

ABSTRACT
The double-stranded telomeric binding protein TRF2 is expressed in many human cancers at elevated levels. Moreover, experimental overexpression of TRF2 in human cells causes replication stalling in telomeric tracts, which leads to drastic telomere shortening and fusion of deprotected chromosome ends. To understand which end joining pathway is involved in mediating these chromosome fusions, we overexpressed TRF2 in human HCT116 cell lines that were deficient for the DNA Ligase 4 (Lig4)-dependent classical non-homologous end joining (C-NHEJ) or the DNA Ligase 3 (Lig3)-dependent alternative non-homologous end joining (A-NHEJ) pathway. Surprisingly, abrogation of either Lig4 or nuclear Lig3 significantly reduced inter-chromosomal fusion of drastically shortened telomeres, suggesting that both the C-NHEJ and A-NHEJ pathways are involved in mediating this type of fusion. Fusion between deprotected sister chromatids, however, only required the Lig3-dependent A-NHEJ pathway. Interestingly, a previous study reported similar end joining pathway requirements for the fusion of critically shortened telomeres during a telomere attrition-based cellular crisis. We speculate that, as in cellular crisis, the same repair pathway(s) may drive clonal and genomic evolution in human cancers containing elevated TRF2 levels.
KEYWORDS: TRF2 overexpression, drastic telomere shortening, inter-chromosomal fusions, sister chromatid fusions, Lig3-dependent A-NHEJ, Lig4-dependent C-NHEJ
Introduction
Mammalian telomeres consist of long stretches of TTAGGG DNA repeats that terminate in a short 3′ single-stranded overhang. The reverse transcriptase telomerase extends telomeres by directing the addition of new telomeric sequences to this overhang [1]. In human somatic cells, where telomerase activity is very low, telomeres progressively shorten during successive S phases due to the end replication problem [2]. Critically short telomeres will then trigger a DNA damage response [3], which normally signals cell cycle arrest or senescence [4]. In checkpoint-deficient cells, however, the deprotected chromosome ends are repaired by NHEJ, causing end-to-end fusions, which, in turn, initiate genome instability [5,6].
Telomeric DNAs are bound by the shelterins, a six-subunit protein complex that plays a major role in protecting the integrity of chromosome ends [3]. Within the shelterin complex, the TPP1-POT1 heterodimer binds the single-stranded telomeric overhang [7,8], while TRF1 and TRF2 bind the duplex telomeric DNA [9,10] and recruit TIN2, TPP1, POT1 and Rap1 along the telomere tract through protein-protein interactions [11–16]. When TRF2 or TPP1-POT1 are depleted, the uncapped telomeres activate a DNA damage response and are repaired by NHEJ pathways [5,17,18].
NHEJ consists of two genetically distinct pathways: Lig4-dependent C-NHEJ and Lig3-dependent A-NHEJ [19,20]. C-NHEJ is thought to mediate the fast repair of double-stranded breaks and does not involve extensive resection at the break point. In contrast, A-NHEJ is generally associated with slower repair that involves deletions of longer stretches of DNA and at least 3 nucleotides of microhomology between the fusion molecules at the break point [21]. Importantly, distinct end joining pathways are utilized to process different types of deprotected telomeres: experimental removal of TRF2 from telomeres elicits C-NHEJ-mediated chromosome fusions [17,18], while telomeres devoid of TPP1-POT1 proteins are fused via the A-NHEJ pathway [5,6,22]. Adding complexity to these observations is the fact that the fusion of critically shortened telomeres at a telomere-driven replicative crisis requires the action of both C-NHEJ and A-NHEJ pathways [23].
While a deficiency of TRF2 causes chromosome fusion, somewhat paradoxically, so does a superabundance of TRF2. Specifically, we have demonstrated that TRF2 overexpression, to physiologically relevant levels that are seen in a subset of human cancers, caused telomere replication stalling and the formation of telomeric ultrafine anaphase bridges (UFBs) [24]. Resolution of these telomeric UFBs led to drastic telomere shortening and increased end-to-end fusions. The vast majority of TRF2 overexpression-induced chromosome fusions possessed features that were characteristic of the A-NHEJ repair process: the fusions were accompanied by large deletions that extended into the adjacent sub-telomeric region; also, 1–4 nucleotides of microhomology were frequently observed between the fused molecules at the break point. While these observations were suggestive of the use of A-NHEJ, they were only correlative. To unequivocally address this issue, we have now genetically determined which NHEJ pathway is involved in mediating chromosome fusions elicited by elevated levels of TRF2. To accomplish this goal, TRF2 was overexpressed in human HCT116 colon cancer cell lines in which either the C-NHEJ or A-NHEJ pathway was inactivated by genetically deleting Lig4 or nuclear Lig3, respectively [25,26]. Unexpectedly, we demonstrate that the deprotected chromosome ends generated by too much TRF2 engaged both Lig4-dependent C-NHEJ and Lig3-dependent A-NHEJ to form inter-chromosomal fusions. Sister chromatid fusions, however, were facilitated solely by Lig3-dependent A-NHEJ. Thus, this study provides clinically-relevant insight into the mechanism(s) of genomic instability caused by TRF2 overexpression and it expands our understanding of the basic knowledge of NHEJ pathway choice selection at dysfunctional telomeres in human somatic cells.
Materials and methods
Cells
Human HCT116 colorectal cancer Lig4−/− and Lig3−/−:mL3 (nuclear Lig3-null cells complemented with a mitochondrial-specific Lig3) derivative cell lines were generated as described [25,26]. All cell lines were grown in DMEM supplemented with 10% fetal bovine serum, and continuously passaged to maintain exponential growth.
Lentiviral plasmids
The pHR’CMV lentiviral expression vector system was kindly provided by Dr. Didier Trono. The TRF2 expression lentiviral vector contains the full-length, untagged, wild-type TRF2 cDNA driven by a CMV promoter, followed by an internal ribosome entry site and a hygromycin resistance gene. The telomerase expression lentiviral vector contains a human telomerase catalytic subunit (hTERT) cDNA driven by a CMV promoter, followed by an internal ribosome entry site and a hygromycin resistance gene, and a telomerase RNA (hTR) cDNA driven by an IU1 promoter [27]. Lentivirus preparation and lentiviral infection were performed as described previously [24,28].
Telomere Restriction Fragment length analysis
5–10 μg of genomic DNA digested with Hinf I and Rsa I was fractionated by 0.6% agarose-TBE gel electrophoresis and transferred to a charged nylon membrane (Hybond XL, GE). Southern blotting was carried out with an end-labeled telomeric probe (C3TA2)4. Blots were then analyzed by the ImageQuant software. Mean telomere lengths were calculated according to the positions of molecular weight markers run on the same gel.
Immunoblotting analysis
Whole cell extracts were resolved with 10% SDS-PAGE and transferred to a PVDG nitrocellulose membrane. Immunoblots were incubated with a mouse monoclonal anti-TRF2 (BD Transduction Laboratories), followed by a horseradish peroxidase-conjugated donkey anti-mouse IgG (Jackson ImmunResearch). ECL Prime reagent (GE Healthcare) was used for signal detection. The same blot was then stripped and re-probed with a mouse monoclonal anti-tubulin antibody (Sigma-Aldrich), which served as a loading control.
Metaphase fluorescence in situ hybridization
Metaphase spreading and fluorescence in situ hybridization was performed as described [29], using Alexa488-OO-5′-(CCCTAA)3-3′ (telomeric sequence) and TMR-OO-5′-CTTCGTTGGAAACGGGA-3′ (centromeric sequence) PNA probes (Panagene). Images were acquired with a Nikon Ti-U microscope using a 60x objective. All image files were mixed and randomly assigned coded names to allow blinded scoring for chromosome fusions, signal-free ends and fragile telomeres.
Results
TRF2 overexpression causes drastic telomere shortening
TRF2 overexpression in human cells causes persistent replication stalling in duplex telomeric repeat tracts, which ultimately leads to stochastic deletion of large chunks of telomeric sequences and the formation of end-to-end fusions [24]. Both cytogenetic evidence and sequencing analyses showed that the vast majority of the fused chromosomes do not contain telomeric sequences at the fusion junction. This feature is conspicuously different from that observed in TRF2 depletion-induced C-NHEJ-mediated fusions, where long tracts of telomeric repeats are often preserved on either side of the fusion point [17,18,30]. To determine which NHEJ pathway is responsible for generating chromosome fusions in cells overexpressing TRF2, we overexpressed TRF2 in HCT116 cells genetically engineered to be deficient for either the C-NHEJ (Lig4−/−) [26] or A-NHEJ (Lig3−/−:mL3; a mitochondrial form of Lig3 is supplemented to the cell line to rescue the lethality of a Lig3 knock-out) [25] pathway and examined telomere length and chromosome morphology. As a negative control, the same cell lines overexpressing GFP were analyzed side-by-each for all subsequent experiments.
The mean bulk telomere length of the HCT116 parental cells was ~5kb, whereas that of Lig4−/− and Lig3−/−:mL3 were only ~2.5kb (Figure 1(a)). Since telomere length is a heterogeneous trait, this difference in mean telomere length was likely due to the stochastic nature of the single-cell sub-cloning required to obtain the cell lines and not biologically significant. The extent of TRF2 overexpression-induced telomere shortening, however, is known to closely correlates with mean telomere length [24]. Therefore, we extended the telomeres in these cell lines to similar lengths. This was accomplished by infecting the cells with a lentiviral vector expressing both the telomerase catalytic and RNA subunit (hTERT+hTR) for ~20 days until mean telomere lengths stabilized at an equivalent length greater than 10kb for all three cell lines (Figure 1(a)). We then overexpressed full-length, untagged wild-type TRF2 to comparable levels in these HCT116 cell lines containing pre-extended telomeres (Figure 1(b)). Cells expressing GFP were used as control. As shown in Figure 1(c), all cell lines were collected for further analysis after they underwent similar number of cell divisions (~8 days since the overexpression of TRF2). As expected, TRF2 overexpression induced stochastic telomere shortening, changing the distribution of bulk telomeres from a tight cluster to a wide smear in all three cell lines (Figure 1(d)).
Figure 1.
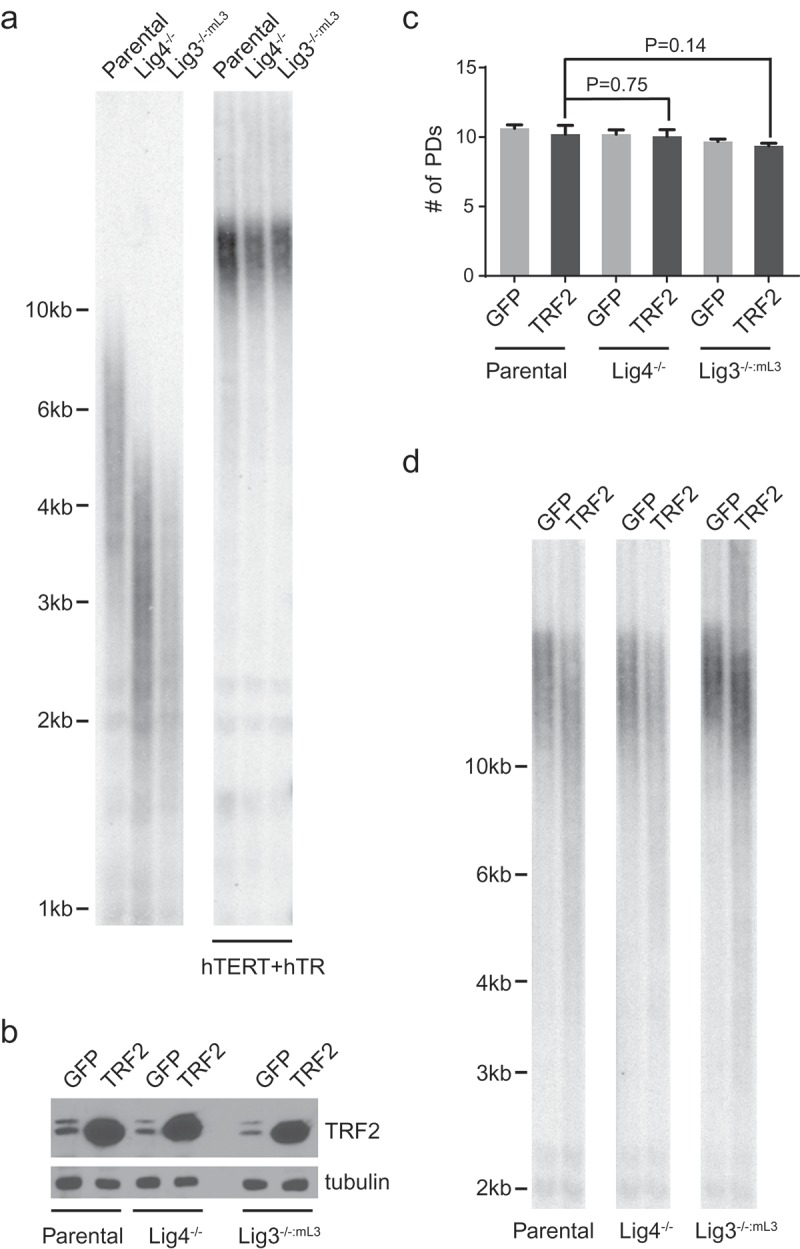
Overexpression of TRF2 causes drastic telomere shortening. (a) Left: Telomere Restriction Fragment (TRF) analysis of the indicated cell lines was performed using a telomeric repeat probe. Right: Telomeres of the respective HCT116 cells were pre-extended to comparable lengths by expression of telomerase (hTERT+hTR). Cells of indicated genotype were infected with lentiviruses expressing hTERT+hTR. Afterwards, the infected cells were pooled and continuously passaged for ~20 days. TRF analysis was then performed using a telomeric repeat probe. (b) Immunoblot analysis assessing TRF2 expression levels in the respective HCT116 cell lines. Note that the endogenous TRF2 appears as a doublet. (c) Population doublings (PDs) undergone by respective HCT116 cell lines when they were collected for TRF or FISH analysis. Data were obtained from three independent sets of experiments. Bars represent the mean and SD. P values calculated by two-tailed Student’s t-tests. (d) TRF analysis demonstrating telomere shortening caused by overexpression of TRF2. Note that all the cell lines used for TRF2 overexpression in this study contained pre-extended telomeres as shown in the right panel of (a). For (c) and (d), cells were infected with lentiviruses expressing either GFP as a control or TRF2 and then analyzed ~8 days post-infection.
Lig3- or Lig4-deficiency does not affect the induction of fragile telomeres and signal-free ends by TRF2 overexpression
To further examine telomere morphology, we performed fluorescent in situ hybridization (FISH) using telomeric and centromeric probes on metaphase chromosomes. Upon TRF2 overexpression, an increase of fragile telomeres (decondensed or multiple split telomeric fluorescence signals whose formation are replication stress-dependent) was observed in all three lines (Figure 2(a)). We also observed an increase of telomere signal-free ends in all three lines (Figure 2(b)). The absence of Lig4 or nuclear Lig3 did not significantly change the level of increase in fragile telomeres or signal-free ends, suggesting that the two ligases do not affect TRF2 overexpression-induced telomere replication fork stalling nor the ensuing telomere shortening.
Figure 2.
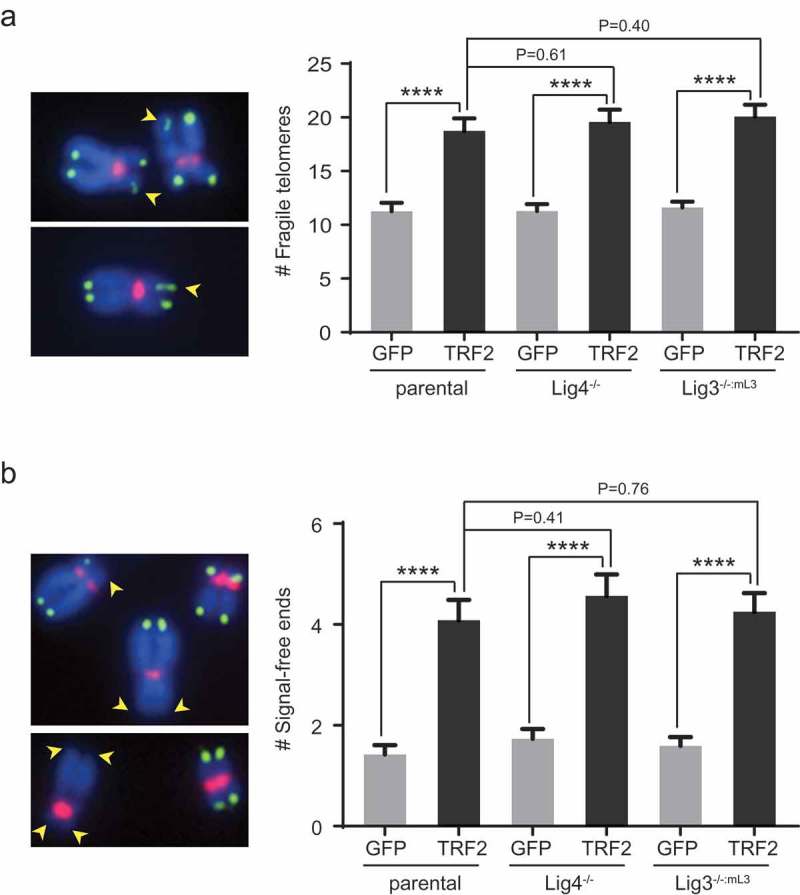
Overexpression of TRF2 leads to increased telomere fragility and signal-free ends in different cell lines. (a) Quantification of fragile telomeres in the indicated cell lines. Bars represent mean values and SEM (~100 metaphases from two independent experiments examined for each line). Representative fragile telomeres from the parental cells overexpressing TRF2 are marked by yellow arrowheads on images at the left panel. (b) Quantification of telomere signal-free ends in the indicated cell lines. Bars represent mean values and SEM (~100 metaphases from two independent experiments examined for each line). Representative signal-free ends from the parental cells overexpressing TRF2 are marked by yellow arrowheads on images at the left panel. For (a) and (b), cells were infected with lentiviruses expressing a GFP control or TRF2 and then analyzed ~8 days post-infection by metaphase spreading followed by FISH analysis. Chromosomes (blue) were hybridized with PNA probes for telomeric sequences (green) or centromeric sequences (red). All quantifications were carried out blindly. ****P < 0.0001; Two-tailed Student’s t-tests were performed to make pairwise comparison for statistical significance.
Lig3- or Lig4-deficiency affects TRF2 overexpression-induced chromosome end fusions
In agreement with previous findings [24], chromosome end-to-end fusions were detected in parental HCT116 cells overexpressing TRF2. The vast majority (>80%) of inter-chromosomal fusions were the chromosome type (Figure 3(a)), in which both chromatids of the involved chromosomes were fused together. Sister chromatid fusions also arose after TRF2 overexpression (Figure 3(a)). Almost all of these fusions were devoid of telomere signals at the fusion sites (Figure 3(b,c), left panels), indicating that the loss of telomeric sequences preceded the chromosome deprotection and fusion events. Interestingly, TRF2 overexpression induced significantly fewer inter-chromosomal fusions in Lig4- or Lig3-deficient cells (Figure 3(a,b)). In contrast, the number of TRF2-induced sister chromatid fusion was reduced by Lig3- but not Lig4-deficiency, to a level indistinguishable from that observed in control cells (Figure 3(a,c)). Taken together, our data show that upon TRF2 overexpression, both the Lig4- and Lig3-dependent end joining pathways are engaged by damaged telomeres, while only the Lig3-dependent pathway plays a major role in mediating sister chromatid fusions.
Figure 3.
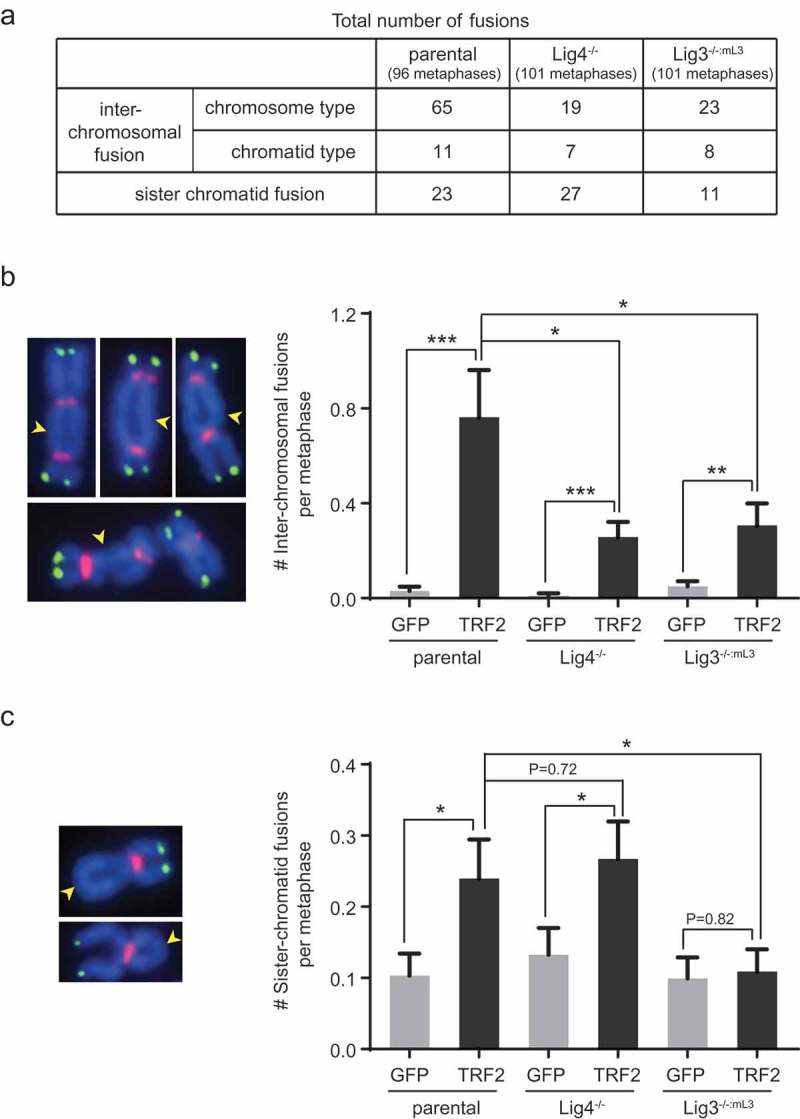
Lig3- and Lig4-deficiency affect TRF2 overexpression-induced inter-chromosomal fusion and sister chromatid fusion differently. (a) Summary of fusion events in the indicated TRF2-overexpressing HCT116 cell lines. ~100 metaphases from two independent experiments were examined for each line. (b) Lig3- or Lig4-deficiency reduced efficiency of TRF2 overexpression-induced inter-chromosomal fusion. Bars represent mean value of fusions and SEM (~100 metaphases from two independent experiments examined for each line). Representative fusions from the parental cells overexpressing TRF2 are marked by yellow arrowheads on images at the left panel. (c) Lig3-, but not Lig4-deficiency abrogated TRF2 overexpression-induced sister chromatid fusions. Bars represent mean value of sister chromatid fusions and SEM (~100 metaphases from two independent experiments examined for each line). Representative fusions from the parental cells overexpressing TRF2 are marked by yellow arrowheads on images at the left panel. Cells were infected with lentiviruses expressing a GFP control or TRF2 and then analyzed ~8 days post-infection by metaphase spreading followed by FISH analysis. Chromosomes (blue) were hybridized with PNA probes for telomeric sequences (green) or centromeric sequences (red). All quantifications were carried out blindly. *P < 0.05; **P < 0.01; ***P < 0.001; Two-tailed Student’s t-tests were performed to make pairwise comparison for statistical significance.
Discussion
Overexpression of TRF2 induces persistent replication stalling and formation of ultrafine anaphase bridges (UFBs) in the subsequent mitosis [24]. Resolution of UFBs leads to stochastic shortening of telomeres and consequently, end deprotection at some chromosome ends [24]. If UFB resolution and end deprotection happens before sister chromatid segregation, NHEJ results in chromatid-type and chromosome-type fusions (collectively called the inter-chromosomal fusions), as well as sister chromatid fusions (Figure 4). If UFB resolution and end deprotection happens after sister chromatid segregation, NHEJ results in formation of dicentric chromosomes which, after DNA replication, appear as chromosome-type fusions in the next mitosis (Figure 4). When examining which end joining pathway is involved in mediating TRF2 overexpression-induced fusions, we found that the depletion of either Lig4 or nuclear Lig3 significantly reduced the efficiency of inter-chromosomal fusion. In contrast, Lig3-dependent A-NHEJ, but not the Lig4-dependent C-NHEJ, was required for mediating sister chromatid fusions (Figure 4).
Figure 4.
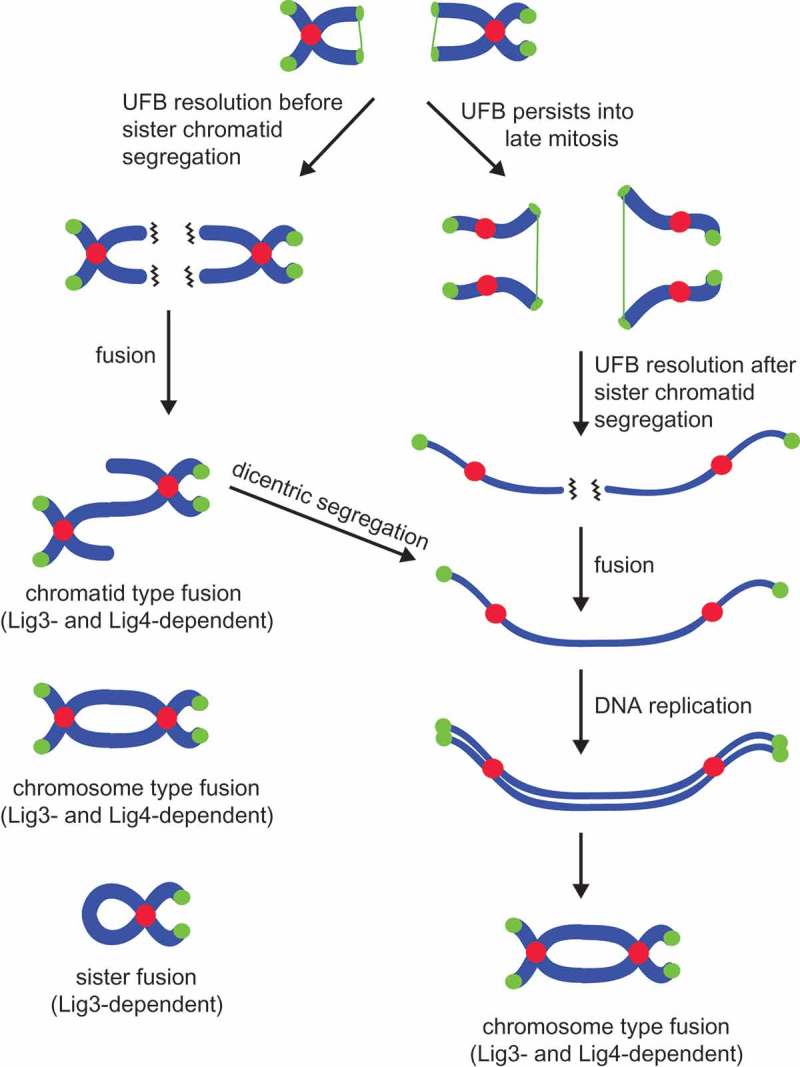
A schematic diagram showing chromosome fusions via NHEJ upon TRF2 overexpression as detected by metaphase spreading. Chromosomes are depicted in blue, centromeres in red, and telomeres in green. Wavy lines indicate deprotected chromosome ends. The green thin thread connecting between sister chromatids represents un-replicated telomeric regions that persist into mitosis to form ultrafine anaphase bridges (UFBs) [24]. Resolution of UFBs leads to critical shortening of telomeres at some chromosome ends. Chromosome-type fusions mainly arise when NHEJ occurs after sister chromatids are well segregated (during G1 or late stages of mitosis) or result from dicentrics generated by a chromatid-type fusion from a previous cell cycle.
Sequence analysis of the TRF2 overexpression-induced fusions in a previous study [24] revealed large deletions of telomeric sequence that extended into the sub-telomeric region. Furthermore, the fused molecules frequently contained 1 to 4 nucleotides of microhomology at the fusion junction [24]. These features were generally considered to be characteristic of A-NHEJ [19,20,23]. Indeed, we showed here a prominent role of A-NHEJ in mediating TRF2 overexpression-induced fusions. The significant involvement of Lig4-dependent C-NHEJ to this repair process was unexpected, as C-NHEJ usually entails very little processing/resection at the break point. One possibility is that the “resection-dependent C-NHEJ” [31], a recently defined slower sub-pathway of C-NHEJ involved in repairing double-stranded breaks within heterochromatin regions, might contribute to the end processing and fusion of a subset of deprotected telomeres induced by TRF2 overexpression. Further experiments are required to examine whether DNA ligase 1 (Lig1) may compensate for the absence of Lig3 and/or Lig4 for TRF2-induced chromosome fusions in mechanistically distinct manners. There is precedence for this in at least the case of crisis-induced fusion events [32,33]. Several constituents of the DNA damage repair pathways, such as the Ku complex [5,34–36], WRN [37], PARP1 [38–40], and DNA polymerase theta [41], have been reported to affect C- and A-NHEJ choices at damaged telomeres. A subject for future study is to understand the contribution of each to TRF2 overexpression-induced fusions.
The fact that most of the TRF2 overexpression-induced inter-chromosomal fusions are of the chromosome type makes us speculate that the majority of chromosome deprotection likely occurs during G1 phase of the cell cycle or at late mitosis when sister chromatids are well segregated. If chromosome ends become deprotected during G2 or any other stage before sister chromatids are segregated from each other, there should instead be a considerable number of chromatid type fusions in which only one of the two chromatids are fused end-to-end (Figure 4). Another interesting finding is that sister chromatid fusions require the A-NHEJ, but not the C-NHEJ pathway. One potential explanation is that sister chromatid fusions happen at a time during mitosis when C-NHEJ is largely suppressed. Indeed, it has been found that human cells apply multiple layers of suppression to the C-NHEJ pathway during mitosis [42].
It is worth noting that similar end joining pathway requirements have been established for fusion of critically shortened telomeres during telomere attrition-based cellular crisis [23]: both the Lig3-dependent A-NHEJ and the Lig4-dependent C-NHEJ pathway contribute to inter-chromosomal fusion of critically shortened telomeres, while only the Lig3-dependent A-NHEJ facilitates intra-chromosomal fusion (i.e. sister chromatid fusion). Intra-chromosomal fusion has been demonstrated to play an important role in initiating certain types of genomic rearrangements that allow cells to escape from crisis [43]. We speculate that the Lig3-dependent A-NHEJ may likewise be indispensable in driving clonal and genomic evolution in the subset of human cancers containing significantly increased TRF2 levels.
Funding Statement
This work was funded by an American Cancer Society Research Scholar Grant RSG-12-069-01-DMC to L.X. Work in the Hendrickson laboratory was funded, in part, through grants from the National Institutes of Health (GM088351) and the National Cancer Institute (CA190492).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Blackburn EH, Collins K.. Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb Perspect Biol. 2011;3:a003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Harley CB, Futcher AB, Greider CW.. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. [DOI] [PubMed] [Google Scholar]
- [3].Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. [DOI] [PubMed] [Google Scholar]
- [4].Campisi J, d‘Adda Di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. [DOI] [PubMed] [Google Scholar]
- [5].Rai R, Zheng H, He H, et al. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. Embo J. 2010;29:2598–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Maser RS, Wong KK, Sahin E, et al. DNA-dependent protein kinase catalytic subunit is not required for dysfunctional telomere fusion and checkpoint response in the telomerase-deficient mouse. Mol Cell Biol. 2007;27:2253–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang F, Podell ER, Zaug AJ, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445:506–510. [DOI] [PubMed] [Google Scholar]
- [8].Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–1175. [DOI] [PubMed] [Google Scholar]
- [9].Broccoli D, Smogorzewska A, Chong L, et al. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 1997;17:231–235. [DOI] [PubMed] [Google Scholar]
- [10].Bilaud T, Brun C, Ancelin K, et al. Telomeric localization of TRF2, a novel human telobox protein. Nat Genet. 1997;17:236–239. [DOI] [PubMed] [Google Scholar]
- [11].Kim SH, Kaminker P, Campisi J. TIN2, a new regulator of telomere length in human cells. Nat Genet. 1999;23:405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Houghtaling BR, Cuttonaro L, Chang W, et al. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr Biol. 2004;14:1621–1631. [DOI] [PubMed] [Google Scholar]
- [13].Liu D, Safari A, O‘Connor MS, et al. PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol. 2004;6:673–680. [DOI] [PubMed] [Google Scholar]
- [14].Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li B, Oestreich S, de Lange T. Identification of human Rap1: implications for telomere evolution. Cell. 2000;101:471–483. [DOI] [PubMed] [Google Scholar]
- [16].Hockemeyer D, Palm W, Else T, et al. Telomere protection by mammalian Pot1 requires interaction with Tpp1. Nat Struct Mol Biol. 2007;14:754–761. [DOI] [PubMed] [Google Scholar]
- [17].Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–718. [DOI] [PubMed] [Google Scholar]
- [18].Smogorzewska A, Karlseder J, Holtgreve-Grez H, et al. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol. 2002;12:1635–1644. [DOI] [PubMed] [Google Scholar]
- [19].Sfeir A, Symington LS. Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? Trends Biochem Sci. 2015;40:701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].McVey M, Lee SE. MMEJ repair of double-strand breaks (director‘s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bhargava R, Sandhu M, Muk S, et al. C-NHEJ without indels is robust and requires synergistic function of distinct XLF domains. Nat Commun. 2018;9:2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jones RE, Oh S, Grimstead JW, et al. Escape from telomere-driven crisis is DNA ligase III dependent. Cell Rep. 2014;8:1063–1076. [DOI] [PubMed] [Google Scholar]
- [24].Nera B, Huang H-S, Lai T, et al. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat Commun. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Oh S, Harvey A, Zimbric J, et al. DNA ligase III and DNA ligase IV carry out genetically distinct forms of end joining in human somatic cells. DNA Repair (Amst). 2014;21:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Oh S, Wang Y, Zimbric J, et al. Human LIGIV is synthetically lethal with the loss of Rad54B-dependent recombination and is required for certain chromosome fusion events induced by telomere dysfunction. Nucleic Acids Res. 2013;41:1734–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li S, Rosenberg JE, Donjacour AA, et al. Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and anti-telomerase short-interfering RNA. Cancer Res. 2004;64:4833–4840. [DOI] [PubMed] [Google Scholar]
- [28].Xu L, Blackburn EH. Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J Cell Biol. 2004;167:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lansdorp PM, Verwoerd NP, van de Rijke FM, et al. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet. 1996;5:685–691. [DOI] [PubMed] [Google Scholar]
- [30].van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. [DOI] [PubMed] [Google Scholar]
- [31].Biehs R, Steinlage M, Barton O, et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell. 2017;65:671–84 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liddiard K, Ruis B, Takasugi T, et al. Sister chromatid telomere fusions, but not NHEJ-mediated inter-chromosomal telomere fusions, occur independently of DNA ligases 3 and 4. Genome Res. 2016;26:588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liddiard K, Ruis B, Kan Y, et al. DNA Ligase 1 is an essential mediator of sister chromatid telomere fusions in G2 cell cycle phase. Nucleic Acids Res. 2018;47:2402–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res. 1996;24:4639–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zellinger B, Akimcheva S, Puizina J, et al. Ku suppresses formation of telomeric circles and alternative telomere lengthening in Arabidopsis. Mol Cell. 2007;27:163–169. [DOI] [PubMed] [Google Scholar]
- [36].Celli GB, Denchi EL, de Lange T. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat Cell Biol. 2006;8:885–890. [DOI] [PubMed] [Google Scholar]
- [37].Shamanna RA, Lu H, de Freitas JK, et al. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat Commun. 2016;7:13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Badie S, Carlos AR, Folio C, et al. BRCA1 and CtIP promote alternative non-homologous end-joining at uncapped telomeres. Embo J. 2015;34:410–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Doksani Y, de Lange T. Telomere-internal double-strand breaks are repaired by homologous recombination and PARP1/Lig3-dependent end-joining. Cell Rep. 2016;17:1646–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Harvey A, Mielke N, Grimstead JW, et al. PARP1 is required for preserving telomeric integrity but is dispensable for A-NHEJ. Oncotarget. 2018;9:34821–34837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mateos-Gomez PA, Gong F, Nair N, et al. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Terasawa M, Shinohara A, Shinohara M. Double-strand break repair-adox: restoration of suppressed double-strand break repair during mitosis induces genomic instability. Cancer Sci. 2014;105:1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Baird DM, Hendrickson EA. Telomeres and chromosomal translocations: there’s a ligase at the end of the translocation. Adv Exp Med Biol. 2018;1044:89–112. [DOI] [PubMed] [Google Scholar]