

ABSTRACT
This study was to investigate the biological function and underlying mechanisms of FENDRR in cholangiocarcinoma (CCA) cell proliferation, migration and invasion. FENDRR and survivin expression in CCA tissues or cell lines were measured by qRT-PCR. In QBC939 and HuCCTl cells, cell proliferation was detected by CCK-8, cell migration and invasion were using transwell assay. RNA pull-down and RIP assay were performed to determine whether FENDRR can combine with SETDB1 in CCA cell. The effect of SETDB1 on survivin and H3K9me1 expression in CCA cells were determined by western blotting. ChIP analysis was performed to analyze the combination of SETDB1 with survivin promoter in CCA cell. The effect of SETDB1 knockdown on survivin and H3K9me1 expression in CCA cells after transfection with FENDRR were determined by western blotting. The results showed that lncRNA FENDRR was downregulated in CCA tissues and cells, and was negatively correlated with survivin expression. Further investigation demonstrated that FENDRR represses CCA cell proliferation, migration and invasion through regulating survivin expression. FENDRR associated with SETDB1 and H3K9 to epigenetically silence survivin and then regulated cell proliferation, migration and invasion. These findings indicate an important role for FENDRR–survivin axis in CCA cell proliferation, migration and invasion, and reveal a novel epigenetic mechanism for survivin silencing. Our data indicated that FENDRR silences survivin via SETDB1-mediated H3K9 methylation, thereby represses CCA cell proliferation, migration and invasion.
KEYWORDS: LncRNA FENDRR, proliferation, survivin, cholangiocarcinoma
Introduction
Cholangiocarcinoma (CCA) is one of the most aggressive and lethal tumors originating from malignant transformation of cholangiocytes and epithelial cells lining the intrahepatic and extrahepatic biliary ducts [1]. It is the second most common primary liver malignant tumor after hepatocellular carcinoma (HCC) and the most common biliary tract malignancy worldwide [2]. CCA is characterized by its resistance to conventional chemotherapy and radiotherapy, in addition to a lack of available methods for early diagnosis and treatment [3]. Unfortunately, the majority of patients diagnosed with CCA present are at an advanced stage [4]. Curative surgery is recommended only for early-stage patients and is not available for advanced stage patients. Systemic chemotherapy with gemcitabine and cisplatin is standard practice for advanced stage patients. However, in spite of the combination of chemotherapy, the 5-year survival rates in CCA patients remains less than 20–40% [5]. Therefore, there is a greater need to identify novel therapeutic targets by deciphering the critical molecular mechanisms regulating CCA in order to improve patient survival times.
Survivin is a 16.5-kDa intracellular protein that is a well-known member of the inhibitor of apoptosis protein (IAP) family, and its expression is elevated in the majority of tumors [6]. Survivin expression is minimal in normal tissues; therefore, it has become a lead target for both as a tumor diagnostic, prognostic and for anti-cancer therapies. Mounting evidence has demonstrated that survivin takes part in the regulation of cell cycle, apoptosis, cell proliferation as well as invasion and play important roles in various tumor carcinogenesis, such as gastric cancer [7], bladder cancer [8], epithelial ovarian cancer [9]. Previous studies have suggested survivin is upregulated in CCA tissues, and inhibition of survivin promoted cell apoptosis [10,11].
Long non-coding RNAs (lncRNAs) are a group of RNAs, greater than 200 nucleotides in length, which are important for the regulation of gene function and various cellular processes [12]. LncRNA has been shown to involve in CCA development [13]. Long non-coding RNA FENDRR expression was downregulated in cancer cancerous tissues and cell lines of breast [14], prostate cancer [15] and gastric cancer [16], and FENDER overexpression suppressed cell proliferation, invasion and migration in vitro. One study also demonstrated that FENDER is significantly down-regulated in CCA tissue, however, report about the role of FENDER in the CCA development is in its fancy, and there is no evidence of a relationship between FENDER and survivin in CCA.
In our study, we indicated that FENDRR recruits SETDB1 to bind to survivin gene promoter through combination with SET domain bifurcated 1(SETDB1), inducing H3K9 methylation, thereby represses survivin expression, resulting in inhibition of CCA cell proliferation, migration and invasion. Therefore, our study has shown that FENDRR/SETDB1/H3K9 methylation/survivin.
Results
Downregulation of lncRNA FENDRR expression in CCA tissues and cells
The differential expression of lncRNA FENDRR in CCA tissues and paired adjacent non-cancerous tissues were examined. We showed that FENDRR expression level was significantly decreased in CCA tissues, as compared to that in normal tissues (Figure 1(a)). FENDRR expression in CCA cell lines, including QBC939, SSP-25, HuCCTl and RBE cells, was lower than that in normal cholangiocytes HIBEC cell (Figure 1(b)). Survivin expression in CCA tissues and adjacent non-cancerous tissues were measured by qRT-PCR. Upregulated survivin expression was observed in CCA tissues relative to non-cancerous tissues (Figure 1(c)), and was negatively correlated with survivin expression (Figure 1(d)).
Figure 1.
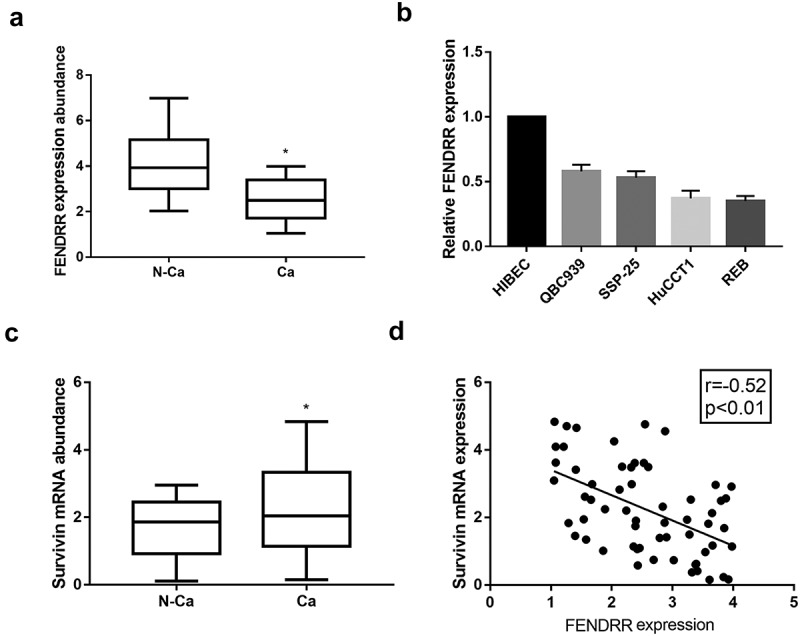
Downregulation of lncRNA FENDRR expression in CCA tissues and cells. (a): FENDRR expression in CCA tissues and paired adjacent non-cancerous tissues were measured by qRT-PCR. (b): FENDRR expression in normal cholangiocytes HIBEC cells and CCA cell lines were measured by qRT-PCR. (c): Survivin expression in CCA tissues and paired adjacent non-cancerous tissues were measured by qRT-PCR. (d): The expression correlation between FENDRR and survivin in CCA tissues (n = 60).
FENDRR repressed CCA cell proliferation, migration and invasion
We further analyzed the biological functions of FENDRR in CCA cells. The stably transfected cell lines with overexpression or knockdown of FENDRR were established in QBC939 and HuCCTl cells and the transfected efficiency of FENDRR was subsequently confirmed by qPCR analysis (Figure 2(a)). Cell proliferation was measured by using CCK-8 in the 48 h after transfection. Results showed that si-FENDRR significantly promoted CCA cells proliferation whereas FENDRR overexpression markedly inhibited cell proliferation (Figure 2(b)). Meanwhile, transwell assay was performed to investigate the role of FENDRR in the regulation of cell migration and invasion in CCA cells and the results demonstrated that the numbers of migrating cells in the FENDRR group were significantly decreased compared with the vector group (Figure 2(c)), and cell invasion showed a similar pattern in QBC939 cells and HuCCTl cells (Figure 2(d)). These observations indicate FENDRR repressed CCA cell proliferation, migration and invasion.
Figure 2.
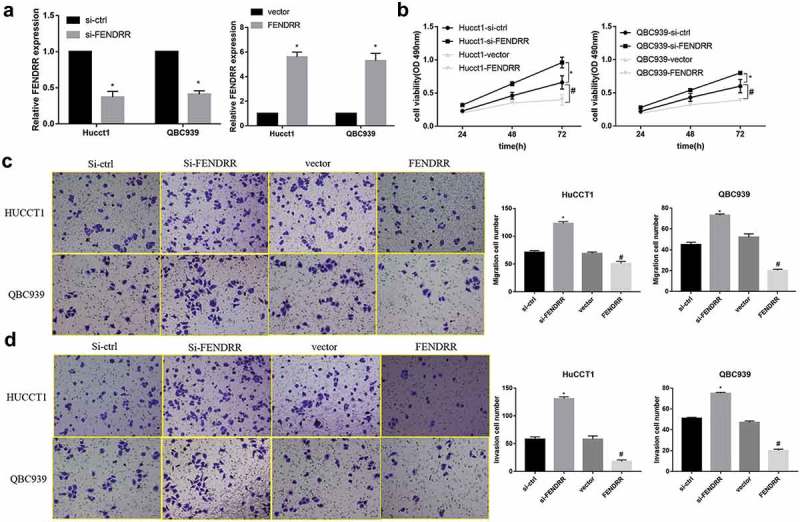
The effect of FENDRR on CCA cell function. (a): Transfection efficiency of FENDRR, si-FENDRR and their respective controls were detected by qRT-PCR in QBC939 and HuCCTl cells. (*p < 0.05 vs. si-ctrl or vector). (b): Cell proliferation analysis using CCK-8 in QBC939 and HuCCTl cells. (c,d): Cell migration and invasion analysis using transwell in QBC939 and HuCCTl cells. (*p < 0.05 vs. si-ctrl; #p < 0.05 vs. vector).
FENDRR repressed CCA cell proliferation, migration and invasion through regulating survivin expression
To identify whether survivin affects FENDRR-regulated cell proliferation, migration and invasion in HuCCTl and QBC939 cells, we first examined the expression level of survivin in transfected CCA cell lines described by qRT-PCR and WB. The results showed that survivin expression both at mRNA level and protein level was significantly up-regulated in HuCCTl and QBC939 cells transfected with si-FENDRR as compared with those in CCA cells transfected with si-ctrl (Figure 3(a,b)). CCK-8 assay showed that cell proliferation rate was significantly decreased in the FENDRR group, when compared to the si-NC group, but this change was rescued by survivin (Figure 3(c)). Meanwhile, transwell assay demonstrated that the ability of FENDRR to repress cell migration and invasion was markedly compromised when survivin overexpression in HuCCTl and QBC939 cells was added (Figure 3(d,e)). To sum up, these data manifest that FENDRR repressed CCA cell proliferation, migration and invasion through regulating survivin expression.
Figure 3.
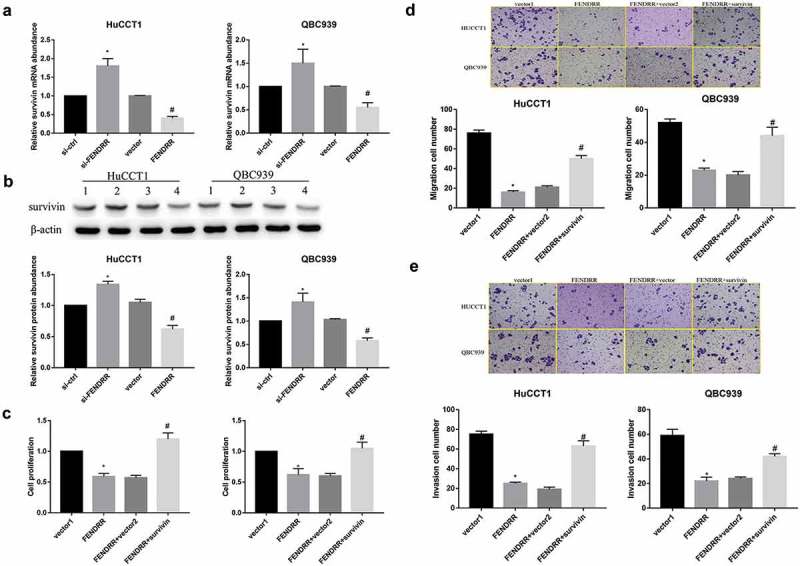
FENDRR repressed CCA cell proliferation, migration and invasion through regulating survivin expression (a): Relative survivin mRNA expression in QBC939 cells and HuCCTl cells after transfection with FENDRR and si-FENDRR was detected by qRT-PCR. (b): Relative survivin protein expression in QBC939 cells and HuCCTl cells after transfection with FENDRR and si-FENDRR was examined by WB. (c): CCK-8 assays were performed to detect the proliferation effect of survivin overexpression on HuCCTl and QBC939 cells after transfection with FENDRR. (d): Transwell assays were performed to measure the migration effect of survivin overexpression on HuCCTl and QBC939 cells after transfection with FENDRR. (e): Transwell assays were performed to examine the invasion effect of survivin overexpression on HuCCTl and QBC939 cells after transfection with FENDRR. (*p < 0.05 vs. si-ctrl or vector1; #p < 0.05 vs. vector or FENDRR+vector2).
FENDRR inhibits survivin expression via SETDB1-mediated H3K9 methylation
To further determine mechanism of FENDRR regulating survivin expression, RNA pull-down and RIP assay were performed to confirm correlation between FENDRR and SETDB1, as seen in Figure 4(a), the results revealed that FENDRR significantly enriched SETDB1, and there was a great enrichment for FENDRR in CCA cell using anti-SETDB1, compared to the anti-IgG group. Western blotting results indicated that the expression of SETDB1 and H3K9me1 were significantly up-regulated in CCA tissues as well as QBC939 and HuCCTl cells, as compared to that in normal tissues or HIBEC cell (Figure 4(b,c)). Besides, no statistically significant difference was observed in survivin expression between the vector and SETDB1 overexpression groups, however, SETDB1 markedly elevated H3K9me1 level both in QBC939 and HuCCTl cells (Figure 4(d)). We also conducted a ChIP assay to examine the occupancy of SETDB1 at the survivin locus. The results indicated that SETDB1 was recruited to the promoter region of survivin in CCA cell after transfection with FENDRR. (Figure 4(e)), and silencing SETDB1 reversed inhibitory effect of FENDRR overexpression on survivin expression (Figure 4(f)). From these results, it is clear that the interaction of FENDRR and SETDB1 is crucial for epigenetic silence of survivin.
Figure 4.
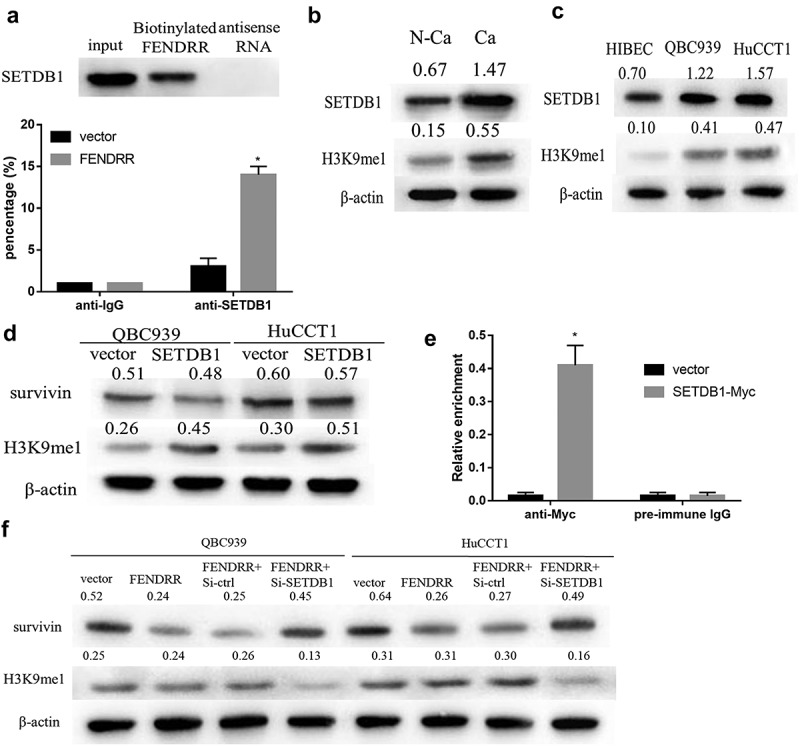
FENDRR inhibits survivin expression in SETDB1-dependent manner. (a): RNA pull-down and RIP assay were performed to determine whether FENDRR can combine with SETDB1. (b): The expression of SETDB1 and H3K9me1 in CCA tissues and paired adjacent non-cancerous tissues were measured by western blotting. (c): The expression of SETDB1 and H3K9me1 in normal cholangiocytes HIBEC cells and CCA cell lines were measured by western blotting. (d): The effect of SETDB1 on survivin and H3K9me1 expression in HuCCTl and QBC939 cells were determined by western blotting. (e): ChIP analysis of SETDB1 binding to the survivin promoter in CCA cell after transfection with FENDRR. (f): The effect of SETDB1 knockdown on survivin and H3K9me1 expression in HuCCTl and QBC939 cells after transfection with FENDRR were determined by western blotting. (*p < 0.05 vs. vector).
Discussion
In the present work, we report that FENDRR represses CCA cell proliferation, migration and invasion through suppression of survivin. And there exists a negative correlation between FENDRR and survivin expression in CCA tissues. Mechanistic investigation demonstrates that FENDRR recruits SETDB1 to bind to survivin gene promoter through combined with SETDB1, inducing H3K9 methylation.
Emerging evidence has revealed the important roles of FENDRR in cell differentiation and tumor progression. FENDRR could promote the apoptosis of human brain microvascular endothelial cells via miR-126 regulating VEGFA in hypertensive intracerebral hemorrhage [17]. Yan Li et al revealed that FENDRR has low expression in breast cancer cell lines and cancerous tissues, and inhibits cell proliferation, promotes cell apoptosis, and is associated with good prognosis in breast cancer [14]. FENDER was shown to be down-regulated in CCA tissue, however, to date, the functional significance of FENDER in CCA cell proliferation, migration and invasion remain elusive. In the present study, we demonstrated that FENDER is significantly decreased in CCA tissues or cell lines compared with adjacent non-cancerous tissues or HIBEC cells, indicating FENDER exerts important function in CCA. Gain- and loss-of-function assays demonstrated that FENDER inhibited cell proliferation, migration and invasion in CCA. FENDER may be an effective target for antimetastatic therapy.
Histone methylation plays a significant role in human carcinogenesis plays a significant role in human carcinogenesis. In the majority of colorectal cancer and liver cancer, the high expression of SMYD3 (H3K4 methyltransferase) have been observed, and down-regulation of SMYD3 significantly inhibit the proliferation of cancer cells [18], EZH2 (H3K27 methyltransferase), regulating BRCA1 gene, is involved in the proliferation and metastasis of breast cancer cells, and over-expression of EZH2 can promote the progression of breast cancer [19]. G9a (H3K9 methyltransferase) promotes the formation of breast cancer stem-like cells through Leptin-STAT3-G9a Signaling [20]. Methylation usually occurs in lysine residues (K) and arginine residues (R) of histone H3 and H4. SETDB1 protein belongs to the SET-domain (Su(var)3–9, E(z), Thrithorax) protein methyltransferase family [21], which is mainly involved in tri-methylation of H3K9 and usually cause gene silencing or transcription inhibition. In recent years, mounting evidence has linked the aberrant high expression of SETDB1 to the human carcinogenesis, including melanoma, lung cancer, breast cancer, ovarian cancer [22–24]. Further investigations are ongoing to explore the underlying mechanism of epigenetic modifications by SETDB1. In this study, we identified the expression of SETDB1 and H3K9me1 were significantly up-regulated in CCA tissues cells, as compared to that in normal tissues or HIBEC cell. These results suggested that SETDB1 were indeed up-regulated in CRC as well as in other solid tumors [22,24]. On the other hand, survivin is an oncogene that regulates the apoptosis, proliferation, and invasion of many cancers [25–27]. Knockdown of survivin by using siRNA could dramatically suppress the proliferation and invasion of bladder cancer cells [8]. Co-silencing of Livin and Survivin can effectively inhibit the cell proliferation and apoptosis of lung cancer cells [28]. In our work, the findings show that FENDRR inhibits survivin expression via SETDB1-mediated H3K9 methylation.
In summary, we demonstrated that FENDRR is down-regulated in CCA tissues. FENDRR represses cell proliferation, migration and invasion in CCA cells through SETDB1-mediated H3K9 methylation and epigenetically silencing survivin. Collectively, our findings provide a new perspective that the regulatory network of different lncRNAs may act as a critical role in CCA progress.
Materials and methods
Patients and tissue samples
A total of 60 patients with CCA who underwent surgical resection at the Henan Provincial People’s Hospital were included in the present study. Patients who were treated with preoperative radiotherapy or chemotherapy were excluded. CCA tissues and matched adjacent non‑cancerous tissues were collected and snap frozen in liquid nitrogen. Matched non‑cancerous tissues were obtained from regions of at least 3 cm distant from the tumor borders. All patients signed informed consent before surgery. This study was approved by the Human Ethics Committee of the Henan Provincial People’s Hospital.
Cell lines and cell culture
The human CCA cell lines (QBC939, SSP-25, HuCCTl, RBE and TFK-1) and normal biliary epithelium cell line HIBEC were purchased from the Shanghai Institute of Cell Biology (Shanghai, China). Cells were cultured in RPMI 1640 (invitrogen, carlsbad, CA, USA) medium supplemented with 10% fetal bovine serum (FBS; invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Beijing Solarbio Science & Technology Co., Beijing, China) in a humidified incubator in an atmosphere of 5% CO2 at 37°C.
Cell transfection
For the cell transfection experiments, plasmid-FENDRR, plasmid-survivin plasmid-control, si-FENDRR, and si-control RNA were synthesized by Invitrogen (Nanjing, China). Plasmid transfection was performed by using Lipofectamine 2000 reagent (Life Technologies Corporation, Carlsbad, USA) according to the manufacturer’s protocol. Finally, 48 h or 72 h after transfection, transfected cells were collected and used in further experiments.
QRT-PCR
Total RNAs were extracted from tumorous and adjacent normal tissues or cultured cells using Trizol reagent (Invitrogen) following the manufacturer’s protocol. RT and qPCR kits (Takara, Dalian, China) were used to evaluate the expression of FENDRR and survivin in tissue samples or cultured cells, the transcripts were quantified using the SYBR Premix Ex TaqTM II reagent (Takara, Japan) and a Lightcycler 480 Real-time PCR System (Roche, America). The primer sequences were designed as follows: FENDRR, 5’- TAAAATTGCAGATCCTCCG −3’ (forward primer) and 5’- AACGTTCGCATTGGTTTAGC-3’ (reverse primer); survivin, 5’-TCA AGG ACC ACC GCA TCT CTA C −3’ (forward primer) and 5’-TGA AGC AGA AGA AAC ACT GGG C-3’ (reverse primer). Real-time PCR was performed in triplicate, and the relative expression of FENDRR and survivin were calculated using the comparative cycle threshold (CT) (2−ΔΔCT) method with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the endogenous control to normalize the data.
Western blotting
Total proteins were extracted from ECC tissues or cell lines using RIPA lysis buffer (Beyotime, China) and detected quantified with the BCA kit (Beyotime Biotechnology). Equal volume of protein was subjected to SDS-PAGE and transferred onto polyvinylidene difluoride membranes. After blocking with 2% Bovine Serum Albumin (BSA) in phosphate buffered saline (PBS)/0.05% Tween for 2 h at room temperature, the membrane was incubated overnight at 4°C with corresponding primary antibodies including anti-survivin antibody (1:500; Santa Cruz Biotechnology, Inc, USA), anti-SETDB1 (1:1000; Santa Cruz Biotechnology, Inc, USA) and anti-H3K9me1 (1:500; Upstate Biotechnology, Lake Placid, NY, USA), furthermore, it was incubated for 2 h with horseradish peroxidase-conjugated secondary antibody (Boster, Wuhan, China) at room temperature and Electrochemiluminescence Detection Kit was used to detect immunoreactive bands according to the manufacturer’s instructions (Thermol Biotech Inc.; Rockford, IL, USA). GAPDH (Boster, Wuhan, China) was used as an internal control.
Cell proliferation
The proliferation of CCA cells was measured using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan). For the cell proliferation assay, after transfection, approximately 2 × 103 cells were seeded in each well of a 96-well plate, and 10 µl of CCK-8 solution (Dojindo, Kumamoto, Japan) was added to 90 µl of the culture medium at the indicated time at 37°C in 5% CO2 according to the manufacturers’ instruction. Finally, the ultraviolet absorbance at 450 nm of each well was measured using a microplate reader (Bio-Rad, USA).
Cell migration and invasion assay
Cells were suspended in 100 μL serum-free medium and were plated in the top chamber of each insert (8-μm pore size, Corning, USA) with a Matrigel-coated membrane (BD Bioscience, San Jose, USA) for the transwell invasion assay and a non-Matrigel-coated membrane for the transwell migration assay. Lower chambers of the inserts were filled with DMEM medium with 10% FBS. After 24 h of incubation, cells that invaded/migrated to the lower surface of the insert were fixed, stained, and counted under a light microscope.
RNA immunoprecipitation
RIP was performed using RIP kits (Millipore, Darmstadt, Germany) according to the manufacturer’s protocol. Briefly, cells were harvested and lysed in RIP lysis buffer. NEAT1 was immunoprecipitated with an SETDB1 antibody (Cell Signaling Technology, Danvers, MA, USA). IgG control was precoated onto protein A Sepharose beads at 4°C overnight. The magnetic bead bound complexes were immobilized with a magnet, and unbound materials were washed off. Finally, RNAs were extracted and stored at −80°C for further qRT-PCR analysis.
CHIP assay
All chromatin immunoprecipitation (ChIP) assays were executed using ChIP Kit according to the manufacturer’s instructions (Millipore, Boston, MA, USA). Briefly, CCA cell after transfection with FENDRR (5 × 106) was cross-linked with 1% formaldehyde for 10 min at room temperature. The crosslinking was then quenched by the addition of 0.125 M glycine. The soluble chromatin was sonicated to fragment DNA by nuclear lysis buffer. The fragmented chromatin samples were aliquoted as genomic input DNA or immunoprecipitated with SETDB1 (Cell Signaling Technology) and IgG (Cell Signaling Technology) and incubated at 4°C overnight. Immunocomplexes, collected by a magnetic separator, were washed and eluted with ChIP elution buffer. DNA was purified on spin columns. The ChIP products and genomic input DNA were analyzed by RT-PCR with SYBR Green PCR Master Mix (Applied Biosystems, Tokyo, Japan).
RNA pull-down assay
A RNA pull-down assay was carried out using 1.5 μg of biotinylated RNA antisense and incubated for 30 min at 25°C. Then, the mixtures were incubated with yeast tRNA (Sigma) which were pre-blocked by streptavidin beads (Invitrogen). After three times of washing with EMSA buffer, the beads were collected and the RNA and protein complexes were eluted. Resolved proteins were visualized using silver stain and analyzed by real-time PCR and western blot. Biotinylated sequence was obtained from PCR-generated templates.
Statistical analysis
All data were analyzed with SPSS 16.0. Data were presented as mean ± standard deviation (SD). Student’s t-test was used to analyze differences between the two groups. One-way ANOVA analysis was used to determine the multi-sample analysis. Differences at P < 0.05 were considered to be statistically significant.
Funding Statement
We acknowledge the Medical Science & Technology Support Project of Henan Province, Provincial & Ministerial Joint Projects (Nos. 201601024), and the Science & Technology Support Project of Henan Province (Nos. 132102310187, 122102310137, and 172102310517), and the “23456” Talent Support Project of Henan people’s hospital.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Wang WT, Ye H, Wei PP, et al. LncRNAs H19 and HULC, activated by oxidative stress, promote cell migration and invasion in cholangiocarcinoma through a ceRNA manner. J Hematol Oncol. 2016;9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bergquist A, von Seth E.. Epidemiology of cholangiocarcinoma. Best Pract Res Clin Gastroenterol. 2015;29:221–232. [DOI] [PubMed] [Google Scholar]
- [3].DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg. 2007;245:755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lau SH, Lau WY.. Current therapy of hilar cholangiocarcinoma. Hbpd Int. 2012;11:12–17. [DOI] [PubMed] [Google Scholar]
- [5].Rizvi S, Borad MJ, Patel T, et al. Cholangiocarcinoma: molecular pathways and therapeutic opportunities. Semin Liver Dis. 2014;34:456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis. 2007;28:1133–1139. [DOI] [PubMed] [Google Scholar]
- [7].Liu Z, Zhang X, Xu X, et al. RUNX3 inhibits survivin expression and induces cell apoptosis in gastric cancer. Eur J Cell Biol. 2014;93:118–126. [DOI] [PubMed] [Google Scholar]
- [8].Yang R, Liu M, Liang H, et al. miR-138-5p contributes to cell proliferation and invasion by targeting Survivin in bladder cancer cells. Mol Cancer. 2016;15:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sankpal UT, Ingersoll SB, Ahmad S, et al. Association of Sp1 and survivin in epithelial ovarian cancer: sp1 inhibitor and cisplatin, a novel combination for inhibiting epithelial ovarian cancer cell proliferation. Tumour Biol. 2016;37:14259–14269. [DOI] [PubMed] [Google Scholar]
- [10].Zhong F, Yang J, Tong ZT, et al. Guggulsterone inhibits human cholangiocarcinoma Sk-ChA-1 and Mz-ChA-1 cell growth by inducing caspase-dependent apoptosis and downregulation of survivin and Bcl-2 expression. Oncol Lett. 2015;10:1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Koprowski S, Sokolowski K, Kunnimalaiyaan S, et al. Curcumin-mediated regulation of Notch1/hairy and enhancer of split-1/survivin: molecular targeting in cholangiocarcinoma. J Surg Res. 2015;198:434–440. [DOI] [PubMed] [Google Scholar]
- [12].Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wangyang Z, Daolin J, Yi X, et al. NcRNAs and cholangiocarcinoma. J Cancer. 2018;9:100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li Y, Zhang W, Liu P, et al. Long non-coding RNA FENDRR inhibits cell proliferation and is associated with good prognosis in breast cancer. Onco Targets Ther. 2018;11:1403–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang G, Han G, Zhang X, et al. Long non-coding RNA FENDRR reduces prostate cancer malignancy by competitively binding miR-18a-5p with RUNX1. Biomarkers. 2018;23:435–445. [DOI] [PubMed] [Google Scholar]
- [16].Xu TP, Huang MD, Xia R, et al. Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J Hematol Oncol. 2014;7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dong B, Zhou B, Sun Z. LncRNA-FENDRR mediates VEGFA to promote the apoptosis of brain microvascular endothelial cells via regulating miR-126 in mice with hypertensive intracerebral hemorrhage. Microcirculation. 2018;25:e12499. [DOI] [PubMed] [Google Scholar]
- [18].Sarris ME, Moulos P, Haroniti A, et al. Smyd3 is a transcriptional potentiator of multiple cancer-promo-ting genes and required for liver and colon cancer development. Cancer Cell. 2016;29:354–366. [DOI] [PubMed] [Google Scholar]
- [19].Granit RZ, Gabai Y, Hadar T, et al. EZH2 promotes a bi-lineage identity in basal-like breast cancer cells. Oncogene. 2013;32:3886–3895. [DOI] [PubMed] [Google Scholar]
- [20].Chang CC, Wu MJ, Yang JY, et al. Leptin-STAT3-G9a signaling promotes obesity-mediated breast cancer progression. Cancer Res. 2015;75:2375–2386. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- [21].Harte PJ, Wu W, Carrasquillo MM, et al. Assignment of a novel bifurcated SET domain gene, SETDB1, to human chromosome band 1q21 by in situ hybridization and radiation hybrids. Cytogenet Cell Genet. 1999;84:83–86. [DOI] [PubMed] [Google Scholar]
- [22].Ceol CJ, Houvras Y, Jane-Valbuena J, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang H, Cai K, Wang J, et al. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells. 2014;32:2858–2868. [DOI] [PubMed] [Google Scholar]
- [24].Regina C, Compagnone M, Peschiaroli A, et al. Setdb1, a novel interactor of DeltaNp63, is involved in breast tumorigenesis. Oncotarget. 2016;7:28836–28848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lu M, Wang C, Wang J. Tanshinone I induces human colorectal cancer cell apoptosis: the potential roles of Aurora A-p53 and survivin-mediated signaling pathways. Int J Oncol. 2016;49:603–610. [DOI] [PubMed] [Google Scholar]
- [26].Zhang J, Wang S, Han F, et al. MicroRNA-542-3p suppresses cellular proliferation of bladder cancer cells through post-transcriptionally regulating survivin. Gene. 2016;579:146–152. [DOI] [PubMed] [Google Scholar]
- [27].Zhou XL, Wang M. Expression levels of survivin, Bcl-2, and KAI1 proteins in cervical cancer and their correlation with metastasis. Genet mol res: GMR. 2015;14:17059–17067. [DOI] [PubMed] [Google Scholar]
- [28].Zhao X, Yuan Y, Zhang Z, et al. Effects of shRNA-silenced livin and survivin on lung cancer cell proliferation and apoptosis. J BUON. 2014;19:757–762. [PubMed] [Google Scholar]