

ABSTRACT
Depletion of T regulatory cells (Tregs) in the tumor microenvironment is a promising cancer immunotherapy strategy. Current approaches for depleting Tregs are limited by lack of specificity and concurrent depletion of anti-tumor effector T cells. The transcription factor forkhead box p3 (Foxp3) plays a central role in the development and function of Tregs and is an ideal target in Tregs, but Foxp3 is an intracellular, undruggable protein to date. We have generated a T cell receptor mimic antibody, “Foxp3-#32,” recognizing a Foxp3-derived epitope in the context of HLA-A*02:01. The mAb Foxp3-#32 selectively recognizes CD4 + CD25 + CD127low and Foxp3 + Tregs also expressing HLA-A*02:01 and depletes these cells via antibody-mediated cellular cytotoxicity. Foxp3-#32 mAb depleted Tregs in xenografts of PBMCs from a healthy donor and ascites fluid from a cancer patient. A TCRm mAb targeting intracellular Foxp3 epitope represents an approach to deplete Tregs.
KEYWORDS: Immunosuppression, Tregs, Foxp3, TCRm mAb, immunotherapy
Introduction
The efficacy of T cell checkpoint blockade therapy has demonstrated the importance of reducing immunosuppressive pathways in tumors as a strategy for successful immunotherapy. Regulatory T cells (Tregs) are powerful inhibitors of anti-tumor immunity and a critical barrier to successful immunotherapy. Tregs expressing CD4, CD25 and Foxp3 are often found abundantly in the tumor microenvironment and decreased ratios of CD8 + T cells to Tregs in tumor-infiltrating lymphocytes (TILs) are an indicator of poor prognosis in various human cancers.1–6 Depletion of Tregs has been shown to enhance spontaneous and vaccine-induced anti-tumor immune responses in animal models and in cancer patients.7,8 These studies demonstrate the prominent tumor-promoting role of Tregs and provide an opportunity for cancer immunotherapy in which inhibition of Treg function in the tumor environment could permit improved antitumor immune effector competency.
Although depletion of Tregs in patients with cancer has received intense interest, effective applications of this strategy have been hampered because Tregs do not express a specific cell surface protein that can be targeted by a mAb; nor do Tregs posses a selectively druggable regulatory pathway. Various strategies have been attempted for depletion of Tregs or inhibition of Treg function, which typically have focused on using mAbs to surface protein expressed on Tregs. These include using mAb (Daclizumab) specific for CD25, the IL-2 receptor, and Dennileukin difitox, a fusion protein of IL-2 and diphtheria toxin.9,10 In addition, elimination of Tregs by a mAb to glucocorticoid-induced TNF-related protein (GITR) and suppression of Treg function by use of anti-CTLA4 antibodies, disruption of tumor homing by Tregs, or modulation of T cell plasticity, also have been attempted.11–14 Clinical studies with various cancers, in which Daclizumab and Dennileukin difitox were combined with vaccines, have demonstrated conflicting results,15,16 perhaps because CD25 and GITR are also expressed on activated CD4 and CD8 effector T cells.17 Therefore, a potential benefit of Treg depletion by targeting these molecules could be lost by concurrent elimination of activated effector lymphocytes. A recent study in vitro showed that a mAb specific for chemokine receptor-4 (CCR4) could selectively deplete effector Tregs expressing a higher level of Foxp3, which resulted in augmentation of CD8 T cell response specific for the NY-ESO-1 peptide.18 CCR4 is expressed on activated T cells, T helper 2, NK cells, macrophages, and dendritic cells, therefore confounding the selective effects of the mAb. A defucosylated, humanized anti-CCR4 mAb, Mogamulizumab, has been in clinical trials in various cancers, but its efficacy remains to be determined.19,20 Similarly, a recent study showed that another chemokine receptor, CCR8, is preferentially expressed on Tregs in breast cancer patients and is associated with poor prognosis.21 This chemokine receptor is also expressed on tissue-resident memory CD8 + T cells and NK-T cells, and therefore, the therapeutic potential of targeting this molecule remains to be investigated. In addition, modulating transforming growth factor (TGF)-beta, a crucial cytokine for Treg function has also been tried.22 Cyclophosphamide has been shown to suppress Tregs as well, but the mechanisms of selectivity are unknown, and its broad cytotoxic actions toward both normal lymphocytes and neoplastic cells, renders attribution of its effects to Treg depletion difficult.23,24
Foxp3 has been identified as a key mediator of Treg function and is also the most definitive marker of CD4 + CD25 + Tregs. Foxp3 is required for Treg cell lineage differentiation, maintenance and importantly, Treg suppressive functions. Apart from naturally occurring Tregs that arise in the thymus, inducible Treg cells have been identified, with predominance in infection and cancer.25,26 Interestingly, in addition to the critical role of Foxp3 in Tregs, many cancer cells also express Foxp3 protein. Foxp3-expressing pancreatic carcinoma cells and cutaneous T cell lymphoma cells have been shown to suppress T cell proliferation.27–29 These studies suggest that in the tumor microenvironment, Treg-like cancer cells can act as suppressor cells, possibly representing a new mechanism of immune evasion.
There is a great need for more specific approaches to suppress Tregs, in order to assess the function of these cells and to pharmacologically regulate the cells for therapeutic effect in a number of clinical settings. Foxp3 would be an ideal target for these attempts. However, Foxp3 is currently not druggable by small molecules and as an intracellular protein, unreachable by traditional mAbs. In principle, peptides from the Foxp3 protein that are degraded and processed for cell surface presentation could serve as targets of TCR recognition. In a melanoma mouse model, mice vaccinated with dendritic cells (DCs) electroporated with Foxp3-encoding mRNA elicited Foxp3-specific CTL responses leading to preferential depletion of Foxp3 + Tregs in tumors. Simultaneous vaccination of mice with the TRP2 melanoma antigen and Foxp3 enhanced the vaccine-induced protection against highly metastatic B6/F10.9 melanoma.30 Although no Foxp3-derived epitopes were identified in these studies, it demonstrated the possibility of using a TCR-based approach to target Foxp3 peptides in Tregs. Encouragingly, a recent in vitro human study has identified Foxp3-derived epitopes in the context of HLA-A*02:01 molecules that induce CTL capable of killing Foxp3-expressing T lymphoma cells.31 We explored the possibility of creating a TCR-mimic mAb (TCRm) specific for Foxp3-derived epitopes as an approach to selective depletion of Tregs. Additional direct anti-cancer effects also would be possible with such a TCRm mAb for cancer cells that express Foxp3. A TCRm mAb is a typical antibody Ig structure that combines two important features of T cells and mAbs. First, it offers TCR-like recognition of a peptide/MHC complex, allowing mAb access to intracellular antigens. Second, it can fully utilize the versatility, functions, and potency of a traditional mAb, allowing engineering and modification of the mAb forms, to enhance the potency and control the dosage. This approach can bypass patient-specific T cell therapy, allowing the therapy to a vast majority of patients. Using phage display technology, we have generated a human TCRm mAb, named Foxp3-#32, that recognizes a human Foxp3-derived epitope in the context of HLA-A*02:01. The mAb binds to and mediates antibody-dependent cellular cytotoxicity (ADCC) against Foxp3-expressing Tregs and Foxp3-expressing malignant T cells. We also engineered the Foxp3-#32 mAb into different forms: a bispecific T cell engager to further enhance its potency and a mouse IgG1 form to use for characterization. Our study provides a proof of concept that a TCRm mAb could be a novel approach to target Tregs and Foxp3-expressing human cancers.
Results
Selection of Foxp3-derived epitopes in the context of HLA-A*02:01
There is little information on the epitopes derived from Foxp3 that could induce T cell responses. Therefore, we identified immunogenic epitopes that could generate cytotoxic CD8 T cells against Foxp3. The entire human Foxp3 protein sequence was screened using three computer-based predictive algorithms BIMAS (http://www-bimas.cit.nih.gov/cgi-bin/molbio/ken_parker_comboform), SYFPEITHI (http://www.syfpeithi.de/) and RANKPEP (http://bio.dfci.harvard.edu/Tools/rankpep.html) to identify potential high-affinity binders to HLA-A*02:01. We selected a number of potential epitopes derived from human Foxp3 for CD8 T cells in the context of HLA-A*02:01 molecule to test if the peptides were able to induce specific CD8 T cell responses (Supplemental Table 1). Importantly, all the selected HLA-A*02:01-binding peptides were predicted to be cleaved at the C-terminal, suggesting a higher probability of being processed by proteasomes.
Peptide-specific T cell response in the context of HLA-A*02:01 molecule
As the computer algorithms are not always predictive of in vitro or in vivo activity, we tested potential presentation of the predicted peptides by HLA-A*02 binding on T2 cells and by their ability to stimulate peptide-specific CD8 T cell responses from HLA- HLA-A*02:01+ donors. Initially, seven peptides were selected to test T cell responses (Supplemental Table 1). Six out of seven peptides (except for peptide 304–312) consistently induced peptide-specific T cell responses in multiple donors. Because human Foxp3 is a member of a large forkhead family of related proteins, to avoid potential off-targets shared within the family proteins, we selected the peptide TLIRWAILEA (position 344–353; “TLI”) from among the sequences as the epitope on which to focus because the TLI peptide has minimal homology with other Foxp family members, such as Foxp1, 2 and 4. Interestingly, this peptide has also been shown to induce strong peptide-specific CD8 + T cell responses, which recognize Foxp3+/HLA-A*02:01+cutaneous T lymphoma cells.31
CD3 + T cells from multiple HLA-A*02:01 + donors were stimulated 3 to 5 times with the TLI peptide, and the peptide-specific T cell response was measured by IFN-γ ELISPOT and 51Cr-release assays. After four rounds of stimulation, T cells recognized autologous CD14+ monocytes pulsed with TLI peptide, but not CD14+ APC alone or pulsed with an irrelevant HLA-A*02:01-binding peptide EW, by IFN-γ ELISPOT assay (Figure 1(a)). Importantly, a T cell response was also observed against HLA-A*02:01 + Foxp3+ cutaneous T lymphoma cell lines MAC-1 and MAC-2A,31 but not the Foxp3 negative/HLA-A* 02:01 negative T leukemia cell line Jurkat, suggesting that TLI-stimulated T cells could recognize a naturally processed Foxp3 epitope presented by HLA-A*02:01 molecule (Figure 1(b)). Consistent with the results of IFN-γ secretion, TLI peptide-stimulated T cells killed T2 cells pulsed with the TLI peptide and MAC-1 cells that had not been pulsed with peptide but did not kill the HLA-A*02:01 negative, Foxp3+ cell line HL-60 (Figure 1(c,d).
Figure 1.
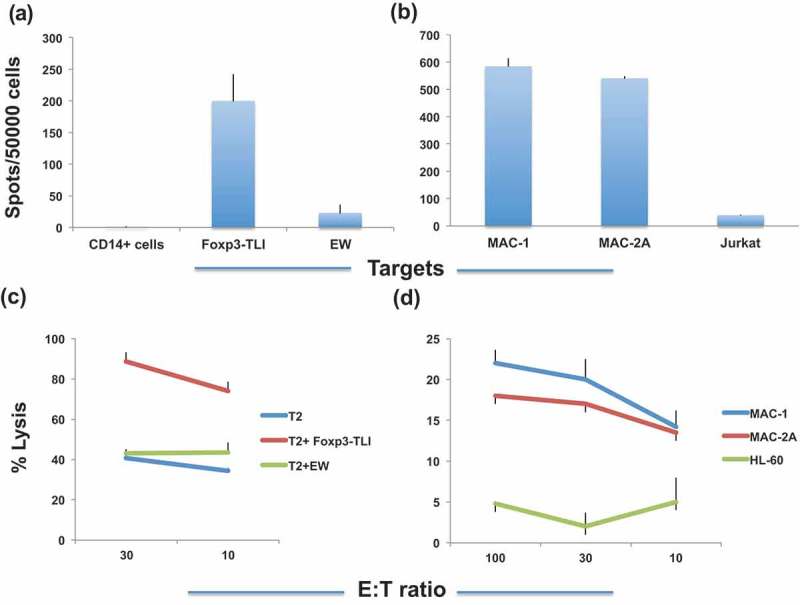
Foxp3-TLI induces peptide-specific T cell response. (a). CD3 T cells from HLA-A*02:01+ donors were stimulated with Foxp3-TLI peptide for four rounds and T cell response (recorded as spots per added effector cells on the Y-axis) was measured against TLI peptide or with irrelevant peptide EW pulsed on T2 cells by IFN-gamma ELISPOT assay. CD14+ APC’s serve as a negative control. (b). TLI-stimulated T cells also recognize MAC-1 and MAC-2A cells but not HLA-A0*201-cell line Jurkat. Each point represents average ± SD from triplicate cultures. Data represent results from five similar experiments. (c). T cells from an HLA-A*02:01+ donor were stimulated for five rounds, and the cytotoxicity was measured by 51Cr-release (Y-axis) against the TLI or EW control peptides pulsed onto T2 cells. Data show the average plus/minus SD from triplicate wells and one of two similar experiments. (d). Similarly, unpulsed T lymphoma cell lines MAC-1 and MAC-2A (HLA-A*02:01 + Foxp3+) cell line or HLA-A*02:01 negative AML cell line HL-60 were used targets for T cell cytotoxicity. Each point represents average ±SD from three similar experiments from three donors.
Selection of a TCR-mimic mAb specific for Foxp3 peptide TIL in the context of HLA- A*02:01 molecule using phage display technology
By confirming that the Foxp3-TLI peptide is able to induce an epitope-specific T cell response that recognizes tumor cells expressing the Foxp3 protein, we proceeded to generate a TCRm mAb specific for the TLI/HLA-A*02:01 complex, by using phage display technology as previously described.32 The selected clones were tested for their binding to live HLA-A*02:01 + T2 cells pulsed with TLI or control peptides. Any clones that showed binding to T2 cells without the TLI peptide or with HLA-A*02:01-complexed with irrelevant peptides were removed. Based on these data and binding to live cells that express Foxp3 and HLA-A*02:01 we selected eight scFv clones for additional characterization.
Characterization of bispecific mAbs specific for Foxp3 TIL/HLA-A*02:01complex
Cell surface epitope density for TCR and TCRm targets is expected to be 50–100 times lower than for typical mAbs recognizing cell surface proteins, which may limit cytolytic activity.32 Therefore, as a strategy to enhance the TCRm cytotoxicity, we generated bi-specific T cell engager constructs (a “BiTE” form of bispecific mAb: one arm recognizes Foxp3 TLI/HLA-A*02:01, and the other arm recognizes human CD3) of the eight selected clones reactive with the Foxp3-TLI peptide/HLA-A*02:01 complex. The bispecific mAbs were tested against T2 cells pulsed with or without Foxp3-TLI or an irrelevant peptide. While all eight bispecific mAb constructs showed binding to T2 cells pulsed with Foxp3-TLI peptide to various degrees, and no binding to T2 cells alone or with control peptides, only mAb Foxp3-#32 bound to the Foxp3+/HLA-A*02:01 positive T cell lymphoma lines MAC-1 and MAC-2A. Figure 2(a,b) show binding of Foxp3-#32 bispecific mAb to T2 pulsed with TLI peptide and MAC-2A with no peptide pulsing. Since MAC-2A does not express CD3, the binding is mediated by the arm of the bispecific mAb to Foxp3p/HLA-A*02:01 complex. To confirm that the arm of anti-CD3 of the bispecific mAb is functional, a CD3+, HLA-A*02:01 and Foxp3 negative cell line Jurkat was used. Foxp3-#32 bispecific mAb was able to bind Jurkat (Figure 2(c)), showing the function of the CD3 reactive arm. To further demonstrate the specificity of the Foxp3-#32 mAb, we used full-length Foxp3-#32 IgG (Fc portion was exchanged to a mouse IgG1 to avoid human FcR mediated binding). The mAb bound to MAC-2A, but not Jurkat (Figure 2(e,f)). This further demonstrated that the binding of the Foxp3-#32 bispecific mAb to Jurkat is purely due to the anti-CD3 scFv in the construct. HLA-A2 expression by both MAC-2A and Jurkat is shown in Figure 2(d), demonstrating the binding of Foxp3-#32 is restricted to HLA-A*02:01 expressing cells.
Figure 2.
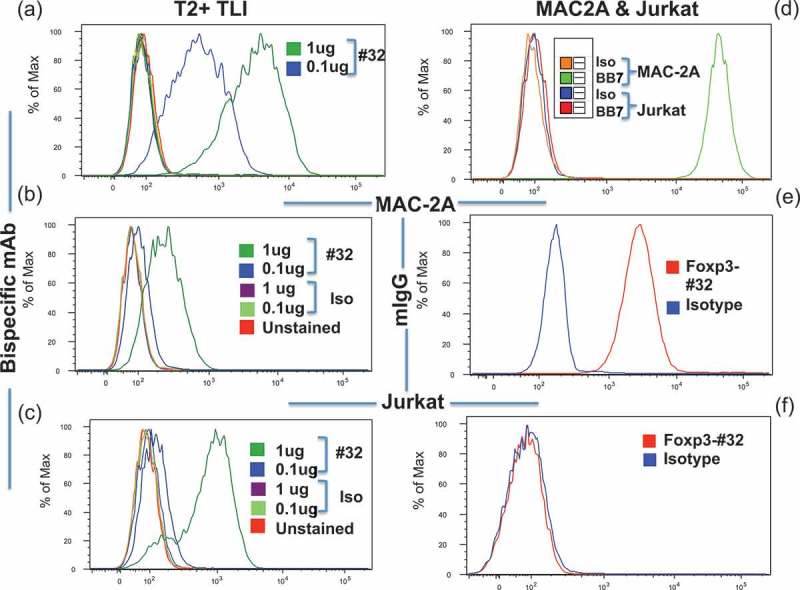
Binding of the Foxp3-#32 mAb. (a) Binding of T2 cells pulsed with TLI peptide by Fox3-#32 bispecific mAb at the concentration of 0.1ug/ml or 1ug/ml. Other cell controls were included: T2 cells alone, or T2 cells pulsed with irrelevant A2-binding peptide CT. The staining included: Secondary mAb against myc tag (GA6xHis, for the bispecific constructs), or the same concentrations of Foxp3-#32 or control irrelevant bispecific mAb (clone NC-16). All the controls showed no binding above the unstained level (histogram plots overlapped). There was dose-dependent binding of Fox3-#32 bispecific mAb. Foxp3-#32 bispecific mAb was used at 1ug (dark green) or 0.1ug/ml (dark blue). (b) and (c). Fox3-#32 bispecific mAb binding to unpulsed MAC-2A (b) and Jurkat (c), respectively. (d). HLA-A02 expression on MAC-2A (green line) or Jurkat (red line) cells was measured by staining the cells with anti-A2 mAb BB7 and/or its isotype control mouse IgG2b (orange for MAC-2A and blue for Jurkat)), as indicated. (e) and (f). Binding of a full-length mouse IgG1 Fox3-#32 mAb (red line) or its isotype control (blue) to MAC-2A (e) and Jurkat (f), respectively. Mabs were used at 1 ug/ml. Binding strength is shown by log median fluorescent intensity on the X-axis and number of cells on the Y-axis.
TCRs typically recognize the middle positions of the peptides presented by MHC class-I. To analyze which amino acids of the TLI peptide were important residues required for recognition by the Foxp3-#32 mAb, we analyzed the binding of both the full length mIgG1 Foxp3-#32 and Foxp3-#32 bispecific mAb to T2 cells pulsed with analog TLI peptides. TLI peptide was substituted with alanine at position 1, 2, 3, 4, 5, 7, 8, and 9, or with glycine at position 10. Position 6 was already alanine and it was left intact. The mutant peptides were loaded onto T2 cells and tested for Foxp3-#32 mAb (mouse IgG1) binding and for HLA-A02 expression. Alanine or glycine substitution at position 2, 4, 8, 9, or 10 strongly reduced the binding of Foxp3-#32 mAb (Figure 3(a), upper panel). HLA-A2 expression was substantially reduced after pulsing with analogs with position 2 and position 10 substitution (Figure 3, lower panel), suggesting, as expected, that these positions are the anchor residues and therefore Foxp3-#32 binding after changes at position 2 and position 10 are most likely due to the lack of binding of these peptides to the HLA-A*02 molecule, rather than binding of the antibody to the complex. Taken together, the data showed that the most crucial residues recognized by Fox3-#32 mAb are positions 4, 8, and 9 and that positions 2 and 10 are needed for the peptide to bind to the HLA. A crystal structure would be needed to more precisely characterize the epitope.33
Figure 3.
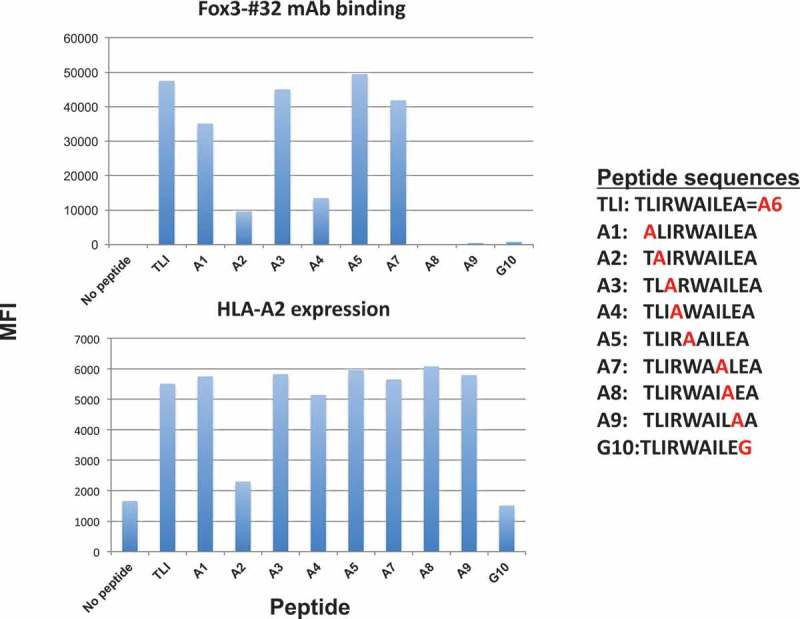
Epitope specificity. The Foxp3-TLI peptide sequence was substituted with alanine at positions 1, 2, 3, 4, 5, 7, 8, 9 or with glycine (G10) at position 10 as shown in the insert. T2 cells were pulsed with indicated peptides at 50ug/ml, and the binding of a full-length Foxp3-#32 mAb (mouse IgG1) conjugated to APC (3ug/ml) was measured by flow cytometry (upper panel). The same cells were simultaneously stained with anti-HLA-A2 mAb, clone BB7.2, to measure the relative binding of the peptides to HLA-A2 molecule (lower panel).
Recognition of human Tregs expressing Foxp3 and HLA-A*02:01 by Foxp3-#32 mAb
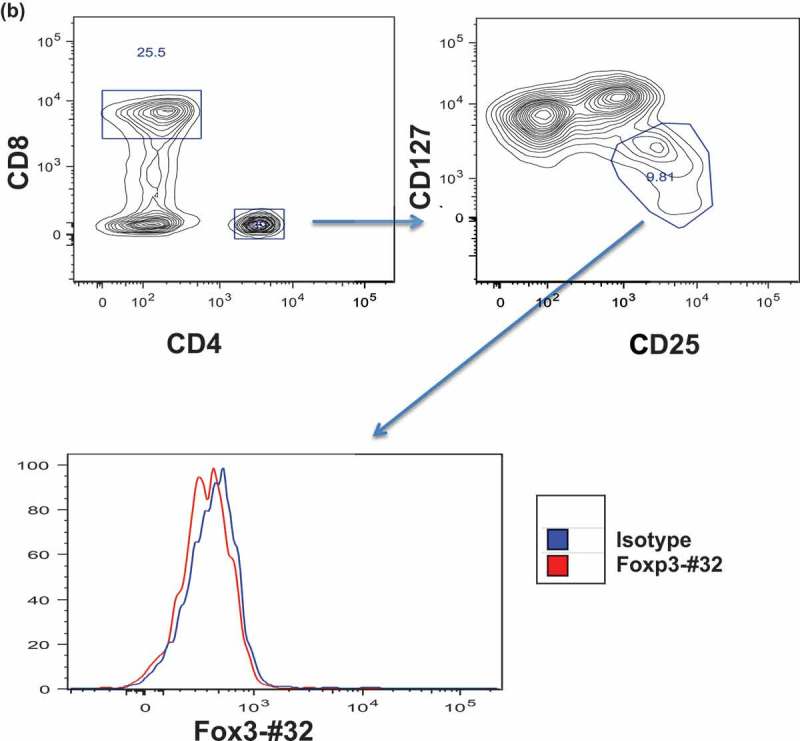
Although the Foxp3-#32 mAb has demonstrated selective binding to T2 cells pulsed with TLI peptide, it was crucial to test if the recognized epitope is processed and presented by HLA-A*02:01 molecule in naturally occurring Tregs and induced Tregs. We compared Foxp3-#32 mAb binding to Tregs from HLA-A*02:01 positive or negative PBMCs from healthy donors. CD4 + T cells were gated on CD25 high/CD127 low population, characteristic markers of natural Tregs. The binding by Foxp3-#32 mAb was predominately seen in the CD4+ CD25hiCD127lo population compared to its isotype control in HLA-A*02:01 + donor (Figure 4(a), lower right histogram), but not to CD4 + CD25int/loCD127hi cells (Figure 4(a), lower left histogram). The mAb Foxp3-#32 did not bind to the same CD4 + CD25hiCD127lo Treg population from an HLA-A*02:01 negative donor (Figure 4(b)), nor to the CD3/CD8 double positive T cells from HLA-A*02:01 positive donor (supplementary Figure 1A).
Figure 4.
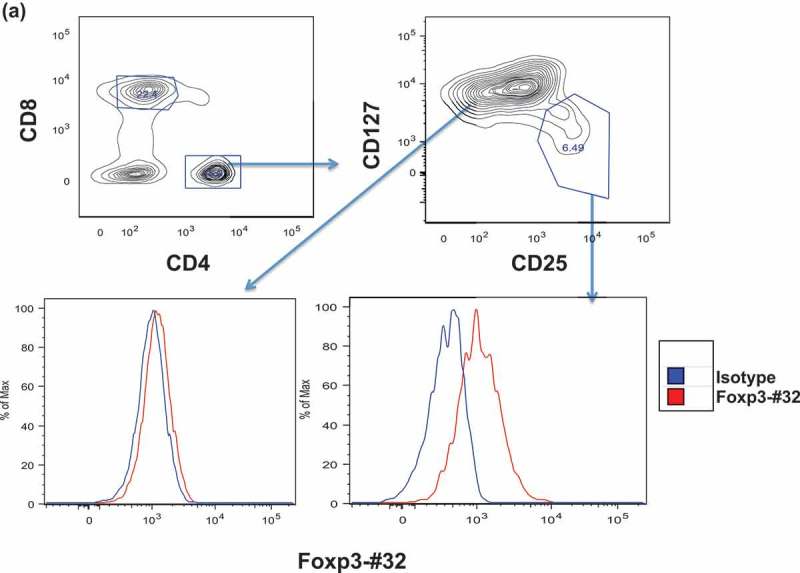
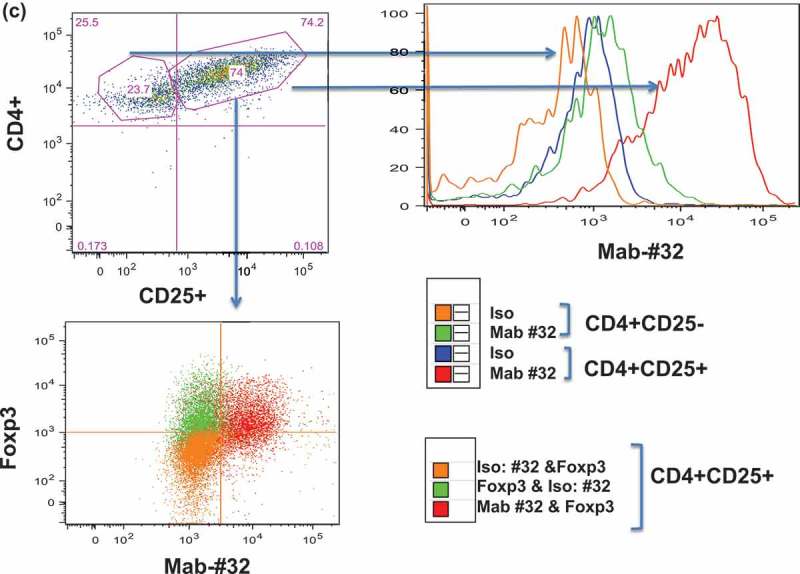
Binding of Foxp3-#32 mAb to Treg cells. Mouse IgG1 version of Foxp3-#32 mAb was used to test the binding, in order to exclude any FcR-mediated binding A-B. Binding of Foxp3-#32 mAb to natural Treg cells in PBMC in healthy donors. PBMCs were stained with mAbs specific for CD4, CD25 CD127, and Foxp3-#32 mouse IgG1. Data show that mAb Foxp3-#32 only bound to CD4+/CD25high/CD127low Tregs, not CD4+/25low/CD127high conventional T cell population (a), nor CD4+/CD25high/CD127low Tegs from an HLA-A0*201 negative donor (b). Data show representative results from three sets of different individuals. C-D. Binding of the Foxp3-#32 mAb to in vitro-generated Treg cells. CD4+ T cells from a HLA-A*02:01+ donor were FACS sorted and stimulated with either MAC-2A cells (c) or allo-PBMC (d) as both stimulator and feeder cells, in the presence of IL-2 (100 unit) and TGF-β (10ng/ml) for three weekly stimulations. Cells were stained with mAbs to surface CD4, CD25, intracellular Foxp3 and mAb Foxp3-#32. MAbs Foxp3-#32 binding was determined by gating on the DAPI-, CD4 and CD25 double positive (upper left panel, upper right quadrant) or CD4 + CD25- (upper left panel, upper left quadrant) cells Dot plot overlay show the Foxp3-#32 plus Foxp3 protein dual staining (lower left panel, red dots), and its isotype control mouse IgG1 and rat isotype control for mAb to Foxp3 (dual controls, orange dots) and mAbs to Foxp3 protein plus isotype control mouse IgG1 for Foxp3-#32 mAb (green dots), in the CD4 + CD25+ population. When the CD4 + CD25+ population was gated, strong binding of mAb Foxp3-#32 (red line) was shown as compared to its isotype control (blue line) (upper right panel) and to the CD4 + CD25 negative population (green line). Similar results were also seen in Tregs generated by allogenic (allo)-PBMC stimulation using a HLA-A*02:01 negative donor (Figure 4(d)). Data represent one of three experiments from three sets of healthy donors.
Figure 4.
(Continued).
There have been a number of methods to generate Tregs in vitro that would yield increased numbers of Tregs to study.34,35 Therefore, to test if the Foxp3-#32 mAbs could recognize these produced Tregs, we generated Treg lines by repetitive stimulation of purified CD4 + T cells from HLA-A*02:01+ donors with either allo-PBMCs or tumor cells MAC-2A in the presence of IL-2 and TGF-β, because tumor cells have been shown to induce Tregs. T cells generated by tumor stimulation resulted in a population of 74% CD4 + CD25 + cells (Figure 4(c), upper left panel) that was dual stained positive for both intracytoplasmic Foxp3 protein and Foxp3-#32 mAb (Figure 4(c), lower left panel, red dots). Isotype for Foxp3-#32 did not stain intracellular Foxp3+ population (green dots). Dual isotype (mAbs for Foxp3 protein and #32 mAb) controls showed no binding to these cells (orange dots). However, strong binding of mAb Foxp3-#32 (red curve) was shown as compared to its isotype control (blue curve) on CD4 + CD25+ population (Figure 4(c), upper right panel). There was far weaker binding of the mAb-Foxp3-#32 to the CD4 + CD25-population (green curve vs orange curve). This low level of positivity may be due to gating imperfections or to some activated cells also expressing low levels of the epitope. It is also possible that Foxp3 may be transiently expressed on activated CD4 T cells, in addition to Tregs bearing the same gated markers. Similar results were also seen in Tregs generated by allogenic (allo)-PBMC stimulation using an HLA-A*02:01 negative donor (Figure 4(d)). Altogether, these results demonstrated that Foxp3-#32 mAb is able to recognize human Treg cells derived from three different methods of preparation.
Figure 4.
(Continued).
Figure 4.

(Continued).
Many types of human cancer cells express Foxp3, which is associated with poor prognosis and greater metastatic potential. Especially, T cell malignancies have been shown to share the characteristics of Tregs, phenotypically and functionally.36,37 Therefore, a Foxp3-targeting mAb could also potentially kill tumor cells expressing Foxp3. In addition to the MAC-2A T lymphoma cell line, we found a T leukemia virus-transduced cell line, C5MJ that also expressed Foxp3 and we transduced C5MJ cell line with HLA-A*02:01. We tested if the Foxp3-#32 mAb could recognize these two cell lines, that could be used as target cells for ADCC by the Foxp3-#32 mAb. The binding of the Foxp3-#32 mAb was demonstrated on MAC-2A and C5MJ/A2 cells (Supplemental Figure 1B).
Foxp3-#32 bispecific mAb-mediated T cell cytotoxicity against Foxp3+ Tregs and tumor cells
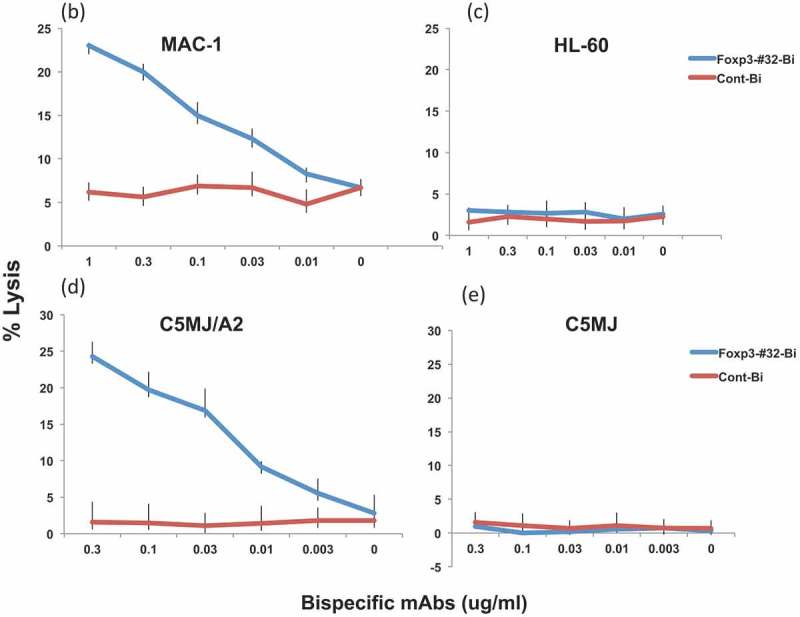
Having demonstrated the binding of the Foxp3-#32 to the Foxp3+ HLA-A2+ cells, we next tested if the Foxp3-#32 bispecific mAb mediates cytolytic activity such as ADCC. First, T2 cells pulsed with TLI or control HLA-A*02:01-binding peptide CT, were incubated with human PBMCs used as effectors, in the presence or absence of the Foxp3-#32-bispecific mAb or its control bispecific mAb. Foxp3-#32 bispecific mAb-mediated specific, effective killing against T2 cells pulsed with TLI peptide, but not T2 cells alone or pulsed with control peptide (Figure 5(a)), nor Foxp3 negative/HLA-A*02:01 negative cell line HL-60 (Figure 5(c)). Similarly, PBMCs in the presence of Foxp3-#32-bispecific mAb showed dose-dependent killing against Treg-like T lymphoma cell lines MAC-1, T leukemia cell line C5MJ transduced with HLA-A*02:01, but not its parental cells HLA-A02 negative C5MJ, in a dose-dependent manner (Figure 5(b,d, and e). The MAC-1 cell line does not express CD3, and T cell cytotoxicity against the cells was not mediated by the scFv arm of the anti-CD3 mAb.
Figure 5.

Foxp3-Foxp3-#32-bispecific mAb-mediated T cell cytotoxicity against Foxp3+/HLA-A*02:01+ cells. PBMCs were incubated with: (a) Foxp3-#32 bispecific mAb against T2 pulsed with TLI peptide, green line. Controls included: TLI-pulsed T2 cells alone, blue line; control T2 cells alone, red line; a control bispecific mAb against T2 alone; control bispecific mAb against T2 pulsed with TLI peptide, purple line; Foxp3-#32 bispecific mAb against T2 pulsed with CT control peptide, light blue line; control bispecific mAb against T2 pulsed with control EW peptide, orange line. Data are representative of mean ± SD from triplicate wells and one of three experiments. Bispecific mAb Fosp3-#32 (blue line) or control (red line) was also tested against unpulsed cell lines MAC-1 (b), HL-60 (c), C5MJ/A2+ (d) or C5MJ (e) at the concentrations ranging from 1ug/ml to 0.003 ug/ml. The data represent the mean value ±SD from three experiments. The cytotoxicity was measured by 5 h 51Cr release assay.
Figure 5.
(Continued).
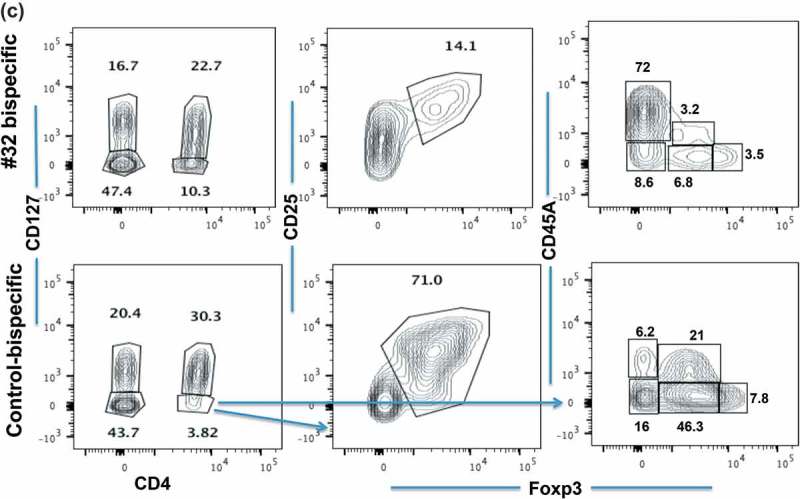
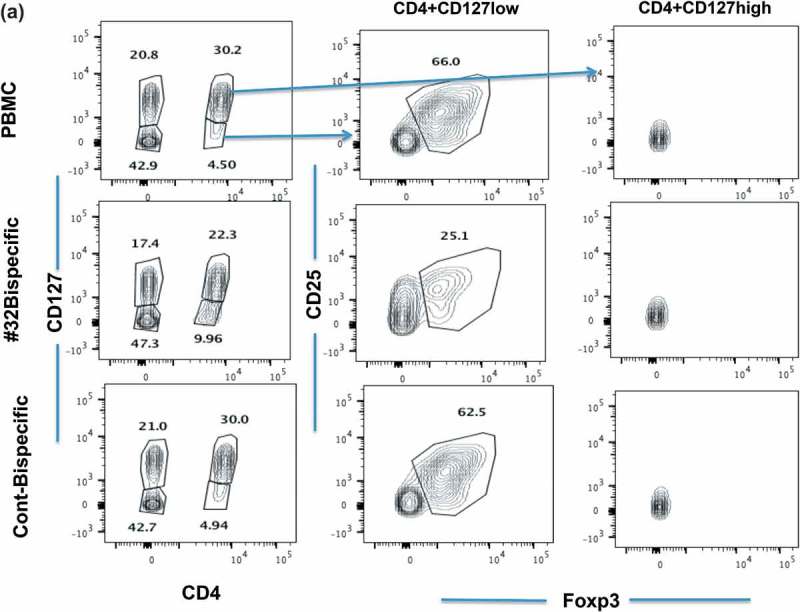
We next asked the most important question of whether the cytolytic function of Foxp3-#32 bispecific mAb is able to selectively deplete natural Tregs from PBMCs by using flow cytometric analyses with a panel of well-defined Treg markers. The lymphocyte population was first gated based on forward and side scatter. Since the mAb is targeting a Foxp3-derived epitope, the reduction of the Foxp3+ population in the cells that express bona fide Treg markers would provide direct evidence for depletion of the Foxp3+ Tregs. PBMCs from both HLA-A*02:01 positive or negative donors were incubated with Foxp3-#32 bispecific mAb or the control bispecific mAb for one to three days. Several gating strategies were employed: first, gating on the lymphocyte population (Supplemental Figure 2A), then on CD4 + CD127high (conventional T cells) or CD127low (Tregs) populations, followed by gating with two sets of markers: CD25 vs intracytoplasmic Foxp3 or CD45RA vs intracytoplasmic Foxp3.18 Representative flow cytometric analysis after incubation two days is shown (Figure 6(a)). PBMCs alone and PBMCs treated with the control bispecific mAb (top row and bottom row, respectively) showed similar patterns with approximately 30% CD4 + CD127 high and 5% CD4 + CD127 low populations. The CD127 high cells are not Tregs as shown by lack of Foxp3 expression in the right column. Cells treated with Foxp3-#32 bispecific mAb (middle row) minimally changed the percentage of these two populations (left column panels). Further, CD25 + intracytoplasmic Foxp3+ cells were only detected in CD4 + CD127low, because resting conventional T cells do not express CD25, nor Foxp3 (see middle panels vs right panels). There was about a 60% reduction in the percentage CD25 + Foxp3+ cells treated with FoxP3-#32 bispecific mAb, compared to the cells treated with control bispecific mAb (middle column, middle row vs middle column, bottom row). The corresponding selective Treg depletion in absolute cell numbers was also calculated (Supplemental Figure 2C and Supplemental Table 2.) This was calculated by multiplying total PBMC numbers (Supplemental Figure 2B) with the percentage of lymphocytes, and the percentage of CD4 + CD127low, CD25 high, Foxp3+ population (Figure 6(a)). The data are consistent with selective depletion of the Treg population from PBMCs.
Figure 6.
(Continued).
Figure 6.
(Continued).
Figure 6.
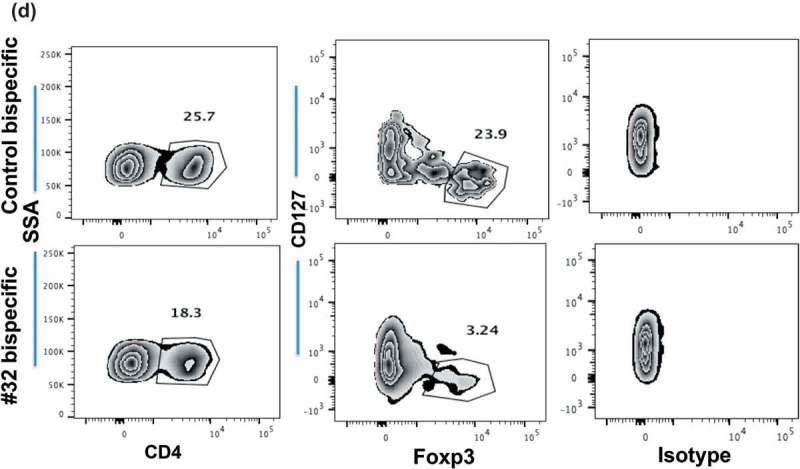
Depletion of natural Treg population by Foxp3-#32. Representative flow cytometry plots of Tregs from healthy donors and patients. (a). The frequency of CD4+/CD127high or CD127low populations from an HLA-A*02:01+ donor after 2 days of culture is shown in the left column. The inserted numbers are the percentages of cells in each gated population shown. CD25+/Foxp3 expression is shown in the middle column on CD4+/CD127 low population. CD25+/Foxp3 expression is shown in the right columns on CD4+/CD127high population. (b). CD4+/CD127 low or high population in Foxp3-#32 or control bispecific mAb-treated group (shown in 6A) were further analyzed based on CD45RA vs Foxp3 expression from the same cells. The percentage of each fraction is indicated as an inserted number, and absolute cell numbers of each fraction were shown in the bar graph. (c). A similar gating strategy was used for the cells after 3 days of culture from the same donor. Data show the CD25+/Foxp3+ cells (middle column) or CD45RA vs Foxp3+ cells (right column) gated from the CD4+/CD127low population (left column). Data represent one of three similar experiments. The percentage of each fraction is indicated as an inserted number. (d). Ascites cells from an HLA-A*02:01+ patient with ovarian cancer treated with Foxp3-#32 bispecific mAb for two days were stained with the Treg markers described in Figure 6(d). Cells were first gated on lymphocytes, excluding large tumor cells and monocyte population, on the side and forward scatter. Then, the CD4+ populations were analyzed with Treg markers: CD127low/Foxp3+. The percentage of each fraction is indicated as an inserted number. Absolute Foxp3+ cell numbers were shown in the bar graph, by calculating total cell numbers times the percentage of lymphocytes, times the percentage of CD4+/CD127 low, within the three Foxp3 fractions (I, II, III) and Foxp3 negative fractions (IV, V; CD45RA+ and negative populations). Data represent one of two similar experiments from the same patient and a total of three patients.
Because the CD4 + CD127low population increased in the Foxp3-#32 bispecific mAb-treated group, to further confirm the cell reduction as an absolute vs relative depletion of Foxp3+ Tregs, we further analyzed these two populations using a set of markers proposed by Sakaguchi18 (Figure 6(b)). The upper panels show the CD127 low cells, and the lower panels show the CD127 high cells. PBMCs treated with control bispecific mAb showed 17.4% fraction I, 10% fraction II and 31% fraction III. (For clarity, these subsets are labeled in the first panel with Roman numerals I to V and the percentage of each cell type within the gate box is indicated by the number). All three populations of FoxP3 positive cells (I, II, III) were depleted when cells were treated with Foxp3-#32 bispecific mAb including fraction I (naïve Tregs) and II (effector and terminally differentiated Tregs) and also fraction III (nonclassic Tregs: CD45RA-, Foxp3 low). Total Tregs from fraction I and II are about 28% in the control group. In contrast, the percentage of CD45RA + T cells increased more than fourfold in the Foxp3-#32 bispecific mAb-treated group compared to the control bispecific mAb group. Absolute numbers of Foxp3 positive cells (fraction I, II and III, respectively) showed approximately 10%, 30%, and 30%. In contrast, increased numbers of cells were found in CD45 RA+/Foxp3-population (fraction V, effector T cells) in the Foxp3-#32 bispecific mAb-treated group relative to the control antibody (bar chart inset). Typically, the action of a bispecific T cell engager mAb would be to activate T cells and kill the target only when it engages both target and T cells. Our data suggest that upon engaging with Treg target cells via Foxp3-#32 bispecific mAb, naive T cells are activated to proliferate. Importantly, the results also suggested that CD45 positive conventional T cells are not depleted by the Foxp3-#32 bispecific mAb treatment.
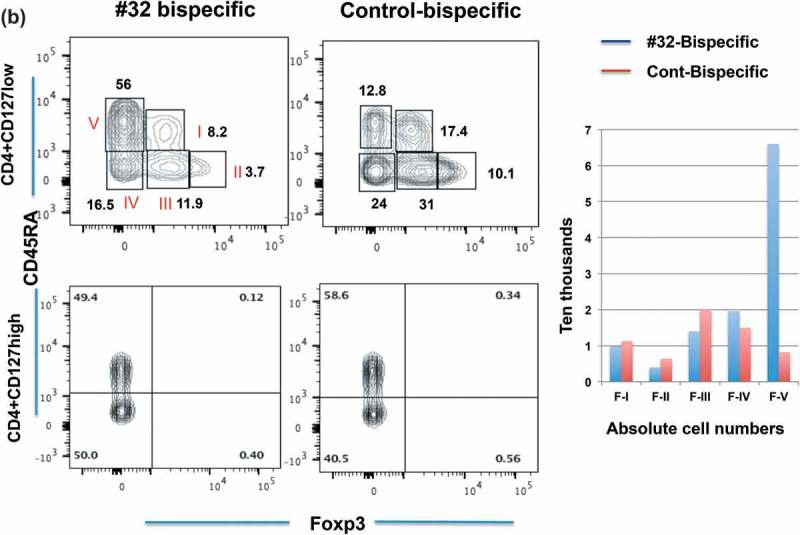
When cells were analyzed in the same manner after three days of treatment, CD4+/CD127low/CD25+/Foxp3+ Treg populations showed further percentage and absolute depletions: 14% remaining in the population in Foxp3-#32 bispecific mAb-treated cells compared to 78% remaining in control bispecific mAb-treated group (an 82% reduction) (Figure 6(c), middle two panels). Furthermore, CD45RA low/Foxp3+ naïve and CD45RA-/Foxp3high effector Tregs were reduced to 7% of the population, compared to 29% remaining with the control bispecific mAb (Figure 6(c), right two panels).
The reduction of the CD4 + CD25 + CD127 low and Foxp3+ cells in the Foxp3-#32 bispecific mAb-treated group was seen as early as the first day after treatment. The CD4 + CD127 low population was about 4% in PBMCs, Foxp3-#32 bispecific mAb-treated and control bispecific mAb-treated groups. However, the CD25 + Foxp3+ cells were 62.3%, 42.5% and 57% in these three groups, showing a 30% reduction. In addition, total PBMC numbers did not show any significant change after one to three days of treatment among the three groups in two separate experiments. However, the percentage of lymphocytes (gated on forward and side shatters) showed only a minimal reduction in the cells treated with Foxp3-#32 bispecific mAb after 2–3 days (Supplemental Figure 2A), which is consistent with a selective depletion of Tregs, which are only a small fraction of lymphocytes. No major effector T cell depletion was observed.
No Foxp3+ Tregs were depleted in HLA-A*02:01 negative donor, in the same experiments (Supplemental Figure 3A). These results demonstrated that the Foxp3-#32 bispecific mAb selectively depleted Foxp3+ cells in the context of HLA-A*02:01molecule.
We performed a similar experiment using ascites from ovarian patients who are HLA-A*02:01 positive. After two days of treatment with Foxp3-#32 or control bispecific mAb, the CD4 + CD127 low/Foxp3+ population decreased from 24% (control) to 3% (#32 bispecific mAb). Isotype control mAb for the intracellular Foxp3 protein was shown in parallel to confirm the specificity of the intracellular Foxp3 protein staining (Figure 6(d), right two panels). We also treated the cells with FoxP3-#32 IgG with a Fc region mutated to improve ADCC, because CD33 + CD14+ monocytes/macrophages infiltration was observed in the ascites of the patient. The depletion of effector Tregs (fraction II) was evident on day 2 after treatment with the specific TCRm (Supplemental Figure 3B, upper panels) and this population decreased to 0.4%, compared to the untreated cells (4.8%) and control mAb-treated cells (3.4%) after three days (Supplemental Figure 3B, lower panels). There was no typical naïve Treg population (fraction I) on day 2. Similar phenotypes have also been shown in other types of cancer, due to heterogeneity of tumor samples.38
To further confirm our results, we used Treg lines from HLA-A*02:01+ donors (phenotype shown in Figure 4(c,d) as Treg targets. Treg lines used as targets were incubated overnight with purified T cells from HLA-A*02:01-negative donors, in the presence or absence of Foxp3-#32 or control bispecific mAb. Following this we measured the percentage of Foxp3+ cells in HLA-A*02:01 + T cell population by staining the cells with mAbs to HLA-A2 and intracellular Foxp3 protein. Since HLA-A*02:01+ cells are only present in the target Treg lines, reduction of the Foxp3+ cells within HLA-A*02:01 positive cells indicated that the Tregs were depleted (Supplemental Figure 4A). While control cell cultures treated with effector PBMCs alone (upper left panel), or effectors with the control bispecific mAb (lower right panel), showed 9–10% HLA-A*02:01/Foxp3 double positive cells in the co-culture, the percentage of HLA-A*02:01+/Foxp3+ T cells decreased more than 60% in the presence of Foxp3-#32-bispecific mAb (lower left panel). Foxp3+/HLA-A*02:01 negative cells (effector T cells, possibly activated by Treg allo-stimulation) were not killed by the Foxp3-#32 mAb, indicating the HLA-A2 restriction for the mAb recognition. Upper right panel shows the specificity of Foxp3 intracellular staining because isotype for Foxp3 mAb did not show any positivity. Similar results were obtained from a second Treg line #2 (Supplemental Figure 4B). These results demonstrated that the Foxp3-#32-bispecific mAb is able to recognize and mediate T cell cytotoxicity against human Tregs in the context of HLA-A*02:01 molecules.
To test long-term cytotoxic effects of Foxp3-#32-bispecific mAb against Foxp3+/HLA-A*02:01+ cells, we incubated GFP/luciferase+ MAC-1 or MAC-2A cells with effector PBMCs from HLA-A*02:01 negative donors, in the presence of the Foxp3-#32- or control bispecific mAb and measured the total bioluminescent intensity (BLI) after three days. Significant cytotoxicity of the Foxp3-#32 bispecific mAb against MAC-2A was seen as there was little target BLI signal left, indicating that the MAC-2A cells were killed in the presence of the Foxp3-#32-bispecific mAb (Supplemental Figure 4C). Similar results were also seen with MAC-1 cell line.
Foxp3-#32 bispecific mAb selectively depletes Tregs in vivo in the context of HLA-A*02:01.
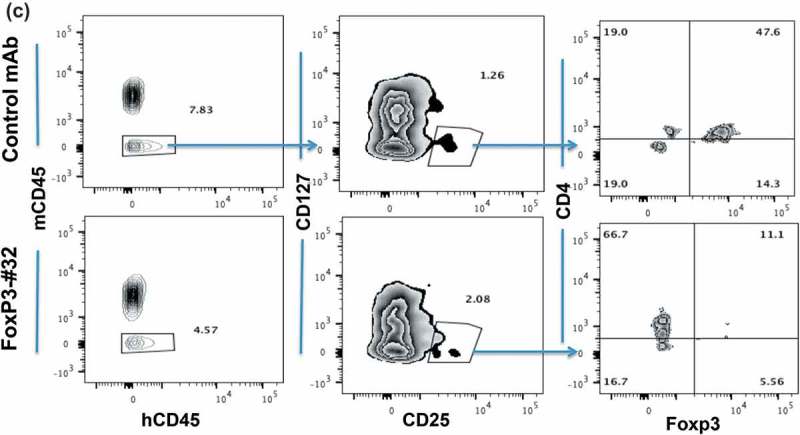
We next sought to further confirm the functional and thus potential therapeutic relevance of Foxp3-#32 mAb by depleting Tregs in vivo. PBMCs from HLA-A*02:01 positive healthy donors were intravenously (iv) injected into NSG mice, followed by iv. injection of the Foxp3-#32 or its isotype control bi-specific mAb 2 h later. Human Treg depletion was measured one and two days after mAb injection, by harvesting human cells from spleen and blood of the mice. The percentage of human Tregs (CD4 + CD25 + CD127lowFoxp3+) was normalized to the amount in untreated mice (Figure 7(a)). While Tregs in both donors did not change significantly after treatment with control mAb, treatment with Foxp3-#32 bispecific mAb reduced Tregs by nearly 60% in three donors. After 2 days post-treatment, Tregs were more than 80% depleted, confirmed by two Treg marker panels (CD25 high/Foxp3+ or CD45RA+/Foxp3+ in the CD4+/CD127 low population) (Figure 7(b), middle three and right three panels). We next tested if the Foxp3-#32 mAb was able to deplete Tregs from cancer patients. Human ovarian cancer ascites cells (a mixture of lymphocytes and tumor cells) from HLA-A*02:01 positive donors were injected into NSG mice intraperitoneally (ip), followed by ip injection of Foxp3-#32 mAb and its isotype control 2 h later. Cells from peritoneal cavity were harvested 1-day post treatment, and the depletion of Tregs was measured by flow cytometry after gating out mouse cells using a mAb for mouse CD45 (Figure 7(c), left two panels). Representative data showing the percentage of human (human CD45 positive) Tregs that were measured by gating on CD127 low/CD25+ cells (middle two panels), and then CD4+/Foxp3+ cells within this population (right two panels) are shown. Cells in the Foxp3-#32 bispecific mAb-treated group showed more than 70% depletion (lower right plot) compared to the control bispecific mAb-treated group (upper right plot). The results demonstrated that Foxp3-#32 bispecific mAb is effective in depleting Tregs in vivo.
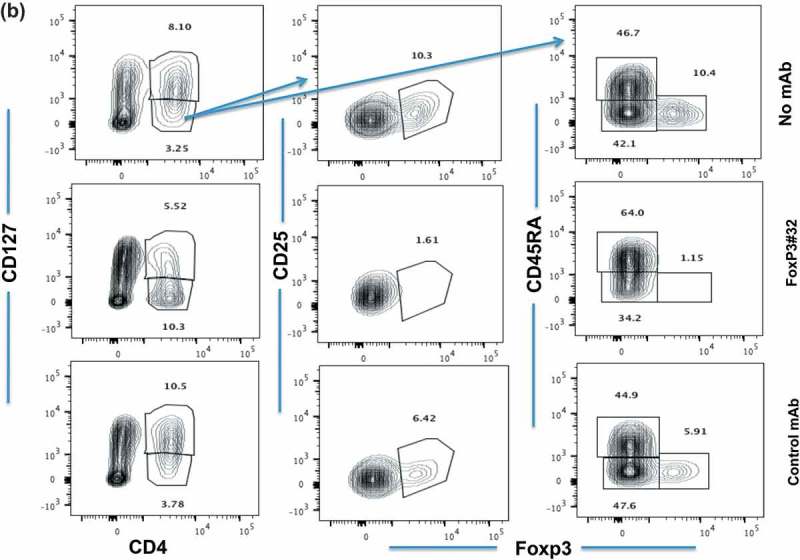
Figure 7.
(Continued).
Figure 7.
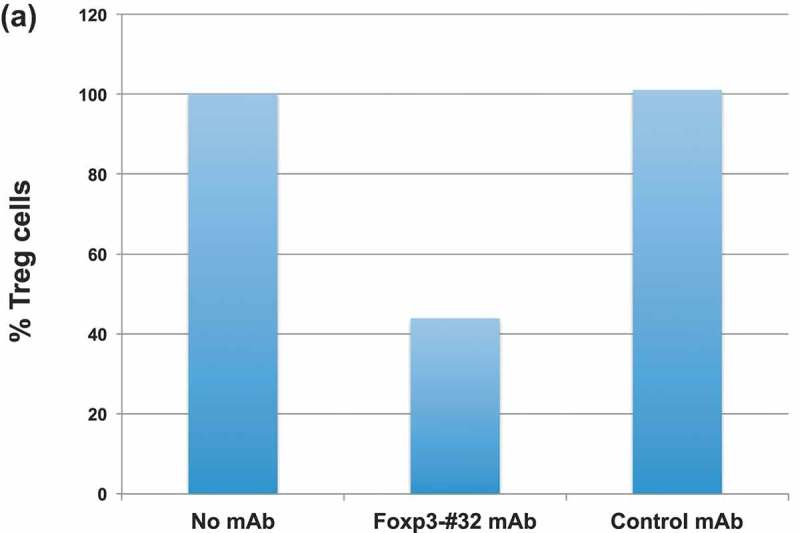
Depletion in vivo of Tregs from human xenografts in NSG mice. Tregs were characterized by markers including CD4+/CD127low/CD25high/Foxp3+ and CD45RA, as described in the in vitro study in Figure 6. (a) Tregs as a percentage of cells (Y-axis) from three HLA-A*02:01 positive donors recovered from mice after PBMC engraftment and treatment with mAbs is normalized to the no mAb untreated cells. Mice engrafted with PBMCs from donors 1 and 3 received 20ug/mouse bispecific mAb Foxp3-#32 or its isotype control. Mice that received PBMCs from donor 2 were injected with 10ug/mouse bispecific Foxp3-mAb #32 or its isotype control. Cells (splenocytes and blood) from three mice per group were pooled and stained with the mAbs to Treg markers and the data represent results from three donors plus/minus SD. P Value for depletion was 0.009 and 0.006 for Foxp3-#32 bispecific mAb vs control bispecific mAb and vs PBMCs alone group, respectively. (b). Mice engrafted with donor 2 were injected with bispecific mAb #32 or its isotype control at 10ug/mouse for consecutive 2 days. The gated CD4+/CD127low population (left column) is next shown as the percentage of CD25 and Foxp3 double positive (middle column) or CD45RA vs Foxp3 cells (right column) to access Treg depletion. All Treg markers were shown after gating on lymphocyte population. (c). NSG mice engrafted with ascites cells from an HLA-A*02:01 positive patient with ovarian cancer were injected ip with 10ug/mouse bispecific mAb #32 or its isotype control. One day after injection, cells were harvested from the peritoneal cavity and stained with human Treg markers as indicated. Human CD45 positive cells (left column) were gated on CD127low/CD25 high population (middle column) and further gated on CD4+/Foxp3+ population to access the depletion of Treg cells (right column). The percentage of each fraction is indicated as an inserted number. Data show a representative plot from one of the three xenografts from three patients (mean+/SD: #32 = 19.5 ± 4.6; control = 51.3 ± 2.9; p value: 0.003).
Potential off targets for mAb Foxp3-#32 in the context of HLA-A*02:01
αβ TCRs are known to have significant cross-reactivities to other peptides/MHC complexes.39,40 Theoretically, TCRm mAb could have, and do have similar properties,41 because both TCR and TCRm mAb recognize short linear peptide epitopes embedded within MHC class I binding groove, and other peptides in the exome may share amino acid homologies or physicochemical features that allow binding. To test if the Foxp-#32 mAb-mediated ADCC was effective against normal hematopoietic cells as a result of any possible expressed off-target epitopes in these cells, we incubated PBMCs from three normal healthy donors that were either HLA-A*02:01positive or negative overnight, in the presence of the Foxp3-#32-bispecific mAb. While control MAC-1 cells were completely killed by Foxp3-#32-bispecific mAb (Supplemental Table 3), no significant reduction of T (CD3+), B (CD19+) and monocytes (CD33+) was detected in either HLA-A*02:01positive or negative donors. In addition, total PBMCs did not show any significant changes (Supplemental Figure 5). These data do not exclude off targets or cross-reactive epitopes on other human cells and tissues.
Discussion
The development of therapeutic strategies to deplete or interfere with the function of Tregs, without compromising anti-tumor immunity has been challenging because there is no Treg-specific cell surface markers, nor a druggable Treg-specific pathway. One of the obstacles for specific depletion of Tregs is that both Tregs and effector T cells may exhibit an activated phenotype, especially in the pattern of expression of key cell surface proteins; both cell types express high levels of CD25, CTLA-4, OX40, and GITR.1,13,42 Although Tregs express CTLA-4, results from clinical studies suggest that the effects of anti-CTLA-4 treatment are due primarily to increased activation of effector T cells.12 Recent studies have shown that C-C chemokine receptor 4 (CCR4), the cognate receptor for CC chemokines CCL17 and CCl22, is predominantly expressed in effector Tregs (eTregs; CD45RA-Foxp3hi CD4+) in TILs in melanoma patients, but also in a variety of other cell types. Depletion in vitro of this population using anti-CCR4 mAb enhanced T cell responses when stimulated with NY-ESO-1 peptides and in a treated patient with T cell leukemia-lymphoma, the Treg fraction is reduced, and the NY-ESO-1-specific CD8 T cell response is augmented.18 Similarly, targeting GITR using cognate ligand or agonistic mAb has been shown to be effective in murine cancer models.42 However, the clinical efficacy of such a strategy remains to be investigated in human trials.
Foxp3+ Treg cells are significantly enriched in the tumor microenvironment, peripheral blood or ascites in cancer patients. In TILs, the ratio of effector T cells to Tregs is prognostic in a variety of cancers.3,12 Interestingly, the immunosuppressive function of Foxp3 may not be limited to Treg cells.36,37 Foxp3 expression in pancreatic cancers induced complete inhibition of T cell proliferation in vitro.27 Immune suppressive function of Foxp3 has also been suggested in CD4+/CD25+ adult T leukemia (ATL) patients.28,29 An earlier study demonstrated the possibility of targeting intracellular Foxp3 by an approach of using peptide-specific CTLs.31 Our results here are consistent with this earlier study and formed a premise for making the TCRm mAb to this epitope.
Activated T cells (non-Tregs) can also transiently express Foxp3.43 In general, Tregs express low levels of CD127, while other T cells express a high level of CD127, an alpha chain of IL-7 receptor.43–46 Therefore, the CD127 low population in conjunction with CD25 and Foxp3 positivity was used to identify the Tregs. Remarkably, we could detect selective depletion of this small Treg population by the Foxp3-#32 bispecific mAb when the PBMCs from HLA-A*02:01+ donors were treated with the mAb. Since the CD4 + CD127 low population increased in the Foxp3-bispecific mAb-treated group, possibly due to a down-regulation of the CD127 in small numbers of conventional T cells upon activation, we used another set of markers, CD45RA vs Foxp3 expression, to exclude the depletion of conventional T cells. We confirmed the Foxp3-selective depletion of cells with this set of markers, including both effector Tregs and naïve (resting) Tregs (fraction I and II) along with fraction III. Importantly, we also demonstrated Foxp3-selective depletion from healthy human PBMC and from tumor-associated lymphocytes or “TILs” in ascites from an HLA-A*02:01 positive patient with ovarian cancer by Foxp3-#32 bispecific mAb and Fc-enhanced IgG1.
Similarly, the mAb Foxp3-#32, could bind to the CD127low CD25high CD4+ natural Tregs and CD4 + CD25hi Foxp3+ population in Tregs, but not the CD25lo/negative Foxp3 negative population. The peptide/MHC epitopes are typically found in extremely low density on target cells, making recognition and cytotoxicity difficult. Therefore, we postulated that a Foxp3 TCRm mAb would only bind to the cells with the highest expression of Foxp3 such as Tregs. Depending on the level of expression in various populations of T cells, this opens a possible therapeutic window. In addition, because the goal of a therapeutic anti-Treg antibody is to upset the balance of T cells to favor anti-cancer activity of CD8 and CD4 T cells, complete elimination of the target Treg cells may not be needed, unlike the situation with an antibody directed to the cancer cell itself; furthermore, absolute specificity may not be required.
The properties of TCRm mAb binding to their targets differ from those of typical antibodies in ways that have the potential to limit clinical utility. The peptides must be processed and presented in sufficient amounts to be recognized by the TCRm; the control of these processes are still poorly understood and may be affected by the activation state of the cell. As the epitope is a linear peptide within the constraints of the HLA groove, binding to similar off-target peptides may be possible, if presented by other cells, as has been seen with both TCR and TCRm mAbs.39–41 While no significant killing was seen against any PBMC by the bispecific mAb format of this TCRm (Supplemental Table 2 and 3), the possible off-targets presented on other cells, at both the molecular and cellular level, will need to be defined better before advancing a TCRm such as this forward to systemic clinical use.
Materials and methods
Peptide synthesis
All peptides used in this study were purchased and synthesized by Genemed Synthesis, Inc. (San Antonio, TX). Peptides were sterile and 80% to >90% pure. The peptides were dissolved in DMSO and diluted in saline at 5 mg/mL and stored at −80°C. Control peptides used for HLA-A*02:01 were Ewing sarcoma-derived peptide EW (QLQNPSYDK) and choline transporter-like protein 4-derived peptide CT (KLLVVGGVGV). Biotinylated single chain Foxp3p/HLA-A*02:01 complexes were synthesized by refolding the peptides with recombinant HLA-A*02 and beta2 microglobulin (β2M) at Eureka Therapeutics, Inc. (Emeryville, CA).
Cytokines, antibodies, and cells
Human granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-1β, IL-2, IL-4, IL-6, IL-15, tumor necrosis factor (TNF)-α and prostaglandin E2 (PGE2), TGF-β were purchased from R&D Systems (Minneapolis, MN). Beta 2-microglobulin (β2-m) and human IFN-γ were purchased from Sigma (St. Louis, MO). Cell isolation kits for CD14 and CD3 were purchased from Miltenyi Biotec. (Bergisch Gladbach, Germany). Human Treg isolation kits were purchased from Stem Cell Technology (Canada). Foxp3+ and HLA- A*02:01+ cutaneous T lymphoma cell lines MAC-1 and MAC-2A were kindly provided by Dr. Mads H. Anderson, at the University of Denmark. The human T leukemia virus (HTLV) positive cell line C5MJ was kindly provided by Dr. Alexander Rudensky laboratory (MSKCC, New York) and the cells were transduced with HLA-A*02:01 molecule as described by Latouche.47 The HLA-A*02:01 SFG vector was a gift from Dr. Michelle Sadelain, at MSKCC. MAC-1 and MAC-2A cell lines were engineered to express a high level of GFP-luciferase fusion protein, using retroviral vectors containing a plasmid encoding the luc/GFP. The cell lines were cultured in RPMI 1640 supplemented with 10% FCS, penicillin, streptomycin, 2 mmol/l glutamine, and 2-mercaptoethanol at 37 C/5% CO2. Leukemia cell lines Jurkat and HL-60 are from ATCC. Cells were checked regularly for mycoplasma. Cell identities were confirmed by mAb phenotype or genotype. Peripheral blood mononuclear cells (PBMC) from healthy donors and tumor samples from patients with ovarian cancer undergoing surgery were obtained after informed consent on Memorial Sloan-Kettering Institutional Review Board approved protocols.
The Foxp3-#32 bispecific mAb of mouse IgG1 (for flow cytometry) and their respective controls were produced at Eureka Therapeutics, Inc.48,49 APC conjugation to mouse IgG1 form of Foxp3-#32 and its control was done by using lightening-link APC antibody labeling kit according to the instructions of the manufacturer (Novus Biologicals). Mabs against human HLA-A*02 (clone BB7.2), its isotype control mouse IgG2b (clone MPC-11), human CD3 (clone HIT3A and OKT3), CD4 (clone RPA-T4), CD8 (clone RPA-T8), CD25 (clone 2A3), CD33 (clone WM53), mouse anti-His tag mAb (clone F24-796) conjugated to FITC or PE, were purchased from BD Biosciences, (San Diego, CA). Mabs specific for human Foxp3 clone PCH101, its isotype control rat IgG2a kappa, clone 236A/E7 and its isotype control mouse IgG1 kappa, CD4 (clone OKT4), CD127 (clone HIL-7R-M21), were purchased from eBioscience. Fixation and permeabilization kit for intracellular staining was also purchased from eBioscience.
Animals
Female, six to eight week old NOD SCID SCID γc−/-(NSG) mice were purchased from Jackson laboratory (Farmington, CT) and were used for in vivo xenograft studies. All animal studies were conducted on IACUC approved protocols.
Flow cytometry analysis
For cell surface staining, cells were incubated with appropriate mAbs for 30 min on ice, washed, and incubated with secondary antibody reagents when necessary. For Foxp3-#32-bispecific mAb staining, human T cells or cancer cells were incubated with different concentrations of Foxp3-#32-bispecific mAb or control bispecific mAb for 30 min on ice, washed, and incubated with secondary mAb against His-Tag. Flow cytometry data were collected on a Beckman Dickinson Fortessa and analyzed with FlowJo 9.8.1 and FlowJo10 software.
In vitro stimulation and human T-cell cultures
PBMCs from HLA-A*02:01+ healthy donors, on MSKCC IRB approved protocols, were obtained by Ficoll density centrifugation. CD14+ monocytes were isolated by positive selection using mAb to human CD14 coupled with magnetic beads and were used for the first stimulation of T cells. The CD14-fraction of PBMC was used for isolation of CD3, by negative immunomagnetic cell separation using a pan T cell isolation kit. The purity of the cells was always more than 98%. T cells were stimulated for 7 days in the presence of RPMI 1640 supplemented with 5% autologous plasma (AP), 20 ug/mL synthetic peptides, 2 ug/mL B2M, and 5–10 ng/mL IL-15. Monocyte-derived dendritic cells (DCs) were generated from CD14+ cells, by culturing the cells in RPMI 1640 medium supplemented with 1% AP, 500 units/mL recombinant IL-4, and 1,000 units/mL GM-CSF. On days 2 and 4 of the incubation, fresh medium with IL-4 and GM-CSF was either added or replaced half of the culture medium. On day 6, maturation cytokine cocktail was added (IL-4, GM-CSF, 500 IU/mL IL-1, 1,000 IU/mL IL-6, 10 ng/ml TNF-α, and 1 ug/mL PGE-2). On day 7 or 8, T cells were re-stimulated with mature DCs at a 30:1, T: APC ratio, with IL-15. T cells were stimulated 3 to 5 times in the same manner, using either autologous DCs or CD14+ cells as antigen-presenting cells (APCs). A week after final stimulation, the peptide-specific T cell response was examined by IFN-gamma (γ) enzyme-linked immunospot (ELISPOT) assay.50 All studies with human cells were done on IRB approved protocols.
IFN-γ ELISPOT assay
HA-Multiscreen plates (Millipore) were coated with 100 uL of mouse anti-human IFN-γ antibody (10 ug/mL; clone 1-D1K; Mabtech) in PBS, incubated overnight at 4 C, washed with PBS to remove unbound antibody, and blocked with RPMI 1640/10% autologous plasma (AP) for 2 h at 37°C. CD3 + T cells were plated with either autologous CD14 + (10:1 E: APC ratio) or autologous DCs (30:1 E: APC ratio). Various test peptides were added to the wells at 20 ug/mL. Negative control wells contained APCs and T cells without peptides or with irrelevant peptides. Positive control wells contained T cells plus APCs plus 20 ug/ml phytohemagglutinin (PHA, Sigma). All conditions were done in triplicates. Microtiter plates were incubated for 20 h at 37°C and then extensively washed with PBS/0.05% Tween and 100 ul/well-biotinylated detection antibody against human IFN-γ (2 ug/mL; clone 7-B6-1; Mabtech) was added. Plates were incubated for an additional 2 h at 37°C and spot development was done as described (7–9). Spot numbers were automatically determined with the use of a computer-assisted video image analyzer with KS ELISPOT 4.0 software (Carl Zeiss Vision).50
51Chromium release assay
The presence of specific CTLs was measured in a standard chromium release assay as described.50 Briefly, target cells are labeled with 50 uCi/million cells of Na2 51CrO4 (NEN Life Science Products, Inc.). After extensive washing, target cells are incubated with T cells at various effector: target (E: T) ratios. All conditions were done in triplicates. Plates were incubated for 4–5 hours at 37°C in 5% CO2. Supernatant fluids were harvested, and radioactivity was measured in a gamma counter. Percentage specific lysis was determined from the following formula: [(experimental release – spontaneous release)/(maximum release – spontaneous release)] x 100%. Maximum release was determined by lysis of radiolabeled targets in 1% SDS.
Phage screening, selection of scFv specific for the Foxp3-derived epitopes
A human ScFv antibody phage display library (7 × 1010 clones) was used for the selection of mAb clones as described previously.32 In brief, biotinylated irrelevant peptide/HLA-A*02:01 complexes were used to remove any clones that potentially bind to HLA-A*02:01. Remaining clones were screened for the Foxp3p/HLA-A*02:01 complex. The selected clones were enriched by 3–4 rounds of panning. Positive clones were determined by standard ELISA method against biotinylated single-chain Foxp3p/HLA-A*02:01 complexes. The positive clones were further tested for their binding to peptide/HLA-A*2 complexes on live cell surfaces by flow cytometry, using a TAP-deficient, HLA-A*02:01+ cell line, T2, which is defective in the presentation of endogenous HLA-associated peptides. T2 cells were pulsed with positive and multiple control peptides (50 ug/ml) in the serum-free RPMI1640 medium, in the presence of 20 ug/ml β2M overnight. The cells were washed, and the staining was performed in the following steps. The cells were first stained with purified scFv phage clones and followed by staining with a mouse anti-M13 (bacteriophage) mAb, and finally the goat Fab2 anti-mouse IgG conjugated to FITC or PE. Each step of the staining was done between 30 −60 minutes on ice and the cells were washed twice between each step of the staining.32,49
Engineering full-length human IgG1 using the selected scFv fragments
Full-length human IgG1 of the selected phage clones were produced in HEK293 and Chinese hamster ovary (CHO) cell lines, as described.32 In brief, antibody variable regions were subcloned into mammalian expression vectors, with matching Lambda or Kappa light chain constant sequences and IgG1 subclass Fc. The molecular weight of the purified full-length IgG antibodies were measured under both reducing and non-reducing conditions by electrophoresis. The mouse IgG1 form of Foxp3-#32 was generated by exchanging a constant region of human IgG1 (CH1, 2, 3 and CL) to a mouse IgG1 form.
Construction, expression and purification of Foxp3-#32 bispecific mAb
The Foxp3-#32 bispecific mAb in the format of a typical bispecific T cell engager was engineered as previously described.49 N-terminal end of mAb Foxp3-#32 scFv was linked to the C-terminal end of an anti-human CD3ϵ scFv of a mouse monoclonal antibody by a flexible linker. The DNA fragments encoding for the scFv of two mAbs were synthesized by GeneArt (InVitrogen) and subcloned into Eureka’s mammalian expression vector pGSN-Hyg using standard DNA technology. A hexhistamine (His) tag was inserted downstream of the Foxp3-#32 bispecific mAb at the C-terminal end for the detection and purification of the bispecific mAb.
Chinese hamster ovary (CHO) cells were transfected with the Foxp3-#32 bispecific mAb expression vector and stable expression was achieved by standard drug selection with methionine sulfoximine (MSX), a glutamine synthetase (GS)-based method. CHO cell supernatants containing secreted Foxp3-#32 bispecific mAb were collected, from which, the #32 bispecific mAb was purified using HisTrap HP column (GE healthcare) by FPLC AKTA system. Briefly, CHO cell culture was clarified and loaded onto the column with low imidazole concentration (20 mM), and then an isocratic high imidazole concentration elution buffer (500 mM) was used to elute the bound Foxp3-bispecific mAb protein. A negative control bispecific mAb antibody was constructed from an irrelevant human IgG1 antibody (Cat#ET901, Eureka Therapeutics), replacing the Foxp3-#32 scFv.
Characterization of the full-length human IgG1 for the Foxp3 peptide/HLA-A*02:01 complex
Specificities of the fully human or mouse IgG1 mAbs or the bispecific mAb’s for the Foxp3 peptide/A2 complex were determined by staining T2 cells pulsed with or without Foxp3 peptides or various analogs or control peptides, using direct or indirect staining. The fluorescence intensity was measured by flow cytometry. The same method was used to determine the binding of the mAb to cell lines.
Treg generation, phenotypic analysis, and Foxp3-#32 mAb binding
CD4 + T cells were purified from PBMCs of healthy HLA-A*02:01 positive donors by FACS sorting, and were stimulated with allo-PBMCs (HLA-A*02:01 negative) as stimulator and feeder cells at ratios of effector: stimulator (E:S) 1:5–10, or with tumor cells (E:S: 1:1) in the presence of recombinant human IL-2 (100 unit) and TGF-β (10 ng/ml) for one to two weeks and the same stimulation was repeated to maintain the Treg cells (37–39). The phenotype of Tregs was determined by surface staining of the cells with mAbs to CD4, CD25+, CD127, CD45RA, mouse Foxp3 mAb-Foxp3-#32 conjugated to APC, for 30 min on ice, washed. Foxp3 expression was measured by intracellular protein staining using mAb to human Foxp3 (clone PCH101, or its isotype control rat IgG2a kappa) and Cytofix/CytoPerm kit (eBiosciences), according to the instructions of the manufacturer. The analysis was done by flow cytometry on a Beckman Dickinson Fortesa.
Cytotoxicity of Foxp3-#32 bispecific mAb specific for Tregs in the context of HLA-A*02:01
Four methods were used to measure the ADCC against Tregs by Foxp3-#32 bispecific mAb. First, for the natural Tregs, PBMCs from healthy donors who are either HLA-A*02:01 positive or negative were incubated with or without Foxp3-#32 bispecific mAb or control irrelevant bispecific mAb at 1 ug/ml for one to three days. Cells were harvested, washed and stained with mAbs to CD4, CD25, CD127, CD45RA, followed by intracellular staining with mAb to Foxp3 or its isotype control. Treg reduction was accessed on the expression of well-defined Treg markers. In brief, lymphocytes were gated based on forward and side scatters, followed by gating on CD4 + CD127 high or CD4 + CD127 low population. The CD4 + CD127 high or CD4 + CD127 low population was further determined by two sets of Treg markers: CD25 vs Foxp3; or CD45RA vs Foxp3.18,38 Second, natural Tregs only represent a few percents of CD4 + T cells; therefore, in order to obtain sufficient readout on Treg killing, we also used Tregs generated in vitro as targets. The killing of Tregs was determined by reduction of Treg population by flow cytometry. In brief, purified CD3T cells by negative selection from HLA-A*02:01 negative donors used as effectors were incubated with Tregs generated from HLA-A*02:01+ donors at an E: T ratio 5:1, in the presence or absence of Foxp3-#32 bispecific mAb (1 ug/ml) or its control bispecific mAb for over night. The cells were washed and stained with mAbs to CD4, CD25, Foxp3, and HLA-A*02. HLA-A*02 positive cells were gated (as Treg targets) and the killing of Tregs was determined by the reduction of the percentage of CD4 + CD25 + Foxp3+ cells in the HLA-A*02:01+ cells, compared to control cultures with effectors alone or with effectors plus control bispecific mAb. Third, Treg-like T lymphoma cell line MAC-2A, or T leukemia cell line C5MJ/A2 (Foxp3+/HLA-A*02:01+) were used as targets in ADCC assay by a standard 51Cr-release assay. Fourth, since 51Cr assay cannot be used to determine a longer term ADCC, we used an in vitro bioluminescence imaging (BLI) method to test the ADCC activity of the Foxp3-#32 bispecific mAb. In brief, PBMCs from HLA-A*02:01 negative donors were incubated with MAC-1, or MAC-2A cells that had been transduced with GFP/luciferase, at an E:T ratio 30:1, in the presence of Foxp3-#32 bispecific mAb or its control bispecific mAb at 1 ug/ml, for 3 days, then 30 ug of luciferin was added to each well, before imaging. Tumor growth was calculated by the average of the luminescence signal of triplicate microwell cultures.
In addition, to test if the mAb shows any non-specific or off-target toxicity to normal cells, PBMCs from HLA-A*02:01 positive or negative healthy donors were incubated in the presence or absence of 0.2 or 1 ug/ml Foxp3-#32 bispecific mAb or its control bispecific mAb overnight. Cells were washed and stained with mAbs to human CD3, CD19 and CD33 to determine whether these cell lineages are killed by the bispecific mAbs. Total cell numbers were measured by trypan blue exclusive staining.
Antibody-dependent cellular cytotoxicity (ADCC)
Target cells used for ADCC were T2 cells pulsed with or without Foxp3-TLIp or irrelevant control peptides, or Foxp3+ and HLA-A*02:01+ or negative cell lines MAC-1, C5MJ/A2, C5MJ, and HL-60 that were not pulsed with peptides. The Foxp3-#32 bispecific mAb or its isotype control, at various concentrations, were incubated with target cells and fresh PBMCs HLA-A*02:01- donors, at different E: T ratio for 4–5 h. Cytotoxicity was measured by the standard 51Cr-release assay.
Depletion in vivo of Tregs by Foxp3-#32 bispecific mAb in xenografts
Two methods were used to access the in vivo depletion of Tregs in immunodeficient NSG mice. First, 30 million PBMCs from HLA-A*02:01 positive donors were injected i.v. into NSG mice. Two hours later, bispecific mAb Foxp3-#32 or its isotype control was injected i.v. into mice. One and two days post-injection, blood and splenocytes from mice were collected and stained with mAbs to Tregs as described in the studies in vitro. Mabs at 10 or 20 ug/mouse were used from three different donors. The cells from three mice in each treatment group were pooled and tested to ensure enough cells were recovered. Second, 30 million ascites cells from HLA-A*02:01 positive patients with ovarian cancer were injected i.p. into mice, followed by injection i.p. of bispecific mAb Foxp3-##32 or its isotype control at 10 ug/mouse. The next day, the cells were harvested from the peritoneal cavity and stained with mAbs to characterize Treg depletion by flow cytometry as described for the studies in vitro.
Funding Statement
This study was supported by grants from NIH R01 CA33049, P30 CA008748, the Experimental Therapeutics Center, the Tudor Foundation, and the lymphoma Foundation.
Acknowledgments
We thank Dr. Mads H. Anderson, at the University of Denmark for kindly providing the T lymphoma cell lines; Catherine Konopacki for her insightful discussions and technical help.
Disclosure of potential conflict of interest
MSKCC has filed for patents on the Foxp3-#32 mAb on behalf of DAS and TD. Zhiyuan Yang, Liangxin Liu, and Cheng Liu are employees of Eureka Therapeutics, Inc, which has shared rights to the FoxP3#32. DAS and TD are consultants for Eureka Therapeutics. The rest of the authors declare no potential conflicts of interest.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B.. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003;9:606–612. [PubMed] [Google Scholar]
- 2.Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuur PS, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. [DOI] [PubMed] [Google Scholar]
- 3.Delleuw RJ, Kost SE, Kakal JA, Nelson RH.. The prognostic value of Foxp3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. 2012;18:3022–3029. doi: 10.1158/1078-0432.CCR-11-3216. [DOI] [PubMed] [Google Scholar]
- 4.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 5.Baumgartner J, Wilson C, Palmer B, Banerjee A, McCarter M. Melanoma induces immunosuppression by up-regulating Foxp3+ regulatory T cells. J Surg Res. 2007;141:72–77. doi: 10.1016/j.jss.2007.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempei TR. Dynamic Treg interaction with intratumoral APCs promote local CTL dysfunction. J Clin Invest. 2014;124(6):2425–2440. doi: 10.1172/JCI66375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulator T cells. J Clin Invest. 2005;115(12):3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210(11):2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, Colligon TA, Trosko JA, Leinbach LI, Pletcher CH, et al. CD25 blockade depletes and selectively reprogram regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4(134):134rd62. doi: 10.1126/scitranslmed.3003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Litzinger MT, Fermando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. IL-2 immunotoxin denileukin diftitox reduces regulator T cells and enhances vaccine-mediated T-cell immunity. Blood. 2007;110(9):3192–3201. doi: 10.1182/blood-2007-06-094615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. [DOI] [PubMed] [Google Scholar]
- 12.Colombo MP, Piconese S. Regulatory T cell inhibition versus: the right choice in cancer immunotherapy. Nat Rev Cancer. 2007;7:880–887. doi: 10.1038/nrc2250. [DOI] [PubMed] [Google Scholar]
- 13.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of CTLA-4 antibodies. J Exp Med. 2009;206:1717–1725. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maker AV, Attia P, Rosenberg SA. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J Immunol. 2005;175:7746–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerenal-Lewis M, Crawford J, Bonomi P, Maddox AM, Hainsworth J, McCune DE, Shukla R, Zeigler H, Hurtubise P, Chowdhury TR, et al. A phase II trial of denileukin diftitox in patients with previously treated advanced Non-Small Cell Lung Cancer. Am J Clin Oncol Cancer Clin Trials. 2009;32:269–273. doi: 10.1097/COC.0b013e318187dd40. [DOI] [PubMed] [Google Scholar]
- 16.Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Difititox. DBA (389) IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother. 2005;28:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cote AL, Zhang P, O’Sullivan JA, Jacobs VL, Clemis CR, Sakaguchi S, Guevara-Patiño JA, Turk MJ. Stimulation of glucocorticoid-induced TNF receptor family-related receptor on CD8 T cells induces protective and high-avidity T cell responses to tumor-specific antigens. J Immunol. 2011;186:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, et al. Anti-CCR4 mAb selectively depletes effector-type Foxp3+CD4+ regulator T cells, evoking antitumor immune responses in humans. Pnas. 2013;110(44):17945–17950. doi: 10.1073/pnas.1316796110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogura M, Ishida T, Hatake K, Taniwaki M, Ando K, Tobinai K, Fujimoto K, Yamamoto K, Miyamoto T, Uike N, et al. Multiple phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol. 2014;32(11):1157–1163. doi: 10.1200/JCO.2013.52.0924. [DOI] [PubMed] [Google Scholar]
- 20.Ishida T, Utsunomiya A, Jo T, Yamamoto K, Kato K, Yoshida S, Takemoto S, Suzushima H, Kobayashi Y, Imaizumi Y, et al. Mogamulizumab for relapsed adult-T-cell leukemia-lymphoma: updated follow-up analysis of phase I and II trials. Cancer Sci. 2017;108(10):2022–2029. doi: 10.1111/cas.13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plitas G, Konopacki C, Wu K, Bos P, Morrow M, Putintseva EV, Chudakov DM, Rudensky AY. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45(5):1122–1134. doi: 10.1016/j.immuni.2016.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konopacki C, Plitas G, Rudensky AY. Reigning in regulatory T-cell function. Nat Biotech. 2015;33(7):718–719. doi: 10.1038/nbt.3285. [DOI] [PubMed] [Google Scholar]
- 23.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56(5):641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Audia S, Nicolas A, Cathelin D, Larmonier N, Ferrand C, Foucher P, Fanton A, Bergoin E, Maynadie M, Arnould L, et al. Increase of CD4+CD25+ regulatory T cells in the peripheral blood of patients with metastatic carcinoma: a Phase I clinical trial using cyclophosmide and immunotherapy to eliminate CD4+CD25+ T lymphocytes. Clin Exp Immunol. 2007;150(3):523–530. doi: 10.1111/j.1365-2249.2007.03521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fonteno JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 26.Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev. 2011;241(1):260–268. doi: 10.1111/j.1600-065X.2011.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B, Grützmann R, Pilarsky C, Ungefroren H, Saeger HD, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion. Cancer Res. 2007;67(17):8344–8350. doi: 10.1158/0008-5472.CAN-06-3304. [DOI] [PubMed] [Google Scholar]
- 28.Heid JB, Schmidt A, Oberle N, Goerdt S, Krammer PH, Suri-Payer E, Klemke CD. Foxp3+CD25-tumor cells with regulatory function in sezary syndrome. J Invest Dermatol. 2009;129:2875–2885. doi: 10.1038/jid.2009.175. [DOI] [PubMed] [Google Scholar]
- 29.Matsubara Y, Hori T, Morita R, Sakaguchi S, Uchiyama T. Phynotypic and functional relationship between adult T-cell leukemia cells and regulatory T cells. Leukemia. 2005;19:482–483. doi: 10.1038/sj.leu.2403628. [DOI] [PubMed] [Google Scholar]
- 30.Nair S, Boczkowski D, Fassnacht M, Pisetsky D, Gilboa E. Vaccination against the forkhead family transcription factor Foxp3 enhances tumor immunity. Cancer Res. 2007;67(1):371–380. doi: 10.1158/0008-5472.CAN-06-2903. [DOI] [PubMed] [Google Scholar]
- 31.Larsen SK, Munir S, Woetmann A, Frosig TM, Odum N, Svane IM, Becker JC, Andersen MH. Functional characterization of Foxp3-specific spontaneous immune responses. Leukemia. 2013;27:2332–2340. doi: 10.1038/leu.2013.196. [DOI] [PubMed] [Google Scholar]
- 32.Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, Scott A, Whitten J, Maslak P, Casey E, et al. Targeting the intracellular WT1 oncoproduct with a therapeutic human antibody. Sci Transl Med. 2013;5(176):176ra33. doi: 10.1126/scitranslmed.3005661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ataie N, Xiang J, Cheng N, Brea EJ, Lu W, SCheinberg DA, Liu C, Ng HL. Structure of a TCR-mimic antibody with target predicts pharmacogenetics. J Mol Biol. 2016;428(1):194–205. doi: 10.1016/j.jmb.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu L, Zhou X, Wang J, Zheng SG, Horwitz DA. Characterization of protective CD4+CD25+Foxp3+ regulatory T cells generated with IL-2, TGF-β and retinoic acid. PLoS One. 2010;5(12):e15150. doi: 10.1371/journal.pone.0015150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Godfrey WR, Ge YG, Spoden DJ, Levine BL, June CH, Blazar BR, Porter SB. In vitro-expanded human CD4+CD25+ T-regulatory cells can markedly inhibit allogeneic dendritic cell-stimulated MLR cultures. Blood. 2004;104(2):453–461. doi: 10.1182/blood-2004-01-0151. [DOI] [PubMed] [Google Scholar]
- 36.Karanikas V, Spletas M, Zamanakou M, Kalala F, Loules G, Kerenidi T, Barda AK, Gourgoulianis KI, Germenis AE. Foxp3 expression in human cancer cells. J Transl Med. 2008;6:19–26. doi: 10.1186/1479-5876-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Truiulzi T, Tagliabue E, Balsari A, Casalini P. Foxp3 expression in tumor cells and implications for cancer progression. J Cell Physiol. 2013;228:30–35. doi: 10.1002/jcp.24125. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oates J, Hassan NJ, Jakobsen BK. ImmTACs for targeted cancer therapy: why, what, how, and which. Mol Immunol. 2015;67:67–74. doi: 10.1016/j.molimm.2015.01.024. [DOI] [PubMed] [Google Scholar]
- 40.Attaf M, Legut M, Cole DK, Sewell AK. The T cell antigen receptor: the Swiss army knife of the immune system. Clin Exp Immunol. 2015;181:1–18. doi: 10.1111/cei.12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang AY, Gejman RS, Brea EJ, Oh CY, Mathias MD, Casey E, Dao T, Scheinberg DA. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin Biol Ther. 2016;16(8):979–987. doi: 10.1080/14712598.2016.1176138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaer D, Murphy JT, Wolchok JD. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol. 2012;24(2):217–224. doi: 10.1016/j.coi.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J, Ioan-Facsinay A, van der Voort EIH, Huizinga J, Toes REM. Transit expression of Foxp3 in human activated nonregulatory CD4+ T cells. EJ Immunol. 2007;37:129–138. doi: 10.1002/eji.200636435. [DOI] [PubMed] [Google Scholar]
- 44.Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, Solomon M, Selby W, Alexander SI, Nanan R, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203(7):1693–1700. doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu W, Putnam AL, Zhou XY, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, Fazekas de St Groth B, et al. CD127 expression inversely correlates with Foxp3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203(7):1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alves NL, van Leeuwen EM, Derks IA, van Lier RA. Differential regulation of human IL-7 receptor alpha expression by IL-7 and TCR signaling. J Immunol. 2008;180:5201–5210. [DOI] [PubMed] [Google Scholar]
- 47.Latouche JB, Sadelain M. Induction of human cytotoxic T lymphocytes by artificial antigen-presenting cells. Nat Biotech. 2000;18:405–409. doi: 10.1038/74455. [DOI] [PubMed] [Google Scholar]
- 48.Veomett N, Dao T, Liu H, Xiang J, Pankov D, Dubrovsky L, Whitten JA, Park SM, Korontsvit T, Zakhaleva V, et al. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin Cancer Res. 2014;20(15):4036–4046. doi: 10.1158/1078-0432.CCR-13-2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, Xiang J, Yan S, de Morais Guerreiro MD, Veomett N, et al. Therapeutic bispecific T cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotech. 2015;33(10):1079–1086. doi: 10.1038/nbt.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.May R, Dao T, Pinilla-Ibarz J, Korontsvit T, Zakhaleva V, Zhang RH, Maslak P, Scheinberg DA. CD4+ peptide epitopes from WT1 oncoprotein stimulate CD4+ and CD8+ T cells that recognize and kill human malignant mesothelioma tumor cells. Clin Cancer Res. 2007;13:4547–4555. doi: 10.1158/1078-0432.CCR-07-0708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.