement with prior NMR relaxation experiments (4). Thus, this region is not shielded from deuterium exchange by secondary structure or other areas of the protein.
Figure 8.

The C-terminal tail in HO2 remains unstructured in the presence of the catalytic core. HDX-MS comparisons of the apo states of HO2 constructs at various residue lengths are shown. A, the impact of residues 1–212 on the HO2(1–288) construct; the deuterium uptake of HO2(213–288) in the apo state was subtracted from the uptake of HO2(1–288) in the apo state. B, the impact of residues 1–212 in the apo state on the HO2(1–316) construct; the deuterium uptake of HO2(213–316) in the apo state was subtracted from the uptake of HO2(1–316) in the apo state. C, the impact of residues 289–316 in the apo state; the deuterium uptake of HO2(213–288) in the apo state was subtracted from the uptake of HO2(213–316) in the apo state.
The same pattern of little protection from deuterium exchange was seen when we labeled the full-length construct apo-HO2(1–316) and its counterpart C-terminal tail region (213–316) in the apo form: the HRM region (241–288) remains unstructured even upon addition of these extra 28 residues in the membrane-spanning region. However, the α-helical region (213–240) of the C-terminal tail construct was not as protected in HO2(1–316) as in HO2(1–288) (Fig. 8B), further indicating that, whereas both full-length and HO2(1–288) constructs promote residues 213–240 to form an α-helical structure, it is not to the same extent with HO2(1–316). There were few differences in dynamics between the apo forms of the two short constructs themselves: the peptide containing HRM2 (Cys282-Pro283) was slightly more protected from exchange in HO2(213–316) than in HO2(213–288) (Fig. 8C).
Discussion
Here, we probed full-length and truncated forms of HO1 and HO2 with HDX-MS, SEC, and SV to better understand and expand upon previous biophysical studies of HO1 and HO2 that have primarily utilized C-terminally truncated, soluble forms of the proteins. Our studies included, for the first time, medium-resolution conformational analyses of the full-length forms of both HO2 and HO1. The use of HDX-MS to study these proteins overcame the challenges (including solubility issues and the tendency to oligomerize) of studying full-length forms of these proteins, which normally associate with membranes. We asked how the N- and C-terminal regions, the regions in which HO1 and HO2 sequence differ the most, affect the structures and dynamics of the proteins. In HO2, the N- and C-terminal regions have similar lengths and were shown to be intrinsically disordered (3, 4). HDX-MS data confirm the disordered nature of these regions in that peptides in both the N- and C-terminal regions rapidly reach a high level of deuterium exchange, a hallmark for regions with little protection due to structure. However, the N- and C-terminal regions have very different impacts on conformation and dynamics of HO2. Based on HDX-MS data, the first 27 amino acids at the N terminus of HO2 do not influence dynamics or conformation of the HO2 catalytic core. In contrast, the very dynamic region containing the 28 most C-terminal residues completely alters protein dynamics within the HO2 core in solution. Full-length HO2 is significantly more dynamic than the truncated forms, including those for which there are crystal structures, incorporating significantly more deuterium in both the apo and heme-bound forms than the truncated constructs. Hence, the seemingly unstructured C-terminal residues surprisingly exert a mobilizing effect over the rest of the protein without decreasing the affinity of the protein for heme or the catalytic activity. As these experiments were performed on the full-length protein without it being anchored in a membrane (see also “Discussion” below), this deprotective effect observed in the HDX-MS experiments may be related to unnatural influences the C-terminal region exerts when the membrane is not present.
The unexpected ability of the membrane anchor region to enhance the dynamics on the entire HO2, but not HO1, is difficult to reconcile from a structural perspective. The fact that heme-bound HO1, but not HO2, was observed in a closed conformation by X-ray crystallography might suggest a difference in dynamics between the two proteins, but it is not clear how the membrane anchor would amplify those differences. One possibility is that differences in the quaternary structure of HO2(1–316) and the truncated forms may account for the observed difference in dynamics. HO2(1–316), but not HO2(1–288), was shown to oligomerize in previous gel-filtration studies when purified, suggesting that the membrane anchor plays a key role in oligomerization even in the absence of the membrane (Fig. 2) (9, 16). However, HO1(1–288) oligomerizes, whereas HO1(1–265) does not (9, 14), consistent with a previous study demonstrating the formation of HO1(1–288) oligomers in the ER using an in vivo FRET method (14). Although both full-length HO2 and HO1 can form oligomers, HO2 appears to form oligomers to a greater extent as compared with HO1. Hence, the fact that such a difference in dynamics was observed between the full-length and truncated forms of HO2, but not HO1, suggests that the results presented here could be at least partly related to differences in the oligomerization dynamics of these proteins.
The greater extent to which HO2 forms oligomers as compared with HO1 appears to be assisted by the involvement of regions outside of the membrane anchor in formation of higher-ordered species. As discussed above, the X-ray crystal structure of C-terminally truncated, heme-bound HO2 shows that helices A1, A4, and A7 are at the interface between the two molecules of HO2 in the asymmetric unit, which could potentially be a dimer interface. Indeed, these areas experience significant deprotection in the full-length apo form of HO2(1–316) compared with the truncated forms, suggesting that the strength of those interactions leads to differences in quaternary structure between the truncated and full-length constructs. Although our data cannot conclusively prove that the potential dimer interface observed in the X-ray crystal structure is relevant in vivo, the repeated observation of differential HDX suggests an important role for these helices. In addition, residues in the C-terminal tail of HO2 play a role in the dimerization of HO2. We observed an abrupt increase in the fraction of HO2 in the monomeric state as the protein was truncated from HO2(1–260) to HO2(1–248) by SEC. Note that the SEC and SV experiments were performed at concentrations (25–40 μm) ∼10–20 times higher than the HDX-MS experiments (2.2 μm). It is unclear whether the formation of oligomers persists at the concentrations used for HDX-MS.
An alternative explanation for the observed influences of the membrane anchor region on HO2 involves the membrane. Given that HO2 is a membrane-anchored protein, do its conformation and dynamics change when membrane is not present? One way to address this question would be to perform HDX-MS experiments for HO2 in the presence of membrane mimetics (e.g. liposomes) and compare with exchange of the protein free in solution. We have conducted preliminary studies in this regard, but a definitive conclusion cannot yet be made without a number of controls, lipid compositions, construct lengths, and analysis of HO1. It will be exciting to see just how the membrane participates, and we hope to report on those results in the future.
Although it is unclear from a structural perspective how the C-terminal residues of HO2 enhance the dynamics of full-length protein, the apparent differences in dynamics between HO1 and HO2 is interesting when asking why humans express both proteins. As more information becomes available regarding cellular heme trafficking (26), it may come to light that HO1 and HO2 receive heme from different sources, necessitating differences in structure and dynamics.
Also of note is that the kinetics of heme binding to the catalytic core of HO2(1–248) were recently shown to be more consistent with a conformational selection model than the more commonly observed two-step mechanism of heme binding (27), possibly distinguishing HO2 from other heme-binding proteins. Whether or not this distinguishes HO2 from HO1, however, has yet to be tested. Regardless, in combination with the dynamic nature of HO2, particularly in its full-length form, we hypothesize that a possible role for HO2 not just in heme degradation but also in heme sensing/trafficking will emerge.
In conclusion, the biophysical characterization of HO1 and HO2 presented here demonstrates key differences between the proteins. Specifically, HO1 and HO2 differ in their dynamics and ability to self-associate. Despite the high similarity between the core regions in which the proteins catalyze the degradation of heme, the dissimilar C-terminal regions of HO1 and HO2 play major roles in controlling structural dynamics, distinguishing these two proteins.
Experimental procedures
Protein expression and purification
The human full-length HO1 and HO2 cDNAs in a pGEX-4T-2 vector were generous gifts from Dr. Paul Ortiz de Montellano (University of California, San Francisco, CA) and Dr. Mahin D. Maines (University of Rochester Medical Center, Rochester, NY), respectively. The R254K mutation was introduced into the HO1-containing vector using the QuikChange protocol (Agilent Technologies) to enhance the stability of the full-length protein (15). HO2(28–316) and HO2(213–316) were constructed using a Q5 Site-Directed Mutagenesis kit (New England Biolabs) to delete the indicated regions from the full-length HO2/pGEX-4T-2 vector. HO2(1–316), HO2(28–316), HO2(213–316), and the R254K variant of HO1(1–288) were expressed from Escherichia coli C41 cells (Lucigen) transformed with the appropriate plasmid and subsequently purified as described previously (15). Briefly, the GSH S-transferase (GST)-tagged proteins were released from the membrane with the addition of 1% N-lauroylsarcosine (Sigma-Aldrich) and bound to GSH-Sepharose 4B (GE Healthcare). After thrombin was added to cleave the affinity tag, smaller-molecular-weight species not containing the C-terminal membrane anchor regions were removed by washing, and then the correct molecular weight proteins were eluted with PBS, pH 7.4, containing 20% glycerol and 2% n-octyl β-d-glucoside (Thermo Fisher Scientific).
HO2(1–288) and HO2(1–248) were expressed from the pET28a vector in BL21(DE) cells (Life Technologies) and purified by Ni-NTA–agarose affinity chromatography (Qiagen) as described previously (10). The N-terminal His6 tag was removed by treatment with thrombin and a second Ni-NTA–agarose chromatography step as described previously (10) for HO2(1–248) only. The treatment of HO2(1–288) with thrombin resulted in the formation of some smaller species that complicated HDX-MS analysis (data not shown), so the affinity tag was left intact for those studies. HO2(28–248) was cloned into pET28a as described for HO2(1–288) previously (10). HO2(28–248) was expressed and purified as described above for HO2(1–248), including the removal of the affinity tag by thrombin cleavage.
HO1(1–265) was cloned by ligation-independent cloning (28) into the pMCSG10 (29) vector and purified by Ni-NTA–agarose affinity chromatography (Qiagen) as described previously (21). The N-terminal affinity tag was removed by treatment with tobacco etch virus (TEV) protease and a second Ni-NTA–agarose chromatography step as described previously (21). Likewise, HO2(1–288) and HO2(28–288) were cloned into the same vector and purified in the same way as HO1(1–265) with removal of the affinity tag by treatment with TEV protease. The HO2(1–288)/pMCSG10 vector was used to create HO2(5–288), HO2(10–288), HO2(15–288), HO2(23–288), HO2(1–260), HO2(1–270), and HO2(1–280) with the Q5 Site-Directed Mutagenesis kit to delete the indicated regions. The truncated proteins were purified in the same way as HO1(1–265) with removal of the affinity tag by treatment with TEV protease.
All proteins were buffer-exchanged into 50 mm Tris buffer (pH 8.0) containing 50 mm KCl before use in subsequent experiments by repeated concentration and dilution in 10-kDa molecular-mass-cutoff Microcon centrifugal filter devices (Millipore). Fe3+-heme was prepared by dissolving hemin in 15% DMSO and 0.1 m sodium hydroxide in 50 mm Tris buffer (pH 8.0) containing 50 mm KCl and passing the solution through a 22-μm filter. The heme concentration was determined using an ϵ385 of 58.4 mm−1 cm−1 (30). For heme-bound protein samples, as-purified proteins were incubated with 1.25 molar eq of heme from freshly prepared Fe3+-heme for 1 h on ice. The protein–heme mixture was passed through a PD-10 desalting column (GE Healthcare) as described previously (12). Unless indicated, for HO2 constructs that spanned the HRM-containing region, the as-purified protein included a disulfide bond between Cys265 and Cys282, preventing heme from binding to this region of the protein. Protein samples in which the disulfide bond was reduced were treated with TCEP in an anaerobic chamber prior to removal of TCEP and subsequent incubation with heme as described previously. Protein concentrations were determined using either Bradford reagent or, for proteins with the membrane anchor, bicinchoninic acid assay (Life Technologies) using BSA as a standard. Heme-bound spectra were collected in 50 mm Tris (pH 8.0), 50 mm KCl as indicated. Free thiol concentrations were measured by a modified DTNB assay (27, 31) in which 50 μl of 5 mm DTNB was added to 950 μl of 6 m guanidine hydrochloride in 100 mm Tris (pH 8.0). 100 μl of sample was added, and the mixture was incubated at room temperature for 15 min before spectra were recorded. DTT was used to standardize the assay.
Heme off-rate measurements
Heme off-rates were measured as described previously (12) with minor modifications using apo-H64Y/V68F-myoglobin (green heme) (17) generously supplied by J. Olson (Rice University, Houston, TX). Briefly, 30 μm apo-H64Y/V68F-myoglobin was mixed with 3 μm heme-bound HO (prepared as described above) in 50 mm Tris (pH 8.0), 50 mm KCl at 20 °C. Absorbance at 600 nm was recorded over time in a Shimadzu UV-2600 UV-visible spectrophotometer. Data were fit to a double-exponential function using GraphPad Prism 7.
Heme oxygenase activity assay
Steady-state activity of HO was measured as described previously (21). Briefly, a 200-μl reaction containing 0.1 μm HO, 15 μm heme, 0.35 μm biliverdin reductase, 0.25 μg/μl BSA, 20 units of catalase, and varying concentrations of truncated CPR in 50 mm Tris (pH 8.0), 50 mm KCl was incubated for 2 min at 37 °C. The reaction was initiated by the addition of 1 μl of 100 mm NADPH. Activity was monitored by following the increase in absorbance at 468 nm due to bilirubin formation. Using a difference extinction coefficient of 48 mm−1 cm−1, activity was calculated as nmol of bilirubin formed/min/mg of HO. Data were fit to a Michaelis–Menten function using GraphPad Prism 7.
CPR used in the assay was a truncated, soluble form lacking the N-terminal amino acids (residues 1–66) that act as the membrane anchor. The protein was purified as described previously with an N-terminal His6 tag (21). Biliverdin reductase was purified as described previously with an N-terminal GSH S-transferase affinity tag (21). Affinity tags were cleaved and separated from the respective proteins before use in the assay, also as described previously (21).
HDX-MS
Labeling was initiated by diluting 3 μl of HO2 or HO1 at 33.3 μm (99.9 pmol in 50 mm Tris and 50 mm KCl at pH 8.0) 15-fold in an identical buffer containing 99% deuterium oxide at room temperature. HDX-MS was performed using the same experimental method for all constructs in the heme-bound and apo forms, regardless of construct length, allowing direct comparisons to be made. For short time-point experiments (5 s to 1 min), labeling was carried out in triplicate. For the longer time-point experiments (10 min to 4 h), labeling was carried out in duplicate or triplicate. The labeling reaction was quenched at seven time points (5 s, 10 s, 20 s, 1 min, 10 min, 1 h, and 4 h) by lowering the pH to 2.5 and the temperature to 0 °C upon the addition of 30 μl of quench buffer (150 mm NaH2PO4, 100 mm tris(2-carboxyethyl)phosphine, (pH 2.19)). Some HDX-MS comparisons were only carried out at five time points (10 s, 1 min, 10 min, 1 h, and 4 h). Once the reactions were quenched, the samples were immediately injected into a Waters nanoACQUITY UPLC equipped with HDX technology (32). The samples were digested using an online pepsin column at 15 °C, and the resulting peptides were trapped with a VanGuard BEH C18 1.7-μm guard column. The peptides were separated using a 5–85% acetonitrile gradient over 10 min at a flow rate of 60 μl/min over a Waters ACQUITY UPLC HSS T3 1.8-μm 1.0 × 50-mm analytical column. Both the acetonitrile and water mobile phases contained 0.1% formic acid. After every three injections of a sample, a wash cycle was performed to minimize carryover. Mass spectral analyses were carried out using a Waters Synapt G2-si with a standard electrospray ionization source and ion mobility. Peptides were identified from undeuterated controls using Waters MSE and Waters ProteinLynx Global Server (PLGS) 3.0. DynamX 3.0 was used to filter peptides in the generation of peptide maps and deuterium incorporation graphs. Parameters used to filter peptides were: 0.2 product/amino acid, two consecutive products, and an error of 10 ppm. Relative deuterium incorporation was calculated by subtracting the centroid of the isotopic distribution of a labeling experiment by the centroid of the isotopic distribution of the undeuterated control. Back-exchange was not calculated for these experiments as all experiments that were compared with one another were performed under the same experimental conditions; results are therefore reported as relative deuterium level (33). Differences in deuterium level equal to or greater than 0.7 Da between two states were considered meaningful (34). For chicletograms, a linear representation of peptides was used; data for every peptide that was able to be followed via HDX-MS can be found in Excel File S1.
Size-exclusion chromatography
For the HO2(1–316) sample, 150 μl of 10 μm heme-bound HO2(1–316) in 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl and 0.1 mm EDTA was used. Sample was run at 1 ml min−1 in the same buffer using a Shodex KW-803 column on a Shimadzu HPLC with an LC-10AT pump system. The column was calibrated with gel-filtration standards (Bio-Rad), and the void volume was determined using blue dextran (2000 kDa; Sigma).
For truncated HO samples, 200 μl of 25 μm heme-bound protein was loaded onto a Superdex 75 Increase 10/300 GL column attached to an ÄKTA Pure chromatography system (GE Healthcare). Samples were run at 0.8 ml min−1 in 50 mm Tris buffer (pH 8.0) containing 50 mm KCl. The column was calibrated using gel-filtration standards (BSA, cytochrome c from equine heart, and carbonic anhydrase from bovine erythrocytes) from Sigma-Aldrich.
Sedimentation velocity experiments
SV experiments were performed at 20 °C in an Optima XL-I analytical centrifuge (Beckman Coulter, Inc.) using an An50Ti rotor as described previously (21, 35). Heme-bound proteins samples were analyzed at 1 mg ml−1 in 50 mm Tris buffer (pH 8.0) containing 50 mm KCl (30–40 μm, dependent on the length of the HO2 construct). Protein samples (400 μl) and reference buffer were loaded into a double-sector cell. The samples were equilibrated at 20 °C for 2 h prior to centrifugation. Samples were then centrifuged at 40,000 rpm, and a total of 240 absorbance (425 and 450 nm) scans were recorded at 4-min intervals. SEDFIT version 10.58d (22) was used for analyzing the data with the continuous c(s) distribution model to generate apparent c(s) distributions for each sample.
Author contributions
B. A. K., A. S. F., T. E. W., J. R. E., and S. W. R. formal analysis; B. A. K. and A. S. F. validation; B. A. K., A. S. F., and D. F. B. investigation; B. A. K., A. S. F., T. E. W., D. F. B., and S. W. R. methodology; B. A. K., A. S. F., J. R. E., and S. W. R. writing-original draft; B. A. K., A. S. F., D. F. B., J. R. E., and S. W. R. writing-review and editing; A. S. F., J. R. E., and S. W. R. conceptualization; A. S. F. and J. R. E. visualization; T. E. W. and S. W. R. data curation; J. R. E. resources; J. R. E. and S. W. R. project administration; S. W. R. supervision; S. W. R. funding acquisition.
Supplementary Material
This work was supported by National Institutes of Health Grants R01-GM123513 (to S. W. R.) and P30GM103335 and GM061068 (to D. F. B.) and a research collaboration with Waters Corp. (to J. R. E.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Excel File S1.
- CPR
- cytochrome P450 reductase
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic acid)
- ER
- endoplasmic reticulum
- HDX
- hydrogen–deuterium exchange
- HO
- heme oxygenase
- HRM
- heme regulatory motif
- Ni-NTA
- nickel-nitrilotriacetic acid
- SEC
- size-exclusion chromatography
- SV
- sedimentation velocity
- TEV
- tobacco etch virus
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Maines M. D., Trakshel G. M., and Kutty R. K. (1986) Characterization of two constitutive forms of rat-liver microsomal heme oxygenase: only one molecular-species of the enzyme is inducible. J. Biol. Chem. 261, 411–419 [PubMed] [Google Scholar]
- 2. Schuller D. J., Wilks A., Ortiz de Montellano P. R., and Poulos T. L. (1999) Crystal structure of human heme oxygenase-1. Nat. Struct. Biol. 6, 860–867 10.1038/12319 [DOI] [PubMed] [Google Scholar]
- 3. Bianchetti C. M., Yi L., Ragsdale S. W., and Phillips G. N. Jr. (2007) Comparison of apo- and heme-bound crystal structures of a truncated human heme oxygenase-2. J. Biol. Chem. 282, 37624–37631 10.1074/jbc.M707396200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bagai I., Sarangi R., Fleischhacker A. S., Sharma A., Hoffman B. M., Zuiderweg E. R., and Ragsdale S. W. (2015) Spectroscopic studies reveal that the heme regulatory motifs of heme oxygenase-2 are dynamically disordered and exhibit redox-dependent interaction with heme. Biochemistry 54, 2693–2708 10.1021/bi501489r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dwyer B. E., Nishimura R. N., De Vellis J., and Yoshida T. (1992) Heme oxygenase is a heat-shock protein and PEST protein in rat astroglial cells. Glia 5, 300–305 10.1002/glia.440050407 [DOI] [PubMed] [Google Scholar]
- 6. Boname J. M., Bloor S., Wandel M. P., Nathan J. A., Antrobus R., Dingwell K. S., Thurston T. L., Smith D. L., Smith J. C., Randow F., and Lehner P. J. (2014) Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J. Cell Biol. 205, 847–862 10.1083/jcb.201312009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hsu F. F., Yeh C. T., Sun Y. J., Chiang M. T., Lan W. M., Li F. A., Lee W. H., and Chau L. Y. (2015) Signal peptide peptidase-mediated nuclear localization of heme oxygenase-1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene 34, 2360–2370 10.1038/onc.2014.166 [DOI] [PubMed] [Google Scholar]
- 8. Schaefer B., Moriishi K., and Behrends S. (2017) Insights into the mechanism of isoenzyme-specific signal peptide peptidase-mediated translocation of heme oxygenase. PLoS One 12, e0188344 10.1371/journal.pone.0188344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Linnenbaum M., Busker M., Kraehling J. R., and Behrends S. (2012) Heme oxygenase isoforms differ in their subcellular trafficking during hypoxia and are differentially modulated by cytochrome P450 reductase. PLoS One 7, e35483 10.1371/journal.pone.0035483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yi L., and Ragsdale S. W. (2007) Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J. Biol. Chem. 282, 21056–21067 10.1074/jbc.M700664200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yi L., Jenkins P. M., Leichert L. I., Jakob U., Martens J. R., and Ragsdale S. W. (2009) Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J. Biol. Chem. 284, 20556–20561 10.1074/jbc.M109.015651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fleischhacker A. S., Sharma A., Choi M., Spencer A. M., Bagai I., Hoffman B. M., and Ragsdale S. W. (2015) The C-terminal heme regulatory motifs of heme oxygenase-2 are redox-regulated heme binding sites. Biochemistry 54, 2709–2718 10.1021/acs.biochem.5b00266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fleischhacker A. S., Carter E. L., and Ragsdale S. W. (2018) Redox regulation of heme oxygenase-2 and the transcription factor, Rev-Erb, through heme regulatory motifs. Antioxid. Redox Signal. 29, 1841–1857 10.1089/ars.2017.7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hwang H. W., Lee J. R., Chou K. Y., Suen C. S., Hwang M. J., Chen C., Shieh R. C., and Chau L. Y. (2009) Oligomerization is crucial for the stability and function of heme oxygenase-1 in the endoplasmic reticulum. J. Biol. Chem. 284, 22672–22679 10.1074/jbc.M109.028001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huber W. J. 3rd, and Backes W. L. (2007) Expression and characterization of full-length human heme oxygenase-1: the presence of intact membrane-binding region leads to increased binding affinity for NADPH cytochrome P450 reductase. Biochemistry 46, 12212–12219 10.1021/bi701496z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huber W. J. 3rd, Scruggs B. A., and Backes W. L. (2009) C-terminal membrane spanning region of human heme oxygenase-1 mediates a time-dependent complex formation with cytochrome P450 reductase. Biochemistry 48, 190–197 10.1021/bi801912z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hargrove M. S., Singleton E. W., Quillin M. L., Ortiz L. A., Phillips G. N., Olson J. S., and Mathews A. J. (1994) His64(E7) → Tyr apomyoglobin as a reagent for measuring rates of hemin dissociation. J. Biol. Chem. 269, 4207–4214 [DOI] [PubMed] [Google Scholar]
- 18. Hargrove M. S., Barrick D., and Olson J. S. (1996) The association rate constant for heme binding to globin is independent of protein structure. Biochemistry 35, 11293–11299 10.1021/bi960371l [DOI] [PubMed] [Google Scholar]
- 19. Carter E. L., Gupta N., and Ragsdale S. W. (2016) High affinity heme binding to a heme regulatory motif on the nuclear receptor Rev-erbβ leads to its degradation and indirectly regulates its interaction with nuclear receptor corepressor. J. Biol. Chem. 291, 2196–2222 10.1074/jbc.M115.670281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Owens C. P., Du J., Dawson J. H., and Goulding C. W. (2012) Characterization of heme ligation properties of Rv0203, a secreted heme binding protein involved in Mycobacterium tuberculosis heme uptake. Biochemistry 51, 1518–1531 10.1021/bi2018305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spencer A. L., Bagai I., Becker D. F., Zuiderweg E. R., and Ragsdale S. W. (2014) Protein/protein interactions in the mammalian heme degradation pathway: heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. J. Biol. Chem. 289, 29836–29858 10.1074/jbc.M114.582783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lad L., Schuller D. J., Shimizu H., Friedman J., Li H., Ortiz de Montellano P. R., and Poulos T. L. (2003) Comparison of the heme-free and -bound crystal structures of human heme oxygenase-1. J. Biol. Chem. 278, 7834–7843 10.1074/jbc.M211450200 [DOI] [PubMed] [Google Scholar]
- 24. Lou X., Zhu Q., Lei Z., van Dongen J. L., and Meijer E. W. (2004) Simulation of size exclusion chromatography for characterization of supramolecular complex: a theoretical study. J. Chromatogr. A 1029, 67–75 10.1016/j.chroma.2003.12.023 [DOI] [PubMed] [Google Scholar]
- 25. Yu C. M., Mun S., and Wang N. H. (2006) Theoretical analysis of the effects of reversible dimerization in size exclusion chromatography. J. Chromatogr. A 1132, 99–108 10.1016/j.chroma.2006.07.017 [DOI] [PubMed] [Google Scholar]
- 26. Reddi A. R., and Hamza I. (2016) Heme mobilization in animals: a metallolipid's journey. Acc. Chem. Res. 49, 1104–1110 10.1021/acs.accounts.5b00553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carter E. L., Ramirez Y., and Ragsdale S. W. (2017) The heme-regulatory motif of nuclear receptor Rev-erbβ is a key mediator of heme and redox signaling in circadian rhythm maintenance and metabolism. J. Biol. Chem. 292, 11280–11299 10.1074/jbc.M117.783118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aslanidis C., and de Jong P. J. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 10.1093/nar/18.20.6069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eschenfeldt W. H., Lucy S., Millard C. S., Joachimiak A., and Mark I. D. (2009) A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498, 105–115 10.1007/978-1-59745-196-3_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dawson R. M., Elliott D. C., Elliott W. H., and Jones K. M. (eds) (1969) Data for Biochemical Research, 2nd Ed., pp. 316–317, Oxford University Press, Oxford, UK [Google Scholar]
- 31. Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 10.1016/0003-9861(59)90090-6 [DOI] [PubMed] [Google Scholar]
- 32. Wales T. E., Fadgen K. E., Gerhardt G. C., and Engen J. R. (2008) High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal. Chem. 80, 6815–6820 10.1021/ac8008862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wales T. E., and Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 10.1002/mas.20064 [DOI] [PubMed] [Google Scholar]
- 34. Houde D., Berkowitz S. A., and Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 10.1002/jps.22432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dam J., Velikovsky C. A., Mariuzza R. A., Urbanke C., and Schuck P. (2005) Sedimentation velocity analysis of heterogeneous protein-protein interactions: Lamm equation modeling and sedimentation coefficient distributions c(s). Biophys. J. 89, 619–634 10.1529/biophysj.105.059568 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.