

Abstract
Capillary zone electrophoresis (CZE)-tandem mass spectrometry (MS/MS) has been recognized as an efficient approach for top-down proteomics recently for its high-capacity separation and highly sensitive detection of proteoforms. However, the commonly used collision-based dissociation methods often cannot provide extensive fragmentation of proteoforms for thorough characterization. Activated ion electron transfer dissociation (AI-ETD), that combines infrared photoactivation concurrent with ETD, has shown better performance for proteoform fragmentation than higher energy-collisional dissociation (HCD) and standard ETD. Here, we present the first application of CZE-AI-ETD on an Orbitrap Fusion Lumos mass spectrometer for large-scale top-down proteomics of Escherichia coli (E. coli) cells. CZE-AI-ETD outperformed CZE-ETD regarding proteoform and protein identifications (IDs). CZE-AI-ETD reached comparable proteoform and protein IDs with CZE-HCD. CZE-AI-ETD tended to generate better expectation values (E-values) of proteoforms than CZE-HCD and CZE-ETD, indicating higher quality of MS/MS spectra from AI-ETD respecting the number of sequence-informative fragment ions generated. CZE-AI-ETD showed great reproducibility regarding the proteoform and protein IDs with relative standard deviations less than 4% and 2% (n=3). Coupling size exclusion chromatography (SEC) to CZE-AI-ETD identified 3028 proteoforms and 387 proteins from E. coli cells with 1% spectrum-level and 5% proteoform-level false discovery rates. The data represents the largest top-down proteomics dataset using the AI-ETD method so far. Single-shot CZE-AI-ETD of one SEC fraction identified 957 proteoforms and 253 proteins. N-terminal truncations, signal peptide cleavage, N-terminal methionine removal and various post-translational modifications including protein N-terminal acetylation, methylation, S-thiolation, disulfide bonds, and lysine succinylation were detected.
Keywords: capillary zone electrophoresis-tandem mass spectrometry, activated ion electron transfer dissociation, top-down proteomics, Escherichia coli, S-thiolation, disulfide bonds, lysine succinylation
Graphical Abstarct
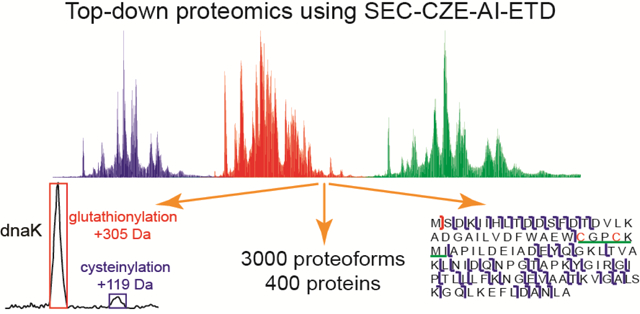
Introduction
Top-down proteomics aims to characterize proteoforms in their intact state and often in complex protein mixtures [1,2]. Most practitioners of the method employ reversed-phase liquid chromatography (RPLC) for proteoform separation followed by electrospray ionization-mass spectrometry and tandem mass spectrometry (ESI-MS and MS/MS) for determining masses of proteoforms and their corresponding fragment ions. The state-of-the-art RPLC-MS/MS-based systems have achieved identification, and even quantification, of thousands of proteoforms from complex samples [3–6]. Much effort has been made to improve the separation of proteoforms with RPLC. Monolithic columns and packed columns with beads having various sizes, different lengths of carbon chains and varied porosity have been investigated for proteoform separation [4, 7–9]. However, several challenges remain for top-down proteomics, including high-capacity separations of proteoforms and achieving informative MS/MS dissociation of proteoforms.
Capillary zone electrophoresis (CZE)-MS and MS/MS has been recognized as an alternative approach for top-down proteomics because of its high efficiency for proteoform separation and high sensitivity for proteoform detection [10–15]. Also, the recent improvements in CE-MS interface have facilitated the CZE-MS/MS for top-down proteomics [16–18]. The McLafferty group reported identifications (IDs) of intact proteins using CZE-MS with attomole amounts of materials in 1996 [10]. The Yates group demonstrated that CZE-MS achieved similar signal-to-noise ratios to RPLC-MS for analysis of a protein complex sample with 100-fold less sample consumption [12]. The Dovichi group has reported 600 proteoform IDs using RPLC-CZE-MS/MS from yeast cells [13]. And, for large protein characterization, the Kelleher group identified 30 proteins with masses in a range of 30–80 kDa from Pseudomonas aeruginosa PA01 cell lysate using CZE-MS/MS [14], demonstrating the potential of CZE-MS/MS for characterization of large proteins.
Challenges remain for large-scale top-down proteomics using CZE-MS/MS, including the narrow separation window (typically 30 min) and low sample loading capacity (low nL) of CZE. Recently, our group achieved a 90-min separation window and a 1-μL sample loading volume using CZE-MS for analysis of an Escherichia coli (E. coli) cell lysate, leading to IDs of 600 proteoforms in a single CZE-MS/MS run [19,20]. We employed a separation capillary with high-quality linear polyacrylamide (LPA) coating on its inner wall to eliminate electroosmotic flow in the capillary, widening the separation window [21]. We used a protein stacking method, dynamic pH junction [22,23], for highly efficient and online concentration of proteins in the capillary, boosting the sample loading volume. We coupled size exclusion chromatography (SEC)-RPLC fractionation to the dynamic pH junction-based CZE-MS/MS for deep top-down proteomics of E. coli cells [24]. Nearly 6,000 proteoforms and 850 proteins were identified using the multi-dimensional system. The dynamic pH junction-based CZE-MS/MS has established the foundation of top-down proteomics using CZE-MS/MS.
Extensive fragmentation of proteoforms in the gas phase is another challenge in top-down proteomics. Collision-based dissociation methods (e.g., higher energy-collisional dissociation, HCD) are widely used for fragmentation of proteoforms [3–8, 11, 13–15]. However, HCD often fails to provide extensive fragmentation of proteoforms, and has preferential cleavage sites [25,26], limiting its utility for thorough characterization of proteoforms. Alternative fragmentation methods are vital for top-down proteomics. Recently, an activated ion electron transfer dissociation (AI-ETD) method that combines infrared photoactivation concurrent with ETD has been developed and systematically evaluated for fragmentation of intact proteins [27–30]. RPLC-AI-ETD has been evaluated for high-throughput top-down characterization of intact proteins (less than 20 kDa) in human colorectal cancer cells with a production of 935 proteoforms and 295 proteins [30]. More importantly, AI-ETD showed better performance than HCD and standard ETD regarding sequence coverage of identified proteoforms and proteoform characterization scores. CZE has also been coupled with AI-ETD for top-down characterization of a standard protein mixture and a bacterial secretome sample [31]. About 40 proteoforms were identified using the CZE-AI-ETD from the secretome sample, and other results have demonstrated a good complementarity of HCD and AI-ETD for intact protein fragmentation.
In this work, for the first time, we coupled the dynamic pH junction-based CZE to the AI-ETD on an Orbitrap Fusion Lumos mass spectrometer for large-scale top-down proteomics. An E. coli cell lysate was employed to evaluate the performance of the system. First, we investigated how the laser power used for the AI-ETD influenced the proteoform IDs. Then we compared CZE-AI-ETD and CZE-ETD, as well as CZE-AI-ETD and CZE-HCD, for top-down proteomics of the E. coli cells. After that, we optimized the electric field and the data dependent acquisition method for the CZE-AI-ETD system. After evaluating the reproducibility of the CZE-AI-ETD system, we coupled SEC fractionation to CZE-AI-ETD for large-scale top-down proteomics of the E. coli cells.
Experimental Procedures
Details of “Materials and Reagents”, “Sample Preparation”, and “SEC Prefractionation” are described in Supporting Information I.
CZE-ESI-MS and MS/MS
An ECE-001 CE autosampler and a commercialized electro-kinetically pumped sheath flow CE-MS interface from CMP Scientific (Brooklyn, NY) were used in all experiments [17,18]. The automated CE system was coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Waltham, MA) via the electro-kinetically pumped sheath flow interface. A 1-meter-long fused silica capillary (50 μm i.d., 360 μm o.d.) with LPA coating on the inner wall was used for CZE separation. The LPA coating was made based on the procedure in references [20] and [21]. One end of the capillary was etched with hydrofluoric acid to reduce the outer diameter of the capillary to ~70–80 μm based on the procedure described in reference [20]. (Caution: use appropriate safety procedures while handling hydrofluoric acid solutions)
The sample was injected into the capillary via applying 5-psi pressure for 95 s corresponding to a 500-nL volume based on the Poiseuille’s law. The separation voltage applied at the injection end was either 10 kV for 240 min, 20 kV for 120 min, or 30 kV for 90 min. Between CZE-MS/MS runs, the separation capillary was flushed with a background electrolyte (BGE) using a 10-psi pressure for 10 min. For optimizing the laser power for AI-ETD and comparing the AI-ETD and ETD, 30 kV for 90 min was applied. For comparing the AI-ETD and HCD, optimizing the data-dependent acquisition method, and evaluating the reproducibility of the CZE-AI-ETD, 20 kV for 120 min was applied. For analyzing the SEC fractions of the E. coli proteome, 20 kV for 120 min was used for the first 9 fractions and 30 kV for 120 min for the last 5 fractions. The ESI voltage was 2–2.3 kV. The ESI spray emitter was pulled from a glass capillary (1.0-mm o.d., 0.75-mm i.d., 10 cm long) with a Sutter P-1000 flaming/brown micropipette puller. The size of the emitter orifice was 20–40 μm. The BGE for CZE was 5% (v/v) acetic acid (pH 2.4) and the sheath buffer for ESI consisted of 0.2% (v/v) formic acid and 10% (v/v) methanol.
An Orbitrap Fusion Lumos mass spectrometer was used for all experiments. For all experiments, data-dependent acquisition (DDA) was utilized with intact protein mode turned on, advanced peak determination set to True, and default charge state set to 10. The ion transfer tube temperature was set to 275 °C. Charge exclusion and exclude isotopes settings were turned on for proteins with charge state between 7–24 able to be fragmented. Include undetermined charges states was set to False and include charge states 25 and higher was set to True. Dynamic exclusion was used with a setting of 30 s. The same MS settings were used for all experiments. Use wide quad isolation was set to True, the orbitrap resolution was 120,000, scan range was 600–200 m/z, maximum injection time was 100 ms, AGC target was 500,000, the number of microscans was 4, and the RF lens (%) was 60.
For optimizing the laser power for AI-ETD (12, 18, 24, and 30 W) and comparing AI-ETD and ETD, a Top 2 DDA method was used. The option for performing dependent scan on single charge state per precursor was set to False. For MS/MS, the isolation window was set to 3, orbitrap resolution was 60,000, maximum injection time was 118 ms, AGC target was 500,000 and the number of microscans was 4. For AI-ETD and ETD, the ETD reaction time was set to 20 ms, ETD reagent target was 700,000, and maximum ETD reagent injection time was 200 ms. For optimizing the DDA methods (Top N) for AI-ETD (18 W laser power), Top 2, Top 4, and Top 5 DDA methods were investigated. The option to perform dependent scan on single charge state per precursor only was set to True. For optimizing the separation voltage, the AI-ETD method (18 W laser power) including a Top 2 DDA method was used. For analyzing the SEC fractions of the E. coli proteome, AI-ETD with 18 W laser power and a Top 4 DDA method were employed. The option to perform dependent scan on single charge state per precursor only was set to True. For comparing the AI-ETD (18 W) and HCD, Top 5 DDA methods were used for both AI-ETD and HCD. The details of MS/MS with AI-ETD were the same as that described above. For MS/MS with HCD, a normalized collision energy 20% was used for fragmentation. Other parameters were the same as that for AI-ETD.
Data Analysis
For the raw files from single-shot analyses of the whole E. coli cell lysate, we employed the Proteome Discoverer 2.2 (Thermo Fisher Scientific) with the ProSight PD Top Down High/High node for database search. Briefly, MS/MS spectra of proteoforms were deconvoluted with Xtract (signal to noise ratio threshold of three) and searched against the whole E. coli database downloaded from the http://proteinaceous.net/database-warehouse-legacy/. A three-tier search was used. Tier one consisted of an absolute mass search with 2.0 Da precursor mass tolerance and 10 ppm fragment ion mass tolerance. Tier two contained a biomarker search with 10 ppm precursor mass tolerance and 10 ppm fragment ion mass tolerance. Tier three had an absolute mass search with 1000 Da precursor mass tolerance and 10 ppm fragment ion mass tolerance. Only b and y types of fragment ions were considered for HCD fragmentation; only c and z types of fragment ions were considered for ETD and AI-ETD fragmentation. The target-decoy approach was used to evaluate the false discovery rates (FDRs) of proteoform spectrum match (PrSM) and proteoform IDs [32,33]. A 1% spectrum-level FDR was used to filter the PrSMs and a 5% proteoform-level FDR was used to filter the proteoform IDs.
For the raw files from the fractionated E. coli sample using SEC, we employed the TopFD (TOP-down mass spectrometry feature detection) and TopPIC (TOP-down mass spectrometry based proteoform identification and characterization) pipeline for database search [34]. The 14 raw files corresponding to the 14 SEC fractions were analyzed. First, the 14 raw files were converted into 14 mzML files with the Msconvert tool [35]. Then, the spectral deconvolution was performed with TopFD to generate msalign files for database search using TopPIC (version 1.2.2). The E. coli (strain K12) UniProt database (UP000000625, 4313 entries, version June 28, 2018) was used for database search. The database search parameters were as follows. The maximum number of unexpected modifications was 2. The precursor and fragment mass error tolerances were 15 ppm. The maximum mass shift of unknown modifications was 500 Da. The FDRs were estimated using the target-decoy approach [32,33]. To reduce the redundancy of proteoform IDs, we reviewed the proteoforms that were identified by multiple MS/MS spectra as one same proteoform ID if these MS/MS spectra corresponded to the same proteoform feature reported by the TopFD or those proteoforms were from one same protein and had smaller than 1.2-Da mass differences.
Two steps of analyses were performed. In the first step, we used TopPIC to search each raw file against the E. coli proteome database separately. In the second step, we combined all the PrSMs identified from the 14 data files, and filtered the PrSM IDs with a 1% spectrum-level FDR. The proteoform IDs were then filtered with a 5% proteoform-level FDR. The identified proteoforms are listed in Supporting Information II.
Results and discussion
Comparing CZE-AI-ETD, CZE-ETD, and CZE-HCD for top-down proteomics
We first optimized the laser power for AI-ETD before comparing it with ETD and HCD. The laser power can significantly affect the performance of AI-ETD based on a very recent RPLC-AI-ETD report [30]. Here, we evaluated the performance of CZE-AI-ETD with four different laser powers: 12 W, 18 W, 24 W, and 30 W, Figure S1. The CZE-MS system obtained a 1-hour separation window and reasonably good signal (NL: 5.8E8) with only 1 μg of E. coli proteins. CZE-AI-ETD with the 18-W laser power produced over 20% and 8% more PrSMs and proteins than other three laser powers. The 18-W laser power generated over 5% more proteoform IDs than 12-W and 24-W laser powers and yielded similar proteoform IDs to the 30-W laser power.
We then compared the performance of CZE-AI-ETD (18 W) and CZE-ETD for top-down characterization of the E. coli proteome. CZE-AI-ETD identified about 12% more proteoforms and proteins compared to CZE-ETD, Figure 1a. More importantly, CZE-AI-ETD tended to obtain better expectation values (E-values) of proteoform IDs than CZE-ETD, Figure 1b. E-value represents a nonlinear transformation of the number of matching fragment ions in a spectrum. The data suggest that AI-ETD can produce better fragmentation of proteoforms compared to ETD. As shown in Figure S2, AI-ETD (18 W) yielded much better residue cleavage and a much higher number of matching fragment ions than ETD (52% vs. 8%; 73 vs. 9 fragment ions) for thioredoxin 1. The disulfide bond was localized accurately based on the fragment ions from AI-ETD. The 18 W laser power was used in all following AI-ETD experiments.
Figure 1.

Summary of the comparisons between CZE-AI-ETD and CZE-ETD as well as CZE-AI-ETD and CZE-HCD. (a) Number of IDs from AI-ETD (18 W) and ETD. (b) Distribution of –log (E-value) of identified proteoforms using AI-ETD (18 W) and ETD. (c) Number of IDs from AI-ETD (18 W) and HCD. (d) Distribution of –log (E-value) of identified proteoforms using AI-ETD (18 W) and HCD.
We further compared the CZE-AI-ETD (18 W) with CZE-HCD. Single-shot CZE-HCD identified 994 PrSMs, 363 proteoforms and 195 proteins from the E. coli sample. CZE-HCD produced a moderate increase in PrSMs and slightly better proteoform and protein IDs than CZE-AI-ETD, Figure 1c. In the experiments, CZE-HCD generated 50% more MS/MS spectra than CZE-AI-ETD per 120 min analysis but resulted in only minor improvement in the number of proteoform and protein IDs. Interestingly, CZE-AI-ETD inclined to gain better E-values of proteoform IDs than HCD, Figure 1d. We need to note that different CZE separation conditions (30 kV for 90 min vs. 20 kV for 120 min) and MS/MS conditions (Top 2 vs. Top 5 DDA methods) were used for the experiments for Figure 1a and Figure 1c, leading to significant differences in the number of IDs from the CZE-AI-ETD.
Optimizing the CZE-AI-ETD method for top-down proteomics
We optimized the CZE separation voltage and the maximum number of MS/MS spectra followed by one MS spectrum in the DDA method (Top N). A high separation voltage shortens the analysis time but produces limited number of MS/MS spectra for proteoform IDs. A low separation voltage slows down the separation, allowing the acquisition of a large number of MS/MS spectra for proteoform IDs. However, the low separation voltage results in wider protein peaks and lower protein signal, which certainly affect the quality of MS/MS spectra. The Top N method in DDA influences the number of proteoform IDs because of the production of different numbers of MS/MS spectra.
When the separation voltage of CZE was changed from 30 kV to 10 kV, the analysis required much longer time, and the protein signal decreased significantly, Figure S3a. CZE with 20 kV separation voltage produced better separation efficiency than 30 kV and 10 kV, Figure S3b. The separation efficiency was up to half a million for one proteoform (m/z 775.05, charge +9). CZE-AI-ETD with 20 kV voltage generated more proteoform IDs than that with 10 kV and 30 kV voltages (292 vs. 278 or 255), Figure S3c. Interestingly, CZE-AI-ETD with 30 kV voltage identified 5% and 12% more proteins than that with 20 kV and 10 kV voltage, respectively. CZE-AI-ETD with 10 kV separation voltage gained the highest number of PrSMs, most likely due to the wider proteoform peaks generated using the lower voltage. The 20 kV separation voltage was employed in the following experiments.
We then optimized the DDA method by comparing the proteoform and protein IDs from Top 2, Top 4, and Top 5 methods. The Top 4 method identified 384 proteoforms and 191 proteins in a single CZE-AI-ETD run, and the number of proteoform IDs was 4% and 9% higher than that from the Top 2 and Top 5 methods. The Top 4 method identified 2% and 7% more proteins than the Top 2 and Top 5 methods. The Top 4 method was used in the following experiments.
We also evaluated the reproducibility of the optimized CZE-AI-ETD method for top-down characterization of the E. coli proteoform, Figure 2. The CZE-AI-ETD system produced reproducible separation profiles and base peak intensity across triplicate analyses, Figure 2a. The relative standard deviations (RSDs) of PrSM IDs, proteoform IDs, and protein IDs were 12%, 3% and 1%, respectively, Figure 2b. We further examined the protein-level and proteoform-level overlaps among the three CZE-AI-ETD runs, Figures 2c and 2d. The overlaps were about 58% (protein-level) and 37% (proteoform-level) among the three runs.
Figure 2.

Evaluation of reproducibility of the optimized CZE-AI-ETD system for top-down proteomics. (a) Base peak and total ion current (TIC) electropherograms of the E. coli sample analyzed by the optimized CZE-AI-ETD in triplicate. (b) Numbers of PrSMs, proteoforms, and proteins identified by the optimized CZE-AI-ETD. The error bars show the standard deviations of the IDs from the triplicate CZE-AI-ETD analyses. (c) The protein-level overlaps among the CZE-AI-ETD runs. (d) The proteoform-level overlaps among the CZE-AI-ETD runs.
SEC-CZE-AI-ETD for large-scale top-down characterization of the E. coli proteome
We fractionated the E. coli proteome into 14 fractions using SEC based on the size of proteoforms. Each SEC fraction was analyzed by the optimized CZE-AI-ETD in 120 min. Analyses of these 14 SEC fractions took 28 hours. As shown in Figure 3a, the SEC fraction 12 was analyzed by the CZE-AI-ETD system, and a 50-min separation window was obtained in the run. The base peak electropherograms of the 14 SEC fractions are shown in Figure S4 and Figure S5. The corresponding raw files have been deposited to the ProteomeXchange Consortium via the PRIDE [36] partner repository with the dataset identifier PXD012247.
Figure 3.

SEC-CZE-AI-ETD for large-scale top-down proteomics of the E. coli cells. (a) Base peak electropherogram of SEC fraction 12 of the E. coli proteome analyzed by the optimized CZE-AI-ETD. (b) Distributions of the PrSM, proteoform and protein IDs across the 14 SEC fractions. (c) Distribution of the number of proteoform IDs per gene. (d) Distribution of the mass of identified proteoforms. (e) Box chart of the number of matching fragment ions of identified proteoforms. (f) Correlation between the proteoform mass and the normalized number of matching fragment ions. The number of matching fragment ions was normalized to the proteoform length that is the number of amino acid residues in a proteoform sequence. (g) Summary of the detected post-translational modifications. (h) Summary of the detected N-terminal methionine (M) removal, potential signal peptide cleavage, and N-terminal truncations.
Nearly 12,000 PrSMs, 3,028 proteoforms, and 387 proteins were identified from the E. coli proteome using the SEC-CZE-AI-ETD system with 1% spectrum-level and 5% proteoform-level FDRs. The list of identified proteoforms is shown in Supporting information II. The data represents the largest top-down proteomics dataset using the AI-ETD method so far. The PrSM, proteoform and protein IDs were not uniformly distributed across the 14 SEC fractions, Figure 3b. The number of proteoform IDs per SEC fraction ranged from as few as 25 proteoforms (Fraction 2) to 957 proteoforms (Fraction 14). On average 216 proteoforms were identified per SEC fraction. Later SEC fractions tended to produce more PrSM, proteoform and protein IDs. We need to note that single-shot CZE-AI-ETD of the SEC fraction 14 identified 957 proteoforms and 253 proteins in 120 min, and the number of proteoform and protein IDs from the fraction accounted for about 32% and 65% of the total proteoform and protein IDs.
The 3,028 proteoforms corresponded to 387 E. coli genes, an average of about 8 proteoforms per gene. The genes were classified into three categories based on the number of identified proteoforms: one proteoform per gene for 191 genes, 2–10 proteoforms per gene for 127 genes, and 10–144 proteoforms per gene for 69 genes, Figure 3c. We identified 144, 130 and 111 proteoforms for genes rbsB, rplL, and mglB, respectively. The mass of identified proteoforms ranged from 1 kDa-35 kDa, and most of the proteoforms (89%) were smaller than 20 kDa, Figure 3d. 325 proteoforms from 51 proteins and 30 proteoforms from 6 proteins were larger than 20 kDa and 30 kDa, respectively. The proteoforms larger than 30 kDa were identified with at least 7 fragment ions, and the average number of matching fragment ions was 18.
The number of matching fragment ions of identified proteoforms ranged from 6 to nearly 100, Figure 3e. The mean was 23, and the median was 17. Roughly, 25% of the proteoforms were identified with fewer than 10 fragment ions. The proteoform mass influenced the number of matching fragment ions, Figure 3f. The number of fragment ions of each proteoform was normalized to the length of each corresponding proteoform, and the normalized number of fragment ions was used to evaluate the performance of AI-ETD for generation of sequence-informative fragment ions. When the proteoform mass increased, the performance of AI-ETD tended to decrease, Figure 3f. However, the normalized number of fragment ions varied obviously for proteoforms with similar masses, suggesting that the performance of AI-ETD for proteoform fragmentation was also influenced by other proteoform features.
Post-translational modifications with SEC-CZE-AI-ETD
We detected several kinds of post-translational modifications (PTMs) from the E. coli proteome, including protein N-terminal acetylation, methylation, S-thiolation, disulfide bonds, and lysine succinylation, Figure 3g. Only a few proteins in the E. coli sample had these PTMs. We detected 28 proteins with N-terminal acetylation, 56 proteins with methylation, 25 proteins with S-thiolation, 15 proteins with disulfide bonds (S-S), and 7 proteins with lysine succinylation. We identified 712 proteoforms from 113 proteins with N-terminal methionine removal, 800 proteoforms from 137 proteins with potential signal peptide cleavage, and 1,041 proteoforms from 206 proteins with N-terminal truncations, Figure 3h.
The N-terminal acetylation was determined by the TopPIC software with a 42-Da mass shift at the N-terminus of one proteoform. The methylation was determined with a 14±1 Da or 28±1 Da mass shift. The S-thiolation was determined with a 305±2 Da mass shift for glutathionylation and a 119±2 Da mass shift for cysteinylation. We also manually checked that there was one cysteine residue in the sequence corresponding to the mass shift. For the S-S, if they are reported in the literature, we confirmed the detection through a – (2±1) Da mass shift and two cysteine residues for one S-S, and through a – (4±1) Da mass shift and four cysteine residues for two S-S. If the S-S were not reported before, we required more accurate masses of the mass shifts (−2 Da for one S-S and −4 Da for two S-S). The lysine succinylation was determined with a 100±2 Da mass shift for one succinylation site, a 200±2 Da mass shift for two succinylation sites, and a 300±2 Da mass shift for three succinylation sites. If the first 7–50 amino acids of a proteoform were cleaved from its N-terminus, we considered the proteoform had a potential signal peptide cleavage based on information from the “Center for Biological Sequence Analysis” (http://www.cbs.dtu.dk/services/SignalP-1.1/sp_lengths.html). If more than 50 amino acids were cleaved from the N-terminus of one proteoform, we reviewed the proteoform as truncated.
S-thiolation is a kind of PTM in which free thiol groups on proteins react with low mass thiols (e.g., glutathione and cysteine) to form disulfides. S-glutathionylation and S-cysteinylation are two kinds of S-thiolation. Protein S-thiolation can occur in response to oxidative stress and protect cysteine from irreversible oxidation, and it can happen under physical conditions to influence protein function [37–39]. Recently, Ansong et al. reported that Gram-negative bacteria cultured in Lysogeny broth (LB) medium preferred to use S-glutathionylation as a way for thiol protection [6]. We cultured the E. coli cells in LB medium for the experiment. We detected 25 proteins with S-glutathionylation PTM and only four proteins with S-cysteinylation PTM. Interestingly, the four cysteinylated proteins had both cysteinylated and glutathionylated proteoforms. Information of these proteins is listed in Supporting Information II. We compared the relative abundance of cysteinylated and glutathionylated proteoforms of two proteins, Figures 4a and 4b. The glutathionylated proteoform showed much higher intensity than the cysteinylated proteoform, suggesting that the E. coli cells cultured in LB medium preferentially used S-glutathionylation as a mechanism for thiol protection. Figure S6 shows the sequences and fragmentation patterns of the cysteinylated and glutathionylated proteoforms of the two proteins.
Figure 4.

Examples of the S-thiolation, disulfide bond, and lysine succinylation PTMs. (a) Extracted ion electropherogram of the dnaK proteoforms with S-glutathionylation and S-cysteinylation. m/z 1022.66 (charge +8) and m/z 944.49 (charge +8) were extracted with a 20-ppm mass tolerance for the S-glutathionylation and S-cysteinylation proteoforms. (b) Extracted ion electropherogram of the ridA proteoforms with S-glutathionylation and S-cysteinylation. m/z 920.02 (charge +15) and m/z 907.55 (charge +15) were extracted with a 20-ppm mass tolerance for the S-glutathionylation and S-cysteinylation proteoforms. (c) The sequence and fragmentation pattern of thioredoxin 1. The sequence underlined with a green line had a −3 Da mass shift corresponding to a disulfide bond between the two cysteine residues. (d) The sequence and fragmentation pattern of DksA. The sequence underlined with a green line had a −4 Da mass shift corresponding to two disulfide bonds between the four cysteine residues. (e) The sequence and fragmentation pattern of DNA-binding protein HU-alpha. The sequence underlined with a green line had a 300 Da mass shift corresponding to succinylations on the three lysine residues. (f) The sequence and fragmentation pattern of DNA-binding protein HU-beta. The sequence underlined with a green line had a 99 Da mass shift corresponding to succinylation on one of the three lysine residues.
We identified 15 proteins with S-S, including 9 proteins with one and 5 proteins with two. Interestingly, we also identified one protein, RNA polymerase-binding transcription factor DksA, which had one proteoform with one S-S and another proteoform with two S-S. Five out of the 15 proteins have been reported as S-S containing proteins in the literature. Information of the 15 proteins is listed in Supporting Information II. Figures 4c and 4d show two examples of these proteins. These two proteins, Thioredoxin 1 and DksA, were well fragmented with AI-ETD. The disulfide bonds were well localized based on the matching fragment ions. Thioredoxin 1 has one S-S between the two cysteine residues highlighted in red in Figure 4c [40]. Figure 4d and Figure S7 show the sequences and fragmentation patterns of DksA proteoforms with two and one S-S between the cysteine residues highlighted in red. DksA does not have S-S based on the literature, and instead, it binds one zinc ion through the four cysteine residues highlighted in Figure 4d [41]. The detected S-S on protein DksA might be endogenous or might form after cell lysis because the E. coli cells were lysed under a denaturing condition and the DksA-zinc complex was most likely destroyed during the process.
We also identified seven proteins with the lysine succinylation PTM, and these seven proteins were reported as succinylated proteins in the literature [42]. Three proteins had one modification site (100 Da mass shift), two proteins had three modification sites (300 Da mass shift), and one protein had two modification sites (200 Da mass shift). Interestingly, the lysine residues on ribose import binding protein RbsB were not succinylated consistently across different proteoforms. Two RbsB proteoforms had two succinylation sites, but the sites were different between the proteoforms. We also identified one RbsB proteoform with only one succinylation site. The information of proteins with lysine succinylation is shown in Supporting Information II. As shown in Figure 4e, the three modification sites on DNA-binding protein HU-alpha were localized based on the fragment ions generated by AI-ETD. Figure 4f shows the sequence and fragmentation pattern of another succinylated protein, DNA-binding protein HU-beta, indicating one succinylation site on one of the three lysine residues highlighted in red.
Conclusions
We demonstrated the first application of CZE-AI-ETD for large-scale top-down proteomics. CZE-AI-ETD outperformed CZE-ETD and CZE-HCD considering the number of proteoform and protein IDs as well as the number of sequence-informative fragment ions generated. Coupling SEC fractionation to CZE-AI-ETD enabled IDs of 3,028 proteoforms and 387 proteins from the E. coli proteome, which represents the largest top-down proteomics dataset using the AI-ETD method so far. The SEC-CZE-AI-ETD system detected various PTMs, including protein N-terminal acetylation, methylation, S-thiolation, disulfide bonds, and lysine succinylation.
Supplementary Material
Acknowledgments
We thank Prof. Heedeok Hong’s group at Michigan State University (Department of Chemistry) for kindly providing the E. coli cells for this project. We thank the support from the National Institute of General Medical Sciences, National Institutes of Health (NIH), through Grant R01GM118470 (X. Liu), R01GM125991 (L. Sun and X. Liu), P41GM108538 (J. Coon), R35GM118110 (J. Lodge and J. Coon).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
The authors declare no competing financial interest.
References
- [1].Toby TK, Fornelli L, Kelleher NL: Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem 9, 499–519 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Smith LM, Kelleher NL: Proteoforms as the next proteomics currency. Science. 359, 1106–1107 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL: Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 480, 254–258 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Durbin KR, Fornelli L, Fellers RT, Doubleday PF, Narita M, Kelleher NL: Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. J. Proteome Res 15, 976–82 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cai W, Tucholski T, Chen B, Alpert AJ, McIlwain S, Kohmoto T, Jin S, Ge Y: Top-Down Proteomics of Large Proteins up to 223 kDa Enabled by Serial Size Exclusion Chromatography Strategy. Anal. Chem 89, 5467–5475 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, Nakayasu ES, Cort JR, Pevzner P, Smith RD, Heffron F, Adkins JN, Pasa-Tolic L: Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc. Natl. Acad. Sci. U S A 110, 10153–8 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shen Y, Tolić N, Piehowski PD, Shukla AK, Kim S, Zhao R, Qu Y, Robinson E, Smith RD, Paša-Tolić L: High-resolution ultrahigh-pressure long column reversed-phase liquid chromatography for top-down proteomics. J. Chromatogr. A 1498, 99–110 (2017) [DOI] [PubMed] [Google Scholar]
- [8].Roth MJ, Plymire DA, Chang AN, Kim J, Maresh EM, Larson SE, Patrie SM: Sensitive and reproducible intact mass analysis of complex protein mixtures with superficially porous capillary reversed-phase liquid chromatography mass spectrometry. Anal. Chem 83, 9586–92 (2011) [DOI] [PubMed] [Google Scholar]
- [9].Zhou Y, Zhang X, Fornelli L, Compton PD, Kelleher N, Wirth MJ: Chromatographic efficiency and selectivity in top-down proteomics of histones. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci 1044–1045, 47–53 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Valaskovic GA, Kelleher NL, McLafferty FW: Attomole Protein Characterization by Capillary Electrophoresis-Mass Spectrometry. Science. 273, 1199–1202 (1996) [DOI] [PubMed] [Google Scholar]
- [11].Han X, Wang Y, Aslanian A, Bern M, Lavallée-Adam M, Yates JR 3rd: Sheathless capillary electrophoresis-tandem mass spectrometry for top-down characterization of Pyrococcus furiosus proteins on a proteome scale. Anal. Chem 86, 11006–12 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Han X, Wang Y, Aslanian A, Fonslow B, Graczyk B, Davis TN, Yates JR 3rd: In-line separation by capillary electrophoresis prior to analysis by top-down mass spectrometry enables sensitive characterization of protein complexes. J. Proteome Res 13, 6078–86 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhao Y, Sun L, Zhu G, Dovichi NJ: Coupling Capillary Zone Electrophoresis to a Q Exactive HF Mass Spectrometer for Top-down Proteomics: 580 Proteoform Identifications from Yeast. J. Proteome Res 15, 3679–3685 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li Y, Compton PD, Tran JC, Ntai I, Kelleher NL: Optimizing capillary electrophoresis for top-down proteomics of 30–80 kDa proteins. Proteomics. 14, 1158–64 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun L, Knierman MD, Zhu G, Dovichi NJ: Fast top-down intact protein characterization with capillary zone electrophoresis-electrospray ionization tandem mass spectrometry. Anal. Chem 85, 5989–95 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moini M: Simplifying CE-MS operation. 2. Interfacing low-flow separation techniques to mass spectrometry using a porous tip. Anal. Chem 79, 4241–6 (2007) [DOI] [PubMed] [Google Scholar]
- [17].Wojcik R, Dada OO, Sadilek M, Dovichi NJ: Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis. Rapid Commun. Mass Spectrom 24, 2554–60 (2010) [DOI] [PubMed] [Google Scholar]
- [18].Sun L, Zhu G, Zhang Z, Mou S, Dovichi NJ: Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. J. Proteome Res 14, 2312–21 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lubeckyj RA, McCool EN, Shen X, Kou Q, Liu X, Sun L: Single-Shot Top-Down Proteomics with Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry for Identification of Nearly 600 Escherichia coli Proteoforms. Anal. Chem 89, 12059–12067 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].McCool EN, Lubeckyj R, Shen X, Kou Q, Liu X, Sun L: Large-scale Top-down Proteomics Using Capillary Zone Electrophoresis Tandem Mass Spectrometry. J. Vis. Exp 140, e58644 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhu G, Sun L, Dovichi NJ: Thermally-initiated free radical polymerization for reproducible production of stable linear polyacrylamide coated capillaries, and their application to proteomic analysis using capillary zone electrophoresis-mass spectrometry. Talanta. 146, 839–43 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aebersold R, Morrison HD: Analysis of dilute peptide samples by capillary zone electrophoresis. J. Chromatogr 516, 79–88 (1990) [DOI] [PubMed] [Google Scholar]
- [23].Britz-McKibbin P, Chen DD: Selective focusing of catecholamines and weakly acidic compounds by capillary electrophoresis using a dynamic pH junction. Anal. Chem 72, 1242–52 (2000) [DOI] [PubMed] [Google Scholar]
- [24].McCool EN, Lubeckyj RA, Shen X, Chen D, Kou Q, Liu X, Sun L: Deep Top-Down Proteomics Using Capillary Zone Electrophoresis-Tandem Mass Spectrometry: Identification of 5700 Proteoforms from the Escherichia coli Proteome. Anal. Chem 90, 5529–5533 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Haverland NA, Skinner OS, Fellers RT, Tariq AA, Early BP, LeDuc RD, Fornelli L, Compton PD, Kelleher NL: Defining Gas-Phase Fragmentation Propensities of Intact Proteins During Native Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom 28, 1203–1215 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Huang Y, Triscari JM, Tseng GC, Pasa-Tolic L, Lipton MS, Smith RD, Wysocki VH: Statistical characterization of the charge state and residue dependence of low-energy CID peptide dissociation patterns. Anal. Chem 77, 5800–13 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rush MJP, Riley NM, Westphall MS, Coon JJ: Top-Down Characterization of Proteins with Intact Disulfide Bonds Using Activated-Ion Electron Transfer Dissociation. Anal. Chem 90, 8946–8953 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Riley NM, Westphall MS, Coon JJ: Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. J. Proteome Res 16, 2653–2659 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Riley NM, Westphall MS, Coon JJ: Sequencing Larger Intact Proteins (30–70 kDa) with Activated Ion Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom 29, 140–149 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Riley NM, Sikora JW, Seckler HS, Greer JB, Fellers RT, LeDuc RD, Westphall MS, Thomas PM, Kelleher NL, Coon JJ: The Value of Activated Ion Electron Transfer Dissociation for High-Throughput Top-Down Characterization of Intact Proteins. Anal. Chem 90, 8553–8560 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhao Y, Riley NM, Sun L, Hebert AS, Yan X, Westphall MS, Rush MJ, Zhu G, Champion MM, Mba Medie F, Champion PA, Coon JJ, Dovichi NJ: Coupling capillary zone electrophoresis with electron transfer dissociation and activated ion electron transfer dissociation for top-down proteomics. Anal. Chem 87, 5422–9 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Keller A, Nesvizhskii AI, Kolker E, Aebersold R: Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 74, 5383–92 (2002) [DOI] [PubMed] [Google Scholar]
- [33].Elias JE, Gygi SP: Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–14 (2007) [DOI] [PubMed] [Google Scholar]
- [34].Kou Q, Xun L, Liu X: TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics. 32, 3495–3497 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kessner D, Chambers M, Burke R, Agus D, Mallick P: ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 24, 2534–6 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H: 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–56 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A: Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem. Sci 34, 85–96 (2009) [DOI] [PubMed] [Google Scholar]
- [38].Hochgräfe F, Mostertz J, Pöther DC, Becher D, Helmann JD, Hecker M: S-cysteinylation is a general mechanism for thiol protection of Bacillus subtilis proteins after oxidative stress. J. Biol. Chem 282, 25981–5 (2007) [DOI] [PubMed] [Google Scholar]
- [39].Chu F, Ward NE, O’Brian CA: PKC isozyme S-cysteinylation by cystine stimulates the pro-apoptotic isozyme PKC delta and inactivates the oncogenic isozyme PKC epsilon. Carcinogenesis. 24, 317–25 (2003) [DOI] [PubMed] [Google Scholar]
- [40].Schultz LW, Chivers PT, Raines RT: The CXXC motif: crystal structure of an active-site variant of Escherichia coli thioredoxin. Acta Crystallogr. D Biol. Crystallogr 55, 1533–8 (1999) [DOI] [PubMed] [Google Scholar]
- [41].Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG: Regulation through the secondary channel--structural framework for ppGpp-DksA synergism during transcription. Cell. 118, 297–309 (2004) [DOI] [PubMed] [Google Scholar]
- [42].Weinert BT, Schölz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C: Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 4, 842–51 (2013) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.