

The Mitochondrial Calcium Uniporter (MCU) complex, the channel responsible for Ca2+ entry inside mitochondria, is involved in a wide range of diseases, including cancer [1]. Although elevated mitochondrial Ca2+ levels have been associated to apoptosis induction, probably by promoting the activation of mitochondrial Permeability Transition Pore (mPTP), a key effector of cell death [2], up-regulation of MCU complex functions have been observed in different cancer contexts [3]. Thus, it is unclear if increased mitochondrial Ca2+ content could sustain cancer progression or augment the susceptibility to apoptosis. Here, we show that high mitochondrial [Ca2+] with concomitant mPTP inhibition could potentiate migratory capacity and invasiveness of cancer cells.
To test the impact of high basal mitochondrial Ca2+ levels on cancer-related features, we used HeLa cells stably silenced for MICU1 gene [4] or PC3 prostate cancer cells stably overexpressing the pore-forming channel subunit MCU [5] (Figure 1(a)). Using a mitochondrial-targeted GCaMP6m probe, which displayed high Ca2+ sensitivity [4], we observed that both MICU1 depletion and MCU up-regulation induced an increase in mitochondrial [Ca2+] at resting conditions (Figure 1(b–c)), due to the accumulation of MCU channel complexes that are not regulated by MICU activity [6,7]. Loss of the gatekeeping functions predisposed to cell death induced by the Ca2+-dependent apoptotic stimulus C2-ceramide (Figure 1(d–e)), which is minimized by pre-treatment with the known mPTP inhibitor Cyclosporine A (CsA), thus suggesting that high MCU Ca2+ affinity sensitizes cells to apoptosis by affecting mPTP opening. Importantly, in our cellular settings, high mitochondrial Ca2+ levels do not promote cell death at basal conditions, indicating that the elevated capacity of mitochondria to accumulate Ca2+ is not toxic per se. Moreover, CsA treatment does not affect mitochondrial Ca2+ levels (Figure 1(b–c)). With our surprise, analysis of cancer cell proliferation, using a crystal violet-based assay, revealed that CsA treatment strongly augmented the proliferative rate of cells with high mitochondrial Ca2+ content, either due to MICU1 depletion (Figure 1(f)) or MCU over-expression (Figure 1(g)), without affecting the growth of the control counterparts.
Figure 1.
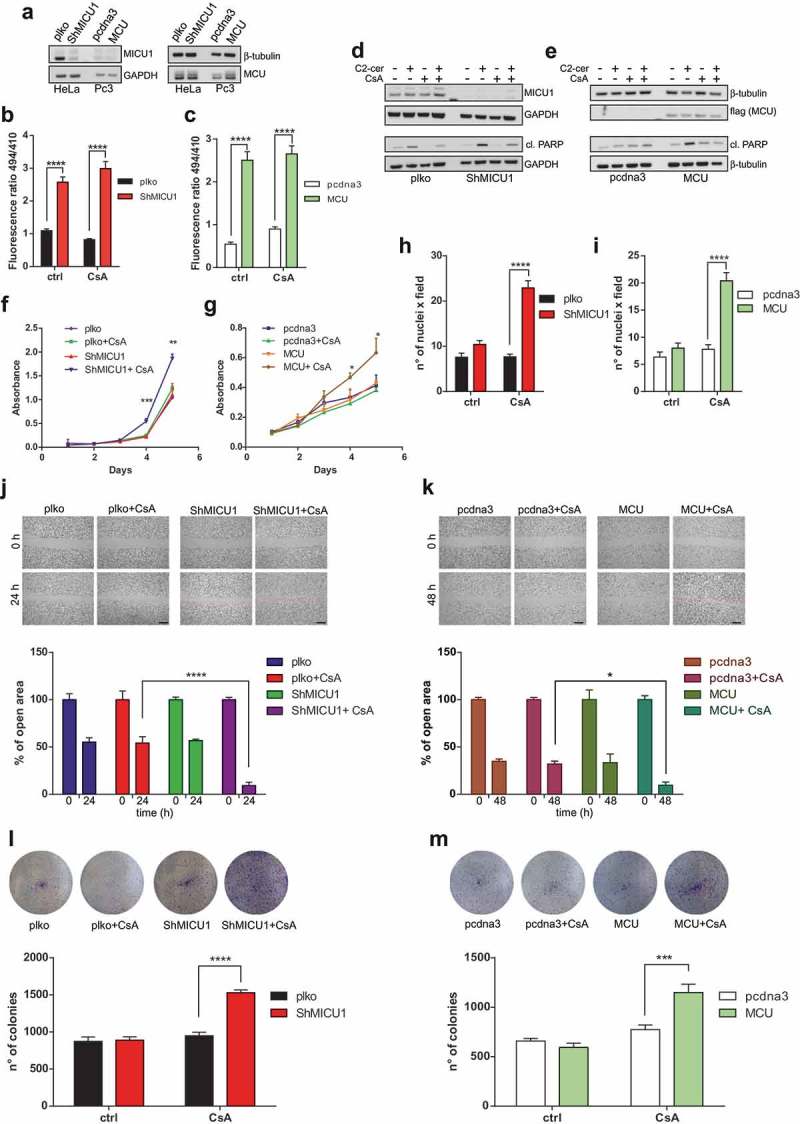
(a) Western blots of HeLa and PC3 cells to assess MICU1 and MCU levels. Antibodies used: GAPDH (#2118) and β-tubulin (#2128) by Cell Signalling; MICU1 (HPA037480) and MCU (HPA016480) by Sigma-Aldrich. (b-c) HeLa (b) and PC3 (c) cells were transiently transfected using Lipofectamine 2000 reagent (Thermo Fisher Scientific) with the mitochondrial-targeted GCaMP6m construct to assess basal mitochondrial Ca2+ levels at resting state. The cells were treated or not with 1μM Cyclosporine A (CsA). N = 4; **** p < 0.0001. Two-way ANOVA. (d–e) Western blots of HeLa (d) or PC3 (e) cells, treated, where indicated, with 40μM C2-ceramide (C2-cer) for 4 h or 1 μM CsA for 4 h (alone or added 1 h before C2-cer). Other antibodies used: cleaved PARP (#9541) by Cell Signalling; flag (F7425) by Sigma-Aldrich. (f–g) 10,000 HeLa (f) or PC3 (g) cells were plated in five sets of five wells of a 12-well plate. Starting from the following day (day 1), cells were treated as indicated, and 1 set of wells was washed once with PBS, fixed in 4% formaldehyde (PFA) solution for 10 min at RT, and then kept in PBS at 4°C. At day 5, all the wells were stained with crystal violet. After lysis with 10% acetic acid, the absorbance was read at 595 nm. N = 3; *p < 0.05; **p < 0.01; ***p < 0.001. Multiple t-test. (h–i) 1.5 × 105 HeLa (H) or 2.5 × 105 PC3 cells (I) were resuspended in medium without serum, with or without CsA, and then seeded on Transwell 8 μm pore size (Corning Incorporated 3422), using 20% FBS as attractant. After 24 h or 48 h, respectively, cells were fixed with PFA and stained with DAPI. N = 3; ****p < 0.0001. Two-way ANOVA. Scale bar: 200 μm. (j-k) HeLa (j) and PC3 (k) cells were grown in 6-well plates to 80–90% confluence in medium supplemented with 10% FBS. The cell monolayer was then scratched with a P200 tip, and then treated or not with CsA. Cells were allowed to close the wound for 24 h or 48 h, respectively. Migration distance was measured using the ImageJ software. N = 3; *p < 0.05; ****p < 0.0001. Two-way ANOVA. (l–m) 3000 HeLa (l) or 6000 PC3 (m) cells grown in 6-well plates, treated or not with CsA. After 10 or 15 days, respectively, cells were fixed with PFA and stained with crystal violet. Colony number was calculated using the ImageJ software. N = 3; ***p < 0.001; ****p < 0.0001; Two-way ANOVA.
To further dissect this event, we examined other cellular processes that are often associated with tumor progression and aggressiveness. Firstly, we measured the ability of both HeLa and PC3 to migrate through a basement membrane following a serum gradient using a Boyden chamber assay. Treatment with CsA significantly increased the capacity to cross the membrane exclusively in MICU1 Knock-Down (KD) HeLa or MCU-overexpressing PC3 cells (Figure 1(h–i)). Next, we assessed the combinatory effect of higher mitochondrial Ca2+ levels and mPTP inhibition on the capability to close the gap in a classical wound-healing assay. As shown in Figure 1(j), CsA treatment positively regulate MICU1 KD cells in the closure of the wound (% of open area after 24 h from the wound: 9.23 ± 3.83), whereas it does not affect the migration of control cells (plko+CsA: 54.23 ± 7.18). Similar results have been obtained in PC3 cells (Figure 1(k)), where CsA promotes the closure of scratched area only in cells that express MCU at higher extent (% of open area after 48 h from the wound: pcdna3+ CsA: 31.93 ± 3.43; MCU+CsA: 9.59 ± 3.64). Finally, we measured the ability to form colonies in vitro, a marker of tumorigenesis. CsA increased the number of colonies of both HeLa cells stably transduced with MICU1-directed shRNA (Figure 1(l)) and PC3 stably expressing MCU (Figure 1(m)), highlighting the pro-cancerous effects of CsA when tumor cell mitochondria can uptake Ca2+ at a higher degree as a consequence of aberrations in the uniporter complex composition.
In conclusion, although elevated mitochondrial Ca2+ entry generally predisposes to cell death, it could potentiate tumor cell proliferation and invasion when mPTP properties appear altered, a condition that frequently occurs in multiple cancer scenarios, either through post-translational events or the inhibitory activity of multiple pro-malignant factors. In this light, it has been recently shown as chronic mitochondrial Ca2+ aberrations, obtained through MCU depletion, could generate adaptive modifications of mPTP functions, suggesting an altered death response in cells with deregulated mitochondrial Ca2+ homeostasis [8]. Thus, a therapeutic strategy based on increasing mitochondrial Ca2+ uptake could be inefficient in cancer cells where the opening of mPTP is abolished. Conversely, blocking the pro-cancerous effects of mitochondrial Ca2+ entry could result in a significant reduction of tumor progression. The identification of the molecular signaling pathways that are regulated by mitochondrial Ca2+, such as ATP or Radical Oxygen Species (ROS) production, as well as metabolic changes induced by mPTP inhibition, will furnish additional steps for the comprehension of the role of mitochondrial Ca2+ and MCU complex in cancerogenesis.
Funding Statement
This work was supported by the Associazione Italiana per la Ricerca sul Cancro [IG‐18624]; Fondazione Telethon [GGP15219/B]; Fondazione Umberto Veronesi; Ministero della Salute [GR-2016-02364602]; Ministero dell'Istruzione dell'Università e della Ricerca [2017E5L5P3].
Acknowledgments
PP is grateful to Camilla degli Scrovegni for continuous support.
PP is supported by the Italian Association for Cancer Research (AIRC: IG‐18624); Telethon (GGP15219/B), the Italian Ministry of Education, University and Research (PRIN/COFIN) (2017E5L5P3) and by local funds from the University of Ferrara.
SM is supported by “Fondazione Umberto Veronesi” and the Italian Ministry of Health (GR-2016-02364602).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Giorgi C, Marchi S, Pinton P.. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018. November;19(11):713–730. PubMed PMID: 30143745. [DOI] [PubMed] [Google Scholar]
- [2].Bonora M, Bononi A, De Marchi E, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013. February 15;12(4):674–683. PubMed PMID: 23343770; PubMed Central PMCID: PMC3594268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Marchi S, Vitto VAM, Danese A, et al. Mitochondrial calcium uniporter complex modulation in cancerogenesis. Cell Cycle. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marchi S, Corricelli M, Branchini A, et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca(2+) levels and tumor growth. Embo J. 2019. January 15;38(2):e99435 PubMed PMID: 30504268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Marchi S, Lupini L, Patergnani S, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol. 2013. January 7;23(1):58–63. PubMed PMID: 23246404; PubMed Central PMCID: PMC3540261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mallilankaraman K, Doonan P, Cardenas C, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012. October 26;151(3):630–644. PubMed PMID: 23101630; PubMed Central PMCID: PMC3486697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Patron M, Checchetto V, Raffaello A, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell. 2014. March 6;53(5):726–737. PubMed PMID: 24560927; PubMed Central PMCID: PMC3988891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Parks RJ, Menazza S, Holmstrom KM, et al. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc Res. 2019. February 1;115(2):385–394. PubMed PMID: 30165576. [DOI] [PMC free article] [PubMed] [Google Scholar]