

Summary
Despite compelling rates of durable clinical responses to PD-1 blockade, advances are needed to extend these benefits to resistant tumors. We found that tumor-bearing mice deficient in the chemokine receptor CXCR3 responded poorly to anti-PD-1 treatment. CXCR3 and its ligand CXCL9 were critical for a productive CD8+ T cell response in tumor-bearing mice treated with anti-PD-1, but were not required for the infiltration of CD8+ T cells into tumors. The anti-PD-1- induced anti-tumor response was facilitated by CXCL9 production from intratumoral CD103+ dendritic cells, suggesting that CXCR3 facilitates dendritic cell-T cell interactions within the tumor microenvironment. CXCR3 ligands within murine tumors and in plasma of melanoma patients were an indicator of clinical response to anti-PD-1, and their induction in non- responsive murine tumors promoted responsiveness to anti-PD-1. Our data suggest that the CXCR3 chemokine system is a biomarker for sensitivity to PD-1 blockade and that augmenting the intratumoral function of this chemokine system could improve clinical outcomes.
eTOC Blurb
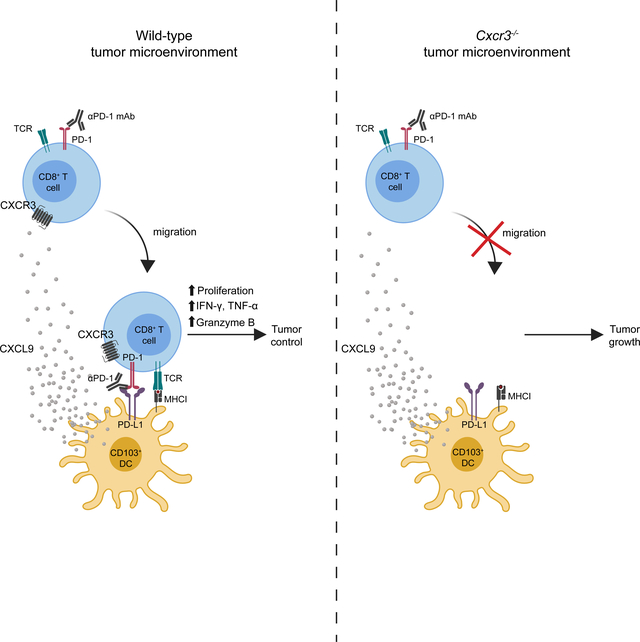
Chow et al. find the CXCR3 chemokine system is not required for CD8+ T cell migration into the tumor, but rather for the enhancement of the intratumoral CD8+ T cell response in the context of PD-1 blockade. The CXCR3 chemokine system may serve as a biomarker for sensitivity to PD- 1 blockade and a target for improving clinical outcomes.
Introduction
CD8+ T cells play a vital role in tumor eradication through the production of cytotoxic molecules, such as perforin and granzyme, and cytokines, such as interferon (IFN)-γ and tumor necrosis factor (TNF)-α (Martínez-Lostao et al., 2015). Indeed, the presence of high densities of CD8+ T cells within tumor tissue is a favorable prognostic indicator in many cancers (Fridman et al., 2012). However, it is well established that the microenvironment of tumors is frequently immunosuppressive, rendering CD8+ T cells dysfunctional and promoting tumor progression (Speiser et al., 2016). In particular, immune checkpoints, such as the programmed cell death (PD)1/PD-L1 pathway, have been exploited by tumors as a critical immunosuppressive mechanism to evade T cell immunity (Hashimoto et al., 2018). In the tumor microenvironment, PD-L1 is upregulated on antigen-presenting cells and/or tumors cells, and its binding to PD-1 on CD8+ T cells dampens their cytokine production, proliferation and migration (Sharpe and Pauken, 2018). PD-1/PD-L1 pathway inhibition can result in robust and durable anti-tumor responses in cancer patients and in preclinical tumor models (Hashimoto et al., 2018). However, only a proportion of patients respond to PD-1 immune checkpoint blockade, emphasizing the need for a better understanding of the underlying mechanisms of PD-1 inhibitor-mediated enhancement of the anti-tumor CD8+ T cell response.
The infiltration of CD8+ T cells and their localization within tumors are critical for PD-1 blockade therapy (Ribas and Wolchok, 2018). Correlative human studies have highlighted the potential importance of chemokines for T cell infiltration into tumors and for patient survival (Bindea et al., 2013; Messina et al., 2012). CXCR3, a chemokine receptor for the interferon- inducible chemokines CXCL9, CXCL10, and CXCL11, is highly expressed on activated T cells and plays essential roles in the spatial distribution, migratory behavior, and function of T cells (Groom and Luster, 2011a; Groom and Luster, 2011b). CXCR3 and its ligands guide the recruitment of effector T cells into the inflamed peripheral tissue in type 1 inflammatory responses (Dufour et al., 2002; Hancock et al., 2001; Hancock et al., 2000; Harris et al., 2012; Khan et al., 2000; Rashighi et al., 2014). The CXCR3 chemokine system also plays important roles in the positioning of T cells within secondary lymphoid organs and peripheral tissue, facilitating the interactions of T cells with antigen-loaded activated dendritic cells (DCs), promoting T cell activation and differentiation, as well as assisting the process of locating and killing virally infected cells (Groom et al., 2012; Hickman et al., 2015; Kastenmuller et al., 2013; Rashighi et al., 2014; Sung et al., 2012). Engineering tumor cells to express CXCL10, a CXCR3 ligand, can induce an anti-tumor immune response (Luster and Leder, 1993), and CXCR3 expression on CD8+ T cells is critical for their entry into tumors in an adoptive cell transfer model (Mikucki et al., 2015). The CXCR3 chemokine system is also relevant in the therapeutic efficacy of chemotherapy (Sistigu et al., 2014). We therefore set out to determine whether the CXCR3 chemokine system participates in anti-tumor immunity induced by PD-1 blockade.
We found that the CXCR3 chemokine system was required for the efficacy of anti-PD-1 therapy in mouse tumor models. CXCR3 was not required for CD8+ T cell migration into the tumor, but rather was required for the enhancement of the intratumoral CD8+ T cell response in the context of PD-1 blockade. Furthermore, experiments with melanoma patient samples suggest that CXCR3 ligands may serve as early biomarkers of response to checkpoint blockade therapy.
Results
CXCR3 is necessary for an effective response to PD-1 blockade therapy
To understand the role of the CXCR3 chemokine system in the efficacy of anti-PD-1 immunotherapy, we used an anti-PD-1-responsive transplantable tumor model - the MC38 tumor cell line (Woo et al., 2012). We inoculated wild-type (WT) and Cxcr3−/− mice subcutaneously with MC38 cells and eight days later, after the tumor was established, treated them with either anti-PD-1 antibody or an isotype-matched control antibody (Figure 1A). As shown in Figure 1B & 1C, tumor growth and mouse survival were comparable between WT and Cxcr3−/− mice treated with the isotype-matched control antibody. By contrast, treatment with the anti-PD-1 antibody markedly reduced tumor growth and led to improved survival in WT mice. Although Cxcr3−/− mice also displayed reduced tumor growth and improved survival in response to anti-PD-1 antibody treatment, these responses were significantly diminished compared with those in WT mice. A similar result was observed in a second tumor model, the D4M.3A.3 UV3 melanoma model (Figure S1A). This result demonstrates a role for CXCR3 in mediating the efficacy of anti-PD-1 antibody treatment.
Figure 1. CXCR3 is necessary for response to anti-PD-1 therapy.

(A) Schematic of the anti- PD-1 treatment schedule. Mice were inoculated subcutaneously with 1×106 MC38 tumor cells, and on days 8, 11 and 14 after tumor inoculation, mice were intraperitoneally treated with 200μg of either isotype control or anti-PD-1 antibodies. Tumor growth and survival were monitored until the experimental endpoints. (B) Tumor growth in WT and Cxcr3−/− mice treated with isotype or anti-PD-1 antibodies (n=5–10 mice per group). Data are presented as the mean ± SEM; ***p<0.001, with statistical significance determined by two-way ANOVA. (C) Survival curves of the percentage of mice whose tumor sizes were <100mm2 at each time point. Statistical differences in survival were assessed by a Mantel-Cox log-rank test (**p<0.01). Representative data are shown from two independent experiments. (D-E) Quantification of CD8+ T cell frequencies (D) and cytokine-producing CD8+ T cells (E) within tumors of WT and Cxcr3−/− mice on day 8 after MC38 tumor inoculation. (F-H) Tumors were excised from mice on day 15 of the schedule presented in Figure 1A, followed by quantification of CD8+ T cell frequencies (F), IFN- γ+ TNF-ɑ+ CD8+ T cells (G) and Ki-67+ granzyme B+ CD8+ T cells (H) within tumors of WT and Cxcr3−/− mice treated with isotype or anti-PD-1 antibodies. (G-H) Contour plots and bar graphs reflect the proportion of IFN-γ+ TNF-ɑ+ (G) or Ki-67+ granzyme B+ (H) cells out of total CD8+ T cells. In (D-H) bar graphs represent the mean value of the indicated data points; error bars represent SEM; *p<0.05, **p<0.01, with statistical significance determined by one-way ANOVA with Tukey’s multiple comparison test. Representative data are shown from two independent experiments. See also Figure S1, S2 and S3.
A functional CD8+ T cell response following PD-1 blockade is dependent on CXCR3
Given the reported role of CXCR3 in T cell recruitment into tumors and the importance of pre- existing intratumoral CD8+ T cells for effective PD-1 blockade therapy (Tumeh et al., 2014), we hypothesized that the reduced therapeutic effect of anti-PD-1 in Cxcr3−/− mice might relate to a decrease in the early infiltration of CD8+ T cells into the tumor. However, an analysis of tumors before beginning anti-PD-1 treatment revealed that the percentage and absolute number of CD8+ T cells within tumors were comparable between WT and Cxcr3−/− mice (Figure 1D & Figure S1B). Intratumoral CD8+ T cells from untreated WT and Cxcr3−/− mice displayed a similar capacity to produce cytokines (Figure 1E) with similar expression of PD-1, LAG-3, CD137, TCF-1, CD28, T-bet. CD39, TIGIT, Tim-3, CD44 and ICOS (Figure S2). We further investigated whether the T cell response against Adpgk, a well-described neoantigen expressed in MC38 tumors (Yadav et al., 2014), was affected in Cxcr3−/− mice. There were no differences in percentage and phenotype of intratumoral tumor antigen-specific CD8+ T cells in WT and Cxcr3−/− mice (Figure S3A & S3B). These data suggest that CXCR3 is not required for the early recruitment and functioning of CD8+ T cells in tumors, at least in this experimental model.
Consistent with these results, the absolute number of intratumoral CD8+ T cells was similar between tumors of WT and Cxcr3−/− mice treated with isotype-matched control antibody (Figure 1F). However, anti-PD-1 treatment significantly increased the number of CD8+ T cells in the tumors of WT mice, whereas the number of intratumoral CD8+ T cells did not change in anti-PD-1-treated Cxcr3−/− mice (Figure 1F). Furthermore, ex vivo activation and intracellular cytokine staining showed an increase in CD8+ T cells expressing IFN-γ and TNF-α in WT mice, but not in Cxcr3−/− mice (Figure 1G). Next, we further evaluated the CD8+ T cell response by examining the expression of the proliferation marker Ki-67 and the cytotoxic protein granzyme B, which have been described as markers of a functional anti-tumor CD8+ T cell response following PD-1 blockade (Twyman-Saint Victor et al., 2015). Anti-PD-1 treatment markedly enhanced the percentage of Ki-67+ granzyme B+ CD8+ T cells in WT mice (Figure 1H). By contrast, there was no significant change in the proportion of Ki-67+ granzyme B+ CD8+ T cells in anti-PD-1-treated Cxcr3−/− mice compared with the isotype control group (Figure 1H).
Anti-PD-1 therapy promotes an intratumoral response
Although the CXCR3 chemokine system did not play a role in the early infiltration of CD8+ T cells into tumors in our model, the increased number and enhanced function of CD8+ T cells in the tumors of WT but not CXCR3−/− mice (Figure 1F–H) upon anti-PD-1 treatment suggested that CXCR3 may instead be involved in T cell recruitment during the course of anti-PD-1 treatment. To address this, first we determined whether anti-PD-1 enhanced the recruitment of CD8+ T cells into tumors from lymphoid organs. To this end, we used FTY720 (a functional antagonist of the S1P1 receptor) to block the egress of T cells from the lymphoid organs (Matloubian et al., 2004), one day before the initiation of anti-PD-1 therapy (Figure 2A). Although FTY720 treatment dramatically reduced T cell numbers in peripheral blood as expected (Figure 2B), anti-PD-1 treatment still resulted in increased numbers of CD8+ T cells within tumors (Figure 2C), indicating an expansion of the CD8+ T cell population that had infiltrated during early tumor development before the initiation of anti-PD-1 treatment. Accordingly, FTY720 administration had no impact on tumor growth suppression in WT mice treated with the anti-PD-1 antibody (Figure 2D), whereas administration of FTY720 before tumor inoculation inhibited anti-PD-1-mediated tumor reduction (Figure 2E). These results indicate that anti-PD-1-induced tumor control was predominantly mediated by T cells that were present within the tumor before initiation of anti-PD-1 treatment. To determine if T cell accumulation within tumors was the result of increased local T cell proliferation, we measured 5-ethynyl-2′-deoxyuridine (EdU) incorporation in CD8+ T cells recovered for the tumor and draining lymph node of MC38 tumor- bearing mice before and after anti-PD-1 treatment. Anti-PD-1 treatment induced the proliferative response of T cells at the tumor site as the percentage of EdU+ CD8+ T cell population was increased within MC38 tumors in WT mice (Figure 2F). By contrast, there was no increased T cell proliferation induced by PD-1 blockade in draining lymph node (Figure S4). In addition, we found that this proliferative response within tumors was abrogated in Cxcr3−/− mice, suggesting that the CXCR3 chemokine system is critical in the expansion of CD8+ T cells following PD-1 blockade in the tumor microenvironment.
Figure 2. Pre-existing intratumoral CD8+ T cell population is sufficient for anti-PD-1- induced tumor control.

(A) Schematic of the anti-PD-1 and FTY720 treatment schedule. Groups of WT mice were inoculated subcutaneously with 1×106 MC38 on day 0. Following tumor inoculation, tumor-bearing mice were intraperitoneally treated with either isotype or anti-PD-1 antibodies (200μg) on days 8, 11 and 14. Administration of DMSO or FTY720 (1mg/kg) was performed at the indicated time points to block the egress of lymphocytes during the course of anti-PD-1 therapy. (B) Representative FACS plots of leukocytes recovered in peripheral blood from mice on day 8 of the schedule presented in Figure 2A (1 day after the 1st dose of FTY720 but before antibody administration). Cells were stained for CD3 and CD45 and analyzed on the gated lymphocyte population. (C) Absolute CD8+ T cell number within tumors of WT mice treated with DMSO control or FTY720 in addition to the anti-PD-1 therapy. Tumors were excised on day 15 of the schedule presented in Figure 2A and FACS analysis of CD8+ T cell number was performed. Data represent the mean ± SEM; *p<0.05, with statistical significance determined by one-way ANOVA with Tukey’s multiple comparison test. Representative data are shown from two independent experiments. (D) Tumor growth and survival of WT mice treated with isotype-matched control or anti-PD-1 antibodies in combination with DMSO or FTY720 as indicated in Figure 2A (n=5–8 mice per group). (E) Groups of WT mice were treated with DMSO or FTY720 (1mg/kg) before tumor inoculation as indicated (n=5 mice per group). 1×106 MC38 was given subcutaneously on day 0, and tumor-bearing mice were intraperitoneally treated with either isotype or anti-PD-1 antibodies (200μg) on days 8, 11 and 14. Tumor growth and survival were monitored until the experimental endpoints. Data represent the mean ± SEM; *p<0.05, ***p<0.001, with statistical significance determined by two- way ANOVA. Survival curves show the percentage of mice whose tumor sizes were <100mm2 at each time point. Statistical differences in survival were assessed by a Mantel-Cox log-rank test (*p<0.05, **p<0.01, ***p<0.001). Data are representative of two independent experiments. (F) Groups of WT and Cxcr3−/− were inoculated subcutaneously with 1×106 MC38 on day 0. Following tumor inoculation, tumor-bearing mice were intraperitoneally treated with FTY720 (1mg/kg) on days 7 and 9 as well as either isotype or anti-PD-1 antibodies (200μg) on day 8. Administration of EdU (50mg/kg) was performed on day 10, followed by quantification of EdU+ CD8+ T cells within tumors on day 11. Contour plots and bar graphs reflect the proportion of EdU+ cells out of total CD8+ T cells in MC38 tumors. Bar graph represents the mean value of the indicated data points; error bars represent SEM; *p<0.05, ***p<0.001, with statistical significance determined by one-way ANOVA with Tukey’s multiple comparison test. Representative data are shown from two independent experiments. See also Figure S4.
CXCR3 on CD8+ T cells is essential for anti-PD-1-mediated CD8+ T cell activation
CXCR3 is expressed not only on T cells but also on other hematopoietic cells, such as innate lymphocytes and plasmacytoid DCs (Griffith et al., 2014). Therefore, we sought to determine the contribution of CXCR3 expressed on CD8+ T cells for anti-PD-1-mediated activation of CD8+ T cells. To this end, irradiated C57BL/6 mice (CD45.1+) were reconstituted with a 1:1 mixture of bone marrow from WT (CD45.1+) and Cxcr3−/− (CD45.2+) mice, followed by tumor induction, FTY720 administration and anti-PD-1 treatment (Figure 3A). The frequency of intratumoral Cxcr3−/− CD8+ T cells was slightly reduced in comparison to WT CD8+ T cells in isotype control- treated animals, suggesting a minimal impairment in the ability of Cxcr3−/− CD8+ T cells to infiltrate tumors in the competitive setting. However, in mice that received anti-PD1 treatment, the frequency of intratumoral Cxcr3−/− CD8+ T cells was reduced to much lower levels compared to WT CD8+ T cells, suggesting preferential expansion of WT CD8+ T cells within the tumor (Figure 3B). We further analyzed the response against the tumor neoantigen Adpgk expressed in MC38 tumors. In isotype control-treated mice, the percent of neoantigen-specific WT and Cxcr3−/− CD8+ T cells were comparable, suggesting similar numbers of WT and Cxcr3−/− neoantigen-specific CD8+ T cells were present in tumors in mice prior to anti-PD-1 treatment. In contrast, the frequency of tumor neoantigen-specific Cxcr3−/− CD8+ T cells was strikingly reduced compared to neoantigen-specific WT CD8+ T cells in the tumor after anti-PD-1 treatment, demonstrating the preferential expansion of WT over Cxcr3−/− neoantigen-specific CD8+ T following anti-PD-1 treatment. (Figure 3C). In addition, anti-PD-1 treatment increased the effector function of WT CD8+ T cells as measured by IFN-γ and TNF-α production but not that of Cxcr3−/− CD8+ T cells within the same tumor (Figure 3D). Taken together, these data demonstrate that CD8+ T cells require intrinsic CXCR3 signaling within the tumor microenvironment for anti-tumor responses during anti-PD-1 treatment.
Figure 3. CXCR3 expressed on CD8+ T-cells is required for anti-PD-1-mediated CD8+ T cell activation.

(A-D) Bone marrow cells from CD45.1+ WT mice and CD45.2+ Cxcr3−/− mice were mixed in a 50:50 ratio and injected into lethally irradiated WT recipient mice. Eight weeks after transplant, 1×106 MC38 cells were injected subcutaneously into chimeric mice. Following tumor inoculation, administration of DMSO control or FTY720 (1mg/kg) started one day before isotype or anti-PD-1 treatment (200μg; day 8, 11 and 14), and continued every second day. Flow plot at the right panel reflects the proportion of CD45.1+ WT and CD45.2+ Cxcr3−/− donor cells in a recipient mouse after reconstitution. Tumors were excised on day 15 and FACS analyses performed to determine the percentages of total WT and Cxcr3−/− CD8+ T cells (B), antigen- specific CD8+ T cells (C) and cytokine-producing CD8+ T cells (D) within tumors of mixed bone marrow chimeric mice (n=5 mice per group). Data are presented as the mean ± SEM; *p<0.05, **p<0.01, ***p<0.001 with statistical significance determined by Student’s t-test. Data are representative of two independent experiments. (E-I) 1×106 MC38 cells were injected subcutaneously into CXCR3-GFP reporter (CIBER) mice and tumors were harvested on day 8 for analysis. (E) Identification of four subsets of dysfunctional CD8+ T cells based on CXCR3 and PD-1 expression. (F) Surface expression of Tim-3, LAG-3, TIGIT, CD44, CD28 and CD137 on different subsets of CD8+ T cells. (G-H) Contour plots and bar graphs reflect the proportion of IFN-γ+ TNF-ɑ+ (G) or granzyme B+ (H) cells within different subsets of CD8+ T cells. Bar graphs represent the mean value of the indicated data points; error bars represent SEM; *p<0.05, ***p<0.001, with statistical significance determined by one-way ANOVA with Tukey’s multiple comparison test. Representative data are shown from two independent experiments. (I) Frequency of CD8+ T cell subsets within tumors at different times following inoculation.
We next sought to characterize the CXCR3-expressing CD8+ T cell populations within MC38 tumors using CXCR3-GFP reporter (CIBER) mice. Based on the expression of CXCR3 and levels of PD-1 expression, we defined the following four subsets of intratumoral CD8+ T cells: CXCR3+ PD-1lo; CXCR3+ PD-1int; CXCR3+ PD-1hi; and CXCR3− PD-1hi (Figure 3E). CXCR3− PD-1hi CD8+ T cells expressed higher levels of inhibitory molecules, including Tim-3, LAG-3 and TIGIT, than the other subsets (Figure 3F), indicating a higher level of dysfunction and T cell exhaustion. In addition, CXCR3+ PD-1int and CXCR3+ PD-1hi CD8+ T cells expressed higher levels of co-stimulatory molecules CD44, CD28 and CD137, than the other subsets. These results are consistent with our observation that CXCR3+ PD-1int and CXCR3+ PD-1hi CD8+ T cells from tumors produced IFN-γ and TNF-α at higher levels than CXCR3− PD-1hi CD8+ T cells (Figure 3G), indicating an important role for CXCR3 in regulating CD8+ T cell function. Previous studies demonstrated that terminally exhausted CD8+ T cells upregulate their expression of granzyme B (Im et al., 2016; Utzschneider et al., 2016). Consistent with this notion, CXCR3− PD-1hi CD8+ T cells expressed the highest level of granzyme B (Figure 3H), suggesting that they could be terminally exhausted. We also found that the percentage of CXCR3-expressing CD8+ T cells declined over time, with lower percentages seen at later stages of tumor growth (>Day 25) (Figure 3I). These results suggest that CXCR3 expression on CD8+ T cells is inversely correlated with tumor growth.
CD103+ DC-derived CXCL9 is required for the efficacy of PD-1 blockade therapy
To further characterize how the CXCR3 chemokine system mediates intratumoral responses to PD-1 blockade, we examined the expression of the CXCR3 chemokine ligands CXCL9 and CXCL10 (CXCL11 is a null mutant in C57BL/6 mice (Sierro et al., 2007) and was therefore not studied). RNA and protein levels of CXCL9 and CXCL10 were markedly increased in tumors after anti-PD-1 treatment (Figure 4A–B). Furthermore, administration of neutralizing monoclonal antibodies against CXCL9 and CXCL10 significantly negated the effectiveness of anti-PD-1 treatment (Figure 4C). Consistent with our earlier findings (Figure 2D), the extent of tumor reduction was the same in the presence or absence of FTY720, substantiating the contribution of the CXCR3 chemokine system to anti-PD-1-mediated activation of pre-existing tumor- associated T cells.
Figure 4. Host-derived CXCL9 is required for response to anti-PD-1.

(A) Quantitative RT- PCR analysis of Cxcl9 and Cxcl10 mRNA expression in MC38 tumors from WT mice 24 hours after treatment with the first dose of isotype or anti-PD-1 antibodies. Data are presented as cDNA copies of indicated gene per copy of GAPDH. Data are mean ± SEM; *p<0.05, with statistical significance determined by Student’s t-test. Data are representative of two independent experiments. (B) Quantitative protein analysis of CXCL9 and CXCL10 in MC38 tumors from WT mice 48 hours after treatment with the first dose of isotype or anti-PD-1 antibodies. Data are mean ± SEM; *p<0.05, with statistical significance determined by Student’s t-test. Data are representative of two independent experiments. (C) Tumor growth and survival of WT mice treated with isotype or anti-PD-1 antibodies in combination with FTY720 or DMSO control and/or antibodies against CXCL9 and CXCL10 as indicated (n=5–10 mice per group). Administration of DMSO control or FTY720 (1mg/kg) started one day before isotype or anti-PD-1 treatment (200μg; days 8, 11 and 14), and continued every second day until day 19. To neutralize CXCR3 ligands, anti-CXCL9 and anti-CXCL10 antibodies were injected intraperitoneally into mice on days 7, 11, 15 and 19 (initial dose: 200μg of each antibody; maintenance dose: 100μg of each antibody). Data represent the mean ± SEM; *p<0.05, with statistical significance determined by two-way ANOVA. Survival curves show the percentage of mice whose tumor sizes were <100mm2 at each time point. Statistical differences in survival were assessed by a Mantel-Cox log-rank test (*p<0.05). Data are representative of two independent experiments. (D) Tumor growth in WT, Cxcl9−/− and Cxcl10−/− mice treated with isotype control or anti-PD-1 antibodies (n=5–10 mice per group) according to the schedule presented in Figure 1A. Data represent the mean ± SEM; *p<0.05, **p<0.01, ***p<0.001, with statistical significance determined by two-way ANOVA. Survival curves show the percentage of mice whose tumor sizes were <100mm2 at each time point. Statistical differences in survival were assessed by a Mantel-Cox log-rank test (**p<0.01). Representative data are shown from two independent experiments. (E) Tumors were excised from mice on day 15 of the schedule presented in Figure 1A, followed by quantification of IFN-γ+ TNF-ɑ+ CD8+ T cells (left panel) and Ki-67+ granzyme B+ CD8+ T cells (right panel) within tumors of WT and Cxcl9−/− mice treated with isotype or anti-PD-1 antibodies. Percentages reflect the proportion of IFN-γ+ TNF-ɑ+ or Ki-67+ granzyme B+ out of total CD8+ T cells. Bar graphs represent the mean values of the indicated data points, and the error bars represent SEM; *p<0.05, **p<0.01, with statistical significance determined by one-way ANOVA with Tukey’s multiple comparison test. Data are representative of two independent experiments. See also Figure S5.
To determine the respective roles of host-derived CXCL9 and CXCL10 in anti-PD-1 induced tumor rejection, mice deficient in Cxcl9 or Cxcl10 were inoculated with MC38 cells and then treated with anti-PD-1 or control antibody. WT, Cxcl9−/− and Cxcl10−/− mice treated with isotype control antibody displayed comparable tumor growth (Figure 4D). Administration of anti-PD-1 antibody suppressed tumor growth in WT and Cxcl10−/− mice and enhanced their survival (Figure 4D). However, the therapeutic benefits of PD-1 blockade was partially lost in Cxcl9−/− mice, indicating a critical role for host-derived CXCL9 in anti-PD-1 immunotherapy (Figure 4D). Moreover, the percentages of IFN-γ+ TNF-α+ CD8+ T cells and Ki-67+ granzyme B+ CD8+ T cells within tumors were increased upon anti-PD-1 treatment in WT, but not in Cxcl9−/− mice (Figure 4E), indicating that the CD8+ T cell anti-tumor response induced by PD-1 blockade was CXCL9-dependent.
DCs play a critical role in orchestrating T cell responses and can generally be divided into two functionally specific subsets: CD103+ DCs are efficient at cross-presentation, which is important in the generation of CD8+ T cell responses, while CD11b+ DCs are superior in promoting CD4+ T helper (Th) cell responses (Durai and Murphy, 2016). Furthermore, CD103+ DCs have been shown to be important in anti-PD-1 treatment, as tumors in mice lacking CD103+ DCs respond poorly to anti-PD-1 treatment (Salmon et al., 2016; Sánchez-Paulete et al., 2016). Therefore, we investigated whether secretion of CXCL9 by CD103+ DCs plays an important role in mediating tumor responses to anti-PD-1 treatment. First, we analyzed the expression profile of the CXCR3 ligands in CD103+ DCs and CD11b+ DCs using our CXCL9- CXCL10 dual reporter mice (REX3) mice. We discovered that myeloid cells were the major intratumoral immune cells that produce CXCL9 and CXCL10 (Figure S5A & S5B). Notably, CD103+ DCs exhibited a distinct chemokine expression profile from CD11b+ DCs as CD103+ DCs produced almost exclusively CXCL9 while CD11b+ DCs were characterized by the production of high level of CXCL10 (Figure 5A). Given that host-derived CXCL9 is important in the efficacy of PD-1 blockade, we next examined the expression of CXCL9 in the CD103+ DC and CD11b+ DC after anti-PD-1 treatment. To this end, REX3 mice were implanted with the MC38 tumor cell line followed by anti-PD-1 administration. We found that CXCL9 production was increased after anti-PD-1 treatment in CD103+ DCs but not CD11b+ DCs (Figure 5B & 5C).
Figure 5. CD103+ DC-derived CXCL9 is important for the efficacy of PD-1 blockade.

(A) Representative flow cytometry gating strategy for determination of CXCL9 and CXCL10 expression in tumor-infiltrating DC subpopulations (CD103+ DC and CD11b+ DC) within MC38 tumors harvested from the CXCL9-CXCL10 dual reporter (REX3) transgenic mice. (B-C) MC38 tumors were excised from REX3 transgenic mice on day 15 following treatment with isotype or anti-PD-1 antibodies according to the schedule presented in Figure 1A and were analyzed for CXCL9-RFP expression in CD103+ DC and CD11b+ DC (gated on CD45+ CD3− CD19− CD11c+ MHCII+ as demonstrated in Figure 5A). Bar graph represent the mean values of the indicated data points, and the error bars represent SEM; ***p<0.001, with statistical significance determined by one-way ANOVA with Tukey’s multiple comparison test. Representative data are shown from two independent experiments. (D-E) Bone marrow cells from CD45.1+ CD45.2+ Zbtb46DTR mice were mixed in a 50:50 ratio with bone marrow cells from CD45.2+ WT, Cxcl9−/− or Cxcl10−/− mice and injected into lethally irradiated CD45.1+ WT recipient mice. Eight weeks after transplant, 1×106 MC38 cells were injected subcutaneously into chimeric mice. Following tumor inoculation, administration of FTY720 (1mg/kg) and DT (20μg/kg) started one day before isotype or anti-PD-1 treatment (200μg; on day 8, 11 and 14), and continued until day 19 as indicated. Tumor growth and survival were monitored until the experimental endpoints (n=5–10 mice per group). Data are given as the mean ± SEM; ***p<0.001, with statistical significance determined by two-way ANOVA. Survival curves show the percentage of mice whose tumor sizes were <100mm2 at each time point. Statistical differences in survival were assessed by a Mantel-Cox log-rank test (**p<0.01). Representative data are shown from two independent experiments. See also Figure S5 and S6.
To determine the functional importance of CD103+ DC-derived CXCL9, we generated mixed bone marrow chimeric mice by reconstituting irradiated WT C57BL/6 mice (CD45.1+) with a mixture of bone marrow from Zbtb46-diphtheria toxin receptor (DTR) (CD45.1+CD45.2+), and either WT control (CD45.2+), Cxcl9−/− (CD45.2+) or Cxcl10−/− (CD45.2+) mice. Following reconstitution, MC38 tumors were inoculated into these mixed BM chimeric mice and then 8 days later treated with FTY720, diphtheria toxin (DT) and anti-PD-1 (Figure 5D). Importantly, CD103+ DCs were preferentially depleted in the tumor upon DT administration (Figure S6). Anti-PD-1 treatment significantly suppressed tumor growth and enhanced survival in mixed BM chimeric mice that had WT DCs or CD103+ Cxcl10−/− DCs in the tumor (Figure 5E). In contrast, mixed BM chimeric mice that had CD103+ Cxcl9−/− DCs exhibited poorer response to anti-PD-1 treatment. Taken together, these data reveal the functional importance of CD103+ DC-derived CXCL9 for response to PD-1 blockade therapy.
Combination of epigenetic modulators and anti-PD-1 convert non-responders to responders
Given the important role of the CXCR3 chemokine system in PD-1 immune checkpoint blockade therapy, we next evaluated whether CXCL9 and CXCL10 could serve as biomarkers for response to anti-PD-1 immunotherapy. We used REX3 transgenic mice to examine the levels of CXCL9 and CXCL10 expression in DCs within multiple distinct solid tumors showing different responsiveness to anti-PD-1. The anti-PD-1-responsive tumors, including the MC38 colon adenocarcinoma, the MCA1956 fibrosarcoma, the D4M.3A.3 UV2 melanoma and the D4M.3A.3 UV3 melanoma, exhibited markedly higher expression of CXCL9 and CXCL10 in CD11c+ MHCII+ DCs compared with the anti-PD-1 resistant tumors, including the B16F10 melanoma, the D4M.3A.3 BRAF-mutant melanoma and the AT-3 breast cancer (Figure 6A). These results suggest that the baseline expression of CXCR3 ligands in tumors may predict responsiveness to anti-PD-1 therapy.
Figure 6. The combination of epigenetic modulators and anti-PD-1 increases CXCR3 ligand expression and converts non-responders to responders.

(A) Expression of CXCL9 and CXCL10 in gated CD11c+ MHCII+ cells within the indicated solid tumors of REX3 transgenic mice. Bar graph represents the mean values of the indicated data points, and the error bars represent SEM. (B) Schematic of experimental design for the treatment schedule for the epigenetic modulators and anti-PD-1. Orthotopic injection of AT-3 tumor cells (1×106) into the mammary fat pad of mice was performed on day 0. Following tumor inoculation, tumor-bearing mice were treated with four different treatment schemes on day 8, 11 and 14: (1) control; (2) DZNeP (5mg/kg) + 5-AZA-Dc (0.2mg/kg); (3) anti-PD-1 (200μg); and (4) DZNeP (5mg/kg) + 5-AZA-Dc (0.2mg/kg) + anti-PD-1 (200μg). (C) Tumors were excised from REX3 transgenic mice on day 15 of the schedule presented in Figure 6B, followed by analysis of CXCL9 and CXCL10 expression in CD11c+ MHCII+ cells from AT-3 tumors of REX3 transgenic mice treated with control, epigenetic modulators, anti-PD-1 or a combination of epigenetic modulators and anti-PD-1 antibody. Bar graph represent the mean values of the indicated data points, and the error bars represent SEM; ***p<0.001, with statistical significance determined by Student’s t-test. Representative data are shown from two independent experiments. (D) AT-3 tumor growth in WT mice that were treated with control, epigenetic modulators, anti-PD-1 or a combination of epigenetic modulators and anti-PD-1 (n=6 mice per group). Data represent the mean ± SEM; ***p<0.001, with statistical significance determined by two-way ANOVA. Survival curves show the percentage of mice whose tumor sizes were <100mm2 at each time point. Statistical differences in survival were assessed by a Mantel-Cox log-rank test (**p<0.01). Data are representative of two independent experiments. (E) 1×106 AT3 tumor cells were injected into the mammary fat pad of chimeric mice, which were generated using the protocol presented in Figure 5D. Following tumor inoculation, administration of DT (20μg/kg) started one day before control or a combination of epigenetic modulators and anti-PD-1 treatment, and continued until day 19. Tumor growth was monitored until the experimental endpoints (n=5–8 mice per group). Data are given as the mean ± SEM; ***p<0.001, with statistical significance determined by two- way ANOVA.
Previous studies have shown that administration of epigenetic modulators to tumor- bearing mice can modulate the expression of CXCR3 ligands in tumors (Peng et al., 2015). We decided to investigate whether we could render AT-3 tumors, which are resistant to anti-PD-1 treatment, responsive to anti-PD-1 treatment by upregulating CXCR3 ligand expression using the epigenetic modulators DZNeP and 5-AZA-dC (Figure 6B). Treatment with the epigenetic modulators alone had minimal effects on the production of CXCL9 and CXCL10 by DCs (Figure 6C). However, treatment with the modulators in combination with anti-PD-1 antibody dramatically increased CXCL9 and CXCL10 expression in CD11c+ MHCII+ DCs. We also observed that treatment with epigenetic modulators only or with anti-PD-1 only had minimal effects on tumor growth, but, importantly, the combination of both agents delayed tumor growth and enhanced mouse survival (Figure 6D). To test whether the therapeutic effect of the combination treatment relies on the DC-derived CXCL9, we performed the treatment in the WT:Zbtb46DTR and Cxcl9−/−:Zbtb46DTR mixed bone marrow chimeric mice. Tumors from Cxcl9−/− :Zbtb46DTR mixed bone marrow chimeric mice were unresponsive to the combination therapy (Figure 6E), underscoring the importance of CXCL9 production by DCs for the efficacy of the combination treatment. These data suggest that PD-1 blockade plus epigenetic modulation of CXCR3 ligand expression could promote responsiveness in tumors that are resistant to anti-PD- 1 therapy.
CXCL9 and CXCL10 expression correlates with clinical response to immunotherapy
The results from our mouse studies prompted us to investigate whether CXCL9 and CXCL10 levels might also correlate with the response to immune checkpoint blockade in human cancer patients. We analyzed chemokine expression in plasma samples from melanoma patients treated with PD-1 blockade therapy alone or in combination with anti-CTLA-4 therapies (Table S1). We observed that the levels of these CXCR3 ligands significantly correlated with treatment outcome. Compared with non-responders, the response group had elevated plasma levels of CXCL9 and CXCL10 within the first few months after immune checkpoint blockade therapy (Figure 7A), suggesting that these CXCR3 ligands could be early biomarkers for response to the immunotherapy. Additionally, consistent with the findings from our mouse tumor model (Figure 3E & 3F), CXCR3− PD-1hi CD8+ T cells isolated from human melanomas exhibited relatively high amounts of Tim-3 and low amounts of CD28 (Figure 7B & S7).
Figure 7. CXCL9 and CXCL10 expression correlates with response to PD-1 blockade therapy.

(A) Levels of CXCL9 and CXCL10 in plasma samples from non-responders (NR) (red) (n=10) and responders (R) (blue) (n=18) before (Pre) and after treatment (Post) with anti-PD-1 (closed symbols) or anti-PD-1 plus anti-CTLA-4 (open symbols). Statistical differences were determined by Wilcoxon matched-pairs signed rank test. (B) Identification of four subsets of dysfunctional CD8+ T cells within tumors based on the cell surface expression of CXCR3 and PD-1 in melanoma patients (top left panel). Surface expression of Tim-3 and CD28 on different subsets of CD8+ T cells (top right panel). Bar graph shows the normalized MFI of Tim-3 and CD28 expression on CD8+ T cell subsets (bottom panel). MFI of Tim-3 and CD28 expression on each subset was normalized to the MFI of Tim-3 or CD28, respectively, on the CXCR3+ PD-1low CD8 T cell population in each individual patient. See also Figure S7 and Table S1.
Discussion
Immunotherapy using PD-1 blockade has emerged as a promising approach for the treatment of a broad range of cancers; however, only a limited proportion of patients treated with immune checkpoint blockade therapy exhibit a durable anti-tumor response. The extent of enhancement of the CD8+ T cell response by PD-1 blockade is one of the critical determinants of the efficacy of anti-PD-1 therapy (Huang et al., 2017). In this study, we provide evidence for the functional relevance of the CXCR3 chemokine system for the anti-tumor CD8+ T cell response in response to PD-1 blockade. In particular, we found that the CXCR3 chemokine system enhanced the proliferation and function of intratumoral CD8+ T cells during anti-PD-1 treatment, and that CD103+ DCs-derived CXCL9 played a pivotal role in this process.
The CXCR3 chemokine system plays an important role in CD8+ T cell recruitment into tumors in adoptive transfer models (Mikucki et al., 2015; Spranger et al., 2017). By contrast, in our study, which involved the endogenous generation and trafficking of tumor-specific T cells, we found that the CXCR3 chemokine system was dispensable for early T cell trafficking into tumors, while it was essential for the anti-PD-1-mediated response of the CD8+ T cell population residing within tumors. The different requirement for the CXCR3 chemokine system for T cell entry into tumors may reflect the differences in the models. In adoptive T cell transfer models, fully differentiated and activated CD8+ T cells are transferred into mice that contain established tumors. In contrast, in our model, endogenously generated tumor-specific CD8+ T cells have the opportunity to traffic into nascent tumors over time as the T cell response is developing and the tumors are forming. Thus, the entry of adoptively transferred fully differentiated and activated CD8+ T cells is dependent on the CXCR3 chemokine system to enter established tumors while the early infiltration of endogenously generated tumor-specific T cells into developing tumors is less dependent on the CXCR3 chemokine system and likely depends on different chemokine systems. In fact, other chemokine systems, such as CCR2, CCR5, CX3CR1, are associated with T cell migration into tumors (Ahrends et al., 2017; Berghuis et al., 2011; Chew et al., 2012; Fridman et al., 2012; González-Martín et al., 2011; Harlin et al., 2009; Liu et al., 2015; Mlecnik et al., 2010).
The importance of recruiting T cells from outside of the tumor for effective PD-1 blockade treatment has been intensively debated. There is rapid proliferation of CD8+ T cells within tumors after PD-1 blockade (Spranger et al., 2014; Wei et al., 2017), suggesting that the pre-existing tumor-infiltrating CD8+ T cell population is sufficient for eliciting a therapeutic response. However, there are some contrasting results regarding the effects of FTY720 administration during therapy on the response to immune checkpoint blockade. It has been suggested that the efficacy of immune checkpoint blockade relies on T cells already present in tumors, as FTY720 administration before treatment did not affect the efficacy of immunotherapy (Garris et al., 2018; Siddiqui et al., 2019; Spranger et al., 2014; Wang et al., 2016). In contrast, another study found that a systemic immune response is required for effective checkpoint blockade therapy (Spitzer et al., 2017). These conflicting results may be due to FTY720 being administered at different time points, and the varying tumor burdens and different degrees of tumor-infiltrating T cells. Nevertheless, these studies show that a significant number of pre- existing T cells within tumors is a fundamental prerequisite for an efficient response to immune checkpoint blockade. Notably, in our studies, we found that induction of the anti-tumor CD8+ T cell response by PD-1 blockade occurred specifically in the tumor microenvironment and was largely dependent on the CXCR3 chemokine system. FTY720 administration did not alter the response to PD-1 blockade, suggesting that reactivation of CD8+ T cells in the reactive lymph node or their recruitment from the draining lymph node is not critical for the response to immunotherapy in our model.
We found that expression of CXCL9 was increased in CD103+ DCs following PD-1 blockade. In addition, CD103+ DC-derived CXCL9 was pivotal for the response to PD-1 blockade. CD103+ DCs excel in the priming and cross-presentation of cell-associated antigens to CD8+ T cells and are critical for the efficacy of anti-PD-1 cancer therapy (Salmon et al., 2016; Sánchez-Paulete et al., 2016). The CXCL9-CXCR3 axis is important in guiding DC-T cell positioning and interactions within lymph nodes for optimal recall response of memory CD8+ T cell in viral infections (Kastenmuller et al., 2013; Sung et al., 2012). Furthermore, CXCL9 expression is increased in CD103+ DC in the context of anti-Tim-3 cancer immunotherapy (de Mingo Pulido et al., 2018). Therefore, the increased expression of CXCL9 in CD103+ DCs induced by PD-1 blockade may enable CD103+ DCs to specifically capture CXCR3-expressing CD8+ T cells, thereby leading to more effective T cell response. In contrast to the importance of host-derived CXCL9 in PD-1 blockade, we found that host-derived CXCL10, another CXCR3 ligand, was not essential for anti-PD-1 therapy-induced tumor control. In our tumor model, CXCL10 was preferentially produced by CD11b+ DCs, which are known to be more important in the CD4+ Th cell response. In fact, CD11b+ DC-derived CXCL10 is essential for the optimal generation of CD4+ Th1 response (Groom et al., 2012). In addition, tumor cells can increase expression of CXCL10 in response to chemotherapy, which is important for anti-tumor T cell responses (Sistigu et al., 2014). Therefore, the role of tumor cell-derived CXCL10 in the efficacy of PD-1 blockade therapy needs to be further elucidated. Our results demonstrated that even though the therapeutic effect of anti-PD-1 treatment was largely dependent on the CXCR3 chemokine system, there was a residual anti-tumor effect of anti-PD-1 in Cxcr3−/− and Cxcl9−/− mice. PD-L1 on tumor cells can directly inhibit the cytotoxic function of CD8+ T cells, and blockade of the PD-1-PD-L1 interaction enhances the direct CD8+ T cell killing of tumor cells (Juneja et al., 2017). Although CD8+ T cells are considered the primary target of anti-PD-1, anti-PD-1 can directly affect macrophages as well (Gordon et al., 2017). Blockade of the PD-1 pathway on macrophages increases their phagocytic activity, thereby reducing tumor growth. Thus, our observation of a residual anti-tumor effect of anti-PD-1 in Cxcr3−/− and Cxcl9−/− mice could be explained by the fact that anti-PD-1 therapy can unleash the killing activity of CD8+ T cells and increase macrophage phagocytosis in the tumor microenvironment.
We extended our observation describing the essential role of the CXCR3 chemokine system for the efficacy of PD-1 blockade in murine models to the clinic by demonstrating that an increase in CXCL9 and CXCL10 plasma levels early after immune checkpoint blockade treatment of human melanoma patients was a positive indicator of a therapeutic response. In addition, we found that baseline levels of CXCL9 and CXCL10 within murine tumors also predicted their response to PD-1 blockade. Based on the preclinical and clinical data reported here, repression of any of the components of the CXCR3 chemokine system may compromise the therapeutic efficacy of PD-1 blockade. In fact, we demonstrated that de-repressing the Cxcl9 and Cxcl10 genes with epigenetic modulators converted anti-PD-1 non-responsive tumors to responsive tumors, and that this correlated with the induction of these chemokines in intratumoral DCs. These preclinical data suggest that tumor-specific induction of these potent chemokines in non-responsive tumors may be a viable therapeutic strategy to extend the benefits of anti-PD-1 therapy. In fact, there are a number of ongoing clinical trials that are testing the efficacy of combining epigenetic modulators with anti-PD-1. These and other studies will be needed to determine the optimal modifier to de-repress the CXCR3 chemokine system in the tumor and its ultimate clinical efficacy when combined with checkpoint blockade. Nonetheless, taken together, our data support the hypothesis that augmenting the intratumoral function of the CXCR3 chemokine system is a viable approach to enhance the efficacy of PD-1 blockade.
STAR Methods
Human studies
IRB approval was obtained prior to study enrollment and written informed consent was obtained from all donors. Patients with metastatic melanoma provided written informed consent for the collection of tissue and blood samples for research and genomic profiling, as approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board (DF/HCC Protocol 11–181). Matched blood samples were obtained from patients at baseline and after treatment with either anti-PD-1 (Pembrolizumab or Nivolumab) or anti-PD-1 + anti-CTLA-4 (Ipilimumab) (see Table S1). Of the cohort, 27 of the 28 patients received anti-PD-1 or anti-PD-1 + anti-CTLA-4 for metastatic stage IV melanoma aside from 1 patient who was given adjuvant anti-PD1 after definitive surgical resection. Blood was collected prior to initiation of therapy (between 9 days prior to therapy and the day of therapy initiation) and post-treatment blood collection occurred between 19 and 93 days (median = 42) after initiation of therapy. Blood was processed at the time of collection and plasma was stored for subsequent cytokine analysis. All patients classified as responders (R) showed clear radiographic decrease in disease and maintained an ongoing response without progression through to last follow-up. Patients classified as non- responders (NR) did not respond to treatment radiographically and/or had clear and rapid progression. In the case of the one patient who received adjuvant therapy, NR was defined as post-treatment recurrence. Progression free survival is given in days from treatment start to radiographic scan when progression was first noted. Human CXCL9 and CXCL10 ELISA kits (R&D Systems) were used to analyze patient plasma samples according to the manufacturer’s instructions.
Fresh tumor specimens were obtained from patients who underwent surgical resection as standard of care. Tumors were harvested in cold PBS, minced into small pieces and digested for 1 hour at 37°C with Collagenase IV (1mg/ml) (Wo rthington) and DNase I (0.02mg/mL) (Sigma-Aldrich). Single cell suspensions were obtained, and standard procedure for flow cytometry analysis was performed (see below).
Mice
C57BL/6 WT, CXCR3-GFP (CIBER) and Zbtb46DTR mice were obtained from Jackson Laboratory. All mice, including REX3-Tg (Groom et al., 2012), Cxcr3−/− (Hancock et al., 2000), Cxcl9−/− (Park et al., 2002), and Cxcl10−/− (Dufour et al., 2002) were in the C57BL/6 background and were housed under specific pathogen-free conditions. All procedures were approved by the Massachusetts General Hospital Subcommittee on Research and Animal Care.
Cell lines
B16F10 melanoma cell line were sourced from American Type Culture Collection. MC38 colon adenocarcinoma, MCA1956 fibrosarcoma, and AT-3 mammary adenocarcinoma cell lines were kindly provided by Dr. Mark Smyth (QIMR Berghofer Medical Research Institute, Herston, Australia). D4M.3A.3 melanoma, D4M.3A.3 UV2 melanoma, and D4M.3A.3 UV3 melanoma cell lines were kindly provided by Dr. David Fisher (Massachusetts General Hospital, Boston, USA). D4M.3A.3 was derived from single cell cloning of D4M.3A, a BRAF(V600E)/Pten−/− melanoma cell line kindly provided by Dr. David Mullins (Dartmouth). To generate the D4M.3A.3 UV2 and UV3 cell lines, which harbor clonal UVB signature mutations, D4M.3A.3 cells were sequentially irradiated in vitro with 25mJ/cm2 UVB 3 times before isolating and culturing single cell clones from the surviving population (manuscript in submission). All cell lines were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% heat-inactivated fetal calf serum (FCS), 2mmol/L glutamax, 100U/mL penicillin, 100μg/mL streptomycin.
Tumor model and in vivo treatments
1×106 MC38 cells were injected subcutaneously into the right flank of mice. Tumor size was measured every 2–3 days with an electronic caliper and reported as tumor size using the formula tumor length x tumor width. For PD-1 immune checkpoint blockade therapy, on day 8, 11 and 14 post tumor inoculation, tumor-bearing mice were treated intraperitoneally with 200 g of isotype control (2A3; BioXcell) or antagonistic anti-PD-1 antibodies (29F.1A12; kindly provided by Dr. Gordon Freeman). For CXCL9 and CXCL10 neutralization, mice were treated intraperitoneally with 200 g of anti-CXCL9 (2A6.9.9) (Christen et al., 2003) and anti-CXCL10 (1F11) (Khan et al., 2000) neutralizing antibodies on day 7 and then treated with 100ug of the antibodies on day 11, 15 and 19. To block T cell egress during the course of anti-PD-1 treatment, 1mg/kg FTY720 (Cayman Chemicals) was given intraperitoneally 1 day prior to the initiation of the anti-PD-1 therapy regimens and continuously every second day until day 19 post tumor inoculation. For the experiment of inhibiting early T cell migration into tumor, 1mg/kg FTY720 was given intraperitoneally one day earlier before tumor inoculation and continuous every second day until day 5. To deplete DC in chimeric mice that were reconstituted with Zbtb46DTR bone marrow, 20ng/g of DT (Sigma-Aldrich) was injected intraperitoneally into mice every third day beginning on day 7.
Cell preparations
After euthanizing mice, primary tumors were removed, minced finely and incubated in an enzyme solution containing 1mg/mL collagenase type IV (Worthington) and 0.02mg/mL DNase I (Sigma-Aldrich) for 45 minutes at 37°C. Digested tu mor was passed through a 70 mol/L cell strainer and washed with PBS. Single-cell suspensions were generated and used for fluorescence-activated cell sorting (FACS) analysis.
Flow cytometry
Single-cell suspensions obtained from human patient tumors or mouse tumors were analyzed using FACS Fortessa flow cytometer (BD Biosciences). Cells were first stained with anti- CD16/32 (2.4G2) for 10 minutes at 4°C, then with specifically conjugated antibodies for 30 minutes at 4°C in the dark. The following anti-mouse or anti-human antibodies were used in the analysis: mouse – CD45 (30F-11), CD3 (17A2), CD8 (53–6.7), IFN-γ (XMG1.2), TNF-α (MP6- XT22), granzyme B (GB11), Ki-67 (B56), PD-1 (RMP1–30), Tim-3 (RMT3–23), CD44 (IM7), LAG-3 (C9B7W), TIGIT (IG9), CD103 (2E7), CD11c (N418), CD11b (M1/70), CD19 (6D5); human – CD28 (CD28.2), CD3 (OKT3), CD45 (HI30), CD8 (SK1), CXCR3 (CEW33D), PD-1 (eBioJ105), Tim-3 (F38–2E2). All antibodies were purchased from BioLegend, Thermo Fisher Scientific or BD Biosciences. Non-viable cells were excluded on the basis of staining with Fixable Viability Dye (Thermo Fisher Scientific or Biolegend). For analysis of polyfunctional T cell responses, cells were incubated with 5μg/ml anti-CD3 antibody (145–2C11; BioLegend), 2μg/ml anti-CD28 antibody (E18; BioLegend), 10μg/ml Brefeldin A (GolgiPlug; BD Biosciences) for 4 hours at 37°C. After surface staining, cells were fixed and permeabilized with Fix & Perm kit (BD Biosciences) and stained for intracellular cytokines. Cells were resuspended in FACS buffer and acquired on a FACS Fortessa flow cytometer (BD Biosciences). Control for all positive staining was based on isotype staining, except for the cytokine staining which was based on unstimulated cells. Datasets were analyzed using FlowJo software (FlowJo, LLC).
Bone marrow chimeric mice
Bone marrow chimeric mice were generated by irradiating recipient mice with 1000 rads. Bone marrow cells were prepared from the tibia and fibia bones of donor mice and 2×106 fresh bone marrow cells resuspended in PBS were then transferred into the irradiated mice intravenously via the lateral tail vein. 8 weeks after engraftment, blood samples from mice were obtained to assess reconstitution efficiency. Mixed bone marrow chimeric mice that had a 1:1 ratio of donor bone marrow cells from the two different strains were selected for experiments.
RNA Isolation and qPCR
Tumors were dissociated and lysed using gentle MACs dissociator (Miltenyi) with RLT buffer and run through QIAshredder spin columns (Qiagen). RNA was isolated using the RNeasy Mini Kit (Qiagen). Following DNAse I treatment (Thermo Fisher Scientific), cDNA was prepared using Multiscribe RT (Thermo Fisher Scientific). qPCR was performed with FastStart Essential DNA Green Master (Roche) using Lightcycler 96 System (Roche). Primer sequences for the detection of CXCL9 were 5’-AATGCACGATGCTCCTGCA-3’ and 5’-AGGTCTTTGAGGGATTTGTAGTGG-3’, and for CXCL10 were 5’-GCCGTCATTTTCTGCCTCA-3’ and 5’-CGTCCTTGCGAGAGGGATC-3’. All samples were normalized using GAPDH detected using 5’-GGCAAATTCAACGGCACAGT-3’ and 5’-AGATGGTGATGGGCTTCCC-3’ primers.
Quantification of chemokine proteins
Tumors were harvested from tumor-bearing mice and homogenized in T-PER tissue protein extraction reagent (Thermo Fisher Scientific). Levels of CXCL9 and CXCL10 in tumor lysates were determined by ELISA kits (Thermo Fisher Scientific), according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism software. The student’s t test and one-way ANOVA with Tukey’s multiple comparison test were used for statistical evaluations as indicated in each experiment. For tumor growth curves, significance was determined via 2-way ANOVA. For mouse survival studies, Mantel-Cox log-rank test was used to evaluate statistical differences in Kaplan-Meier analysis. For human studies, Wilcoxon matched-pairs signed rank test was used for data analysis. Data are generally shown as mean ± SEM unless otherwise stated. Value of p<0.05 was considered significant.
Supplementary Material
Highlights.
Anti-PD-1 efficacy depends on intratumoral activity of the CXCR3 chemokine system.
CD103+ dendritic cell-derived CXCL9 and CXCR3 on CD8+ T cells are required.
CXCR3 ligands are positive indicators of responsiveness to anti-PD-1 therapy.
Inducing CXCR3 ligands in non-responsive tumors restores sensitivity to anti-PD-1.
Acknowledgements
This work was supported by grants from the National Institutes of Health CA204028 and the Cancer Research Institute to A.D.L., the American Association Cancer Research Genentech Immuno-oncology Research Fellowship 17–40-18-CHOW and the MGH Tosteson and Fund for Medical Discovery Postdoctoral Fellowship Awards to M.T.C., the American Association Cancer Research Basic Science Fellowships Program 18–40-01-OZGA and the Swiss National Science Foundation Early Postdoctoral Mobility Award to A.J.O., National Institutes of Health grants AR043369, CA222871, CA163222, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the Melanoma Research Alliance to D.E.F., and the National Institutes of Health grant AI112521 to G.J.F.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
A.D.L. has consulted for Merck, Lilly, Sanofi, Idera, Receptos, and Celtaxsys. D.E.F. has a financial interest in Soltego, Inc., a company developing SIK inhibitors for topical skin darkening treatments that might be used for a broad set of human applications. Dr. Fisher’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. G.J.F. has patents/pending royalties on the PD-1 pathway from Roche, Merck, Bristol-Myers-Squibb, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, Dako and Novartis and has served on advisory boards for Roche, Bristol-Myers- Squibb, Xios, and Origimed.
References
- Ahrends T, Spanjaard A, Pilzecker B, Bąbała N, Bovens A, Xiao Y, Jacobs H, and Borst J (2017). CD4+ T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity 47, 848–861. [DOI] [PubMed] [Google Scholar]
- Berghuis D, Santos SJ, Baelde HJ, Taminiau AH, Maarten Egeler R, Schilham MW, Hogendoorn PC, and Lankester AC (2011). Pro‐inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8+ T‐lymphocyte infiltration and affect tumour progression. J. Pathol 223, 347–357. [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, Weber A, Slankamenac K, Poon RT, Yang H, et al. (2012). Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut 61, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen U, McGavern DB, Luster AD, von Herrath MG, and Oldstone MB (2003). Among CXCR3 chemokines, IFN-γ-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-γ (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J. Immunol 171, 6838–6845. [DOI] [PubMed] [Google Scholar]
- de Mingo Pulido Á, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, Coussens LM, and Ruffell B (2018). TIM-3 regulates CD103+ dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell 33, 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, and Luster AD (2002). IFN- gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol 168, 3195–3204. [DOI] [PubMed] [Google Scholar]
- Durai V, and Murphy KM (2016). Functions of murine dendritic cells. Immunity 45, 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman WH, Pages F, Sautes-Fridman C, and Galon J (2012). The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer 12, 298–306. [DOI] [PubMed] [Google Scholar]
- Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 49, 1148–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Martín A, Gómez L, Lustgarten J, Mira E, and Mañes S (2011). Maximal T cell–mediated antitumor responses rely upon CCR5 expression in both CD4+ and CD8+ T cells. Cancer Res. 71, 5455–5466. [DOI] [PubMed] [Google Scholar]
- Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. (2017). PD-1 expression by tumor-associated macrophages inhibits phagocytosis and tumor immunity. Nature 545, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JW, Sokol CL, and Luster AD (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol 32, 659–702. [DOI] [PubMed] [Google Scholar]
- Groom JR, and Luster AD (2011a). CXCR3 in T cell function. Exp. Cell Res 317, 620–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, and Luster AD (2011b). CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, and Luster AD (2012). CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37, 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, and Luster AD (2001). Donor-derived IP-10 initiates development of acute allograft rejection. J. Exp. Med 193, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, Ling M, Gerard NP, and Gerard C (2000). Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med 192, 1515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, and Gajewski TF (2009). Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69, 3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TH, Banigan EJ, Christian DA, Konradt C, Tait Wojno ED, Norose K, Wilson EH, John B, Weninger W, Luster AD, et al. (2012). Generalized Levy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 486, 545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, Araki K, and Ahmed R (2018). CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu. Rev. Med 69, 301–318. [DOI] [PubMed] [Google Scholar]
- Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, and Yewdell JW (2015). CXCR3 chemokine receptor enables local CD8+ T cell migration for the destruction of virus-infected cells. Immunity 42, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, and Sharpe AH (2017). PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med 214, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenmuller W, Brandes M, Wang Z, Herz J, Egen JG, and Germain RN (2013). Peripheral prepositioning and local CXCL9 chemokine-mediated guidance orchestrate rapid memory CD8+ T cell responses in the lymph node. Immunity 38, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IA, MacLean JA, Lee FS, Casciotti L, DeHaan E, Schwartzman JD, and Luster AD (2000). IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity 12, 483–494. [DOI] [PubMed] [Google Scholar]
- Liu J, Li F, Ping Y, Wang L, Chen X, Wang D, Cao L, Zhao S, Li B, Kalinski P, et al. (2015). Local production of the chemokines CCL5 and CXCL10 attracts CD8+ T lymphocytes into esophageal squamous cell carcinoma. Oncotarget 6, 24978–24989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster AD, and Leder P (1993). IP-10, a -C-X-C- chemokine, elicits a potent thymus- dependent antitumor response in vivo. J. Exp. Med 178, 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Lostao L, Anel A, and Pardo J (2015). How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer. Res 21, 5047–5056. [DOI] [PubMed] [Google Scholar]
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, and Cyster JG (2004). Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427, 355–360. [DOI] [PubMed] [Google Scholar]
- Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, Schell MJ, Sondak VK, Weber JS, and Mule JJ (2012). 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci. Rep 2, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. (2015). Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun 6, 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, Camus M, Gillard M, Bruneval P, Fridman WH, et al. (2010). Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology 138, 1429–1440. [DOI] [PubMed] [Google Scholar]
- Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A, Rabin RL, Zhang HH, Gebeyehu S, Wright TM, et al. (2002). The CXC chemokine murine monokine induced by IFN-γ (CXC chemokine ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. J. Immunol 169, 1433–1443. [DOI] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su M-W, Zhou Y, Deng A, Hunter CA, and Luster AD (2014). CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci. Transl. Med 6, 223ra223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. (2016). Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44, 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Paulete AR, Cueto FJ, Martínez-López M, Labiano S, Morales-Kastresana A, Rodríguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, et al. (2016). Cancer immunotherapy with immunomodulatory anti-CD137 and anti–PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 6, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AH, and Pauken KE (2018). The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol 18, 153–167. [DOI] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211. [DOI] [PubMed] [Google Scholar]
- Sierro F, Biben C, Martínez-Muñoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H, Groom J, Batten M, et al. (2007). Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 104, 14759–14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, et al. (2014). Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med 20, 1301–1309. [DOI] [PubMed] [Google Scholar]
- Speiser DE, Ho PC, and Verdeil G (2016). Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol 16, 599–611. [DOI] [PubMed] [Google Scholar]
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic immunity is required for effective cancer immunotherapy. Cell 168, 487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, and Gajewski TF (2014). Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J. Immunother. Cancer 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JH, Zhang H, Moseman EA, Alvarez D, Iannacone M, Henrickson SE, de la Torre JC, Groom J, Luster AD, and von Andrian UH (2012). Chemokine guidance of central memory T cells is critical for anti-viral recall responses in lymph nodes. Cell 150, 1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. (2015). Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Wang S, Campos J, Gallotta M, Gong M, Crain C, Naik E, Coffman RL, and Guiducci C (2016). Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc. Natl. Acad. Sci. USA 113, E7240–E7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-AA, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. (2017). Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 170, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et al. (2012). Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 72, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.