

ABSTRACT
Protein expression levels depend on the balance between their synthesis and degradation rates. Even quiescent (G0) cells display a continuous turnover of proteins, despite protein levels remaining largely constant over time. In cycling cells, global protein levels need to be precisely doubled at each cell division in order to maintain cellular homeostasis, but we still lack a quantitative understanding of how this is achieved. Recent studies have shed light on cell cycle-dependent changes in protein synthesis and degradation rates. Here we discuss current population-based and single cell approaches used to assess protein synthesis and degradation, and review the insights they have provided into the dynamics of protein turnover in different cell cycle phases.
KEYWORDS: Protein synthesis, protein degradation, cell cycle, single cell approaches, population-based studies
Introduction
More than 50% of cellular dry weight consists of proteins [1], which act as building blocks as well as main executors of most cellular functions. Their continuous synthesis and degradation allow to replace old proteins with new ones and to alter the composition of the proteome during cell differentiation or in response to intra- or extracellular cues. The complex interplay of protein synthesis, folding, trafficking and degradation is referred to as protein homeostasis (or proteostasis). The precise execution of each step involved in the regulation of protein levels is crucial to the function of the cell and thus to the health of the whole organism, as their mis-regulation can lead to a variety of diseases [2].
The birth of a new protein is the end-result of several consecutive steps and can be regulated at various levels. It is initiated by the onset of transcription, followed by pre-mRNA maturation, nuclear export of the mRNA, translation and protein folding. Newly synthesized proteins can also be subject to a myriad of posttranslational modifications, such as phosphorylation, glycosylation, ubiquitination and many others [3]. Importantly, while often used as a synonym for protein translation, in this review we will employ “protein synthesis” as the net result of all gene expression steps leading to the synthesis of a protein, including transcription, pre-mRNA maturation, mRNA export, and translation.
Protein degradation generally follows first order decay kinetics, with half-lives varying widely across different proteins depending on their function and the cell type/organism in which they are expressed. Regulatory proteins such as transcription factors are often short-lived, which allows for rapid adjustment of their levels in response to internal or external stimuli. In contrast, housekeeping proteins are not subject to short-term fluctuations in their levels and are thus generally long-lived [4]. In addition, protein lifetimes in fast-cycling cell types, such as prokaryotes or yeast, are usually shorter than in cell types with a longer cell cycle [5,6].
The major route for selective protein degradation is the ubiquitin-proteasome pathway [4]. To target a protein for degradation, a chain of ubiquitin molecules is added to the side chain of one of its lysine residues. Polyubiquitination is a multistep process, mediated by 3 main classes of enzymes: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin ligases (E3). E3 enzymes mediate substrate specificity [7] and are consequently the most heterogeneous class of enzymes in the ubiquitination pathway [8]. Polyubiquitinated proteins are then recognized and degraded by the proteasome, a large multisubunit protease complex consisting of two major subcomplexes: the core particle (CP), also referred to as the 20S subunit, which executes the degradation reaction, and the regulatory particle (RP), also referred to as the 19S subunit, which mediates target recognition [9].
Each individual step involved in protein synthesis and degradation needs to be tightly regulated to control protein expression levels. How this is achieved is not fully understood. In non-dividing (G0) cells, the levels of the vast majority of proteins are kept constant, even though proteins are still continuously turned over. In contrast, cycling cells such as embryonic stem cells [10,11], intestinal stem cells, transit amplifying cells [12] and hematopoietic stem cells [13] continuously need to increase their biomass, thus more proteins need to be synthesized than degraded. Importantly, cycling cells need to be able to precisely double their global protein levels at each cell cycle, as imbalances in protein homeostasis might broadly compromise cellular functions.
Cycling mammalian cells transit through four consecutive phases: G1, S, G2 and M, each of which consists of a specific series of events that lead to the doubling of protein content to generate two new cells [4]. The transition from one cell cycle phase to the next is regulated by complex molecular programs whose main executors are specific pairs of cyclins and cyclin-dependent kinases (CDKs) [14]. In addition, the cell cycle contains several checkpoints, which ensure that the cell is fully prepared to enter the next phase [15]. The cell cycle poses several major challenges to gene expression and thus global protein turnover, such as DNA duplication during S phase and chromosome condensation during M phase. How cells control the rates of protein synthesis and degradation throughout the cell cycle to allow protein level doubling is not understood.
In this review we start by describing the most commonly used methods to monitor protein synthesis and degradation. We distinguish between population-based bulk biochemical approaches and fluorescence-based single cell methods, which both have their specific advantages and disadvantages (Table 1). In addition, some approaches allow to study synthesis or degradation of specific proteins, whereas other methods measure synthesis and degradation rates on a global level. Furthermore, we discuss what studies based on those methods have revealed about cell cycle-dependent changes and the interplay between protein synthesis and degradation.
Table 1.
Overview of the most commonly used methods to measure protein synthesis and degradation.
| Method | Measures | Specificity | Advantages | Disadvantages |
|---|---|---|---|---|
| Bulk biochemical methods | ||||
| Metabolically labeled amino acids or nucleotides | Synthesis OR degradation | Global OR protein-specific |
|
|
| Incorporation of puromycin (PUNCH-P) | Synthesis | Global AND protein-specific | ||
| Ribosome profiling | Synthesis | Global AND protein-specific | ||
| Cycloheximide chase & Western Blot | Degradation | Global OR protein-specific | ||
| SILAC in combination with MS | Synthesis AND degradation | Global AND protein-specific | ||
| Fluorescence-based approaches | ||||
| Incorporation of puromycin (OP-Puro) | Synthesis | Global |
|
|
| Controllable destabilization domain (e.g. ecDHFR) | Synthesis | Protein- or regulatory domain- specific | ||
| SunTag system | Synthesis | Protein-specific | ||
| Cycloheximide & fluorescence microscopy | Degradation | Protein-specific | ||
| Bleach chase | Degradation | Protein-specific | ||
| Fluorescently labeled protein tags (SNAP/Halo/Clip-tags) | Degradation | Protein-specific | ||
| Fluorescent protein timers | Synthesis AND degradation | Protein-specific | ||
Methods to monitor protein turnover
Bulk biochemical approaches
Protein synthesis rates
Protein levels are often indirectly inferred from quantifications of mRNA levels, which are relatively straightforward using well-established methods such as qPCR, RNA-seq or microarrays. However, the relatively poor correlations between mRNA and protein levels suggest that mRNA quantification alone may not be a reliable proxy for protein synthesis rates [16–18]. Thus, measuring translation rates directly or in combination with transcription rates is required to obtain a more accurate assessment of protein synthesis rates. Traditionally, global protein synthesis has been measured using the incorporation of radioactively or isotopically labeled amino acids into nascent polypeptide chains. More recent studies have also used the translation inhibitor puromycin, which is incorporated into the C-terminus of a nascent polypeptide and induces premature chain termination. The use of biotin-conjugated puromycin allows the puromycylated polypeptides to be captured, purified and further analyzed by mass spectrometry [19]. Another approach to study protein synthesis rates is to directly quantify mRNAs undergoing translation by ribosome profiling, which relies on the isolation and deep sequencing of ribosome-protected mRNA fragments [20,21]. This method enables a transcriptome-wide assessment of ribosome occupancy, thus providing a snapshot of the global translational landscape at a sub-codon resolution. It has the additional advantage to allow for mapping translation start sites and thereby identifying novel open reading frames (ORFs).
Protein degradation rates
Similar to approaches allowing to monitor protein synthesis, early studies quantifying protein degradation mainly relied on metabolic labeling with radioactively or isotopically labeled elements [22]. This approach allows for determining either global or protein-specific degradation rates, the latter including a pull-down step with a specific antibody. Another widely used method to monitor protein degradation is cycloheximide chase, in which protein synthesis is blocked by cycloheximide and the decay of the remaining protein pool is monitored by western blotting at different time-points [23]. In contrast to radioactive or isotopic labeling, this method does not require immunoprecipitation of a specific protein of interest. However, cycloheximide induces considerable cellular stress and ultimately cell death, which limits its application to short-lived proteins.
Combined measurements of protein synthesis and degradation rates
Technological advances in the field of mass spectrometry have considerably increased the throughput and resolution of pulse-chase experiments and thereby provided a more quantitative and global picture of gene expression. In 2011, Schwanhäusser and colleagues published the first genome-wide study of both RNA and protein turnover [6]. They used stable isotope labeling by amino acids in cell culture (SILAC) followed by liquid chromatography and online tandem mass spectrometry (LC-MS/MS) to monitor protein synthesis and degradation, as well as pulse labeling with the nucleoside 4-thiouridine (4sU) to simultaneously measure RNA turnover. This allowed to quantitatively dissect the contribution of different steps of gene expression in determining protein levels on a genome-wide scale. They found that cellular protein abundance is mostly controlled at the level of protein translation and that functionally related proteins often show similar dynamics of RNA and protein turnover. Complementing these findings, Jovanovic et al. have studied the changes in gene expression upon stimulation of mouse immune bone marrow-derived dendritic cells (DC) with lipopolysaccharide (LPS), using a similar genome-wide approach allowing to monitor both RNA and protein turnover simultaneously [24]. They observed that the acquisition of novel cellular functions related to the stimulation response is mainly controlled at the level of transcription. In contrast, they showed that proteins performing basic cellular functions such as ribosomal proteins or components of the cytoskeleton are rather regulated by changes in translation or protein degradation.
One major drawback of bulk biochemical approaches is their low temporal resolution due to the limited number of time-points that can be acquired. Thus, short-term fluctuations in protein turnover cannot be assessed using those methods. More importantly, they only provide population averages, thus masking cell-to-cell variability in proteostasis and unsynchronized, single cell fluctuations in protein synthesis and degradation rates. Finally, they require the use of cell cycle synchronizing drugs to investigate cell cycle-related processes, which often results in incomplete synchronization and can compromise certain steps of gene expression, such as interference with translation by the microtubule-destabilizing agent nocodazole [25].
Single cell imaging approaches
Protein synthesis rates
In contrast to population-based biochemical approaches, fluorescence or luminescence microscopy-based methods provide single cell resolution. Several approaches have been established to study transcription, both in fixed cells, e.g. by single molecule fluorescence in situ hybridization (smFISH) [26], and in live cells, e.g. by using the MS2 system [27] or luminescence imaging of a destabilized luciferase reporter [28]. In contrast, measuring translation rates in single cells has been more challenging. Similarly to the PUNCH-P approach described above, puromycin can also be used as a fluorescence-based approach, since puromycilated polypeptides can be visualized and quantified by conjugating them to a fluorescent dye (OP-Puro) [29]. However, this only allows for studying global translation rates and does not provide protein specificity. In contrast, an approach based on the E. coli destabilization domain from dihydrofolate reductase (ecDHFR) that can be stabilized by its ligand trimethoprim (TMP) [30] allows to monitor translation rates of specific mRNAs in single live cells. The ecDHFR can be conjugated to a translation regulatory domain and fused to a fluorescent reporter, which leads to the immediate degradation of the fusion protein and a reduction of the reporter fluorescence to a basal level in the absence of TMP. Upon addition of TMP, the accumulation of fluorescence can be used to monitor real-time translation rates [30].
Finally, the visualization of translation in vivo with single mRNA resolution has been rendered possible by the recently developed SunTag system. Briefly, this approach is based on the fusion of a protein of interest to an array of SunTag peptides, which can be recognized specifically by a GFP-tagged single-chain intracellular antibody (scFv-GFP) [31,32]. As soon as the newly translated SunTag peptides exit the ribosome, they are labeled by the scFv-GFP, which allows for in vivo monitoring of translation. This approach has been applied to study various aspects of translation, such as measurements of ribosome numbers, initiation and elongation rates, and changes in translation rates in response to various stimuli [31,32].
Protein degradation rates
Inhibition of protein synthesis with cycloheximide can also be used in combination with time-lapse fluorescence microscopy to monitor the decay of fluorescently labeled proteins, but this is only suitable to study short-lived proteins because of cytotoxicity. In contrast, the “bleach-chase” approach does not require translational inhibitors [33]. This method requires cells expressing an endogenous protein fused to a fluorescent reporter. These are then partially bleached by a short pulse of light, which irreversibly renders a fraction of the fluorophores non-fluorescent. After the pulse, the bleached protein population decays and is replaced by newly synthesized fluorescent proteins, which can be observed by time-lapse microscopy. At the same time, a population of unbleached cells is imaged. The protein half-life can then be inferred by subtracting the fluorescence intensities of bleached and unbleached cells. However, since this approach relies on the comparison between the averages of two different populations of cells (bleached vs. unbleached) it does not allow to determine protein degradation rates of individual cells.
Time-lapse imaging of pulse-labeled fluorescent tags, such as SNAP-tags [34], Halo-tags [35] or Clip-tags [36] allow measuring protein degradation rates in single cells. The tags are fused to a protein of interest and can be covalently bound by a fluorescent ligand (Figure 1(a)). Upon washing of the residual ligand or blocking with a non-fluorescent version of the ligand, the decay of the labeled protein population can be monitored, which allows for assessing protein half-lives (Figure 1(a,b)). This approach has been successfully established to measure protein decay of cell populations in vivo [37] and single cell protein half-lives in in vitro cultured adherent cells [38–40]. Importantly, fluorescently labeled protein tags can not only be used to assess cell-to-cell variability in protein half-lives, but also to monitor transient changes in protein degradation [39] (Figure 1(c)), as they can provide a higher temporal resolution than bulk biochemical approaches.
Figure 1.
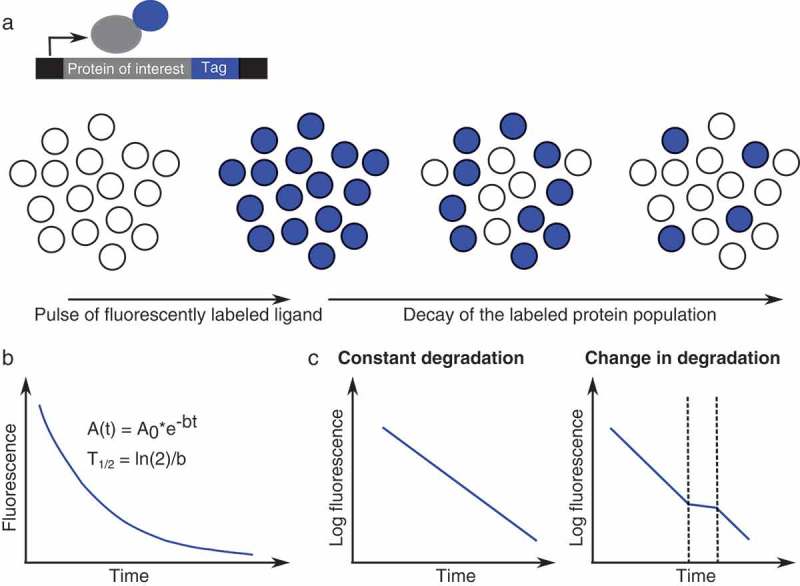
Fluorescently labeled protein tags to monitor protein degradation. (a): A protein of interest is fused to a SNAP/Halo/Clip-tag, which can be pulse-labeled with a fluorescent ligand. Upon washing of the residual dye, the decay of the labeled protein population can be observed. (b): Protein degradation follows an exponential decay. A0: Initial fluorescence intensity, b: decay rate, T1/2: half-life. (c): Schematic representation of how SNAP/Halo/Clip-tags can be used to identify changes in protein degradation.
Tandem fluorescent timers: simultaneous measurements of protein synthesis and degradation rates in single live cells
The fluorescence-based approaches described above do not allow for simultaneous measurements of protein synthesis and degradation rates. Thus, until very recently it has not been possible to determine how synthesis and degradation interplay to control protein expression levels in individual cells. However, a new study reported the development of mammalian cell-optimized fluorescent timers, allowing to disentangle changes in protein synthesis and degradation in single live cells [39]. Fluorescent timers are fluorescent proteins that change their emission spectrum over time, which provides a measure of the age of a protein pool. Traditional timers consist of only one fluorophore that switches its emission spectrum due to a series of chemical reactions, but their application has been impeded by their low brightness and tendency to form oligomers [41,42]. These limitations were overcome by the engineering of tandem fluorescent protein timers (tFTs), which consist of a fusion of the fast maturing green fluorescent protein superfolder GFP (sfGFP) and a slower maturing red fluorescent protein such as mCherry, TagRFP, Td-Tomato, mKate2 or mOrange2 [39,43,44] (Figure 2(a)). Tandem fluorescent timers have mainly been used to assess changes in protein stability or age-dependent protein localization [43–45], as the lag in maturation time between the two fluorophores allows for distinguishing old from newly made proteins and thereby provides an estimate for the average age of a protein pool. However, it is in fact possible to use fluorescent timers to gain quantitative insights into protein synthesis and degradation rates. Alber et al. recently leveraged on this idea by combining single cell time-lapse imaging of timer-tagged proteins with computational modeling, using a set of differential equations to infer both protein synthesis and degradation in single cells [39]. Figure 2(b) illustrates how tandem fluorescent timers can be used to distinguish changes in protein synthesis from changes in protein degradation. An increase in protein synthesis leads to a fast increase in green fluorescence, followed by a slower increase in red fluorescence due to the slower maturation of the red fluorescent protein. In contrast, a decrease in protein degradation leads to an increase in red fluorescence that is more pronounced than the increase in green fluorescence, and thus the green/red ratio decreases. In combination with computational modeling, this allows to extract temporal changes in protein synthesis and degradation rates in single live cells.
Figure 2.
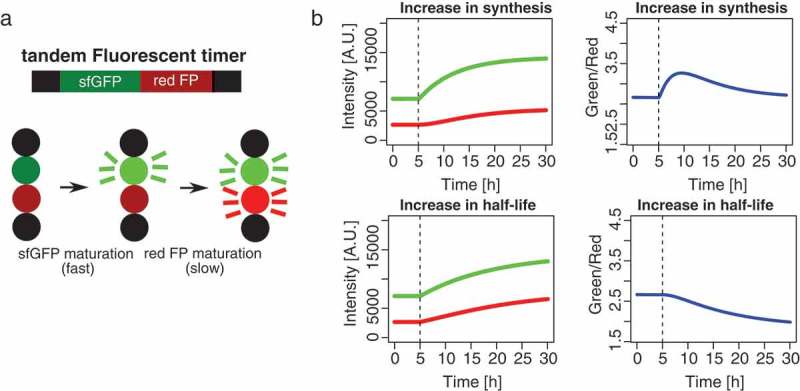
Tandem fluorescent timers allow to simultaneously monitor protein synthesis and degradation. (a): Upon production of the fusion protein, superfolder GFP (sfGFP), matures fast, whereas the red fluorescent protein (FP) matures more slowly. (b): Simulations showing how tandem fluorescent timers can be used to distinguish changes in protein synthesis from changes in protein degradation. Figure reused from [38], Figure 1(d), with permission from Molecular Cell (License number: 4472970734996).
In summary, while population-based bulk biochemical approaches enable proteome-wide studies, mostly without genetic manipulation, fluorescence-based approaches allow for single cell studies and provide a high temporal resolution. However, fluorescence-based approaches can be affected by phototoxicity or bleaching. Fluorescent tags and fusion proteins might also alter endogenous protein turnover, especially when fused to the N-terminus of the protein of interest (N-end rule) [46]. In addition, expression of fluorescent fusion proteins from strong exogenous promoters may lead to changes in turnover, for example due to altered stoichiometry relative to interaction partners [47]. Thus, it is advisable to use endogenous, C-terminal fusions and to carefully test for potential alterations in protein degradation, e.g. by using bulk biochemical methods to directly compare the half-life of a fluorescent fusion protein and its endogenous version. Despite these potential limitations, single cell fluorescence-based approaches are of particular interest for studying cell cycle related processes, as cells can be in silico synchronized and cell cycle-associated processes can therefore be studied without the use of cell cycle-synchronizing drugs.
Dynamics of protein turnover through the cell cycle
Cell cycle-dependent changes in protein synthesis rates
While cell cycle-related changes in the expression of key regulators of cell cycle progression have been known for a long time [19,48–50], how global transcriptional and translational rates are altered in different cell cycle phases has only started to be addressed recently. The most pronounced changes in global transcriptional activity take place during mitosis. Due to the condensation of chromosomes, the RNA polymerase II machinery was suggested to be physically excluded from chromatin, causing a complete and global shutdown of transcriptional activity [51,52]. However, a more recent study showed that transcription is retained at low levels during mitosis [53]. During early G1 phase, 50% of transcribed genes display a hyperactive transcriptional state that leads to a spike in transcription [54]. A further perturbation to transcription occurs when each gene copy is duplicated during S phase. While this should in principle lead to an increase in mRNA synthesis, there is evidence for buffering mechanisms involving H3K56 methylation on newly deposited histones [55].
Translational activity has also been shown to globally decrease during mitosis, which was suggested to facilitate the selective translation of certain specific mRNAs by internal ribosomal entry sites [56–60]. However, these studies mostly relied on mitotic synchronization of cells by microtubule-destabilizing drugs such as nocodazole, which can interfere with translation [25] and thus complicate the interpretation of experimental results. More recently, a study re-addressed global regulation of translation during mitosis using the potentially less perturbing CDK1 inhibitor RO-3306 for synchronization in G2 phase [61]. Metabolic labeling revealed a global decrease in translation by 35%, in line with previous findings. Furthermore, mitosis-specific ribosome profiling revealed a subset of 200 mRNAs that are strongly up- or down- regulated at the translational level during M-phase [61]. In contrast to mitosis, population-based studies revealed no major changes in global protein synthesis rates during G1, S and G2 phase.
Alber et al. have recently monitored changes in protein synthesis of several proteins in single cells throughout the cell cycle. While synthesis rates increased over the cell cycle for several proteins they studied, degradation rates essentially remained constant [39]. This indicates that between the beginning of G1 phase and the end of G2 phase, synthesis rates can change in a protein-specific manner and result in different protein accumulation profiles. They then further focused on the time interval around cell division, and in agreement with previous studies found a decrease in protein synthesis for a large number of proteins [39].
In summary, both population-based and single cell studies suggest that the most pronounced global changes in protein synthesis occur during cell division, which can be attributed to large changes in both transcription and translation rates.
Cell cycle dependent changes in protein degradation rates
While the regulation of transcriptional and translational efficiency throughout the cell cycle is relatively well studied on a global and transcript-specific level, our knowledge about cell cycle dependent changes in protein degradation has long been limited to very specific cases such as cyclins and other key cell cycle regulatory proteins [62] .
More recently, two studies have provided evidence for more global cell cycle-dependent changes in protein degradation rates. In several cultured cell lines, the periodic activation of the Wnt-signaling cascade leads to inhibition of glycogen synthase kinase 3 (GSK3) during mitosis. Since GSK3 kinase-mediated phosphorylation is often leading to target protein degradation, this causes a stabilization of GSK3 target proteins specifically during M phase, which is referred to as Wnt-dependent stabilization of proteins (Wnt/STOP) [63]. Wnt/STOP might promote cell growth, as its inhibition was shown to decrease in the size of G1 cells [63]. Since 20% of all cellular proteins are predicted GSK3 target proteins, Wnt/STOP might affect a substantial fraction of the proteome [64]. Using their fluorescent timer approach, Alber and colleagues recently made the surprising finding that about 40% of proteins are stabilized during cell division [39]. These results were further confirmed by fluorescent pulse-labeling of SNAP-tagged proteins. Importantly, the culture conditions that were used maintain the Wnt signaling pathway constantly on, bypassing a potential Wnt/STOP mechanism and thus suggesting that the observed stabilization of proteins is caused by a different mechanism.
Protein homeostasis at the single cell level
The combination of tandem fluorescent timers with computational modeling and the use of fluorescently labeled protein tags has also provided the unique opportunity to simultaneously quantify protein synthesis and degradation in single live cells [39]. One outstanding question in the field was to what extent protein synthesis and degradation vary between cells, and whether their rates correlate in single cells. Alber et al. discovered that protein degradation rates vary substantially between individual cells, and that these differences are accompanied by corresponding changes in protein synthesis rates [39]. Simulations suggested that cell-to-cell variability in protein expression levels is higher in cells where protein synthesis and degradation are uncorrelated, indicating that a positive correlation between protein synthesis and degradation allows to buffer protein expression variability. Furthermore, degradation rates in single cells were correlated for different proteins, suggesting that the observed cell-to-cell variability is global rather than protein-specific. Taken together, these findings indicate that cells with similar protein levels might actually have quite different underlying global turnover rates, which may affect a variety of cellular processes, such as stress response or differentiation.
Perspectives
Elucidating how protein synthesis and degradation are regulated and how they interplay is of fundamental interest to understand how cells control protein levels. Recent technological advances have allowed for studying protein synthesis and degradation simultaneously, both at the population and single cell level [6,24,39]. The development of live cell approaches allowing time-resolved measurements of several steps of gene expression will be key to gain a quantitative understanding of the information flow from DNA to protein. The use of fluorescent timers to disentangle changes in protein synthesis and degradation represents a first step towards this goal. While CD-tagging allows for rapid generation of cell libraries by non-targeted protein fusion with a fluorescent timer or other tags [65–67], the rapid development of higher throughput CRISPR-Cas9 knock-in approaches will allow for more precise targeting of a large numbers of proteins. The development of brighter fluorescent timers will also hopefully enable studying homeostasis of lowly expressed proteins.
Recent advances have greatly increased our understanding of global cell cycle-dependent changes in protein synthesis and degradation rates (Figure 3). While a reduction in transcriptional activity can be explained by the condensation of chromatin during mitosis, the global decreases in translation and protein degradation rates are more intriguing. In particular, the mechanisms causing a decrease in protein degradation rates independently from Wnt/STOP are not known. Finally, what controls and coordinates protein synthesis and degradation to allow doubling of protein levels at each cell cycle also remains to be investigated. Fortunately, these fascinating new questions should now be within experimental reach thanks to methodological advances that raised them in the first place, and will hopefully be addressed in the near future.
Figure 3.
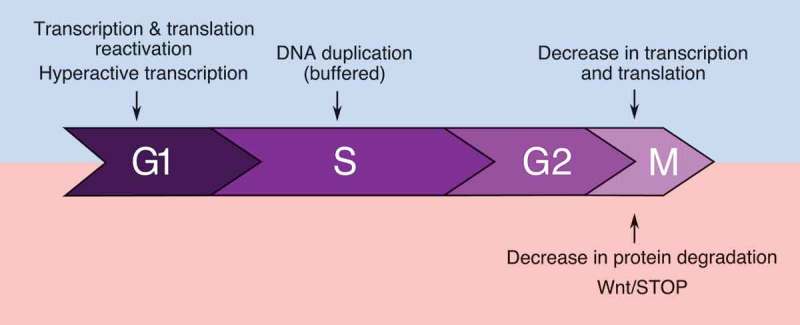
Summary of currently known global changes in protein synthesis (blue) and degradation (red) over the cell cycle.
Funding Statement
This work was supported by the Teofilo Rossi di Montelera e di Premuda Foundation and an anonymous donor, both advised by CARIGEST SA [NA];Fondation Pierre Mercier [NA].
Acknowledgments
This work was supported by the Pierre Mercier Foundation (to D.M.S.), and the Teofilo Rossi di Montelera e di Premuda Foundation and an anonymous donor, both advised by CARIGEST SA (to D.M.S).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Milo R, Phillips R.. Cell biology by the numbers. New York (NY): Garland Science, Taylor & Francis Group; 2016. [Google Scholar]
- [2].Powers ET, Morimoto RI, Dillin A, et al. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. [DOI] [PubMed] [Google Scholar]
- [3].Prabakaran S, Lippens G, Steen H, et al. Post-translational modification: nature’s escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip Rev Syst Biol Med. 2012;4:565–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cooper GM. The cell: a molecular approach. 2. ed. Washington (DC): ASM Press [u.a.]; 2000. [Google Scholar]
- [5].Belle A, Tanay A, Bitincka L, et al. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci U S A. 2006;103:13004–13009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schwanhäusser B, Busse D, Li N, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. [DOI] [PubMed] [Google Scholar]
- [7].Schrader EK, Harstad KG, Matouschek A. Targeting proteins for degradation. Nat Chem Biol. 2009;5:815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci. 2012;125:531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ballabeni A, Park I-H, Zhao R, et al. Cell cycle adaptations of embryonic stem cells. Proc Natl Acad Sci U S A. 2011;108:19252–19257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].White J, Dalton S. Cell cycle control of embryonic stem cells. Stem Cell Rev. 2005;1:131–138. [DOI] [PubMed] [Google Scholar]
- [12].Itzkovitz S, Blat IC, Jacks T, et al. Optimality in the development of intestinal crypts. Cell. 2012;148:608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pietras EM, Warr MR, Passegué E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195:709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol. 2008;9:910–916. [DOI] [PubMed] [Google Scholar]
- [15].Lukas J, Lukas C, Bartek J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst). 2004;3:997–1007. [DOI] [PubMed] [Google Scholar]
- [16].Maier T, Güell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. [DOI] [PubMed] [Google Scholar]
- [17].de Sousa Abreu R, Penalva LO, Marcotte EM, et al. Global signatures of protein and mRNA expression levels. Mol Biosyst. 2009;5:1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aviner R, Geiger T, Elroy-Stein O. Novel proteomic approach (PUNCH-P) reveals cell cycle-specific fluctuations in mRNA translation. Genes Dev. 2013;27:1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ingolia NT, Ghaemmaghami S, Newman JRS, et al. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yewdell JW, Lacsina JR, Rechsteiner MC, et al. Out with the old, in with the new? Comparing methods for measuring protein degradation. Cell Biol Int. 2011;35:457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou P. Determining protein half-lives In: Signal Transduct. Protoc. [Internet]. (NJ): Humana Press; 2004. p. 067–078. DOI: 10.1385/1-59259-816-1:067 [DOI] [Google Scholar]
- [24].Jovanovic M, Rooney MS, Mertins P, et al. Dynamic profiling of the protein life cycle in response to pathogens. Science. 2015;347:1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Coldwell MJ, Cowan JL, Vlasak M, et al. Phosphorylation of eIF4GII and 4E-BP1 in response to nocodazole treatment: a reappraisal of translation initiation during mitosis. Cell Cycle. 2013;12:3615–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Raj A, van den Bogaard P, Rifkin SA, et al. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Querido E, Chartrand P. Using fluorescent proteins to study mRNA trafficking in living cells. Methods Cell Biol. 2008;85:273–292. [DOI] [PubMed] [Google Scholar]
- [28].Suter DM, Molina N, Gatfield D, et al. Mammalian genes are transcribed with widely different bursting kinetics. Science. 2011;332:472–474. [DOI] [PubMed] [Google Scholar]
- [29].Liu J, Xu Y, Stoleru D, et al. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc Natl Acad Sci U S A. 2012;109:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Han K, Jaimovich A, Dey G, et al. Parallel measurement of dynamic changes in translation rates in single cells. Nat Methods. 2014;11:86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang C, Han B, Zhou R, et al. Real-time imaging of translation on single mRNA transcripts in live cells. Cell. 2016;165:990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yan X, Hoek TA, Vale RD, et al. Dynamics of translation of single mRNA molecules in vivo. Cell. 2016;165:976–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Geva-Zatorsky N, Issaeva I, Mayo A, et al. Using bleach-chase to measure protein half-lives in living cells. Nat Protoc. 2012;7:801–811. [DOI] [PubMed] [Google Scholar]
- [34].Keppler A, Gendreizig S, Gronemeyer T, et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. [DOI] [PubMed] [Google Scholar]
- [35].Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. [DOI] [PubMed] [Google Scholar]
- [36].Gautier A, Juillerat A, Heinis C, et al. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. [DOI] [PubMed] [Google Scholar]
- [37].Bojkowska K, Santoni de Sio F, Barde I, et al. Measuring in vivo protein half-life. Chem Biol. 2011;18:805–815. [DOI] [PubMed] [Google Scholar]
- [38].Alber AB, Suter DM. Single-cell quantification of protein degradation rates by time-lapse fluorescence microscopy in adherent cell culture. J Vis Exp. 2018;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Alber AB, Paquet ER, Biserni M, et al. Single live cell monitoring of protein turnover reveals intercellular variability and cell-cycle dependence of degradation rates. Mol Cell. 2018;71:1079–1091.e9. [DOI] [PubMed] [Google Scholar]
- [40].Mandic A, Strebinger D, Regali C, et al. A novel method for quantitative measurements of gene expression in single living cells. Methods San Diego Calif. 2017;120:65–75. [DOI] [PubMed] [Google Scholar]
- [41].Subach FV, Subach OM, Gundorov IS, et al. Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat Chem Biol. 2009;5:118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Terskikh A, Fradkov A, Ermakova G, et al. “Fluorescent timer”: protein that changes color with time. Science. 2000;290:1585–1588. [DOI] [PubMed] [Google Scholar]
- [43].Barry JD, Donà E, Gilmour D, et al. TimerQuant: a modelling approach to tandem fluorescent timer design and data interpretation for measuring protein turnover in embryos. Development. 2016;143:174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Khmelinskii A, Keller PJ, Bartosik A, et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol. 2012;30:708–714. [DOI] [PubMed] [Google Scholar]
- [45].Donà E, Barry JD, Valentin G, et al. Directional tissue migration through a self-generated chemokine gradient. Nature. 2013;503:285. [DOI] [PubMed] [Google Scholar]
- [46].Varshavsky A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011;20:1298–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Boisvert F-M, Ahmad Y, Gierliński M, et al. A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol Cell Proteomics. 2012;11:M111.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cho RJ, Huang M, Campbell MJ, et al. Transcriptional regulation and function during the human cell cycle. Nat Genet. 2001;27:48–54. [DOI] [PubMed] [Google Scholar]
- [49].Whitfield ML, Sherlock G, Saldanha AJ, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Stumpf CR, Moreno MV, Olshen AB, et al. The translational landscape of the mammalian cell cycle. Mol Cell. 2013;52:574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Parsons GG, Spencer CA. Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol Cell Biol. 1997;17:5791–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Prescott DM, Bender MA. Synthesis of RNA and protein during mitosis in mammalian tissue culture cells. Exp Cell Res. 1962;26:260–268. [DOI] [PubMed] [Google Scholar]
- [53].Palozola KC, Donahue G, Liu H, et al. Mitotic transcription and waves of gene reactivation during mitotic exit. Science. 2017;358:119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hsiung CC-S, Bartman CR, Huang P, et al. A hyperactive transcriptional state marks genome reactivation at the mitosis-G1 transition. Genes Dev. 2016;30:1423–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Voichek Y, Bar-Ziv R, Barkai N. Expression homeostasis during DNA replication. Science. 2016;351:1087–1090. [DOI] [PubMed] [Google Scholar]
- [56].Pyronnet S, Dostie J, Sonenberg N. Suppression of cap-dependent translation in mitosis. Genes Dev. 2001;15:2083–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Qin X, Sarnow P. Preferential translation of internal ribosome entry site-containing mRNAs during the mitotic cycle in mammalian cells. J Biol Chem. 2004;279:13721–13728. [DOI] [PubMed] [Google Scholar]
- [58].Ramírez-Valle F, Badura ML, Braunstein S, et al. Mitotic raptor promotes mTORC1 activity, G(2)/M cell cycle progression, and internal ribosome entry site-mediated mRNA translation. Mol Cell Biol. 2010;30:3151–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sivan G, Elroy-Stein O. Regulation of mRNA Translation during cellular division. Cell Cycle. 2008;7:741–744. [DOI] [PubMed] [Google Scholar]
- [60].Sivan G, Kedersha N, Elroy-Stein O. Ribosomal slowdown mediates translational arrest during cellular division. Mol Cell Biol. 2007;27:6639–6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tanenbaum ME, Stern-Ginossar N, Weissman JS, et al. Regulation of mRNA translation during mitosis. eLife [Internet]. 2015. [cited 2018 November15];4 Available from: https://elifesciences.org/articles/07957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shaik S, Liu P, Fukushima H, et al. Protein degradation in cell cycle In: John Wiley & Sons, Ltd , editor. eLS [Internet]. Chichester (UK): John Wiley & Sons, Ltd; 2012. DOI: 10.1002/9780470015902.a0023158 [DOI] [Google Scholar]
- [63].Acebron SP, Karaulanov E, Berger BS, et al. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell. 2014;54:663–674. [DOI] [PubMed] [Google Scholar]
- [64].Taelman VF, Dobrowolski R, Plouhinec J-L, et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jarvik JW, Adler SA, Telmer CA, et al. CD-tagging: a new approach to gene and protein discovery and analysis. BioTechniques. 1996;20:896–904. [DOI] [PubMed] [Google Scholar]
- [66].Harikumar A, Edupuganti RR, Sorek M, et al. An endogenously tagged fluorescent fusion protein library in mouse embryonic stem cells. Stem Cell Rep. 2017;9:1304–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sigal A, Danon T, Cohen A, et al. Generation of a fluorescently labeled endogenous protein library in living human cells. Nat Protoc. 2007;2:1515–1527. [DOI] [PubMed] [Google Scholar]