

Supplemental Digital Content is Available in the Text.
Longitudinal positron emission tomography of translocator protein-18 kDa revealed early central, and persistent peripheral, myeloid activation in a mouse tibial fracture model of complex regional pain syndrome.
Keywords: Complex regional pain syndrome, Microglia, Myeloid cells, Innate immune system, Positron emission tomography, TSPO
Abstract
Complex regional pain syndrome (CRPS) is a severely disabling disease characterized by pain, temperature changes, motor dysfunction, and edema that most often occurs as an atypical response to a minor surgery or fracture. Inflammation involving activation and recruitment of innate immune cells, including both peripheral and central myeloid cells (ie, macrophages and microglia, respectively), is a key feature of CRPS. However, the exact role and time course of these cellular processes relative to the known acute and chronic phases of the disease are not fully understood. Positron emission tomography (PET) of translocator protein-18 kDa (TSPO) is a method for noninvasively tracking these activated innate immune cells. Here, we reveal the temporal dynamics of peripheral and central inflammatory responses over 20 weeks in a tibial fracture/casting mouse model of CRPS through longitudinal TSPO-PET using [18F]GE-180. Positron emission tomography tracer uptake quantification in the tibia revealed increased peripheral inflammation as early as 2 days after fracture and lasting 7 weeks. Centralized inflammation was detected in the spinal cord and brain of fractured mice at 7 and 21 days after injury. Spinal cord tissue immunofluorescent staining revealed TSPO expression in microglia (CD11b+) at 7 days but was restricted mainly to endothelial cells (PECAM1+) at baseline and 7 weeks. Our data suggest early and persistent peripheral myeloid cell activation and transient central microglial activation are limited to the acute phase of CRPS. Moreover, we show that TSPO-PET can be used to noninvasively monitor the spatiotemporal dynamics of myeloid cell activation in CRPS progression with potential to inform disease phase–specific therapeutics.
1. Introduction
Acute pain after injury or surgery is common and treatable; however, an estimated 5% to 30% of patients transition from acute to chronic pain and the mechanisms underlying this transition represent a major knowledge gap.38 Although there are several theories explaining the progression from acute to chronic pain,16 there has been a recent shift from a neuron-centered hypothesis to one that includes neuroimmune interactions both peripherally and centrally.22 For example, peripheral macrophages may be key players in the initial activation of primary afferent neurons46 and subsequent central microglial release of sensitizing factors, such as interleukin-1 beta (IL-1β) and tumor necrosis factor–alpha (TNF-α), can alter the central nervous system (CNS) synaptic milieu in a process termed neuroinflammation.19,40
Complex regional pain syndrome (CRPS) is a debilitating disease affecting 50,000 new patients in the United States every year and occurring most often after a limb fracture or minor surgery.42 Complex regional pain syndrome consists of two distinct phases: an acute/peripheral and a chronic/central phase.54 The acute phase is generally characterized by a warm, edematous limb with local immune cell infiltration and elevated proinflammatory cytokines in the affected skin. By contrast, the chronic phase demonstrates loss of limb warmth and peripheral inflammation13 but includes persistent pain, cognitive deficits,8,31,56 central glial cell activation,4,20 and cytokine elevations.32 This clinical progression is closely recapitulated in a rodent tibial fracture/casting model of CRPS.9 The transition from the acute/peripheral phase to the chronic/central phase of CRPS occurs around 12 months after the inciting event in humans13 and around 5 weeks after injury in the mouse tibial fracture/casting model.9 In prior studies using this model, peripherally restricted biologic agents against IL-1β, nerve growth factor, and TNF-α reduced nociceptive sensitization in the acute but not chronic phase.47 Treatment in the chronic phase, by contrast, may require centrally acting agents such as glial cell modulators. This underscores the need for a better cellular understanding of the spatiotemporal interactions between peripheral and central innate immune activation to most effectively tailor therapy to the underlying disease process. Moreover, mechanisms underlying the transition from acute to chronic CRPS may extend to other forms of chronic pain.
One major difficulty with clinical use of immune modulators is that the peripheral vs central innate immune cell activation status of patients is unknown and likely changes over the course of disease. Recent developments in clinical imaging may provide invaluable techniques to guide and monitor innate immune–targeted interventions in real time. Positron emission tomography (PET) is one such powerful imaging tool that allows for noninvasive tracking of molecular processes in vivo and enables whole-body longitudinal imaging of the same living subject with high sensitivity.28,41 Translocator protein-18 kDa (TSPO)-PET imaging agents have been widely investigated for studying innate immune activation. Importantly, TSPO radiotracers have previously been used in patients with chronic pain3,29,36 with a focus on the CNS. Here, we use a second-generation TSPO-PET tracer, [18F]GE-180,53 to investigate whole-body spatiotemporal dynamics of peripheral and central myeloid cell activation in the mouse tibial fracture/casting model of CRPS.
2. Methods
2.1. Animals
Male, C57BL/6 wild-type mice (10-11 weeks) with average weight of 25 g were purchased from Jackson Laboratories (Jax #00664) and acclimatized for 1 week before experiments. Mice were housed 2 to 5 per cage under a 12-hour light/dark schedule in a temperature-controlled environment with ad libitum access to food and water. Isoflurane 2.0% to 3.0% for induction and 1.5% to 2.5% for maintenance was used for all experiments involving anesthesia, except where noted. All experiments described here involving animals were approved by the Stanford Administrative Panel on Laboratory Animal Care, which is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC International) and by the Veterans Affairs Palo Alto Health Care System Institutional Animal Care and Use Committee in accordance with American Veterinary Medical Association guidelines and the International Association for the Study of Pain. The procedures uphold all federal and state regulations governing the humane care and use of laboratory animals. To comply with the Stanford Administrative Panel on Laboratory Animal Care guidelines on minimizing animal numbers, for each experimental paradigm we used the minimum number of mice required to obtain significance after conducting a power analysis based on the expected mean and SD from existing data for a given assay.
2.2. Complex regional pain syndrome model generation
Mice were anesthetized with isoflurane and underwent a closed right distal tibial fracture followed by casting, as previously described.51 Briefly, the right hind limb was wrapped in gauze and the distal tibia was fractured using a hemostat. The hind limb was then wrapped in casting tape from the metatarsals of the hind paw up to a spica formed around the abdomen to ensure that the cast did not slip off. The cast over the paw was applied only to the plantar surface; a window was left open over the dorsum of the paw and ankle to prevent constriction when postfracture edema developed. At 21 days after fracture, mice were briefly anesthetized, and casts were removed with cast shears. For behavior, mice were tested at baseline before fracture, and then beginning after cast removal at 21 days (acute phase), 7 weeks (chronic phase), and 20 weeks (resolution phase) after fracture.
2.3. Study design
A CRPS mouse model was generated in n = 38 mice by right tibial fracture followed by 21 days of casting. A group of n = 30 uninjured/naive mice were used as controls. Mice were separated into behavior (n = 6 control and n = 6 CRPS), in vivo dynamic PET imaging (n = 3 control and n = 3 CRPS), in vivo longitudinal PET imaging (n = 8 control and n = 8 CRPS), ex vivo tissue gamma counting/autoradiography (BioD/ARG) (n = 8 control, n = 6 CRPS 7 days, and n = 4 CRPS 7 weeks), and immunohistochemistry cohorts (n = 5 control, n = 5 CRPS 7 days, and n = 6 CRPS 7 weeks). Behavior (mechanical allodynia and weight bearing) and CRPS signs (temperature changes and edema) were assessed in the behavioral cohort at baseline and after cast removal at 21 days after fracture, 7 weeks and 20 weeks after fracture. The time course of myeloid cell activation was longitudinally evaluated in vivo by PET/computed tomography (CT) using the TSPO radiotracer, [18F]GE-180, for the PET imaging cohort at 2, 7, 21 days, 7 weeks, and 20 weeks after fracture. To confirm in vivo PET findings and localization of tracer uptake, ex vivo gamma counting and autoradiography were performed in the BioD/ARG group. Finally, immunofluorescence was performed on spinal cord sections to visualize cellular expression of TSPO in the immunohistochemistry cohort (Fig. 1). Sample size for imaging and BioD/ARG groups was limited by the number of successful tail vein cannulations for radiotracer injection on each day. Mice were excluded from these studies if their tail vein could not be cannulated on that given day, as no radiotracer could be injected. They were, however, included in subsequent timepoints if the vein was successfully catheterized.
Figure 1.
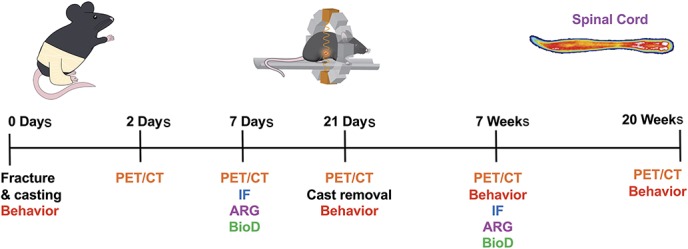
Study design for longitudinal evaluation of behavior and myeloid lineage cell activation in a mouse model of CRPS. Mice underwent right tibial fracture/casting and were followed over 20 weeks after injury for behavioral and imaging studies using TSPO-PET imaging. Immunofluorescent (IF) staining, autoradiography (ARG), tissue gamma counting for biodistribution (BioD), and behavior were also evaluated throughout the study. CRPS, complex regional pain syndrome; CT, computed tomography; PET, positron emission tomography.
2.4. Behavior
To ensure experimental rigor, female researchers (E.H. and V.L.T.) performed all behavioral testing in a blinded fashion. All testing was conducted between 7:00 am and 1:00 pm in an isolated, temperature- and light-controlled room. Mice were acclimated for 30 to 60 minutes in the testing environment within custom clear plastic cylinders (4″ D) on a raised metal mesh platform (24″ H). Mice were randomized by simple selection from their home cage (4 mice per cage) before testing and placed in a cylinder (n = 6 control and n = 6 CRPS). After testing, mouse numbers were recorded on the data sheet. All 12 mice were followed longitudinally for 20 weeks, with 1 CRPS mouse mortality before the 20-week timepoint.
2.4.1. Mechanical nociception assays
To evaluate mechanical reflexive hypersensitivity, we used a logarithmically increasing set of 8 von Frey filaments (Stoelting), ranging in gram force from 0.007 to 6.0 g. These were applied perpendicular to the plantar hind paw with sufficient force to cause a slight bending of the filament. A positive response was characterized as a rapid withdrawal of the paw away from the stimulus filament within 4 seconds. Using the up-down statistical method,15 the 50% withdrawal mechanical threshold scores were calculated for each mouse and averaged across the experimental groups of longitudinally followed cohorts at baseline, 21 days, 7 weeks, and 20 weeks.
2.4.2. Unweighting, temperature, and paw edema measurements
An incapacitance device (IITC, Inc, Life Science, Woodland Hills, CA) was used to measure hind paw unweighting in n = 6 control and n = 6 CRPS mice at baseline and 21 days. The mice were manually held in a vertical position over the apparatus with the hind paws resting on separate metal scale plates, and the entire weight of the mouse was supported on the hind paws. Six consecutive 6-second measurements were taken at 60-second intervals and averaged to calculate the overall hind paw weight-bearing values using the following formula: 2 × (right paw weight bearing)/(right paw weight bearing + left paw weight bearing).
The temperature of the hind paw was measured using a fine-gauge thermocouple wire (Omega, Stamford, CT) in n = 6 control and n = 6 CRPS at baseline and 21 days. Temperature testing was performed over the hind paw dorsal skin between the first and second metatarsals (medial), the second and third metatarsals (central), and the fourth and fifth metatarsals (lateral). The measurements for each hind paw were averaged for the mean paw temperature and displayed as the difference in temperature between right (injured) and left (uninjured) paws.
Hind paw edema was determined by measuring the hind paw dorsal–ventral thickness over the midpoint of the third metatarsal with a LIMAB laser measurement sensor (LIMAB, Goteborg, Sweden) while the mouse was briefly anesthetized with isoflurane in n = 6 control and n = 6 CRPS mice at baseline and 21 days. Hind paw thickness data are expressed as the difference between the right (injured) side and the left (uninjured) side.
2.5. Positron emission tomography tracer synthesis and imaging
2.5.1. [18F]GE-180 synthesis
[18F]GE-180 was synthesized as previously described.10 No carrier-added aqueous [18F]fluoride ion was produced on a PETtrace cyclotron (GE Healthcare, Uppsala, Sweden) by irradiation of a 1.6-mL water target using a 16-MeV proton beam on 95% enriched [18O]H2O by the [18O(p,n)18F] nuclear reaction. [18F]F− in [18O]H2O was transferred to a GE TRACERlab FXFN synthesizer and trapped on an anion-exchange resin (30-PS-HCO3, pretreated with 1-mL EtOH and 1-mL H2O). [18F]F− was then eluted into the reactor using 0.9 mL of an eluent solution containing 4-mg KHCO3 and 10-mg Kryptofix 2.2.2 (K2.2.2: 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosan) in acetonitrile (0.75 mL) and water (150 mL). The solution was azeotropically dried at 65°C under helium flow and vacuum, followed by heating at 88°C under vacuum. Mesylate precursor ((S)-2-(4-(diethylcarbamoyl)-5-methoxy-1,2,3,4-tetrahydro-9H-carbazol-9-yl)ethyl methanesulfonate, 1 mg in MeCN, 1 mL, anhydrous) was added to the dry Kryptofix 2.2.2/K+[18F]F− complex, and the reaction mixture reacted for 10 minutes at 90°C before being cooled to 40°C and diluted with 1 mg/mL ascorbic acid in water (1.5 mL). The diluted product was purified by semipreparative HPLC (column inner diameter 100 × 10 mm, particle size 5 µm, 0.1 mg/mL ascorbic acid in H2O/MeCN (65:35)) and monitored by gamma detection and UV at 254 nm at a flow rate of 4 mL/minute. The radioactive peak corresponding to [18F]GE180 (tR = 25 minutes) was collected and diluted with 1 mg/mL ascorbic acid in water (20 mL), after which it was trapped on a C18 Sep-Pak Plus Light cartridge preconditioned with EtOH (5 mL) and 1 mg/mL ascorbic acid in water (10 mL). The trapped, purified [18F]GE180 was eluted from C18 Sep-Pak Plus Light cartridge using EtOH (0.8 mL) and saline (4.5 mL). The formulated product was transferred under helium flow to a sterile, pyrogen-free 30-mL vial. The radiochemical yield was 7 ± 4% (n = 10, decay uncorrected) at the end of synthesis. Chemical and radiochemical purity were determined by reverse-phase analytical HPLC (Phenomenex Luna column, 150 × 4.6 mm, particle size 3 µm, and pore size 100 Å) using an 11-minute 50% to 95% gradient (0.1 mg/mL ascorbic acid in H2O:MeCN, monitored by gamma detection and UV at 254 and 280 nm).
2.5.2. Pilot dynamic positron emission tomography/computed tomography imaging
Dynamic PET images were acquired over 60 minutes using a dual microPET/CT scanner (Inveon, Siemens, Mountain View, CA) at 7 days after fracture (n = 3 control and n = 3 CRPS) under isoflurane anesthesia after intravenous tail vein injections of 160 to 220 µCi of [18F]GE-180, as previously described.14 Time activity curves (TACs) were plotted for the duration of each 60-minute scan to determine the timepoint for subsequent static scans based on an optimal signal-to-background ratio.
2.5.3. Longitudinal static positron emission tomography/computed tomography imaging
Eight mice per group were anesthetized using isoflurane, and tail vein catheters were inserted for radiotracer injection. Mice that were successfully catheterized and injected were scanned; mice that could not be catheterized were excluded. The total number of mice scanned per timepoint was as follows: 2 days (n = 8 control and n = 5 CRPS), 7 days (n = 8 control and n = 8 CRPS), 21 days (n = 5 control and n = 5 CRPS), 7 weeks (n = 6 control and n = 5 CRPS), and 20 weeks (n = 3 control and n = 4 CRPS) after injury. Mice were injected with 160 to 220 µCi of [18F]GE-180 intravenously, and 10-minute static PET images were acquired 50 to 60 minutes after injection of [18F]GE-180. A CT image was acquired just before each PET scan to provide an anatomical reference frame for the respective PET data. Raw CT images were acquired at 80 kVp at 500 μA, 2-bed position, half-scan 220° of rotation, and 120 projections per bed position with a cone beam micro-X-ray source (50-μm focal spot size) and a 2048 × 3072-pixel X-ray detector. On the basis of attenuation correction from the CT measurements, each 10-minute static PET scan was acquired with default settings of coincidence, a timing window of 3.4 ns, and an energy window of 350 to 650 keV. Positron emission tomography images were reconstructed by performing 2 iterations of a 3-dimensional ordered subsets expectation maximization algorithm (12 subsets) and then 18 iterations of the accelerated version of 3-dimensional maximum a posteriori, with a matrix size of 128 × 128 × 159.
2.5.4. Positron emission tomography analysis and quantification
Computed tomography and PET images were coregistered, and quantification of radiotracer uptake in spinal cord, tibia, and specific brain regions was performed using VivoQuant software (Version 3.0, inviCRO). Cortex, hippocampus, medulla, midbrain, pons, striatum, thalamus, and whole brain regions were chosen a priori to ensure nonbiased results based on human studies5 and limited by size of a mouse brain. A 3D mouse brain atlas was fitted to the skull CT of each mouse to quantify regional uptake values of each region. Spinal cord regions of interest (ROIs) were drawn after removal of spinal vertebrae signal through Otsu thresholding. A 3D ROI was created for full spinal cord quantification before segmenting cervical/thoracic and lumbar spinal cords at T12 and L5 vertebrae to evaluate spatial differences in TSPO activation in the spinal cord. Otsu thresholding was additionally used to quantify tracer uptake in the ipsilateral (right) and contralateral (left) tibia. The ROI radioactivity measurement (nCi/cc) was divided by the decay-corrected dose for each mouse at the time of imaging, and then multiplied by 100 to obtain percent injected dose per gram (%ID/g) values. Peripheral analysis was plotted as a ratio of ipsilateral to contralateral %ID/g for each mouse. For evaluation of CNS tissues, the fold difference in tracer uptake was calculated by creating a ratio of the %ID/g from each mouse to the average %ID/g of all controls imaged the same day. Fold differences in fracture vs control spinal cord and brain tissue at each timepoint were calculated and plotted.
2.6. Ex vivo tissue gamma counting and autoradiography
2.6.1. Ex vivo tissue gamma counting for biodistribution analysis
Mice were deeply anesthetized with 2.0% to 3.0% isoflurane and perfused with phosphate-buffered saline (PBS) after a blood sample was taken through cardiac puncture for biodistribution analysis (n = 8 control, n = 6 CRPS 7 days, and n = 4 CRPS 7 weeks). Radioactivity within each dissected tissue (spinal cord, brain, blood, and spleen) was quantified using a gamma counter (Cobra II Auto-Gamma counter; Packard Biosciences Co, Wellesley, MA). Tissue-associated radioactivity was normalized to tissue weight and the decay-corrected radioactive dose at injection time, yielding the %ID/g for each tissue. Fold differences in tracer uptake in the spinal cord, brain, blood, and spleen were calculated by creating a ratio of the %ID/g from each tissue to the average %ID/g of the corresponding control tissues counted the same day.
2.6.2. Ex vivo autoradiography
Spinal cords and brain tissues from mice used for biodistribution were further analyzed through digital autoradiography. After perfusion and gamma counting, brain tissue was frozen in optimum cutting temperature compound (Sakura Finetek, Inc, Torrance, CA), and 20-µm-thick sections were cut using a cryostat microtome HM500 (Microm, Walldorf, Germany) at −20°C. Whole spinal cords and slides with brain slices were laid in a cassette and exposed to a 18F-sensitive storage phosphor film imaging plate (Fujifilm; GE Healthcare) for 20 hours at −20°C. Plates were scanned using a typhoon phosphorimager (Amersham Biosciences, Piscataway, NJ) and analyzed using ImageJ (image processing software, version 2.0.0). Brain section anatomy was confirmed by staining the exact same sections with Nissl (cresyl violet acetate; Sigma Aldrich, St. Louis, MO), as previously described.25
2.7. Immunohistochemistry
Mice (12-14 weeks) were anesthetized and transcardially perfused with PBS followed by 10% formalin in PBS. Spinal cord segments (L3-L5) were dissected and cryoprotected in 30% sucrose in PBS (n = 5 control, n = 5 CRPS 7 days, and n = 6 CRPS 7 weeks). Tissues were frozen in optimum cutting temperature, sectioned coronally (40 μm) on a cryostat (Leica Biosystems, Wetzlar, Germany), and incubated in blocking buffer (5% donkey serum and 0.3% Triton X-100 in PBS) for 1 hour at room temperature. Sections were incubated overnight at 4°C with primary antibodies rat anti-CD11b (Abd Serotec, Hercules, CA, #MCA711G, 1:500), rabbit anti-TSPO (Abcam, ab109497, 1:500; Abcam, Cambridge, United Kingdom), mouse anti-GFAP (Sigma #G3893, 1:1000), and/or rat anti-CD31/PECAM1 (BD Pharmingen, Franklin Lakes, NJ, #550274, 1:1000). After washing (1% donkey serum and 0.3% Triton X-100 in PBS), sections were incubated with appropriate Alexa Fluor–conjugated secondary antibodies for 2 hours at room temperature. Images were acquired using a Keyence BZ-X800 through the sectioning module to remove nonfocused light. We acquired 3 to 5 dorsal horn images per mouse with n = 5 to 6 mice/group, as outlined above, to evaluate the relative cellular expression of TSPO, CD11b, GFAP, and PECAM1 in the spinal cord dorsal horn. Each image was taken using identical settings including exposure, with 0.4-μm-step z-stacks of 19 slices at ×20 or ×63 objective magnification.
2.8. Quantitative histological analysis
Images were analyzed using Adobe Photoshop CS6 by manual counting of individual CD11b+ cells and then colocalized with TSPO expression. Specifically, CD11b+ cells in which TSPO staining followed microglial cell body and at least one process were considered double positive. Given the extensive and confluent staining pattern of TSPO on endothelial PECAM1+ cells, it was not possible to count all TSPO+ cells in a given image. All counts are therefore expressed as % CD11b+ cells. No cells were found to be TSPO+ GFAP+, consistent with other publications.7,52
2.9. Statistical analysis
Statistical analysis was performed using IBM SPSS for mixed model analysis of TACs. Type III tests of fixed effects were used to determine differences in tracer kinetics of TACs from pilot dynamic PET imaging. Statistical analysis of all other data was performed using GraphPad Prism (version 7). Data are represented as mean + SEM in addition to individual values plotted for all bar graphs for full data transparency. For imaging and behavioral data, two-way analysis of variance with the Sidak multiple comparisons post hoc test, one-way analysis of variance with the Tukey multiple comparisons post hoc test, or unpaired t tests were used when appropriate to compare data in CRPS vs control mice, as indicated in supplemental Table 1 (available at http://links.lww.com/PAIN/A794) and individual figure legends. P values < 0.05 in each set of data comparisons were considered statistically significant.
3. Results
3.1. Tibial fracture/casting recapitulates the signs of complex regional pain syndrome
Mice that underwent right hind limb tibial fracture followed by 21 days of casting developed the cardinal signs of CRPS including mechanical allodynia, increased paw edema and temperature, and decreased weight bearing on the injured paw (Fig. 2). Injured mice exhibited a decrease in the mechanical threshold on the ipsilateral paw compared with controls at 21 days (control 1.90 ± 0.16 g vs CRPS 0.22 ± 0.06 g; F(7,39) = 15.83; ****P < 0.0001) and 7 weeks (control 1.79 ± 0.09 g vs CRPS 0.34 ± 0.04 g; F(7,39) = 15.83; ****P < 0.0001) that resolved by 20 weeks after fracture (Fig. 2A). At the time of cast removal, ipsilateral paws were 0.5 mm thicker than contralateral paws while control mice had no difference in paw thickness (Fig. 2B, control 0.002 ± 0.02 mm vs CRPS 0.53 ± 0.03 mm; df = 10; ****P < 0.0001). The temperature difference between the ipsilateral and contralateral paws was significantly greater for injured mice compared with control mice (Fig. 2C, control 0.05 ± 0.08°C vs CRPS 1.12 ± 0.08°C; df = 10; ****P < 0.0001). The percent weight bearing on the ipsilateral paw was significantly decreased for fractured mice compared with controls at 21 days (Fig. 2D, control 99.7 ± 1.0% vs CRPS 54.5 ± 1.8%; df = 10; ****P < 0.0001).
Figure 2.

Tibial fracture/casting recapitulates the cardinal signs of CRPS. At the time of cast removal, injured mice exhibit a decreased threshold to mechanical stimulation (A), which is significantly different from uninjured controls at 21 days and 7 weeks after injury (****P < 0.0001) and recovered to baseline at 20 weeks. Data are presented as mean ± SEM (n = 6 control and n = 6 CRPS followed longitudinally). (B) After cast removal, mice demonstrate increased ipsilateral paw thickness (****P < 0.0001) and (C) increased ipsilateral limb temperature (****P < 0.0001), relative to uninjured control mice. (D) Mice further display decreased weight bearing on their affected limb compared with uninjured control (****P < 0.0001). Individual data points are shown in addition to mean + SEM (n = 6 control and n = 6 CRPS at 21 days after injury). One-way ANOVA with the Tukey multiple comparisons post hoc test (A) and unpaired t tests (B–D) were used. ANOVA, analysis of variance; CRPS, complex regional pain syndrome.
3.2. Dynamic positron emission tomography time activity curves show differences in tracer uptake after injury
Time activity curves from dynamic scans of control and fractured mice at 7 days after fracture revealed markedly increased uptake of [18F]GE-180 in the spinal cord and brain of fractured mice compared with control littermates but with no significant differences in trajectory of tracer kinetics between the groups (supplemental Figure 1, available at http://links.lww.com/PAIN/A794). The 50- to 60-minute timepoint following tracer administration was chosen for subsequent static scans to ensure tracer uptake values were not influenced by nonspecific binding or blood pool.
3.3. Peripheral inflammation from myeloid cell activation can be tracked longitudinally and persists after injury
Whole-body PET/CT images enabled investigation of the peripheral TSPO-PET signal, specifically in the fractured tibia. Positron emission tomography image quantification revealed marked peripheral inflammation in the ipsilateral tibia of mice at multiple timepoints after fracture. The ratio of tracer uptake in the ipsilateral over contralateral tibia in fractured mice was significantly elevated compared with control mice at 2 days (control 0.91 ± 0.05 vs CRPS 1.21 ± 0.07; df = 10; *P < 0.05), 7 days (control 1.00 ± 0.06 vs CRPS 1.57 ± 0.13; df = 14; **P < 0.01), 21 days (control 0.99 ± 0.04 vs CRPS 1.54 ± 0.04; df = 8; ****P < 0.0001), and 7 weeks (control 0.96 ± 0.07 vs CRPS 1.51 ± 0.09; df = 9; **P < 0.01). No significant differences in tracer uptake were observed at 20 weeks (control 0.89 ± 0.05 vs CRPS 1.31 ± 0.15; df = 5; P = 0.07) (Fig. 3).
Figure 3.

Peripheral myeloid cell activation starts in the acute phase and persists into the chronic phase after fracture. (A) 3D PET/CT images of lower limbs show a significantly increased ratio of tracer uptake in the ipsilateral (right) vs contralateral (left) tibia for mice (B) 2 days (*P < 0.05), (C) 7 days (**P < 0.01), (D) 21 days (****P < 0.0001), and (E) 7 weeks (**P < 0.01) after injury compared with control mice at each timepoint, with no significant difference observed at (F) 20 weeks after fracture (P = 0.0711). Individual data points are shown in addition to mean + SEM (n = 8 mice per group were followed longitudinally; however, only successfully tracer-injected mice on a given day were scanned: 2 days: n = 5 control, n = 8 CRPS; 7 days: n = 8 control, n = 8 CRPS; 21 days: n = 5 control, n = 5 CRPS; 7 weeks: n = 5 control, n = 6 CRPS; and 20 weeks: n = 3 control, n = 4 CRPS). Unpaired t tests were used at each timepoint. CRPS, complex regional pain syndrome; CT, computed tomography; PET, positron emission tomography.
3.4. Spinal cord and brain glial cell activation occurs early and persists for weeks after injury
Whole-body PET/CT images acquired at 2 days, 7 days, 21 days, 7 weeks, and 20 weeks after fracture enabled tracer quantification in the spinal cord (Fig. 4) and brain regions (Fig. 5). The cervical/thoracic spinal cord exhibited a 1.25- and 1.26-fold increase in tracer uptake in fractured mice compared with control mice at 7 and 21 days, respectively (Figs. 4C and D, 7 days control 1.0 ± 0.03 vs CRPS 1.25 ± 0.08; F(1,28) = 18.29; *P < 0.05; 21 days control 1.0 ± 0.03 vs CRPS 1.26 ± 0.08; F(1,16) = 21.65; *P < 0.05). Lumbar spinal cord tracer uptake was 1.27- and 1.28-fold higher in fractured mice than in control mice at 7 and 21 days, respectively (Figs. 4C and D, 7 days control 1.0 ± 0.03 vs CRPS 1.27 ± 0.08; F(1,28) = 18.29; *P < 0.05; 21 days control 1.0 ± 0.04 vs CRPS 1.28 ± 0.06; F(1,16) = 21.65; **P < 0.01). At 7 days after fracture, significant fold increases in tracer uptake of fractured vs control mice were observed in the cortex (control 1.0 ± 0.03 vs CRPS 1.26 ± 0.06; F(1,112) = 83.45; *P < 0.05), hippocampus (control 1.0 ± 0.04 vs CRPS 1.24 ± 0.08; F(1,112) = 83.45; *P < 0.05), midbrain (control 1.0 ± 0.03 vs CRPS 1.23 ± 0.08; F(1,112) = 83.45; *P < 0.05), pons (control 1.0 ± 0.02 vs CRPS 1.28 ± 0.07; F(1,112) = 83.45; **P < 0.01), striatum (control 1.0 ± 0.03 vs CRPS 1.29 ± 0.07; F(1,112) = 83.45; **P < 0.01), thalamus (control 1.0 ± 0.04 vs CRPS 1.28 ± 0.09; F(1,112) = 83.45; **P < 0.01), and whole brain (control 1.0 ± 0.02 vs CRPS 1.26 ± 0.07; F(1,112) = 83.45; *P < 0.05) regions (Fig. 5C), but no differences were observed in the medulla. Tracer uptake in the pons of fractured mice remained significantly elevated (control 1.0 ± 0.03 vs CRPS 1.21 ± 0.09; F(1,64) = 20.04; **P < 0.05, *P < 0.05) at 21 days after fracture (Fig. 5D). No significant differences were observed in the cervical/thoracic spinal cord, lumbar spinal cord, or brain at 2 days, 7 weeks, or 20 weeks after fracture.
Figure 4.

Longitudinal imaging of myeloid cell activation using TSPO-PET demonstrates elevated spinal cord signal during the acute but not chronic phase. (A) 2D sagittal PET/CT images of longitudinal [18F]GE-180-PET shows elevated spinal cord glial activation in mice after injury; white box highlights the lumbar spinal cord. (B–F) Positron emission tomography quantification of tracer uptake in the cervical/thoracic spinal cord (C/T SC) and lumbar spinal cord (LSC) at (B) 2 days, (C) 7 days, (D) 21 days, (E) 7 weeks, and (F) 20 weeks after fracture. Data are shown as fold change in tracer uptake over the average control signal on the respective day. Two-way ANOVA with the Sidak multiple comparisons post hoc test reveals fold increases in tracer uptake in the cervical/thoracic (*P < 0.05) and lumbar (**P < 0.01) SC at 7 and 21 days after injury. Individual data points are shown in addition to mean + SEM (n = 8 mice per group were followed longitudinally; however, only mice that were successfully injected with tracer after tail vein cannulation were scanned: 2 days: n = 5 control, n = 8 CRPS; 7 days: n = 8 control, n = 8 CRPS; 21 days: n = 5 control, n = 5 CRPS; 7 weeks: n = 5 control, n = 6 CRPS; and 20 weeks: n = 3 control, n = 4 CRPS). ANOVA, analysis of variance; CRPS, complex regional pain syndrome; CT, computed tomography; PET, positron emission tomography.
Figure 5.

Widespread brain glial cell activation is apparent in the acute phase after injury. (A) Representative PET/CT coronal brain images reflect PET quantification. Positron emission tomography quantification of select brain regions (cortex, hippocampus, medulla, midbrain, pons, striatum, thalamus, and whole brain) at (B) 2 days, (C) 7 days, (D) 21 days, (E) 7 weeks, and (F) 20 weeks after fracture reveals significant increases in the TSPO-PET signal at 7 and 21 days after fracture by two-way ANOVA with the Sidak multiple comparisons post hoc test (*P < 0.05 and **P < 0.01). All individual data points are shown, except for one data point for (F) thalamus (fold difference 1.66), which was outside of the y-axis, in addition to mean + SEM (n = 8 mice per group were followed longitudinally; however, only mice that were successfully injected with tracer after tail vein cannulation were scanned: 2 days: n = 5 control, n = 8 CRPS; 7 days: n = 8 control, n = 8 CRPS; 21 days: n = 5 control, n = 5 CRPS; 7 weeks: n = 5 control, n = 6 CRPS; and 20 weeks: n = 3 control, n = 4 CRPS). ANOVA, analysis of variance; CRPS, complex regional pain syndrome; CT, computed tomography; PET, positron emission tomography.
3.5. Ex vivo autoradiography and tissue gamma counting confirm tracer biodistribution
Data from ex vivo autoradiography and gamma counting of tissues correspond with PET quantification. Ex vivo autoradiography illustrates a marked increase in tracer uptake in the spinal cord (supplemental Figure 2A available at http://links.lww.com/PAIN/A794) and brain (supplemental Figure 2B available at http://links.lww.com/PAIN/A794) after fracture at 7 days compared with control, but not at 7 weeks. Ex vivo tissue gamma counting of spinal cords revealed a significant increase of 1.31-fold from fractured mice 7 days after fracture compared with control mice (control 1.0 ± 0.08 vs CRPS 7-day 1.31 ± 0.06; F(1,64) = 7.085 *P < 0.05). Fold changes in spinal cord tracer uptake of fractured mice at 7 days vs 7 weeks were also found to be significantly different (CRPS 7-day 1.31 ± 0.06 vs CRPS 7-week 0.88 ± 0.04; F(1,64) = 7.085 **P < 0.01, supplemental Figure 2C available at http://links.lww.com/PAIN/A794). At 7 days and 7 weeks after fracture, ex vivo gamma counting of the brain, blood, and spleen showed no significant fold differences compared with each other or with control mice (supplemental Figures 2D and F available at http://links.lww.com/PAIN/A794).
3.6. Immunohistochemistry reveals translocator protein-18 kDa expression by central nervous system microglia after injury
Double immunofluorescence staining of lumbar spinal cord sections was performed at baseline, 7 days, and 7 weeks after injury using TSPO in conjunction with microglial (CD11b), astrocytic (GFAP), and endothelial (CD31/PECAM1) markers. At baseline, TSPO staining had a characteristic vascular pattern and was predominantly expressed by PECAM1+ vascular endothelial cells (supplemental Figure 3 available at http://links.lww.com/PAIN/A794). Approximately 13% of CD11b+ cells expressed low levels of TSPO (Fig. 6; and supplemental Figure 4 available at http://links.lww.com/PAIN/A794), and no appreciable colocalization of TSPO was found with GFAP+ astrocytes (Fig. 7). At 7 days after fracture, TSPO expression was seen in PECAM1+ vascular endothelial cells, albeit to a lesser extent than at baseline, with overall less prominent vasculature noted throughout the spinal cord (supplemental Figure 3 available at http://links.lww.com/PAIN/A794). At 7 days after fracture, microglial activation was noted by a change in cells to an amoeboid morphology with shorter processes and increased CD11b expression (Fig. 6). At this timepoint, TSPO dramatically shifted to colocalize with CD11b+-activated microglia, and quantitative analysis of colocalization revealed that 57% of CD11b+ cells also expressed high levels of TSPO (control 15 ± 0.93% vs CRPS 7-day 57 ± 2.9%; F(2,13) = 89.82; ****P < 0.0001, Fig. 6 and supplemental Figure 4 available at http://links.lww.com/PAIN/A794). By contrast, although astrocyte expression of GFAP was increased at 7 days and morphology suggested an activated phenotype, there was no appreciable colocalization of TSPO with GFAP+ cells (Fig. 7). At 7 weeks after fracture, the major cell type expressing TSPO was again PECAM1+ vascular endothelial cells. Microglial and astrocytic activation had resolved, with a return to baseline levels of CD11b and GFAP, respectively. Translocator protein-18 kDa expression by CD11b+ cells at 7 weeks remained elevated above control (control 15 ± 0.93% vs CRPS 7-week 26 ± 2.4%; F(2,13) = 89.82, **P < 0.01) but was significantly lower than at 7 days (CRPS 7-day 57 ± 2.9% vs CRPS 7-week 26 ± 2.4%; F(2,13) = 89.82, ****P < 0.0001, supplemental Figure 4, available at http://links.lww.com/PAIN/A794).
Figure 6.
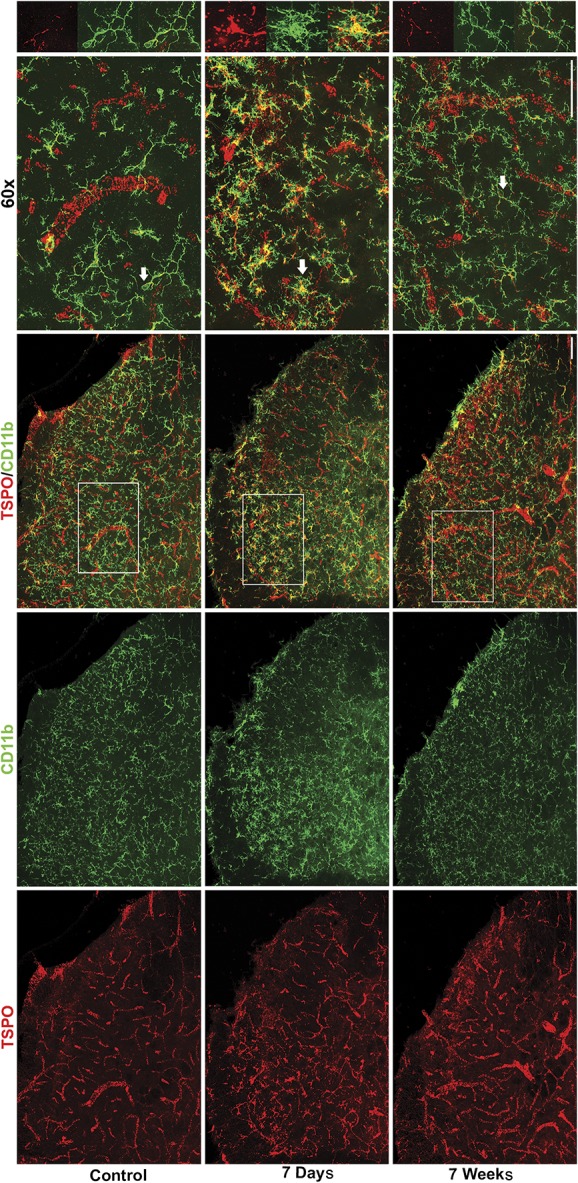
Translocator protein-18 kDa colocalizes with activated microglia in the spinal cord at 7 days after fracture. Images of control and CRPS spinal cord tissue show extensive colocalization of TSPO in activated microglia (CD11b+) at 7 days with only low levels of colocalization at baseline or 7 weeks after fracture. Scale bars are 50 µm (×20) and 50 µm (×60). White arrows in ×60 images indicate cells that are magnified in right hand panels from region outlined by white box in ×20 overlay. Representative images are shown from n = 5 control, n = 5 CRPS 7 days and n = 6 CRPS 7 weeks mice, with 3 to 5 spinal cord sections examined per mouse. CRPS, complex regional pain syndrome.
Figure 7.
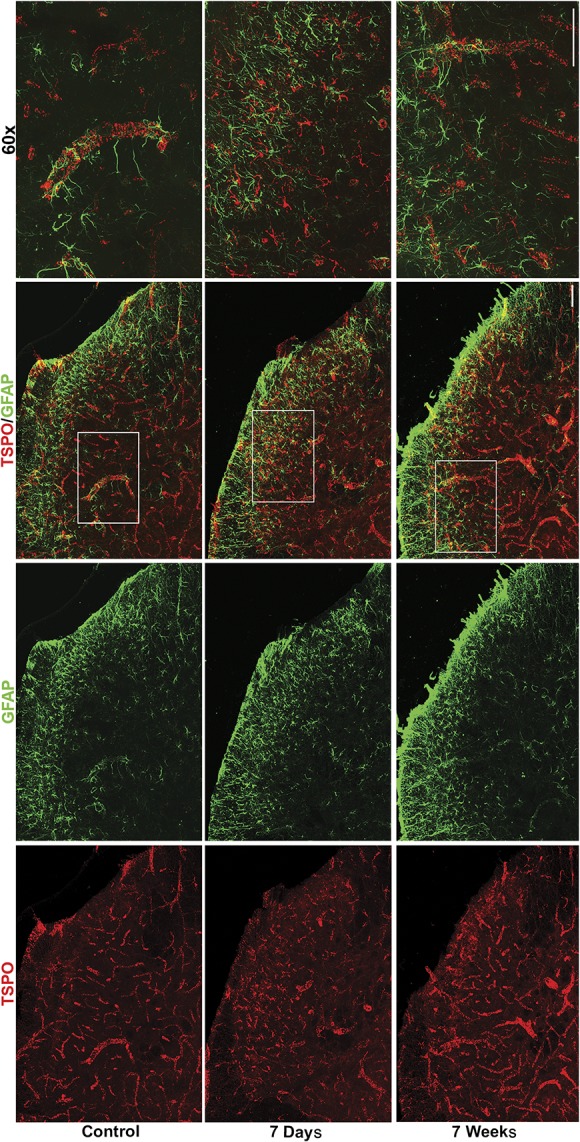
Translocator protein-18 kDa is not found in spinal cord astrocytes. Images of control and CRPS spinal cords at 7 days and 7 weeks after fracture show lack of colocalization of TSPO with GFAP+ astrocytes. Scale bars are 50 µm (×20) and 50 µm (×60). White box in ×20 overlay indicates area that is magnified to ×60. Representative images are shown from n = 5 control, n = 5 CRPS 7 days, and n = 6 CRPS 7 weeks mice, with 3 to 5 spinal cord sections examined per mouse. CRPS, complex regional pain syndrome.
4. Discussion
Our findings highlight the ability of TSPO-PET to longitudinally track peripheral and central myeloid cell activation in the transition from acute to chronic pain. Specifically, we identified unique spatiotemporal dynamics of the innate immune response to injury with early and persistent involvement of peripheral myeloid cells at the site of injury from 2 days through 7 weeks, and early and transient activation of CNS microglia distant from the injury site at 7 and 21 days. Taken together, these data suggest clear differences in the time course of peripheral and central myeloid cell involvement in CRPS disease progression.
4.1. Longitudinal imaging with translocator protein-18 kDa–positron emission tomography is a sensitive technique that can track innate immune activation
Understanding the contribution of myeloid cells to the transition from acute to chronic CRPS is crucial to developing mechanism-based treatments. This study provides preclinical validation of TSPO-PET for tracking these cells peripherally and centrally in the same mice over time. Positron emission tomography is a sensitive imaging technique that is routinely used in both preclinical and clinical research settings to investigate the molecular underpinnings of disease. Translocator protein-18 kDa is a highly hydrophobic protein normally expressed on the outer mitochondrial membrane that holds several functions, including cholesterol transport and steroid hormone synthesis.6 Although TSPO is highly expressed in peripheral organs (ie, kidney, liver, and lung), basal expression in the CNS is primarily restricted to endothelial cells, and it is minimally expressed on resting peripheral and central myeloid lineage cells.6,7,43 On injury, TSPO expression is significantly upregulated on activated myeloid cells, indicating the presence of inflammation and making it a useful biomarker of innate immune activation in several diseases.6,43
The previous gold standard and first-generation TSPO-PET tracer [11C]PK11195 has been used to quantify neuroinflammation in the context of numerous diseases, including pain.26,39 Consistent with our results, one study using [11C]PK11195 revealed significant increases in tracer uptake in a number of brain regions known to be associated with the pain matrix (ie, caudate nucleus, putamen, nucleus accumbens, and thalamus) in patients with CRPS compared with controls.29 Peripheral inflammation, however, was not investigated in this study and has not been evaluated in any clinical TSPO-PET study of chronic pain. Moreover, because several second-generation TSPO-PET tracers (eg, [18F]GE-180, [11C]PBR28, and [11C]DPA713) have been shown to be more specific and sensitive than [11C]PK11195,10,48 studies using second-generation tracers might reveal more subtle alterations in inflammation that can occur over time and/or after therapeutic interventions. Here, we report for the first time the application of a second-generation TSPO-PET tracer to longitudinally track peripheral and central innate immune activation in a mouse model of CRPS.
4.2. Longitudinal translocator protein-18 kDa–positron emission tomography imaging reveals early and persistent peripheral and central innate immune activation in the complex regional pain syndrome model
4.2.1. Peripheral inflammation
In this study, we detected peripheral macrophages at the site of injury using TSPO-PET as early as 2 days and lasting through 7 weeks after fracture. Such peripheral innate immune cells play a crucial role in the initiation of pain symptoms after injury. Local neutrophil activation peaks within 24 hours after injury and is implicated in the recruitment of local and circulating macrophages that likely express high levels of TSPO. The ensuing cascade of proinflammatory cytokines, such as IL-1, IL-6, and TNF-α,37 stimulates sensory neurons and encourages further macrophage invasion. Previous studies have noted that peripheral macrophage activation is maintained in clusters around dorsal root ganglia to phagocytose debris from 7 days to several months after injury.37 This detection of sustained myeloid cell activation in our study is consistent with the previous work37 that suggests continuous activation and cytokine release may be important in the perpetuation of pain symptoms and transition to chronic pain. Importantly, because peripherally restricted biologic agents lack efficacy in the chronic phase of CRPS, the functional contribution of this persistent peripheral myeloid cell activation warrants further exploration.
4.2.2. Central nervous system inflammation and cellular specificity of translocator protein-18 kDa
Previous studies have shown involvement of microglia and astrocytes in the acute (0-4 weeks) vs chronic (5-20 weeks) phases of CRPS, respectively.8,32,47 In this study, we established a time course of central myeloid cell activation after peripheral injury. We were able to detect elevated TSPO expression, indicative of microglial activation, in the spinal cord and brain through the use of longitudinal in vivo PET imaging at 7 and 21 days after fracture. This increased TSPO-PET signal returned to baseline by 7 weeks after injury and was confirmed by ex vivo CNS autoradiography and spinal cord gamma counting. One discrepancy in our study was that we observed a significantly higher signal in mice 7 days after fracture in PET whole brain analysis, but not in the ex vivo gamma counting of whole brain tissue. This is likely due to the partial volume effect, a known limitation of small animal PET,11,30 in which spillover from blood pool or neighboring regions can impact the in vivo brain PET signal. Such a limitation would not be expected to influence imaging in humans, given the relative difference in size of brain structures.
Innate immune cell activation in the CNS appears to be organized independently from the peripheral response but with similar mechanisms. A host of mediators released from injured neurons have been proposed to initiate microglial activation including damage-associated molecular patterns such as high mobility group box 1 (HMGB1) acting through toll-like receptor 4 (TLR4),38 colony stimulating factor 1 (CSF1) through microglial colony stimulating factor 1 receptor (CSF1R),23 and adenosine triphosphate through microglial purinergic receptor 2 (P2XR).27 Mirroring peripheral macrophage infiltration around dorsal root ganglia in the periphery, microglia appear to cluster around central terminals of injured primary afferent and motor neurons in the spinal cord dorsal and ventral horns, respectively.44 These clusters of activated microglia locally release cytokines that can trigger subsequent astrocyte-mediated alterations in neuronal signaling.2,33,49 In the CRPS model, our data illustrate that microglial activation, defined by TSPO expression, is restricted to the acute phase of the disease—temporally positioning microglia as triggers for the subsequent chronic phase transition. Our time course findings are consistent with the previous work that identified microglial involvement in the initiation, but not the maintenance, of several pain conditions.44,58
Interestingly, our double-label immunostaining revealed that before injury, basal spinal cord expression of TSPO is almost entirely attributable to PECAM1+ vascular endothelial cells, consistent with a recent study investigating cellular expression of TSPO throughout the normal mouse brain.7 We further observed that after injury, the spinal cord vasculature was less prominent with overall decreased PECAM1 expression and smaller caliber of vessels. Such CRPS-induced CNS vasoconstriction has yet to be described but is consistent with the sympathetic hyperactivity theory of the disease.12 Furthermore, after injury, we saw upregulation of TSPO exclusively in CD11b+ microglia and not GFAP+ astrocytes. As comprehensively reviewed by Guilarte,24 TSPO may be expressed by either microglia1,14,34 or astrocytes35,55 depending on the timing, location, and nature of the injury studied. This implies that there is something distinct about the astrocytic activation caused by a peripheral injury such as fracture, which induces GFAP+ TSPO− astrocytes, compared with the phenotype caused by neurodegenerative models, which induces GFAP+ TSPO+ astrocytes.24 Taken together, these observations highlight the complexity of glial heterogeneity that is just starting to become clear through the use of next-generation sequencing techniques.21,57
4.3. Illuminating spatiotemporal dynamics of peripheral vs central innate immune activation: translational potential
Here, we illustrate the dynamic capability of whole-body TSPO-PET, a highly translatable technique, to enable visualization and quantification of peripheral and central myeloid lineage activation through disease progression. Previous studies have investigated the potential use of drugs, such as low-dose ketamine50 and low-dose naltrexone,17 that may attenuate glial cell activation to treat CRPS. However, the efficacy of these treatments may be improved if treatments could be targeted and personalized based on the disease stage. Human trials using other PET ligands that may “illuminate pain” are underway at our institution (ClinicalTrials.gov NCT02753101). Such studies not only have the potential to focus treatment efforts on the disease stage but may also highlight pain generators that were previously unrecognized.18,45 Taken together, our results demonstrate that TSPO-PET provides critical information on the spatiotemporal dynamics of the innate immune system response to injury and can be exploited for therapeutic decision-making.
Conflict of interest statement
The authors have no conflict of interest to declare.
Supplementary Material
Acknowledgments
The authors are very grateful to Dr Timothy Doyle from the SCi3 small animal imaging facility at Stanford University for his unfailing support and kind assistance with PET/CT. The authors also thank the radiochemistry facility for use of the cyclotron in the production of [18F]GE180.
V.L. Tawfik acknowledges support from the Department of Anesthesiology, Perioperative and Pain Medicine at Stanford University as well as from the National Institute for Neurological Disorders and Stroke (K08NS094547) and a Foundation for Anesthesia Education and Research (FAER) Mentored Research Training Grant. M.L. James acknowledges support from the Departments of Radiology and Neurology and Neurological Sciences at Stanford University.
This work was previously presented in abstract form at the World Molecular Imaging Congress in Seattle in September 2018.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/A794.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
H.C. Cropper and E.M. Johnson contributed equally to this work. M.L. James and V.L. Tawfik contributed equally to this work.
References
- [1].Abourbeh G, Theze B, Maroy R, Dubois A, Brulon V, Fontyn Y, Dolle F, Tavitian B, Boisgard R. Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [(1)(8)F]DPA-714. J Neurosci 2012;32:5728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Agulhon C, Sun MY, Murphy T, Myers T, Lauderdale K, Fiacco TA. Calcium signaling and gliotransmission in normal vs. reactive astrocytes. Front Pharmacol 2012;3:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Albrecht DS, Ahmed SU, Kettner NW, Borra RJH, Cohen-Adad J, Deng H, Houle TT, Opalacz A, Roth SA, Melo MFV, Chen L, Mao J, Hooker JM, Loggia ML, Zhang Y. Neuroinflammation of the spinal cord and nerve roots in chronic radicular pain patients. PAIN 2018;159:968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Alexander GM, Perreault MJ, Reichenberger ER, Schwartzman RJ. Changes in immune and glial markers in the CSF of patients with complex regional pain syndrome. Brain Behav Immun 2007;21:668–76. [DOI] [PubMed] [Google Scholar]
- [5].Apkarian AV, Hashmi JA, Baliki MN. Pain and the brain: specificity and plasticity of the brain in clinical chronic pain. PAIN 2011;152(3 suppl):S49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Banati RB. Visualising microglial activation in vivo. Glia 2002;40:206–17. [DOI] [PubMed] [Google Scholar]
- [7].Betlazar C, Harrison-Brown M, Middleton RJ, Banati R, Liu GJ. Cellular sources and regional variations in the expression of the neuroinflammatory marker translocator protein (TSPO) in the normal brain. Int J Mol Sci 2018;19:E2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Birklein F, Drummond PD, Li W, Schlereth T, Albrecht N, Finch PM, Dawson LF, Clark JD, Kingery WS. Activation of cutaneous immune responses in complex regional pain syndrome. J Pain 2014;15:485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Birklein F, Ibrahim A, Schlereth T, Kingery WS. The rodent tibia fracture model: a critical review and comparison with the complex regional pain syndrome literature. J Pain 2018;19:1102.e1101–1102.e1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Boutin H, Murray K, Pradillo J, Maroy R, Smigova A, Gerhard A, Jones PA, Trigg W. 18F-GE-180: a novel TSPO radiotracer compared to 11C-R-PK11195 in a preclinical model of stroke. Eur J Nucl Med Mol Imaging 2015;42:503–11. [DOI] [PubMed] [Google Scholar]
- [11].Brendel M, Delker A, Rötzer C, Böning G, Carlsen J, Cyran C, Mille E, Gildehaus FJ, Cumming P, Baumann K, Steiner H, Haass C, Herms J, Bartenstein P, Rominger A. Impact of partial volume effect correction on cerebral β-amyloid imaging in APP-Swe mice using [18F]-florbetaben PET. NeuroImage 2014;84:843–53. [DOI] [PubMed] [Google Scholar]
- [12].Bruehl S. An update on the pathophysiology of complex regional pain syndrome. Anesthesiology 2010;113:713–25. [DOI] [PubMed] [Google Scholar]
- [13].Bruehl S, Maihöfner C, Stanton-Hicks M, Perez RSGM, Vatine JJ, Brunner F, Birklein F, Schlereth T, Mackey S, Mailis-Gagnon A, Livshitz A, Harden RN. Complex regional pain syndrome. PAIN 2016;157:1674–81. [DOI] [PubMed] [Google Scholar]
- [14].Chaney AM, Johnson EM, Cropper HC, James ML. PET imaging of neuroinflammation using [11C]DPA-713 in a mouse model of ischemic stroke. J Vis Exp 2018:e57243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55–63. [DOI] [PubMed] [Google Scholar]
- [16].Chapman CR, Vierck CJ. The transition of acute postoperative pain to chronic pain: an integrative overview of research on mechanisms. J Pain 2017;18:359–e38. [DOI] [PubMed] [Google Scholar]
- [17].Chopra P, Cooper MS. Treatment of complex regional pain syndrome (CRPS) using low dose naltrexone (LDN). J Neuroimmune Pharmacol 2013;8:470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cipriano PW, Lee SW, Yoon D, Shen B, Tawfik VL, Curtin CM, Dragoo JL, James ML, McCurdy CR, Chin FT, Biswal S. Successful treatment of chronic knee pain following localization by a sigma-1 receptor radioligand and PET/MRI: a case report. J Pain Res 2018;11:2353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].De Leo JA, Tawfik VL, LaCroix-Fralish ML. The tetrapartite synapse: path to CNS sensitization and chronic pain. PAIN 2006;122:17–21. [DOI] [PubMed] [Google Scholar]
- [20].Del Valle L, Schwartzman RJ, Alexander G. Spinal cord histopathological alterations in a patient with longstanding complex regional pain syndrome. Brain Behav Immun 2009;23:85–91. [DOI] [PubMed] [Google Scholar]
- [21].Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 2016;19:504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol 2014;14:217–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, Abboud-Werner SL, Lanier LL, Lomvardas S, Basbaum AI. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 2016;19:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guilarte TR. TSPO in diverse CNS pathologies and psychiatric disease: a critical review and a way forward. Pharmacol Ther 2019;194:44–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hoehne A, James ML, Alam IS, Ronald JA, Schneider B, D'Souza A, Witney TH, Andrews LE, Cropper HC, Behera D, Gowrishankar G, Ding Z, Wyss-Coray T, Chin FT, Biswal S, Gambhir SS. [18F]FSPG-PET reveals increased cystine/glutamate antiporter (xc-) activity in a mouse model of multiple sclerosis. J Neuroinflammation 2018;15:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Imamoto N, Momosaki S, Fujita M, Omachi S, Yamato H, Kimura M, Kanegawa N, Shinohara S, Abe K. [11C]PK11195 PET imaging of spinal glial activation after nerve injury in rats. Neuroimage 2013;79:121–8. [DOI] [PubMed] [Google Scholar]
- [27].Inoue K, Tsuda M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci 2018;19:138–52. [DOI] [PubMed] [Google Scholar]
- [28].James ML, Gambhir SS. A molecular imaging primer: modalities, imaging agents, and applications. Physiol Rev 2012;92:897–965. [DOI] [PubMed] [Google Scholar]
- [29].Jeon SY, Seo S, Lee JS, Choi SH, Lee DH, Jung YH, Song MK, Lee KJ, Kim YC, Kwon HW, Im HJ, Lee DS, Cheon GJ, Kang DH. [11C]-(R)-PK11195 positron emission tomography in patients with complex regional pain syndrome: a pilot study. Medicine (Baltimore) 2017;96:e5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lehnert W, Gregoire MC, Reilhac A, Meikle SR. Characterisation of partial volume effect and region-based correction in small animal positron emission tomography (PET) of the rat brain. Neuroimage 2012;60:2144–57. [DOI] [PubMed] [Google Scholar]
- [31].Lenz M, Uçeyler N, Frettlöh J, Höffken O, Krumova EK, Lissek S, Reinersmann A, Sommer C, Stude P, Waaga-Gasser AM, Tegenthoff M, Maier C. Local cytokine changes in complex regional pain syndrome type I (CRPS I) resolve after 6 months. PAIN 2013;154:2142–9. [DOI] [PubMed] [Google Scholar]
- [32].Li WW, Guo TZ, Shi X, Sun Y, Wei T, Clark DJ, Kingery WS. Substance P spinal signaling induces glial activation and nociceptive sensitization after fracture. Neuroscience 2015;310:73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541:481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu B, Le KX, Park MA, Wang S, Belanger AP, Dubey S, Frost JL, Holton P, Reiser V, Jones PA, Trigg W, Di Carli MF, Lemere CA. In vivo detection of age- and disease-related increases in neuroinflammation by 18F-GE180 TSPO MicroPET Imaging in Wild-Type and Alzheimer's transgenic mice. J Neurosci 2015;35:15716–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu X, Liu H, Xu S, Tang Z, Xia W, Cheng Z, Li W, Jin Y. Spinal translocator protein alleviates chronic neuropathic pain behavior and modulates spinal astrocyte-neuronal function in rats with L5 spinal nerve ligation model. PAIN 2016;157:103–16. [DOI] [PubMed] [Google Scholar]
- [36].Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, Hill E, Hsu S, Izquierdo-Garcia D, Ji RR, Riley M, Wasan AD, Zürcher NR, Albrecht DS, Vangel MG, Rosen BR, Napadow V, Hooker JM. Evidence for brain glial activation in chronic pain patients. Brain 2015;138(pt 3):604–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci 2005;6:521–32. [DOI] [PubMed] [Google Scholar]
- [38].Medicine M. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Mil Med 2016;181:397–9. [DOI] [PubMed] [Google Scholar]
- [39].Miller TR, Wetter JB, Jarvis MF, Bitner RS. Spinal microglial activation in rat models of neuropathic and osteoarthritic pain: an autoradiographic study using [3H]PK11195. Eur J Pain 2013;17:692–703. [DOI] [PubMed] [Google Scholar]
- [40].Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 2009;10:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci USA 2000;97:9226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. PAIN 2003;103:199–207. [DOI] [PubMed] [Google Scholar]
- [43].Scarf AM, Kassiou M. The translocator protein. J Nucl Med 2011;52:677–80. [DOI] [PubMed] [Google Scholar]
- [44].Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 2007;10:1361–8. [DOI] [PubMed] [Google Scholar]
- [45].Shen B, Behera D, James ML, Reyes ST, Andrews L, Cipriano PW, Klukinov M, Lutz AB, Mavlyutov T, Rosenberg J, Ruoho AE, McCurdy CR, Gambhir SS, Yeomans DC, Biswal S, Chin FT. Visualizing nerve injury in a neuropathic pain model with [(18)F]FTC-146 PET/MRI. Theranostics 2017;7:2794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shepherd AJ, Mickle AD, Golden JP, Mack MR, Halabi CM, de Kloet AD, Samineni VK, Kim BS, Krause EG, Gereau RWt, Mohapatra DP. Macrophage angiotensin II type 2 receptor triggers neuropathic pain. Proc Natl Acad Sci U S A 2018;115:E8057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shi X, Wang L, Li X, Sahbaie P, Kingery WS, Clark JD. Neuropeptides contribute to peripheral nociceptive sensitization by regulating interleukin-1beta production in keratinocytes. Anesth Analg 2011;113:175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sridharan S, Lepelletier F-X, Trigg W, Banister S, Reekie T, Kassiou M, Gerhard A, Hinz R, Boutin H. Comparative evaluation of three TSPO PET radiotracers in a LPS-induced model of mild neuroinflammation in rats. Mol Imaging Biol 2016;19:77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature 2006;440:1054–9. [DOI] [PubMed] [Google Scholar]
- [50].Tajerian M, Leu D, Yang P, Huang TT, Kingery WS, Clark JD. Differential efficacy of ketamine in the acute versus chronic stages of complex regional pain syndrome in mice. Anesthesiology 2015;123:1435–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tajerian M, Sahbaie P, Sun Y, Leu D, Yang HY, Li W, Huang TT, Kingery W, David Clark J. Sex differences in a murine model of complex regional pain syndrome. Neurobiol Learn Mem 2015;123:100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Venneti S, Lopresti BJ, Wang G, Hamilton RL, Mathis CA, Klunk WE, Apte UM, Wiley CA. PK11195 labels activated microglia in Alzheimer's disease and in vivo in a mouse model using PET. Neurobiol Aging 2009;30:1217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wadsworth H, Jones PA, Chau WF, Durrant C, Fouladi N, Passmore J, O'Shea D, Wynn D, Morisson-Iveson V, Ewan A, Thaning M, Mantzilas D, Gausemel I, Khan I, Black A, Avory M, Trigg W. [(1)(8)F]GE-180: a novel fluorine-18 labelled PET tracer for imaging Translocator protein 18 kDa (TSPO). Bioorg Med Chem Lett 2012;22:1308–13. [DOI] [PubMed] [Google Scholar]
- [54].Wei T, Guo TZ, Li WW, Kingery WS, Clark JD. Acute versus chronic phase mechanisms in a rat model of CRPS. J Neuroinflammation 2016;13:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wei XH, Wei X, Chen FY, Zang Y, Xin WJ, Pang RP, Chen Y, Wang J, Li YY, Shen KF, Zhou LJ, Liu XG. The upregulation of translocator protein (18 kDa) promotes recovery from neuropathic pain in rats. J Neurosci 2013;33:1540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wesseldijk F, Huygen FJ, Heijmans-Antonissen C, Niehof SP, Zijlstra FJ. Six years follow-up of the levels of TNF-alpha and IL-6 in patients with complex regional pain syndrome type 1. Mediators Inflamm 2008;2008:469439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang Y, Barres BA. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol 2010;20:588–94. [DOI] [PubMed] [Google Scholar]
- [58].Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. PAIN 2005;114:149–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.