

Abstract
The extracellular matrix is perturbed in tumors. The tumor matrix promotes the growth, survival and invasion of the cancer and modifies fibroblast and immune cell behavior to drive metastasis and impair treatment. Here, we discuss how the tumor matrix regulates metastasis by fostering tumor cell invasion into the stroma and migration towards the vasculature. We describe the role of the tumor matrix on cancer cell intravasation and vascular dissemination. We examine the impact of the matrix on disseminated tumor cell extravasation and on tumor dormancy and metastatic outgrowth. Finally, we discuss the clinical outcome of therapeutics that normalize tumor-matrix interactions.
Keywords: extracellular matrix, metastasis, mechanics, angiogenesis, pre-metastatic niche, dormancy, outgrowth, metastasis, tissue tension, intravasation, extravasation, integrin, invasion, migration, mechanotransduction
Introduction
The extracellular matrix (ECM) is composed of a myriad of fibrous proteins, proteoglycans and matricellular associated proteins (Mouw et al., 2014). Far from being an inert physical scaffold, the composition, posttranslational modifications and organization the ECM is dynamically regulated and this tunes the ECM’s biochemical and biomechanical properties. Cells sense the biochemical and mechanical properties of the ECM through specialized transmembrane receptors that include integrins, discoidin domain receptors (DDRs) and syndecans. Once activated these ECM receptors recognize specific motifs within the ECM molecule that induce conformational changes within the receptor that promote molecular associations between the receptor and intracellular adhesion plaque proteins that thereafter activate signaling to influence cell behavior. ECM composition, density, organization and posttranslational modifications including cleavage and crosslinking dictate the material property or viscoelasticity e.g. stiffness of the stroma. Cells sense the stiffness of the ECM through the enforced clustering of their transmembrane ECM receptors and via actin and actomyosin-mediated adhesion plaque protein unfolding and enhanced intermolecular associations of intracellular adhesion plaque proteins such as talin that foster the recruitment and retention of other scaffolding proteins such as vinculin and various signaling molecules. Accordingly, the biochemical and mechanical properties of the ECM exquisitely tune the growth, survival, migration, invasion and differentiation of cells within the tissue to influence their fate, and regulate tissue development, phenotype and homeostasis. Not surprisingly, diseases such as cancer that are defined by disrupted tissue homeostasis and loss of a differentiated phenotype are accompanied by alterations in the composition, organization and mechanical properties of the ECM that contribute not only to malignant transformation but also to tumor progression and metastasis.
Tumors are frequently desmoplastic. Tumor desmoplasia is defined as a cancer condition that is characterized by chronic inflammation, fibroblast expansion and activation, elevated angiogenesis and increased levels of remodeled and cross-linked ECM protein that stiffen the tissue stroma (Pickup et al., 2014). Solid cancers develop a desmoplastic response in which the tissue contains higher amounts of ECM proteins such as fibronectin, tenascin and hyaluronic acid (HA) and other proteoglycans and matricellular proteins as well as greater quantities of reorganized and cross-linked type I collagen that collectively stiffen the tissue stroma (Acerbi et al., 2015). Features of a desmoplastic ECM have been detected in clinical biopsies of preneoplastic tissue and experimental findings using three dimensional organoid cultures and in vivo murine models suggest this altered ECM may play a causal role in promoting malignant transformation (Levental et al., 2009). A desmoplastic ECM has also been implicated in metastasis, which is the dissemination of tumor cells from primary tumors to distant tissues and is the leading cause of mortality in most cancer patients.
The development of invasive tumors and their metastatic dissemination involves a series of discrete biological steps each of which associates with distinct changes in ECM composition, posttranslational modifications, organization and biomechanics (Figure 1). To begin with, cancer cells must remodel the basement membrane to invade into the parenchyma to qualify as an invasive cancer. Imaging analysis has revealed that the basement membrane surrounding premalignant lesions is thinner and has lost significant amounts of the critical basement membrane protein laminin-111 (Gudjonsson et al., 2002). Furthermore, more and thicker “bundled” interstitial collagen has been detected surrounding DCIS lesions (Acerbi et al., 2015).
Figure 1.
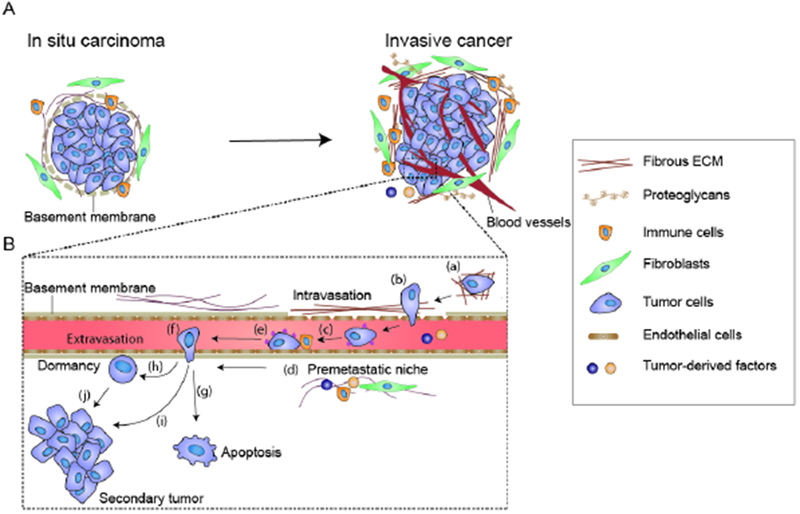
Schematic showing the steps of cancer metastasis. (A) A normal epithelial acini is surrounded by a contiguous laminin-rich basement membrane and the whole structure is embedded within an interstitial extracellular matrix (ECM) that is characterized by a preponderance of curly and loosely organized collagenous proteins. Upon transformation, the acinar lumen of an “in situ” benign carcinoma progressively fills with proliferating tumor cells, basement membrane thickness gradually decreases and laminin levels drop. Furthermore, there is evidence that the surrounding interstitial ECM collagens become remodeled, reorganized and thickened. Malignant transformation to an “invasive carcinoma” is accompanied by further metalloproteinase-mediated ECM remodeling and lysyl oxidase (LOX) and lysyl hydroxlase-mediated collagen crosslinking and stiffening that provide linearized, thickened collagen-rich fibrils upon which the tumor cells migrate and invade into the surrounding parenchyma. ECM remodeling and stiffening occur in tandem with increased proliferation and activation of stromal fibroblasts and infiltration of immune cells including macrophages and neutrophils and induction of angiogenesis. (B) (Step a) Mechanical stresses such as compression stress and ECM stiffening foster tumor cell migration through the parenchyma towards the vasculature. (Step b) ECM stiffness also facilitates tumor cell intravasation into the vasculature by compromising vascular integrity and increasing tumor cell deformability through induction of an epithelial to mesenchymal transition. (Step c) Once within the circulation, the circulating tumor cells (CTCs) encounter hemodynamic shear stress. CTC survival can be potentiated by platelets through their ability to shield the tumor cells from shear stress and through integrin-dependent adhesion signaling activation. (Step d) Primary tumor cells also secrete soluble factors, ECM proteins and exosomes that create a premetastatic niche by incorporating into secondary “distal” tissues that prime the recruitment and retention of immune cells and disseminating tumor cells that foster tumor colonization. (Step e) Tumor cells find a favorable site for extravasation. With the assistance of platelets, CTCs adhere to the endothelium and migrate across the endothelial layer (Step f). The extravasated CTCs may either undergo apoptosis (step g), enter a dormant state (step h), or proliferate to form secondary metastatic lesions (step i). The dormant cells retain their proliferative ability and may eventually re-enter cell cycle and form metastatic lesions (step J).
Preventing metalloproteinase (MMP)-dependent basement membrane cleavage inhibits the invasion of transformed cells in vitro, and reducing lysyl oxidase (LOX)-mediated collagen cross-linking and stiffening delay malignant transformation and modify the frequency of invasive carcinomas in an experimental model of Her2/Neu mammary cancer (Levental et al., 2009; Rizki et al., 2008). Once tumor cells have invaded into the parenchyma their metastatic dissemination is favored by a remodeled interstitial ECM that has been reorganized into stiffened, linearized bundles of type I collagen that is often decorated with fibronectin (Miroshnikova et al., 2017). These linearized ECM tracts favor the directed migration of the tumor cells towards the vasculature where the cells can efficiently intravasate into the circulation (Han et al., 2016). Consistently, the presence of perpendicular, linearized, and stiffened collagen bundles is predictive of poor breast cancer patient prognosis (Conklin et al., 2011). Furthermore, a stiffer invasive stroma has been detected in the more aggressive human HER2+ and triple negative breast cancer lesions where greater quantities of linearized collagens have been detected (Acerbi et al., 2015). Once the tumor cells have intravasated, the circulating tumor cells (CTCs; cancer cells within the bloodstream) travel within the vasculature where interactions with ECM fibrin-fibronectin clots (emboli) or platelets ensure their survival by insulating them from hemodynamic fluid shear stress (Micalizzi et al., 2017). Once the CTCs extravasate, their attachment and survival at the distal site is ensured by a tissue microenvironment in which a hospitable ECM already exists or has been prepared through tumor exosome secreted and deposited or stromal-mediated ECM conditioning and remodeling (Peinado et al., 2017). The nature of the ECM at the secondary tissue site also dictates whether the disseminated cancer cell will undergo apoptosis, enter into a dormant state, or proliferate to form an overt metastatic lesion (Goddard et al., 2018) . Associations between the tumor ECM and the metastatic cascade not only provide intriguing evidence of a clinical association between a desmoplastic ECM and malignant transformation and metastasis, but suggest there is likely a causal link that could be used for predictive biomarkers or be targeted with therapeutics to reduce patient mortality. In this review, we describe how the desmoplastic ECM fosters malignant transformation of tumor cells and we describe its role in regulating tumor metastasis. We end with a discussion of pros and cons of therapeutic approaches targeting the modified ECM to potentiate tumor treatment.
The ECM and adhesion receptors
Biochemical composition and biomechanical properties of the ECM
The ECM is composed of a three-dimensional network of fibrous proteins (e.g. collagen, laminin, and fibronectin, elastin), proteoglycans, and matricellular proteins. Fibrous ECM proteins serve as a physical scaffold for cells and impart mechanical properties to the tissue. For example, type I collagen provides tissues with tensile strength and resistance to deformation. Type I collagen can be cross-linked into fibrillar structures, which involves a series of post-translational modifications within cells and oxidative deamination reactions that occur in the extracellular space (Mouw et al., 2014). Specific lysine residues in procollagen are hydroxylated to generate hydroxylysines by intracellular lysyl hydroxylases. Once secreted the specific lysine or hydroxylysine residues in the extracellular collagen undergo oxidative deamination catalyzed by LOX, leading to the formation of reactive aldehyde residues on collagen. The reactive aldehyde residues subsequently initiate spontaneous condensation reactions to form various covalent intermolecular crosslinks between neighboring collagen molecules. Extensively cross-linked collagen fibers exhibit greater tensile properties and can enhance the stiffness of the collagen. Collagen also exhibits strain stiffening behavior, whereby the material stiffness increases when strained (Cassereau et al., 2015). Laminin and type IV collagen form a web-like structure that provides tensile strength to the basement membrane, the specialized ECM that physically segregates endothelial or epithelial layers from the interstitial matrix. Fibronectin can either exist in soluble form or be assembled into highly extensible fibrillar structures that are required for the assembly of collagen fibers (Sottile and Hocking, 2002). Collectively, the fibrous ECM proteins within the interstitial stroma are embedded within a hydrated, viscous microenvironment enriched in proteoglycans and polysaccharides (e.g. HA), that maintains the hydration state of the ECM and serves to cushion the tissue so that it can resist external compressive loading. The ECM also serves as a reservoir of matrix-bound nanovesicles, growth factors, and cytokines, and thereby can regulate their distribution, activation, and presentation to cells (Huleihel et al., 2016; Hynes, 2009). Unlike fibrous ECM proteins, matricellular proteins are a family of non-structural matrix proteins, such as tenascin-C, thrombospondin-1 (TSP-1) and periostin, which modulate cell behavior by altering interactions between cellular transmembrane receptors and fibrous ECM molecules. For example, tenascin-C promotes glioma cell migration on fibronectin-coated substrates by modulating interactions between αvβ5 integrin and fibronectin (Deryugina and Bourdon, 1996).
Adhesion receptor signaling and mechanotransduction (Figure 2)
Figure 2.
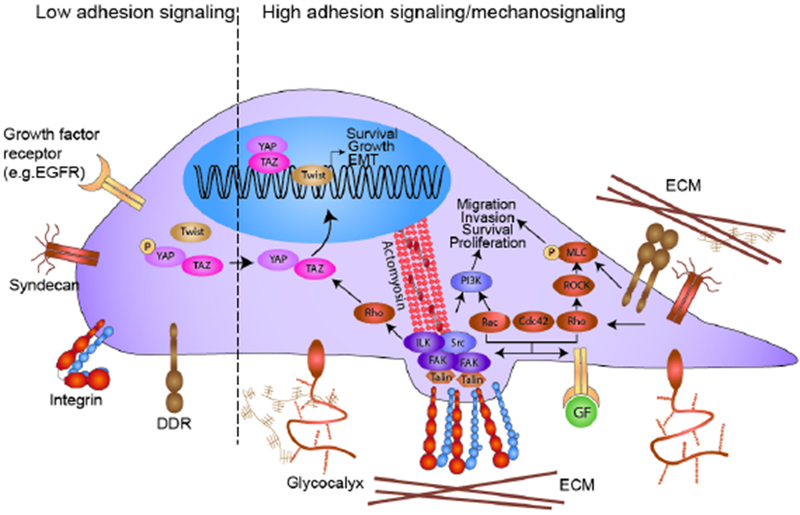
Integrin-dependent adhesion and mechanotransduction pathways. Cells constantly sample the biochemical composition of the surrounding ECM using cell surface receptors such as integrins, discoidin domain receptors (DDRs) and syndecans and modulate intracellular signaling pathways accordingly. Integrins crosstalk with multiple transmembrane proteins including growth factor receptors (GFRs). The crosstalk between integrins and adJacent transmembrane molecules can synergize to potentiate Rho GTPase activity, focal adhesion assembly, kinase signaling and stimulate gene transcription to induce tumor cell growth, survival and motility and may even induce differentiation. (Left) When adhesion signaling is low, integrins remain in an inactive conformation. (Right) Binding of integrins to ECM ligands can trigger the recruitment and activation of talin and the subsequent association of molecules such as vinculin and paxillin and integrin-linked kinase (ILK) and the activation of focal adhesion kinase (FAK) that promote the assembly of adhesion complexes. For example, paxillin and FAK form a complex with Src kinase to activate the PI3K-Akt pro-survival signaling pathway. The crosstalk between integrins and GFR pathways can also potentiate Rho GTPase signaling. RhoA stimulates ROCK kinase activity, which increases the level of phosphorylated myosin light chain (MLC) to stimulate actomyosin contraction. Rac, Rho, and Cdc42 GTPases also promote the formation of invasive cellular protrusions, such as lamellipodia, filopodia, and invadosomes. Upon ECM ligand ligation, DDRs and syndecans can recruit diverse signaling molecules such as myosin IIA to promote cell contractibility and migration. Cells additionally interrogate the mechanical properties of the ECM using integrins through a process termed mechanotransduction. Stiff substrates promote integrin clustering, adhesion signaling, and TWIST1 and YAP translocation into the nucleus to activate transcription programs that promote epithelial mesenchymal transition. In addition, a bulky glycocalyx on the plasma membrane can directly exert stress on integrins to enhance their activation and once ligated induces a kinetic trap that physically clusters integrins to promote their assembly into focal adhesions that enhance adhesion signaling.
Cells bind to the ECM via transmembrane receptors including integrins, DDRs and syndecans that recognize specific residues/moieties within the ECM molecule. Integrins are the best studied ECM binding receptor and comprise a large family of heterodimers (Sun et al., 2016). Integrins exist in an inactive or active form. Once activated, integrins mediate bidirectional signaling between the ECM and cell. Integrins can be activated through two different mechanisms, either inside-out or outside-in signaling. Intracellular biochemical signals can promote talin and kindlin recruitment to the cytoplasmic tails of the integrin, which induces a conformational change in their extracellular domain to facilitate ECM ligand binding. Alternately, ECM ligands bind to the extracellular domain of integrins and promote the recruitment of talin and kindlin to their cytoplasmic tail. Talin plays a central role in coupling integrins to the actin cytoskeleton. A talin molecule contains several domains, including multiple actin and vinculin binding sites. Upon binding to the integrin, talin connects the actin cytoskeleton to the integrin and recruits diverse scaffolding proteins such as focal adhesion kinase (FAK), vinculin and paxillin that foster the formation of nascent integrin adhesions that can mature into focal adhesion. Focal adhesion assembly can also be fostered by a thick glycocalyx, which can promote integrin activation and integrin clustering (Paszek et al., 2014). Focal adhesions serve as signaling platforms. For instance, binding between FAK and Src at adhesion sites activates small Rho GTPases, including RhoA, Rac, and Cdc42, which are master regulators of actin cytoskeleton dynamics and cell growth, survival and motility.
The DDRs are a family of transmembrane receptor tyrosine kinases comprised of DDR1 and 2 that bind to structural collagens. DDR1 is primarily bound and activated by type I and IV collagen whereas DDR2 binds predominantly fibrillar type I and III collagen (Rammal et al., 2016). Upon collagen binding via their extracellular regions, the cytoplasmic tails of the DDRs undergo autophosphorylation and create docking sites for signaling molecules, such as PI3K, Erk and myosin IIA, that regulate diverse cell behaviors such as proliferation and migration. Syndecans are another family of transmembrane proteoglycans that interact with ECM ligands including fibronectin and collagen via their extracellular domains (Afratis et al., 2017). Syndecans can function independently or as co-receptors with integrins, tetraspanins, or growth factor receptors. Upon ECM ligand ligation, the cytoplasmic domain of syndecan interacts with diverse kinases and signaling molecules to foster focal adhesion assembly and actin cytoskeleton rearrangement
Cells constantly sample the biomechanical properties of the ECM and tune intracellular signaling pathways through a process termed mechanotransduction (Sun et al., 2016). ECM stiffness is a material property that describes the extent to which a material is able to resist deformation in response to an external applied force. Cells use actomyosin contractibility to deform their ECM to probe for its material properties or stiffness. Cells interacting with a stiff ECM exert higher actomyosin force that deforms the underlying matrix and nucleates focal adhesion assembly that stimulates mechanotransduction. Contractile forces exerted by cells can also align and deform collagen fibers to strain stiffen the ECM (van Helvert and Friedl, 2016). The higher actomyosin-mediated cellular tension exposes cryptic vinculin binding sites in talin that facilitate the recruitment and retention of vinculin and other scaffolding proteins to strengthen the link between the integrins and the actin cytoskeleton that supports further adhesion growth and maturation (Ciobanasu et al., 2014). By this mechanism, a stiff ECM will foster the maturation of nascent adhesions towards focal adhesions to amplify integrin adhesion signaling. By contrast, when cells interact with a compliant ECM, the actomyosin contractile force they stimulate is lower and consequently is unable to elicit the unfolding of the talin molecule critical for vinculin recruitment and focal adhesion assembly.
Receptor adhesion signaling regulates diverse cell behaviors including cell survival, growth, motility, invasion, and differentiation. Cell-ECM interaction is required to maintain cell survival so that inhibition of cell-ECM interactions using integrin function blocking antibodies will lead to anoikis, which is an apoptotic pathway induced by ECM detachment or “homelessness” (Boudreau et al., 1995). The binding of an integrin to its ECM ligand promotes FAK/Src complex assembly at the cytoplasmic tails of integrins, which recruits various downstream effectors to activate PI3K/Akt and Ras/MEK/Erk pro-survival signaling (Vachon, 2011). Integrins also partner with receptor tyrosine kinases such as epidermal growth factor receptor to amplify PI3K/Akt pro-survival signaling (Schwartz and Assoian, 2001). Given that a stiff ECM encourages integrin clustering, it is not surprising that cells cultured on rigid matrix exhibit sustained PI3K/Akt and ERK signaling and survive better than cells interacting with a softer matrix (Paszek et al., 2005). Indeed, TGF-β-treated normal murine mammary cells on a soft matrix apoptose and have attenuated Akt signaling whereas those on a stiff matrix survive and exhibit sustained PI3K activity (Leight et al., 2012). Integrin-mediated adhesion signaling also promotes cell proliferation. During cell proliferation, ECM ligation regulates the levels of various cyclin-dependent kinases to control cell cycle progression. For example, integrin-mediated adhesion to the ECM activates ERK signaling, which induces the expression of cyclin-D1 that permits G1 phase cell cycle progression (Roovers et al., 1999). Consistently, cells on the stiff ECM which have larger focal adhesions exhibit elevated ERK signaling and are more proliferative compared to cells cultured on the softer matrix (Paszek et al., 2005). In addition, integrin signaling can regulate cell migration by modulating focal adhesion assembly and Rho GTPase signaling. RhoA GTPase stimulates ROCK phosphorylation of myosin light chain and induces actomyosin contraction, which encourage integrin signaling and the recruitment of more adhesion proteins that strengthen the interaction between integrins and the actin cytoskeleton. Rac and Cdc42 GTPases promote the assembly of lamellipodia and filopodia, respectively, to facilitate cell migration. These small GTPases also drive the formation of invadosomes, the actin-rich subcellular structures that mediate focal degradation of the ECM through MMP secretion to facilitate ECM remodeling (Kai et al., 2016). Integrin signaling can influence transcriptome of cells that sustain cell phenotype alterations and regulate cell fate specification by regulating nucleocytoplasmic shuttling of diverse transcription factors. For example, increased cell-ECM ligation promotes β-catenin nuclear translocation and enhanced transcriptional activation of Wnt/ β-catenin target genes such as protooncogene myc (Renner et al., 2016). Consistently, elevated ECM stiffness promotes integrin clustering and TWIST1 dissociation from G3BP1 in the cytoplasm, leading to nuclear translocation of TWIST1 that drive epithelial-mesenchymal transition (EMT) (Wei et al., 2015). A stiff ECM stimulates pathways that alter the phosphorylation and nuclear trafficking of YAP which is a transcriptional coactivator of Hippo pathway that enhances cell growth (Dobrokhotov et al., 2018). ECM-activated DDRs and syndecans also regulate cell behavior. For example, DDR1 activation by fibrillar type I collagen promotes DDR clustering and myosin IIA recruitment to stimulate cell migration and collagen fiber alignment (Coelho and McCulloch, 2018). Activation of DDR2 promotes Erk2-dependent phosphorylation of Snail1 to promote an epithelial-mesenchymal transition (Zhang et al., 2013). Syndecan-4, as a co-receptor of α5β1 integrin, can also promote directional cell migration by localizing Rac activation and membrane protrusions to the cells leading edge (Bass et al., 2007). Not surprisingly, a fibrotic tumor ECM can contribute to malignant transformation and tumor progression by modifying diverse cellular functions and compromising differentiation. In the following sections, we discuss how alterations in the composition, organization and mechanical properties of the ECM foster tumor aggression and promote metastasis.
The ECM and the metastatic journey
Interplay between a fibrotic ECM and malignant transformation and progression
The tissue stroma undergoes dramatic changes prior to malignant transformation and coincident with cancer progression (Figure 1). To begin with, a normal epithelial ductal structure, such as is found in the mammary gland or in the pancreas, is surrounded by a contiguous laminin-rich basement membrane and is embedded within an interstitial ECM that is characterized by curly and loosely organized compliant collagenous proteins. A laminin-rich basement membrane is essential for establishing and maintaining apical basal polarity of epithelial ductal structures where the basal surface of the polarized epithelial cells interact with the basement membrane and the apical “secretory” surface faces the lumen. During the early stages of malignant transformation, the basement membrane is gradually compromised as a result of the loss of basement membrane production or increased MMP degradation and the interstitial ECM simultaneously undergoes visible changes in its organization and biomechanical properties. In the absence of basement membrane, tumor cells lose their apicobasal polarity and more readily respond to interstitial stromal signals that stimulate their invasion into the parenchyma. Thus, the interstitial stroma in experimental pancreatic and breast tumors shows increased levels of ECM proteins including collagen, HA, and tenascin that are progressively remodeled and stiffened prior to the invasion of the tumor cells into the parenchyma (Acerbi et al., 2015; Laklai et al., 2016). Coincident with malignant transformation, the interstitial collagen fibers become progressively thicker and linearized and begin to align parallel to the tumor boundary (Provenzano et al., 2006). For example, two photon imaging revealed that breast cancer patients with linearized and thickened collagen fibers that are oriented radially surrounding the breast tumor mass have a higher predisposition to tumor aggression; possibility because these perpendicular tracks facilitate cell invasion into the parenchyma and fosters their migration through the stroma and subsequent dissemination. Similarly, poorly differentiated patient pancreatic cancers are surrounded by a thicker, stiffer and type XII collagen- and tenascin-rich stroma (Laklai et al., 2016). Abundant quantities of other stromal ECM proteins, such as fibronectin and collagen, also support tumor cell migration and invasion and favor the development of an invasive tumorigenic lesion. Fibronectin promotes migration of epithelial cells (Park and Schwarzbauer, 2014) whereas radially oriented linearized collagen bundles surrounding the tumor serve as migratory tracks to facilitate fibroblast, immune and cancer cell migration and invasion into the parenchyma (Egeblad et al., 2010). Syndecan-1 can also promote assembly and alignment of fibronectin- and collagen-fibers to facilitate directional migration and invasion of cancer cells (Yang et al., 2011). Collagen fibers are potent inducers of invadosome formation in macrophages, fibroblasts, and tumor cells. Invadosomes are structures that facilitate the infiltration of macrophages into the tumor and favor the invasion of tumor cells into the stroma to foster malignant transformation (Eddy et al., 2017) .
Malignant transformation to an invasive carcinoma is accompanied by further interstitial ECM remodeling and LOX-mediated collagen crosslinking and stiffening. Cancer cells frequently exhibit a haptotactic response, in which cell migration is guided by gradients of surface-bound molecules such as the ECM (Oudin and Weaver, 2016). For example, human breast cancer cells tend to exhibit directional movement towards higher concentration of fibronectin (Oudin et al., 2016). Similarly, pancreatic cancer cells exhibit haptotactic behavior towards higher concentration of type I collagen (Lu et al., 2014). Given that the tumor stroma is associated with increased levels of fibronectin and collagen, it is likely that haptotaxis is an important mechanism that enhances cancer and immune cell invasion in the tumor stroma. ECM stiffening fosters the aggressive behavior of cancer cells such that most cells migrate faster on stiffer substrates and their persistent migration can be directed up a stiffness gradient, through a process termed durotaxis (Kai et al., 2016). Durotaxis could explain why stiffer tumors show higher infiltration of immune cells and a higher frequency of fibroblasts and tumor cells interacting at the invasive front. A stiff ECM can promote cell migration and invasion by enhancing integrin clustering, FAK phosphorylation and Rho GTPase activation, which favor the assembly of filopodia, lamellipodia and invadosomes. Filopodia and lamellipodia are required for efficient tumor cell migration through the ECM, whereas invadosomes are essential for tumor cells to breach the dense interstitial ECM, particularly when the nuclei are too large to squeeze through the small matrix pore size (Wolf et al., 2013). In addition, stiff substrates can induce a mesenchymal/basal-like invasive phenotype in tumor cells (Figure 3). Basal-like tumor cells exert increased integrin-dependent contractile force, more readily assemble mature focal adhesions with a unique cassette of adhesion plaque proteins implicated in tumor invasion (Mekhdjian et al., 2017). Tumor cells that exhibit a basal-like phenotype often have a more deformable nucleus that together with the higher integrin adhesion force likely facilitates their migration through confined spaces within the dense interstitial tumor stroma. The importance of ECM stiffness in cancer metastasis is reinforced by studies using transgenic mice engineered to develop mammary or pancreatic tumors. Inhibition of ECM stiffening using LOX inhibitor β-aminopropionitrile (BAPN) or a LOX function-blocking antibody decreased breast tumor cell migration and invasion into the parenchyma (Levental et al., 2009).
Figure 3.
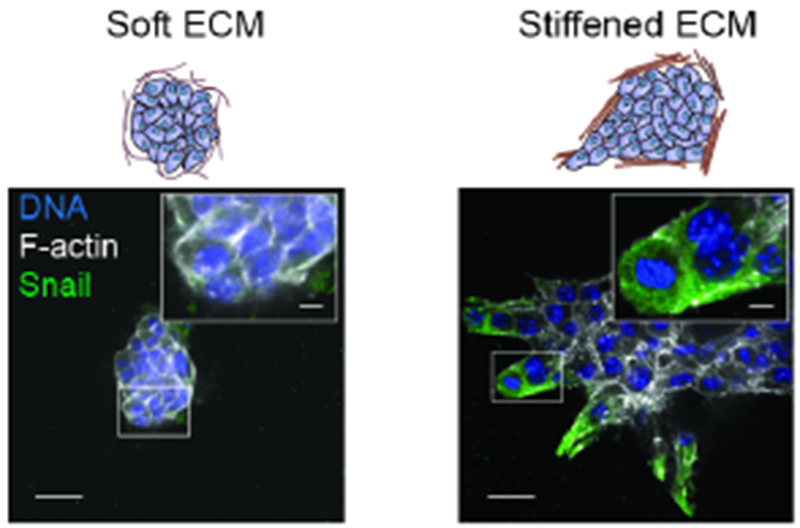
A stiffened ECM promotes the epithelial to mesenchymal transition and invasion of PyMT breast cancer cells. Representative images showing PyMT tumor organoid expressing a SNAIL-YFP reporter embedded within a soft collagen gels or a strain-stiffened collagen gel. Magnified images show the invasive front of the tumor organoids antibody stained for the YFP-EMT-marker SNAIL (green), a transcription factor upregulated during EMT. Cells are also costained for actin (white) and nuclei (blue). Figure is modified, with permission, from (Mekhdjian et al., 2017)
A stiffened ECM increases solid stress within the tumor such that as the tumor mass expands it encounters greater resistance from the desmoplastic, stiffened stroma ECM. Although long-term compressive stress can impede tumor spheroid proliferation, the mechanical stress induced by solid stress can also promote collective migration of cancer cells and reinforce cell-ECM interactions (Tse et al., 2012). Interestingly, transient compression of freshly isolated mammary tissue embedded within three dimensional (3D) collagen matrix using a piston induces sustained activation of Rho/ROCK mechanosignaling (Boyle et al., 2018). Compressive stress can also activate stromal cancer-associated fibroblasts to secrete more type I collagen and Growth Differentiation Factor 15 to enhance pancreatic cancer cell migration and proliferation (Kalli et al., 2018). Importantly, compressive stress is sufficient to promote tumor growth in vivo. The application of compressive stress to mouse colon tissue using ultramagnetic liposomes in the mesenchymal cells of connective tissues activated Ret receptor tyrosine kinase and downstream oncogenic beta-catenin and myc signaling (Fernandez-Sanchez et al., 2015). Notably, the magnetically induced deformation may also change the tension in a different plane and therefore it is possible that the oncogenic signaling is activated by the tension instead of the compression stress. Thus, a fibrotic stiffened ECM can directly foster tumor migration and progression and can indirectly promote cancer aggression by activating stromal fibroblasts and by stimulating immune cell infiltration.
Fibrotic, stiffened tumor stroma fosters the formation of dysfunctional tumor vasculature and facilitates intravasation
A major conduit for metastatic dissemination is via the vasculature. The tissue vasculature is tightly regulated by dynamic interactions between the ECM and the endothelial cells that control the process of angiogenesis. The ECM is a reservoir of diverse pro- and anti-angiogenic signaling molecules that can promote or suppress angiogenesis by modulating endothelial cell growth, survival and migration/invasion (Neve et al., 2014). In the presence of pro-angiogenic signals, endothelial cells proliferate, secrete MMPs to remodel the basement membrane surrounding the vasculature, and migrate into the interstitial matrix. During angiogenic sprouting, the endothelial cells secrete and remodel basement membrane proteins including laminins and collagen type IV and recruit pericytes to co-assemble a mature basement membrane that inhibits nascent vessel regression. The ECM also relays anti-angiogenic signals to restrain or counteract pro-angiogenic signals that limit blood vessel development. Thus, ECM composition and remodeling maintain vascular homeostasis by balancing the level of pro- and anti-angiogenic factors. Not surprisingly given its central role in regulating tissue angiogenesis, the fibrotic tumor ECM exerts a major impact on the tissue vasculature. To begin with the tumor ECM contains higher levels of tenascin and fibronectin and remodeled type I and HI collagens that stimulate vascular outgrowth (Neve et al., 2014). The higher endothelial sprouting associated with tumor angiogenesis also generates proteolytic ECM products that can either promote or inhibit angiogenesis. For example, arresten is a proteolytic fragment of type IV collagen that inhibits angiogenesis by suppressing endothelial cell migration and proliferation (Nyberg et al., 2008). The stiffened, fibrotic tumor ECM also stimulates angiogenesis by increasing the expression of pro-angiogenic factors, potentiating the responsiveness of the endothelium to soluble factors and compromising vascular integrity. Thus, endothelial cells cultured on 2D substrates showed that a “stiff” substrate promotes vascular endothelial growth factor receptor (VEGFR) expression and VEGFR internalization in endothelial cells and sustains ERK signaling that promotes endothelial cell survival (LaValley et al., 2017). Endothelial cells cultured on stiffer 2D hydrogels also secrete higher amounts of MMPs possibly because the stiffer ECM promotes invadosome formation and enhances basement membrane degradation (Hanjaya-Putra et al., 2010). The tension-induced increase in secreted MMPs could also explain the propensity of endothelial cells to sprout and exhibit sustained, directed migration when embedded within a stiffened 3D collagen matrix (Mason et al., 2013).
Although a stiff ECM can stimulate angiogenesis, chronically elevated mechanical stress induced by an extensively stiffened ECM and high solid stress can also impair vascular function. Thus, a fibrotic, highly cross-linked and stiffened ECM can induce the collapse of pre-existing vessels and even occlude blood flow to promote hypoxia (Jain et al., 2014). Treating mice with the pharmacological inhibitor BAPN, which inhibits LOX activity and prevents collagen crosslinking restores blood flow while simultaneously decreasing the number of blood vessel branching within the tumor (Bordeleau et al., 2017). ECM stiffness also regulates vascular integrity to modulate the patency of the endothelial barrier or endothelial permeability, and this effect may have a significant impact on tumor intravasation that is the process by which tumor cells cross the endothelial barrier to enter the blood circulation. Advanced microfluidic systems and intravital multiphoton imaging now permit the visualization of intravasation in real time, and has excellent potential to clarify how the biochemical and biophysical properties of the ECM influences intravasation. A stiff ECM may directly promote tumor cell intravasation by increasing the formation of invadosomes in the tumor cells or macrophages to facilitate degradation of endothelium basement membrane and facilitate transendothelial migration. This is consistent with the observation that invadosomes are not only found in invading cells located at the tumor edge but also those located near the blood vessels (Gligorijevic et al., 2012). A stiff ECM may additionally facilitate intravasation by promoting a basal-like or mesenchymal-like phenotype in the invading tumor cells. Cancer cells with a basal-like phenotype are more contractile and deformable, which facilitates their efficient migration through a dense and stiffened ECM (Mekhdjian et al., 2017). These deformable basal-like cells may also be more capable of squeezing through junctional gaps between endothelial cells. Consistently, high-resolution intravital imaging of the intravasating tumor cells revealed that the tumor cells acquire an hourglass shape when traversing through endothelial cells (Harney et al., 2015). Thus, a dense and stiffened ECM can promote cancer aggression by stimulating migration through the interstitial matrix and fostering tumor cell dissemination into the vasculature.
CTCs prime integrin-dependent signaling pathways to promote their survival in the circulation
Following intravasation, tumor cells encounter a fluid environment that is dramatically different from the primary tumor site. The majority of CTCs perish within hours as a result of hemodynamic shear stress, immune attack, and anoikis (Rejniak, 2016). CTCs can bypass anoikis by enhancing expression of proteins involved in integrin-dependent adhesion formation. For example, focal adhesion proteins such as talin, vinculin, and integrin-linked kinase (ILK) are enriched in colorectal CTCs compared with primary tumor cells (Barbazan et al., 2012). Fibronectin expression is also increased in pancreatic cancer CTCs when compared to primary tumor cells (Yu et al., 2012). These findings suggest that CTCs may secrete fibronectin to reinforce integrin-dependent adhesion survival signaling. Expression of bulky glycoproteins is also frequently upregulated in CTCs (Paszek et al., 2014). A thick glycocalyx can activate integrins that favor integrin clustering that enhances FAK, ERK and Akt signaling that promotes cell survival and proliferation (Paszek et al., 2014; Woods et al., 2017). Furthermore, CTCs can also stimulate pro-survival adhesion signaling through heterotypic interactions with other non-tumor cell types such as platelets and macrophages. For example, co-incubation of platelets with ovarian and colon tumor cells cultured in suspension (anoikis condition) induce Rho activation and YAP translocation into the nucleus (Haemmerle et al., 2017). Thus, CTCs have multiple avenues whereby CTCs can acquire integrin-dependent survival to successfully disseminated in the circulation.
The endothelial ECM can promote arrest and extravasation of circulating tumor cells
Extravasation is the process by which CTCs exit the circulation and enter the distal tissue. During extravasation, CTCs arrest within microcapillaries and associate with the vascular endothelial cells where they can undergo transendothelial migration. CTCs can either become physically trapped in the capillary bed due to its small diameter or they can actively adhere to the endothelium through transmembrane receptors on the CTCs, endothelial cells, immune cells, and platelets (Strilic and Offermanns, 2017). Interaction between αvβ3 integrin of CTCs with plasma fibronectin complexed with fibrin can facilitate tumor cell attachment to the endothelium to foster extravasation (Malik et al., 2010). The ECM associated with the endothelium may also arrest CTCs and facilitate their extravasation into the metastatic tissue (Barbazan et al., 2017). To this end, fibronectin deposits are frequently found on the luminal side of hepatic blood vessels in colon cancer patients and have been observed in a mouse model that spontaneously develops intestinal tumors that metastasize. Co-culture experiments has also revealed that human endothelial cells can secrete and assemble fibrillar fibronectin, which promotes the adhesion of colon carcinoma cells to the endothelium and favors tumor cell extravasation in a talin1-dependent manner. Consistently, talin1-depleted cancer cells that cannot form integrin-dependent adhesions were retained within the capillary and failed to extravasate, suggesting that adhesion signaling is required for cancer cell adhesion to the endothelium and efficient extravasation. Fibronectin deposition is likely triggered remotely by factors secreted by the primary tumor because the ECM deposits are not detectable in the absence of a primary tumor. This concept is consistent with experimental metastasis models that show tumor-derived factors can prepare the pre-metastatic niche for metastasis.
ECM deposition and remodeling in the premetastatic niche foster a favorable microenvironment for disseminated tumor cells
Primary tumors condition potential metastatic sites prior to tumor cell arrival by secreting various bioactive protein and lipid factors that modify the distal tissue to generate a receptive microenvironment that fosters the retention, survival and growth of the disseminated tumor cells. These changes in tissue composition that permit the growth of overt metastases are collectively known as the premetastatic niche. In particular, modifications of ECM composition and organization in the premetastatic niche play key roles in determining the metastatic potential of a tumor (Peinado et al., 2017). Secreted proteins from the primary tumor, such as proinflammatory cytokines and exosomes, can both directly activate tissue resident cells to synthesize and remodel the ECM in distant tissues as well as stimulate recruitment of ECM modifying cell populations, such as bone marrow derived myeloid cells. In addition to fostering a hospitable niche for disseminated tumor cells, ECM remodeling in the premetastatic niche may influence organotropism through organ specific integrin-ECM interactions.
A number of ECM proteins in the premetastatic niche have been implicated as crucial mediators of metastasis. Deposition of periostin promotes the formation of lung metastases and increases LOX activity, likely modifying collagen and elastin crosslinking in the lungs of tumor bearing mice (Wang et al., 2016). Periostin also enhances the accumulation of immunosuppressive myeloid-derived suppressor cells at the lung premetastatic niche, facilitating tumor cell escape from immune detection. Fibronectin levels are frequently elevated in the premetastatic lungs from a variety of primary tumor types and increase metastasis by enhancing myeloid cell recruitment and through direct interactions with disseminated tumor cells. For example, primary skin tumors stimulate fibronectin synthesis in resident pulmonary fibroblasts that facilitate the recruitment of VEGFR1+ bone marrow-derived hematopoietic progenitor cells and tumor cells to the lung premetastatic niches (Kaplan et al., 2005). Pancreatic ductal carcinoma (PDAC) tumors also foster the formation of fibronectin-rich premetastatic niches in the liver to facilitate their metastasis. Specifically, PDAC-derived exosomes are taken up by Kupffer cells in the liver stimulating TGF-β production, which subsequently activates fibronectin production by hepatic stellate cells. Consequently, increased fibronectin deposition in the liver recruits bone marrow-derived macrophages to establish the premetastatic niche (Costa-Silva et al., 2015). Extensive ECM remodeling also coincides with the formation of metastatic lesions. Mass spectrometry conducted on decellularized lungs containing macrometastases revealed considerable changes in ECM composition and the abundance of ECM-associated proteins (Mayorca-Guiliani et al., 2017). Similarly, the ECM appears to be highly citrullinated in liver metastases as compared with primary colorectal tumors. Citrullination of the ECM by tumor-derived peptidylarginine deiminase-4 can promote adhesion of colorectal cancer cells and metastasis in a murine model (Yuzhalin et al., 2018). Although citrullination of the ECM is elevated in the metastatic site, it remains to be investigated if the modification occurs prior to the arrival of the tumor cells or is coincident with formation of metastases. Importantly, fibronectin deposition and citrullination of the ECM are consistently found elevated in biopsies of human metastatic tumors.
The role of altered ECM mechanics in the formation of the premetastatic niche is unclear. For instance, bleomycin-induced lung injury that is characterized by fibrosis favors the metastasis of intravenously injected tumor cells (Orr et al., 1986). Moreover, experimental lung metastases contain more fibrillar linearized type I collagen (Mayorca-Guiliani et al., 2017). Whether these fibrotic changes stiffen the lung parenchyma and if this stiffening occurs prior to and fosters metastatic colonization remains an open question.
Integrin-dependent adhesion signaling governs the switch from tumor dormancy to metastatic outgrowth
The majority of cancer cells that extravasate into a distal “foreign” tissue either undergo apoptosis or enter into a state of dormancy. Dormancy manifests when disseminated tumor cells enter into a state of sustained quiescence, exhibit an equilibrium between growth and death and/or fail to stimulate an angiogenic response (Sosa et al., 2014). Dormant tumor cells pose a significant clinical challenge because they are able to evade immune surveillance and their quiescent state renders them resistant to most chemotherapies that predominately target dividing cells. Indeed, recent studies showed that latent disseminated pancreatic cancer cells down-regulate MHC-I molecules and this permits them to evade immune detection. By contrast, the pancreatic cells within the primary tumor were found to retain high plasma membrane expression of MHC-1 molecules which permitted efficient T cell killing (Pommier et al., 2018). Their critical importance resides in their latent ability to grow into lethal macroscopic lesions. Tumor dormancy is also one of the reasons why some patients remain disease-free after primary tumor resection, only to relapse after prolonged periods of remission (Sosa et al., 2014). Given its relevance to chemoresistance and clinical relapse, understanding how cancer cells decide to apoptose, remain dormant, or form metastases within a foreign microenvironment is paramount for the eradication of cancer.
The ECM composition of the metastatic microenvironment and its effect on adhesion signaling may dictate whether dormant cancer cells apoptose, remain quiescent or undergo outgrowth. Metastatic tumor cells are frequently found associated with the basement membrane surrounding the vascular niche in distal organs (Ghajar et al., 2013). The association of dormant metastatic tumor cells with the laminin-rich basement membrane implicates adhesion in their survival and has been shown to mediate their chemoresistance (Carlson et al., 2019), while increased integrin focal adhesion signaling appears to regulate their switch from a dormant state to an overt metastatic lesion. For example, work conducted using the dormancy-prone Her2 positive breast carcinoma line HCC1954-LCC1 showed that the switch from a dormant to a metastatic tumor is accompanied by pericyte-like cellular spreading along the endothelium and activation of integrin β1-ILK-YAP signaling (Er et al., 2018). The tumor glycocalyx may also regulate metastasis versus dormancy. The level of bulky glycoproteins within the primary tumor predicts metastasis and CTCs frequently express high levels of bulky glycoproteins (Paszek et al., 2014). Increasing the bulkiness of the cell surface glycocalyx using synthetic glycomimetics not only promoted PI3K-dependent cyclin D1-mediated cell cycle progression but fostered metastatic lesion outgrowth of dormant disseminated tumor cells (Woods et al., 2017). The biochemical property of the ECM also regulates dormancy through its effect on cell adhesion and FAK activation where fibronectin ligation of α5β1 integrin was able to promote the outgrowth of dormant cells and its inhibition prevented metastatic outgrowth in the lung (Barkan et al., 2008; Shibue and Weinberg, 2009). Importantly, DDRs can also collaborate with other transmembrane receptors such as tetraspanins to promote tumor metastasis. For instance, collagen-ligated DDR1/TM4SF1 activation of PKC-a-JAK-Stat3 increases Nanog and SOX2 in disseminated breast cancer cells that permits their metastatic outgrowth in lung, bone and brain tissue (Gao et al., 2016). These findings causally implicate ECM-induced adhesion in the survival of dormant tumor cells and adhesion signaling in their metastatic outgrowth and suggest there may be a role for specific ECM ligands and adhesion receptors in regulating this dynamic.
Given the critical role of ECM-mediated adhesion signaling in metastatic growth versus dormancy it is perhaps not surprising that chronic inflammation, stimulated either by exposing the lung parenchyma to inhaled tobacco smoke, or via lipopolysaccharide exposure, was able to induce the metastatic outgrowth of dormant breast cancer cells by stimulating ECM remodeling and promoting integrin-dependent focal adhesion signaling (Albrengues et al., 2018). Specifically, activated neutrophils recruited to the tissue by the inflammatory insult secreted proteolytic enzymes including elastase and MMP9 that induced ECM remodeling to expose a laminin epitope in the basement membrane associated with the dormant tumor cells. The dormant cells surrounded by the cleaved, remodeled laminin then proliferated to form metastatic outgrowths that exhibited increased integrin focal adhesion signaling and elevated actomyosin contractility and depended upon increased YAP activity. Similarly, the matricellular protein TSP-1 which is also localized to the perivascular ECM, has been implicated in maintaining the dormancy of metastatic tumor cells. Induction of ECM remodeling, presumptive cleavage of basement membrane laminins and deposition of molecules including periostin induced in response to angiogenic sprouting was found to promote the switch from a dormant to a proliferating metastatic tumor (Ghajar et al., 2013). Thus, extensive ECM remodeling in the metastatic niche can awaken the dormant cells and promote metastatic outgrowth through the activation of integrin focal adhesion signaling.
Targeting the extracellular matrix: Could this be the metastatic tumor’s Achilles’ heel?
Curing metastatic disease in cancer patients is clinically challenging. Local therapies such as surgery and radiotherapy that target the primary lesion are efficient at shrinking and removing the original tumor but are less effective at eradicating disseminated cancer cells. Conventional chemotherapies that target systemic disease efficiently ablate rapidly proliferating tumor cells but exert a negligible impact on slow-growing or dormant metastatic cells. Given the central role of the ECM in tumor metastasis, strategies that ameliorate fibrosis and integrin adhesion signaling offer an attractive auxiliary approach to reduce tumor aggression, improve cancer treatment and potentially cure metastatic disease (Figure 4).
Figure 4.
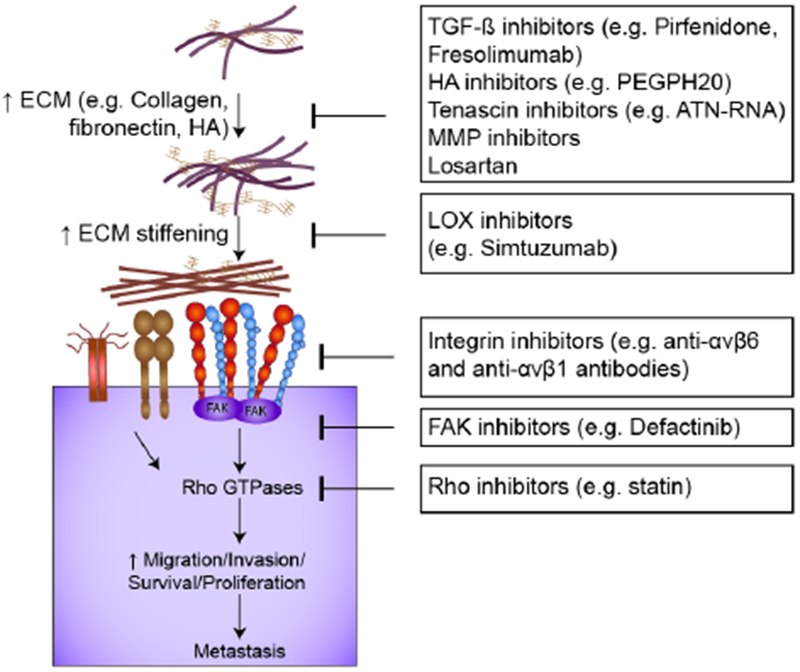
Overview of inhibitors that perturb cell-ECM interactions and/or adhesion signaling and may have applications for the treatment of metastatic cancers.
A number of therapies have been developed that reduce tissue fibrosis with the objective of decreasing tumor aggression and potentiating cancer treatment response. The anti-fibrotic drug Pirfenidone, originally developed to treat idiopathic pulmonary fibrosis, has been proposed as a tractable “anti-tumor, anti-fibrosis” therapy to treat solid cancers. The drug functions in part by suppressing TGF-β activity and has shown promising efficacy in experimental models of murine mammary cancer including “normalizing” the fibrotic stroma, as indicated by reduced levels of fibrillar collagen and HA, suppressed angiogenesis and inhibited lung metastasis (Polydorou et al., 2017; Takai et al., 2016). However, Pirfenidone has off-target effects including reducing production of reactive oxygen species and inflammatory cytokines that may compromise antitumor immunity and thus preclude its immediate clinical adoption until further safety testing can be completed (Bando, 2016). Anti-fibrotic drugs that selectively target TGF-β have also been developed including Fresolimumab, a TGF-β neutralizing antibody that prevents ligand-receptor interactions and hence is able to inhibit its profibrotic activity (Neuzillet et al., 2015). Anti-TGF-β treatments, such as Fresolimumab, efficiently decreased tumor tissue collagen and improved vascular integrity in experimental models of mammary cancer and has also been shown to improve checkpoint inhibitor response by restoring anti-tumor macrophage function and increasing tumor CD8 T cell activity (Liu et al., 2012; Mariathasan et al., 2018). Although phase II clinical trials are in the process of testing whether combinations of Fresolimumab and local radiotherapy can promote cancer regression in metastatic breast cancer patients and glioblastoma (GBM), TGF-β inhibitors can exert deleterious effects on heart function potentially compromising their widespread clinical application (NCT01401062) (Smith et al., 2012). To address this issue, more specific methods to inhibit pathological TGF-β activation have also been developed including therapeutics that specifically inhibit αv-integrin heterodimers. These include the αvβ1 inhibitor c8 that suppresses TGF-β activation and successfully attenuated chemical-induced pulmonary and liver fibrosis in preclinical models (Reed et al., 2015). Similarly, STX-100, a humanized monoclonal antibody that specifically targets αvβ6 integrin efficiently inhibits TGF-β activation to attenuate chemical-induced pulmonary fibrosis (NCT01371035) (Horan et al., 2008).
Therapies have been developed to target individual tumor fibrosis associated ECM molecules including tenascin and HA. Tenascin-C expression has been targeted using ATN-RNA, an RNA-based technology that triggers tenascin-C mRNA degradation (Wyszko et al., 2008). ATN-RNA therapy has been clinically tested in glioblastoma patients. Unfortunately, although the anti-TNC treatment was able to delay tumor growth and recurrence, the therapy only modestly prolonged overall GBM patient survival by a few weeks, precluding its universal clinical adoption (Rolle et al., 2010). Furthermore, it has been questioned whether the ATN-RNA anti-tumor effect in these GBM patients was due to downregulation of tenascin expression or innate immune responses against this foreign RNA (Spenle et al., 2015). By contrast, HA which is an ECM molecule that is strongly upregulated in fibrotic tumors, including those of the gastrointestinal tract and the pancreas, as well as those of the central nervous system, including GBMs, show encouraging clinical promise. HA is a highly negative charged molecule that absorbs large quantities of water, and is thus key for maintaining tissue hydration and compression resistance. Elevated HA levels in the tumor stroma increase interstitial fluid pressure that can impede vascular function and compromise drug delivery. Therapies that target and degrade HA including PEGPH20, a PEGylated hyaluronidase that degrades HA, efficiently reduced interstitial pressure in experimental models of PDAC and human PDACs, that normalized the tumor vasculature, suppressed the aggressive behavior of cancer cells and enhanced Gemcitabine chemotherapeutic drug efficacy (Hingorani et al., 2016; Provenzano et al., 2012). Combinations of PEGH20 with Paclitaxel/Gemcitabine are currently in phase III clinical trials for patients diagnosed with Stage IV PDAC and show promising responses (NCT02715804). Intriguingly, GBM patients and experimental models of human and murine GBM suggest that the abundant HA in these central nervous system tumors binds to tenascin-C to stiffen the tumor stroma and foster GBM aggression and recurrence, suggesting HA targeting may also prove useful for the treatment of GBM tumors (Barnes et al., 2018). Nevertheless, HA fragments can stimulate the infiltration of diverse immune cells such as macrophages, neutrophils, and T cells that foster inflammation and may thus repress anti-tumor immunity to compromise anti-tumor treatment (Lee-Sayer et al., 2015).
ECM remodeling is required for cancer cell invasion, angiogenesis, and transendothelial migration to facilitate vascular intravasation. Not surprisingly, various compounds have been developed to inhibit the activity of MMPs to limit ECM remodeling and prevent tumor cell invasion and angiogenesis and metastasis (Vandenbroucke and Libert, 2014). Consistently, experimental studies have shown impressive anti-invasion, anti-fibrosis and anti-metastatic effects through knockdown or inhibition of various MMPs including collagenases (Page-McCaw et al., 2007). Unfortunately, however, most clinical trials using MMP inhibitors thus far have been disappointing yielding negative and sometimes deleterious results that have prompted the rejection of this attractive set of targets as viable clinical therapeutics. Explanations for the failure of these MMP inhibitor clinical trials include the inappropriate use of broad spectrum inhibitors, selection of late stage patients with metastatic disease, inadequate assessment of the therapeutic window and reliance on monotherapy approaches. Indeed, the MMP family is composed of 24 members with experimental studies illustrating that some members exert antitumor effects while others exhibit pro-tumor activity, arguing that more selective MMP inhibitors need to be developed before their anti-tumor treatment clinical efficacy should be assessed.
ECM remodeling is also accompanied by posttranslational modifications to interstitial collagens that stabilize fibrillar structures and confer mechanical strength to the tissue. Given the florid fibrotic response and ECM remodeling observed in solid tumors it is not surprisingly, LOX, the major fibroblast-expressed stromal enzyme that regulates fibrillar collagen crosslinking is increased in several solid tumors including those of the breast and lung (Erler and Weaver, 2009). Furthermore, credible studies using experimental models of lung, breast and pancreatic cancer attest to their causal role in tumor progression and metastasis through amelioration of stromal fibrosis and inhibition of tumor cell invasion and migration (Gao et al., 2010; Levental et al., 2009; Miller et al., 2015). Therapeutics that reduce ECM stiffening may also impact metastasis by preventing conditioning of the premetastatic niche through modulation of exosome production and secretion by cells within the primary tumor. Regardless, the pharmacological inhibitor of LOX frequently used in these experimental model studies attesting to the utility of inhibiting LOX-mediated collagen cross-linking and stiffening as a viable anti-tumor treatment, BAPN, is contraindicated for use in treating cancer patients due to unmanageable toxicity as revealed when the drug was used in clinical trials to treat patients with scleroderma (Keiser and Sjoerdsma, 1967). Although anti-LOX antibodies do exist their clinical adoption is precluded by the failure of early clinical trials; possibility because current antibodies do not appear to effectively inhibit the enzymatic activity of the enzyme. Furthermore, similar to the clinical trials assessing the utility of MMP inhibitors LOX inhibitors have only been tested in patients with late stage cancers. Importantly, the 18kDa LOX pro-peptide that is released when LOX is activated has anti-Ras function and was shown to induce the phenotypic reversion of cultured Ras-transformed cells (Palamakumbura et al., 2004). Accordingly, reducing levels of the pro-peptide through inhibition of LOX activity could counter any impact of reducing stromal LOX-mediated collagen crosslinking on tumor inhibition and would be particularly contraindicated in Ras-driven tumors such as those of the pancreas and some lung cancers (Wu et al., 2007). LOX-like 2 (LOXL2) is another LOX family enzyme that is elevated in human tumors, although this enzyme is primarily detected in the tumor cells; likely in response to hypoxia-induced HIF1a (Schietke et al., 2010). Once again experimental evidence in model systems suggested inhibiting this enzyme would be clinically tractable. Thus, reducing LOXL2 in tumor cells transplanted into immune compromised mice reduced tumor aggression that was attributed to reduced fibrosis that was hypothesized to reflect decreased collagen crosslinking (Barry-Hamilton et al., 2010; Chang et al., 2017). Consistently, Simtuzumab, a LOXL2 function blocking antibody, successfully reduced tissue tension and delayed disease progression in experimental metastasis model of pancreatic cancer and colorectal cancer. However, Simtuzumab did not result in clinically meaningful improvement in advanced pancreatic and colorectal cancer patients and was terminated in 2016 following phase II clinical trials (Benson et al., 2017; Hecht et al., 2017). Explanations for the failure of the Simtuzumab clinical trials include poor stromal penetration of the Simtuzumab therapeutic and the possibility that intracellular LOXL2 also contributes to tumor aggression. Indeed, PyMT mice in which LOXL2 was genetically ablated only in the mammary epithelium showed a significant inhibition of lung metastasis while genetically increasing mammary epithelial tumor cell LOXL2 significantly enhanced lung metastasis, without any detectable impact on the ECM; possibly because cellular LOXL2 can induce an EMT (Salvador et al., 2017). Alternately, lysyl hydroxylases which are primarily stromal fibroblast-expressed enzymes that modify the hydroxyl groups on intracellular fibrillar collagen have emerged as potentially attractive therapeutic anti-tumor targets (Gilkes et al., 2013).
Clearly, additional studies are needed to clarify the role of LOX and lysyl hydroxylase family members in tumor fibrosis and to design improved therapeutics.
The compressive solid stress that develops in solid tumors can collapse blood vessels and induce hypoxia, elevate interstitial fluid pressure, and impair drug delivery. To address these issues vascular normalization has been proposed as a therapeutic strategy aimed at reducing mechanical stresses in the tumor stroma and ultimately preventing metastasis (Goel et al., 2012). Towards this goal, Bevacizumab monotherapy, was developed as the first anti-VEGF monoclonal antibody approved for clinical use that could effectively inhibit angiogenesis. However, although Bevacizumab can transiently normalize the tumor vasculature to decrease interstitial fluid pressure and thereby improve the efficacy of chemotherapy, it has limited clinical use as a monotherapy because it fails to sustain long term tumor regression (Mollard et al., 2017). Moreover, Bevacizumab and similar anti-angiogenic drugs may also be contraindicated because the treatment selectively targets cancers that rely on angiogenesis for their growth and survival leading to the emergence of cancer cells that can vascularize by vessel co-options, a process whereby cancer cells incorporate pre-existing blood vessels (Frentzas et al., 2016). Alternately, Losartan, which is an angiotensin II receptor antagonist used for the treatment of hypertension, has demonstrated anti-contractility activity that was shown to relieve solid stress in tumors and restore vascular integrity (Chauhan et al., 2013). Losartan inhibits type I collagen synthesis in cancer-associated fibroblasts, has been shown to delay breast, pancreatic, and skin tumor progression in preclinical models and is currently being tested in the clinic as a means to potentiate anti-tumor treatment in pancreatic patients (NCT01821729) (Diop-Frimpong et al., 2011).
Integrin-mediated adhesion signaling is elevated in many cancers and is required for tumor cell invasion into the parenchyma, CTC survival within the circulation, transendothelial migration, and efficient metastatic outgrowth. Although clinical trials that specifically targeted integrin adhesion using monotherapies in patient tumors have proven disappointing, drugs that inhibit integrin adhesion signaling have shown more therapeutic promise (Goodman and Picard, 2012). For instance, recent studies also showed that lipophilic statins, a drug that is frequently prescribed to lower cholesterol levels in patients, also reduces tumor aggression and decreases the metastatic dissemination of various cancer cells, possibly through its off-target effect on inhibiting geranyl-geranylation to impair Rho activation and reduce integrin adhesion signaling (Lampi et al., 2016). Aspirin can also exert anti-tumor and metastasis suppressor functions. Clinical studies show that patients who routinely take low dose aspirin have a decreased risk of cancer and cancer metastasis (Rothwell et al., 2012). Although, aspirin likely suppresses metastasis by decreasing the proangiogenic and proinflammatory PGE2 production in platelets, platelets also promote CTC survival by activating Rho/YAP-mediated adhesion signaling, CTC adhesion to the endothelium, and CTC extravasation (Haemmerle et al., 2017; Xu et al., 2018). Thus, it is also feasible that some of the anti-metastatic effect of aspirin is mediated by its ability to inhibit platelet-tumor cell crosstalk that supports cancer cell dissemination. More specific inhibitors that have been designed to reduce adhesion signaling have also been developed including drugs targeting FAK which is upregulated in cancer cells in response to tissue fibrosis and implicated in tumor metastasis (Lv et al., 2018). Several FAK inhibitors have been developed and tested in both preclinical and clinical trials and have shown encouraging antitumor clinical efficacy when used in combinatorial treatment regimens. For instance, the FAK inhibitor Defactinib has shown a good safety profile and robust biological activity in mouse models and in early clinical trials. In an experimental model of PDAC, Defactinib was also shown to decrease the number of tumor-infiltrating immunosuppressive cells, tumor fibrosis, and the formation of liver metastases (Jiang et al., 2016). Defactinib is currently in Phase II clinical trials for the treatment of advanced PDAC and has thus far been found to sensitize previously unresponsive patients to chemo- and immunotherapy (NCT02546531). Along these lines, designing inhibitors that interfere with talin-vinculin binding might be a promising option to suppress mechanosignaling pathways (personal communication with Pere Roca-Cusach). Accordingly, identification of the mechanotransduction pathways that are unique to metastatic cells may help identify novel targets for anti-metastasis therapy.
Concluding Remarks and Future Directions
Metastasis is an inefficient process, in which only a very small proportion of the primary tumor cells disseminate and survive and grow to form overt metastases that ultimately compromise patient survival. Despite the clinical importance of metastasis however, the steps of the cascade between primary tumor formation and metastases remain poorly understood. What is clear is that the ECM which undergoes dramatic changes during cancer progression plays a central role in the metastatic process. The desmoplastic reaction and mechanical stresses at the primary tumor foster the aggressive behavior of cancer cells and stimulate their migration through the interstitial ECM and vascular dissemination. Adhesion signaling helps tumor cells efficiently intravasate and survive against all odds within the circulation. Once the tumor cells arrive and extravasate at the distal tissue site during their metastatic journey, the ECM and cell adhesion signaling dictate metastatic outgrowth or dormancy. Recent advances in high-resolution intravital microscopy now permit assessment of the spatial and temporal relationship played by the ECM and adhesion signaling during the various steps during the metastatic cascade. This includes ECM-adhesion dependent invasion, migration, intravasation, and extravasation and clarification of what ECM and adhesion signaling factors drive the tumor cells that embed within the distal target tissue site to grow to form metastatic lesions or restrain the tumors to drive them into a state of dormancy. These optimized optical systems can be readily exploited to clarify the interplay between the ECM, cell adhesion and tumor metastasis that will permit the identification of new therapeutic targets. In addition, quantitative proteomic analysis and tissue decellularization approaches have been optimized to the extent that it is now possible not only to characterize the composition of the ECM proteome, or matrisome in the normal and cancerous tissue but also within localized tissue regions at the primary and metastatic site that are key for supporting each of the metastatic steps and to visualize their structure/function (Mayorca-Guiliani et al., 2017). Sophisticated engineered 3D tissue reconstructions that incorporate the tumor, its associated vasculature and immune cells now permit identification of specific protein-protein interactions that mediate efficient intravasation and extravasation of tumor cells. Tumor tissue on a chip and tumor metastasis models on a chip permit high throughput testing of novel therapeutics that could identify a unique role for ECM adhesion receptor interactions (Huh et al., 2011). Empowered by new technological advances we are now prepared to develop new therapies that can modulate the structure/function and biophysical properties of the ECM and regulate adhesion signaling to reduce tumor aggression, limit tumor metastasis and optimize treatment responses to improve cancer patient outcome.
The extracellular matrix is perturbed in tumors. Kai et al. present a Review discussing the tumor extracellular matrix and its effect on tumor cell invasion into the stroma and vascular dissemination.
Acknowledgments
The authors apologize to all colleagues whose work was not cited owing to space limitations. This work was supported by funds from the DOD grant BCRP BC122990, and US National Institutes of Health NCI R01 grants CA222508-01, CA192914, CA 174929, CA08592, U01 grant CA202241, U54 grant CA163155, and R33 grant CA183685 (to VMW) and a University of California at San Francisco Predoctoral Fellowship in Cancer Research (to A.P.D.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, and Weaver VM (2015). Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb) 7, 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afratis NA, Nikitovic D, Multhaupt HA, Theocharis AD, Couchman JR, and Karamanos NK (2017). Syndecans - key regulators of cell signaling and biological functions. FEBS J 284, 27–41. [DOI] [PubMed] [Google Scholar]
- Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Kuttner V, et al. (2018). Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bando M (2016). Pirfenidone: Clinical trials and clinical practice in patients with idiopathic pulmonary fibrosis. Respir Investig 54, 298–304. [DOI] [PubMed] [Google Scholar]
- Barbazan J, Alonso-Alconada L, Elkhatib N, Geraldo S, Gurchenkov V, Glentis A, van Niel G, Palmulli R, Fernandez B, Viano P, et al. (2017). Liver Metastasis Is Facilitated by the Adherence of Circulating Tumor Cells to Vascular Fibronectin Deposits. Cancer Res 77, 3431–3441. [DOI] [PubMed] [Google Scholar]
- Barbazan J, Alonso-Alconada L, Muinelo-Romay L, Vieito M, Abalo A, Alonso-Nocelo M, Candamio S, Gallardo E, Fernandez B, Abdulkader I, et al. (2012). Molecular characterization of circulating tumor cells in human metastatic colorectal cancer. PLoS One 7, e40476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, Liu ZY, Costes SV, Cho EH, Lockett S, et al. (2008). Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res 68, 6241–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes JM, Kaushik S, Bainer RO, Sa JK, Woods EC, Kai F, Przybyla L, Lee M, Lee HW, Tung JC, et al. (2018). A tension-mediated glycocalyx-integrin feedback loop promotes mesenchymal-like glioblastoma. Nat Cell Biol 20, 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, Mikels A, Vaysberg M, Ghermazien H, Wai C, et al. (2010). Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med 16, 1009–1017. [DOI] [PubMed] [Google Scholar]
- Bass MD, Roach KA, Morgan MR, Mostafavi-Pour Z, Schoen T, Muramatsu T, Mayer U, Ballestrem C, Spatz JP, and Humphries MJ (2007). Syndecan-4-dependent Rac1 regulation determines directional migration in response to the extracellular matrix. J Cell Biol 177, 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson AB 3rd, Wainberg ZA, Hecht JR, Vyushkov D, Dong H, Bendell J, and Kudrik F (2017). A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 22, 241–e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, LaValley DJ, Huynh J, et al. (2017). Matrix stiffening promotes a tumor vasculature phenotype. Proc Natl Acad Sci U S A 114, 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, and Bissell MJ (1995). Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science 267, 891–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle ST, Kular J, Nobis M, Ruszkiewicz A, Timpson P, and Samuel MS (2018). Acute compressive stress activates RHO/ROCK-mediated cellular processes. Small GTPases, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, Shor RE, Goddard ET, Dai J, Schweitzer EM, et al. (2019). Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol 21, 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassereau L, Miroshnikova YA, Ou G, Lakins J, and Weaver VM (2015). A 3D tension bioreactor platform to study the interplay between ECM stiffness and tumor phenotype. J Biotechnol 193, 66–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Lucas MC, Leonte LE, Garcia-Montolio M, Singh LB, Findlay AD, Deodhar M, Foot JS, Jarolimek W, Timpson P, et al. (2017). Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 8, 26066–26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, et al. (2013). Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 4, 2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciobanasu C, Faivre B, and Le Clainche C (2014). Actomyosin-dependent formation of the mechanosensitive talin-vinculin complex reinforces actin anchoring. Nat Commun 5, 3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho NM, and McCulloch CA (2018). Mechanical signaling through the discoidin domain receptor 1 plays a central role in tissue fibrosis. Cell Adh Migr 12, 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, and Keely PJ (2011). Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol 178, 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, et al. (2015). Pancreatic cancer exosomes initiate premetastatic niche formation in the liver. Nat Cell Biol 17, 816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, and Bourdon MA (1996). Tenascin mediates human glioma cell migration and modulates cell migration on fibronectin. J Cell Sci 109 (Pt 3), 643–652. [DOI] [PubMed] [Google Scholar]
- Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, and Jain RK (2011). Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A 108, 2909–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrokhotov O, Samsonov M, Sokabe M, and Hirata H (2018). Mechanoregulation and pathology of YAP/TAZ via Hippo and non-Hippo mechanisms. Clin Transl Med 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy RJ, Weidmann MD, Sharma VP, and Condeelis JS (2017). Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol 27, 595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Rasch MG, and Weaver VM (2010). Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol 22, 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, Griscom B, Rosenblum M, Boire A, Brogi E, et al. (2018). Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol 20, 966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler JT, and Weaver VM (2009). Three-dimensional context regulation of metastasis. Clin Exp Metastasis 26, 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sanchez ME, Barbier S, Whitehead J, Bealle G, Michel A, Latorre-Ossa H, Rey C, Fouassier L, Claperon A, Brulle L, et al. (2015). Mechanical induction of the tumorigenic beta-catenin pathway by tumour growth pressure. Nature 523, 92–95. [DOI] [PubMed] [Google Scholar]
- Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E, Nathan M, Wotherspoon A, Gao ZH, Shi Y, et al. (2016). Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med 22, 1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Chakraborty G, Zhang Z, Akalay I, Gadiya M, Gao Y, Sinha S, Hu J, Jiang C, Akram M, et al. (2016). Multi-organ Site Metastatic Reactivation Mediated by Non-canonical Discoidin Domain Receptor 1 Signaling. Cell 166, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Xiao Q, Ma H, Li L, Liu J, Feng Y, Fang Z, Wu J, Han X, Zhang J, et al. (2010). LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc Natl Acad Sci U S A 107, 18892–18897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, et al. (2013). The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15, 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkes DM, Bajpai S, Wong CC, Chaturvedi P, Hubbi ME, Wirtz D, and Semenza GL (2013). Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol Cancer Res 11, 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, and Condeelis J (2012). N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci 125, 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard ET, Bozic I, Riddell SR, and Ghajar CM (2018). Dormant tumour cells, their niches and the influence of immunity. Nat Cell Biol 20, 1240–1249. [DOI] [PubMed] [Google Scholar]
- Goel S, Wong AH, and Jain RK (2012). Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med 2, a006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman SL, and Picard M (2012). Integrins as therapeutic targets. Trends Pharmacol Sci 33, 405–412. [DOI] [PubMed] [Google Scholar]
- Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, and Petersen OW (2002). Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci 115, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle M, Taylor ML, Gutschner T, Pradeep S, Cho MS, Sheng J, Lyons YM, Nagaraja AS, Dood RL, Wen Y, et al. (2017). Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun 8, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Chen S, Yuan W, Fan Q, Tian J, Wang X, Chen L, Zhang X, Wei W, Liu R, et al. (2016). Oriented collagen fibers direct tumor cell intravasation. Proc Natl Acad Sci U S A 113, 11208–11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanjaya-Putra D, Yee J, Ceci D, Truitt R, Yee D, and Gerecht S (2010). Vascular endothelial growth factor and substrate mechanics regulate in vitro tubulogenesis of endothelial progenitor cells. J Cell Mol Med 14, 2436–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, Oktay MH, Pollard JW, Jones JG, and Condeelis JS (2015). Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov 5, 932–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht JR, Benson AB 3rd, Vyushkov D, Yang Y, Bendell J, and Verma U (2017). A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab in Combination with FOLFIRI for the Second-Line Treatment of Metastatic KRAS Mutant Colorectal Adenocarcinoma. Oncologist 22, 243–e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, Tjulandin SA, Gladkov OA, Holcombe RF, Korn R, et al. (2016). Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin Cancer Res 22, 2848–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, Simon KJ, Hahm K, Allaire NE, Rinaldi NJ, et al. (2008). Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med 177, 56–65. [DOI] [PubMed] [Google Scholar]
- Huh D, Hamilton GA, and Ingber DE (2011). From 3D cell culture to organs-on-chips. Trends Cell Biol 21, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huleihel L, Hussey GS, Naranjo JD, Zhang L, Dziki JL, Turner NJ, Stolz DB, and Badylak SF (2016). Matrix-bound nanovesicles within ECM bioscaffolds. Sci Adv 2, e1600502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO (2009). The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK, Martin JD, and Stylianopoulos T (2014). The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng 16, 321–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. (2016). Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai F, Laklai H, and Weaver VM (2016). Force Matters: Biomechanical Regulation of Cell Invasion and Migration in Disease. Trends Cell Biol 26, 486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli M, Papageorgis P, Gkretsi V, and Stylianopoulos T (2018). Solid Stress Facilitates Fibroblasts Activation to Promote Pancreatic Cancer Cell Migration. Ann Biomed Eng 46, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser HR, and Sjoerdsma A (1967). Studies on beta-aminopropionitrile in patients with scleroderma. Clin Pharmacol Ther 8, 593–602. [DOI] [PubMed] [Google Scholar]
- Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, et al. (2016). Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med 22, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampi MC, Faber CJ, Huynh J, Bordeleau F, Zanotelli MR, and Reinhart-King CA (2016). Simvastatin Ameliorates Matrix Stiffness-Mediated Endothelial Monolayer Disruption. PLoS One 11, e0147033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaValley DJ, Zanotelli MR, Bordeleau F, Wang W, Schwager SC, and Reinhart-King CA (2017). Matrix Stiffness Enhances VEGFR-2 Internalization, Signaling, and Proliferation in Endothelial Cells. Converg Sci Phys Oncol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Sayer SS, Dong Y, Arif AA, Olsson M, Brown KL, and Johnson P (2015). The where, when, how, and why of hyaluronan binding by immune cells. Front Immunol 6, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leight JL, Wozniak MA, Chen S, Lynch ML, and Chen CS (2012). Matrix rigidity regulates a switch between TGF-beta1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell 23, 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, Ancukiewicz M, Boucher Y, Jain RK, and Xu L (2012). TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U S A 109, 16618–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Zhou S, Siech M, Habisch H, Seufferlein T, and Bachem MG (2014). Pancreatic stellate cells promote hapto-migration of cancer cells through collagen I-mediated signalling pathway. Br J Cancer 110, 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv PC, Jiang AQ, Zhang WM, and Zhu HL (2018). FAK inhibitors in Cancer, a patent review. Expert Opin Ther Pat 28, 139–145. [DOI] [PubMed] [Google Scholar]
- Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E, and Pilch J (2010). Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res 70, 4327–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason BN, Starchenko A, Williams RM, Bonassar LJ, and Reinhart-King CA (2013). Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothelial cell behavior. Acta Biomater 9, 4635–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayorca-Guiliani AE, Madsen CD, Cox TR, Horton ER, Venning FA, and Erler JT (2017). ISDoT: in situ decellularization of tissues for high-resolution imaging and proteomic analysis of native extracellular matrix. Nat Med 23, 890–898. [DOI] [PubMed] [Google Scholar]
- Mekhdjian AH, Kai F, Rubashkin MG, Prahl LS, Przybyla LM, McGregor AL, Bell ES, Barnes JM, DuFort CC, Ou G, et al. (2017). Integrin-mediated traction force enhances paxillin molecular associations and adhesion dynamics that increase the invasiveness of tumor cells into a three-dimensional extracellular matrix. Mol Biol Cell 28, 1467–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micalizzi DS, Maheswaran S, and Haber DA (2017). A conduit to metastasis: circulating tumor cell biology. Genes Dev 31, 1827–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, et al. (2015). Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med 7, 1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroshnikova YA, Rozenberg GI, Cassereau L, Pickup M, Mouw JK, Ou G, Templeman KL, Hannachi EI, Gooch KJ, Sarang-Sieminski AL, et al. (2017). alpha5beta1-Integrin promotes tension-dependent mammary epithelial cell invasion by engaging the fibronectin synergy site. Mol Biol Cell 28, 2958–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollard S, Ciccolini J, Imbs DC, El Cheikh R, Barbolosi D, and Benzekry S (2017). Model driven optimization of antiangiogenics + cytotoxics combination: application to breast cancer mice treated with bevacizumab + paclitaxel doublet leads to reduced tumor growth and fewer metastasis. Oncotarget 8, 23087–23098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouw JK, Ou G, and Weaver VM (2014). Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol 15, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, and de Gramont A (2015). Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther 147, 22–31. [DOI] [PubMed] [Google Scholar]
- Neve A, Cantatore FP, Maruotti N, Corrado A, and Ribatti D (2014). Extracellular matrix modulates angiogenesis in physiological and pathological conditions. Biomed Res Int 2014, 756078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg P, Xie L, Sugimoto H, Colorado P, Sund M, Holthaus K, Sudhakar A, Salo T, and Kalluri R (2008). Characterization of the anti-angiogenic properties of arresten, an alpha1beta1 integrin-dependent collagen-derived tumor suppressor. Exp Cell Res 314, 3292–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr FW, Adamson IY, and Young L (1986). Promotion of pulmonary metastasis in mice by bleomycin-induced endothelial injury. Cancer Res 46, 891–897. [PubMed] [Google Scholar]
- Oudin MJ, Jonas O, Kosciuk T, Broye LC, Guido BC, Wyckoff J, Riquelme D, Lamar JM, Asokan SB, Whittaker C, et al. (2016). Tumor Cell-Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic Progression. Cancer Discov 6, 516–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudin MJ, and Weaver VM (2016). Physical and Chemical Gradients in the Tumor Microenvironment Regulate Tumor Cell Invasion, Migration, and Metastasis. Cold Spring Harb Symp Quant Biol 81, 189–205. [DOI] [PubMed] [Google Scholar]
- Page-McCaw A, Ewald AJ, and Werb Z (2007). Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 8, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamakumbura AH, Jeay S, Guo Y, Pischon N, Sommer P, Sonenshein GE, and Trackman PC (2004). The propeptide domain of lysyl oxidase induces phenotypic reversion of ras-transformed cells. J Biol Chem 279, 40593–40600. [DOI] [PubMed] [Google Scholar]
- Park J, and Schwarzbauer JE (2014). Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 33, 1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, et al. (2014). The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511, 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254. [DOI] [PubMed] [Google Scholar]
- Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, et al. (2017). Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 17, 302–317. [DOI] [PubMed] [Google Scholar]
- Pickup MW, Mouw JK, and Weaver VM (2014). The extracellular matrix modulates the hallmarks of cancer. EMBO Rep 15, 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydorou C, Mpekris F, Papageorgis P, Voutouri C, and Stylianopoulos T (2017). Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 8, 24506–24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, Auffray C, Albrengues J, Egeblad M, Iacobuzio-Donahue CA, et al. (2018). Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, and Hingorani SR (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, and Keely PJ (2006). Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammal H, Saby C, Magnien K, Van-Gulick L, Garnotel R, Buache E, El Btaouri H, Jeannesson P, and Morjani H (2016). Discoidin Domain Receptors: Potential Actors and Targets in Cancer. Front Pharmacol 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed NI, Jo H, Chen C, Tsujino K, Arnold TD, DeGrado WF, and Sheppard D (2015). The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci Transl Med 7, 288ra279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rejniak KA (2016). Circulating Tumor Cells: When a Solid Tumor Meets a Fluid Microenvironment. Adv Exp Med Biol 936, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner G, Noulet F, Mercier MC, Choulier L, Etienne-Selloum N, Gies JP, Lehmann M, Lelong-Rebel I, Martin S, and Dontenwill M (2016). Expression/activation of alpha5beta1 integrin is linked to the beta-catenin signaling pathway to drive migration in glioma cells. Oncotarget 7, 62194–62207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizki A, Weaver VM, Lee SY, Rozenberg GI, Chin K, Myers CA, Bascom JL, Mott JD, Semeiks JR, Grate LR, et al. (2008). A human breast cell model of preinvasive to invasive transition. Cancer Res 68, 1378–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolle K, Nowak S, Wyszko E, Nowak M, Zukiel R, Piestrzeniewicz R, Gawronska I, Barciszewska MZ, and Barciszewski J (2010). Promising human brain tumors therapy with interference RNA intervention (iRNAi). Cancer Biol Ther 9, 396–406. [DOI] [PubMed] [Google Scholar]
- Roovers K, Davey G, Zhu X, Bottazzi ME, and Assoian RK (1999). Alpha5beta1 integrin controls cyclin D1 expression by sustaining mitogen-activated protein kinase activity in growth factor-treated cells. Mol Biol Cell 10, 3197–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, and Mehta Z (2012). Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 379, 1591–1601. [DOI] [PubMed] [Google Scholar]
- Salvador F, Martin A, Lopez-Menendez C, Moreno-Bueno G, Santos V, Vazquez-Naharro A, Santamaria PG, Morales S, Dubus PR, Muinelo-Romay L, et al. (2017). Lysyl Oxidase-like Protein LOXL2 Promotes Lung Metastasis of Breast Cancer. Cancer Res 77, 5846–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietke R, Warnecke C, Wacker I, Schodel J, Mole DR, Campean V, Amann K, Goppelt-Struebe M, Behrens J, Eckardt KU, et al. (2010). The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: insights into cellular transformation processes mediated by HIF-1. J Biol Chem 285, 6658–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, and Assoian RK (2001). Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci 114, 2553–2560. [DOI] [PubMed] [Google Scholar]
- Shibue T, and Weinberg RA (2009). Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A 106, 10290–10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Robin TP, and Ford HL (2012). Molecular pathways: targeting the TGF-beta pathway for cancer therapy. Clin Cancer Res 18, 4514–4521. [DOI] [PubMed] [Google Scholar]
- Sosa MS, Bragado P, and Aguirre-Ghiso JA (2014). Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile J, and Hocking DC (2002). Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell 13, 3546–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spenle C, Saupe F, Midwood K, Burckel H, Noel G, and Orend G (2015). Tenascin-C: Exploitation and collateral damage in cancer management. Cell Adh Migr 9, 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strilic B, and Offermanns S (2017). Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell 32, 282–293. [DOI] [PubMed] [Google Scholar]
- Sun Z, Guo SS, and Fassler R (2016). Integrin-mediated mechanotransduction. J Cell Biol 215, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai K, Le A, Weaver VM, and Werb Z (2016). Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 7, 82889–82901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse JM, Cheng G, Tyrrell JA, Wilcox-Adelman SA, Boucher Y, Jain RK, and Munn LL (2012). Mechanical compression drives cancer cells toward invasive phenotype. Proc Natl Acad Sci U S A 109, 911–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon PH (2011). Integrin signaling, cell survival, and anoikis: distinctions, differences, and differentiation. J Signal Transduct 2011, 738137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helvert S, and Friedl P (2016). Strain Stiffening of Fibrillar Collagen during Individual and Collective Cell Migration Identified by AFM Nanoindentation. ACS Appl Mater Interfaces 8, 21946–21955. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke RE, and Libert C (2014). Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov 13, 904–927. [DOI] [PubMed] [Google Scholar]
- Wang Z, Xiong S, Mao Y, Chen M, Ma X, Zhou X, Ma Z, Liu F, Huang Z, Luo Q, et al. (2016). Periostin promotes immunosuppressive premetastatic niche formation to facilitate breast tumour metastasis. J Pathol 239, 484–495. [DOI] [PubMed] [Google Scholar]
- Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, et al. (2015). Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol 17, 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, and Friedl P (2013). Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 201, 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods EC, Kai F, Barnes JM, Pedram K, Pickup MW, Hollander MJ, Weaver VM, and Bertozzi CR (2017). A bulky glycocalyx fosters metastasis formation by promoting G1 cell cycle progression. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Min C, Wang X, Yu Z, Kirsch KH, Trackman PC, and Sonenshein GE (2007). Repression of BCL2 by the tumor suppressor activity of the lysyl oxidase propeptide inhibits transformed phenotype of lung and pancreatic cancer cells. Cancer Res 67, 6278–6285. [DOI] [PubMed] [Google Scholar]
- Wyszko E, Rolle K, Nowak S, Zukiel R, Nowak M, Piestrzeniewicz R, Gawronska I, Barciszewska MZ, and Barciszewski J (2008). A multivariate analysis of patients with brain tumors treated with ATN-RNA. Acta Pol Pharm 65, 677–684. [PubMed] [Google Scholar]
- Xu XR, Yousef GM, and Ni H (2018). Cancer and platelet crosstalk: opportunities and challenges for aspirin and other antiplatelet agents. Blood 131, 1777–1789. [DOI] [PubMed] [Google Scholar]
- Yang N, Mosher R, Seo S, Beebe D, and Friedl A (2011). Syndecan-1 in breast cancer stroma fibroblasts regulates extracellular matrix fiber organization and carcinoma cell motility. Am J Pathol 178, 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, et al. (2012). RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature 487, 510–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzhalin AE, Gordon-Weeks AN, Tognoli ML, Jones K, Markelc B, Konietzny R, Fischer R, Muth A, O’Neill E, Thompson PR, et al. (2018). Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat Commun 9, 4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, Keely PJ, and Longmore GD (2013). The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol 15, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]