

Abstract
Autophagy is a highly conserved and regulated process that targets proteins and damaged organelles for lysosomal degradation to maintain cell metabolism, genomic integrity, and cell survival. The role of autophagy in cancer is dynamic and depends, in part, on tumor type and stage. Although autophagy constrains tumor initiation in normal tissue, some tumors rely on autophagy for tumor promotion and maintenance. Studies in genetically engineered mouse models support the idea that autophagy can constrain tumor initiation by regulating DNA damage and oxidative stress. In established tumors, autophagy can also be required for tumor maintenance, allowing tumors to survive environmental stress and providing intermediates for cell metabolism. Autophagy can also be induced in response to chemotherapeutics, acting as a drug-resistance mechanism. Therefore, targeting autophagy is an attractive cancer therapeutic option currently undergoing validation in clinical trials.
Keywords: autophagy, cancer, oncogenic Kras, pancreatic cancer, cancer metabolism
1. INTRODUCTION
As cancers develop and progress, fundamental alterations in basic cellular processes are required to maintain tumor growth. These hallmarks of cancer have been identified during the past 30 years of cancer biology research (Hanahan & Weinberg 2011). More recently, autophagy, a conserved cellular degradation pathway, has been shown to be important for multiple aspects of cancer biology, including cell metabolism, protein and organelle turnover, and cell survival. The role of autophagy in cancer is complex, as demonstrated by studies describing situations in which autophagy can either promote or inhibit tumorigenesis (Kimmelman 2011). The most likely explanation is that the role of autophagy in cancer is dynamic. Although autophagy constrains tumor initiation through its role in tissue homeostasis by maintaining cellular and genomic integrity, it is clearly required for tumor progression and, depending on the tissue of origin and tumor type, can also be required for tumor maintenance. Here we review the role of autophagy in cancer and highlight recent advances, first, as it pertains to constraining tumor initiation and, second, as a protumorigenic mechanism.
2. MOLECULAR MECHANISMS OF AUTOPHAGY
Macroautophagy (referred to as autophagy) is a conserved catabolic cellular pathway that degrades macromolecules and organelles via the lysosome to maintain cellular homeostasis and fitness at a basal state, as well as during periods of stress (Kimmelman 2011, Yang & Klionsky 2010). Autophagy involves the coordinated activity of more than 30 autophagy-related (Atg) proteins that sequester cargo in double-membrane vesicles (autophagosomes) that fuse to lysosomes (autolysosomes), leading to the degradation of cargo, such as toxic protein aggregates, damaged organelles, lipids, and nucleic acids, as well as pathogens, such as Salmonella (Figure 1). The breakdown products of lysosomal degradation (nucleotides, amino acids, and fatty acids) are basic molecular building blocks that can be used in anabolic and bioenergetic pathways (Mizushima & Komatsu 2011, Noda & Inagaki 2015). Two additional forms of autophagy, microautophagy and chaperone-mediated autophagy, which differ from macroautophagy in function and how cargo is delivered to the lysosome, are not discussed but are reviewed in detail elsewhere, including their potential roles in cancer (Cuervo & Wong 2014, Li et al. 2012).
Figure 1.
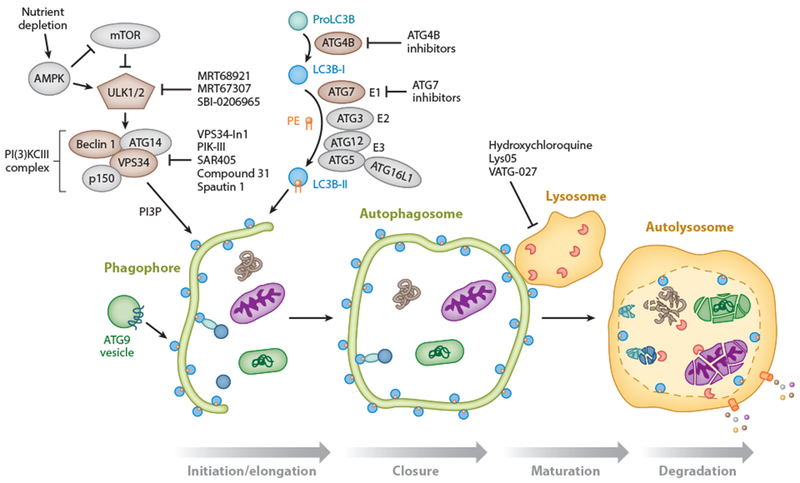
Molecular mechanisms of autophagy. The stages of autophagy (initiation/elongation, closure, maturation, and degradation) are shown. (Bottom) Cargo, such as mitochondria (purple ellipsoid), bacteria (green rounded rectangle), and protein aggregates (brown tangle), is sequestered in selective and bulk degradative manners via a double-membrane phagophore that fuses onto itself to form the autophagosome (closure). The autophagosome subsequently fuses to the lysosome (autolysosome), where the cargo is degraded by lysosomal enzymes and degradation products are recycled to the cytosol by lysosomal transporters. (Top left) During nutrient-replete conditions, mTOR (mammalian target of rapamycin) is activated, and autophagy is inhibited through repression of ULK (unc-51-like autophagy activating kinase) 1 and 2 [ATG (autophagy-related) 1]. Upon nutrient depletion, the ULK1–2 complex is activated and can promote autophagy initiation. ULK1–2 is also activated at low energy states by phosphorylation via AMPK (5′ AMP-activated protein kinase), as well as by repression of mTORC1 activity. New inhibitors of ULK1 and ULK2 include MRT68921, MRT67307 (Petherick et al. 2015), and SBI-0206965 (Egan et al. 2015). Autophagy initiation is also regulated by the production of phosphatidylinositol-3-phosphate (PI3P) by the class III PI3K complex composed of VPS34, ATG14, ATG6 (Beclin 1), and p150 (Vps15). Inhibitors of VPS34 include VPS34-In1 (Bago et al. 2014), PIK-III (Dowdle et al. 2014), SAR405 (Ronan et al. 2014), compound 31 (Pasquier et al. 2014), and Spautin-1 (Liu et al. 2011). ATG9-containing vesicles contribute membrane to the growing autophagosome. (Top middle) Pro-LC3B (one of seven mammalian ATG8 homologs) is converted to LC3B-I by ATG4B, a potential therapeutic target. LC3B-I is subsequently conjugated to phosphatidylethanolamine (PE) via a ubiquitin conjugation-like E1-E2-E3 series of enzymes [ATG7 (a potential therapeutic target), ATG3, and the ATG12-ATG5-ATG16L1 complex]. This produces the lipidated LC3B-II form that then associates with autophagosomal membranes and has roles in autophagosome membrane elongation. LC3B-II is present on the outer and inner surfaces of the autophagosome (blue circle with orange PE moiety). Of note, membrane-associated LC3B-II is converted back to the cytosolic LC3B-I form via the action of the ATG4B enzyme (not shown) for repeated use in autophagosome formation. Autophagosome maturation/lysosomal inhibitors include hydroxychloroquine, Lys05, and VATG-027.
Although autophagy was initially thought to be a bulk, nonselective degradative pathway stimulated in response to stressors, including starvation, more recent research has identified selectivity in the autophagic pathway for the identification of specific cargo for degradation (Khaminets et al. 2015a, Mancias & Kimmelman 2016). Coincident with its importance in maintaining cellular homeostasis, the disruption of autophagic pathways has been shown to play a part in diverse disease processes, including neurodegeneration, atherosclerosis, and cancer (Choi et al. 2013, Kenific & Debnath 2015, Kimmelman 2011, Mizushima & Komatsu 2011, White 2015).
2.1. Molecular Mechanisms of Autophagy Initiation and Autophagosome Formation
Autophagy was initially characterized in mammalian cells as an adaptive response to starvation (De Duve & Wattiaux 1966, Yang & Klionsky 2010). However, it is now clear that autophagy is active at some basal level in all cells and can be further activated by a variety of stressors, including hypoxia, reactive oxygen species (ROS), chemotherapeutics, and radiotherapy (Amaravadi et al. 2007, Degenhardt et al. 2006, Scherz-Shouval & Elazar 2007). The initial studies of the autophagy activation molecular signaling apparatus were performed in yeast, and these identified a complex set of more than 30 different Atg genes, of which many are conserved in higher eukaryotes (Kabeya 2000, Mizushima et al. 1998). The process of autophagy can be broken into several discrete steps: (a) initiation and nucleation of the pre-autophagosomal membrane (phagophore), (b) autophagosome closure, (c) maturation via autophagosome–lysosome fusion, and (d) degradation via lysosomal enzymes (Figure 1). The canonical autophagy initiation pathway is controlled by multiple signaling complexes, including those that interpret the cellular energy or oxidation levels [5′ AMP-activated protein kinase (AMPK)] (Hardie et al. 2012) and nutrient or amino acid levels [mammalian target of rapamycin (mTOR)] (Galluzzi et al. 2014, Jung et al. 2010). These pathways converge on the unc-51-like autophagy activating kinase 1 (ULK1) (Atg1 ortholog) complex that mediates autophagy induction (Egan et al. 2011, Kim et al. 2011a). There are also noncanonical modes of autophagy activation not involving ULK1 or other core autophagy machinery that reflect the diverse mechanisms by which the autophagy program can be initiated (Cheong et al. 2011, Nishida et al. 2009, Scherz-Shouval et al. 2007). The mechanisms for autophagy activation in cancer cells and whether they are conserved are unclear, but recent studies in pancreatic cancer cell lines have identified distinct modes for promoting high basal levels of autophagy. Pancreatic ductal adenocarcinoma (PDAC) cell lines, in part, activate basal autophagy via protein phosphatase 2A-B55α activity toward ULK1, thereby stimulating ULK1-dependent autophagy (Wong et al. 2015). Human PDAC cells can also induce autophagy via a MiT/TFE transcriptional program that increases not only autophagy but also lysosome biogenesis (Perera et al. 2015). Further work is required to clarify the upstream pathways that lead to increased basal autophagy in distinct cancer types.
Following autophagy induction, the class III phosphatidylinositol 3-kinase [PI(3)KCIII] complex, consisting of VPS34, p150, ATG14, and Beclin 1 [BECN1 (Atg6 ortholog)], nucleates autophagosome formation (Figure 1). Subsequently, the ATG9 transmembrane protein mediates the trafficking of source membrane—including from the endoplasmic reticulum, Golgi complex, mitochondria, endosome, and plasma membrane—for autophagosome elongation (Papinski et al. 2014). Two ubiquitin-like conjugation systems (described below) participate in autophagosome closure, maturation, and the recruitment of additional autophagy machinery (Noda & Inagaki 2015).
2.2. Role of ATG8s in Autophagosome Maturation and Selective Autophagy
The primary component of the autophagosome maturation apparatus is the ubiquitin-like protein lipidation system that conjugates phosphatidylethanolamine to the C terminus of ATG8, thereby facilitating incorporation of ATG8 proteins into growing autophagosomal membranes (Klionsky & Schulman 2014, Slobodkin & Elazar 2013). ATG7 acts as an E1 enzyme and ATG10 as an E2 to conjugate the ubiquitin-like ATG12 protein to ATG5. This ATG12–ATG5 conjugate then acts in an E3-like complex with ATG16L1 to facilitate ATG8 lipidation. ATG8s are synthesized in a pro-ATG8 form that is cleaved by ATG4B, leaving a C-terminal glycine residue. In concert, ATG7 (E1), ATG3 (E2), and the ATG12–ATG5–ATG16L1 (E3) complex catalyze the conjugation of phosphatidylethanolamine to the C-terminal glycine of ATG8s (Figure 1) (Noda & Inagaki 2015). This lipidated form of ATG8 is tightly associated with autophagosomal membranes.
Numerous studies have indicated that ATG8 proteins can function as adaptors to recruit further regulatory proteins important for autophagosomal maturation and as adaptors for selective autophagy receptors (Behrends et al. 2010, Slobodkin & Elazar 2013) that physically link their cargo to the forming autophagosomal membrane for lysosomal degradation. Although yeast contain a single ATG8 protein, mammals have seven ATG8 proteins in two structurally related subfamilies [MAP1LC3A, B or B2, and C and GABARAP, GABARAPL1, and GABARAPL2 (also known as GATE-16)], suggesting a complex diversification of their functions (Slobodkin & Elazar 2013). Selective autophagic pathways are generally named for the cargo destined for degradation and include mitophagy (mitochondria), aggrephagy (protein aggregates), ferritinophagy (ferritin), ER-phagy (endoplasmic reticulum), and xenophagy (pathogens, including bacteria), among many (Khaminets et al. 2015b, Mancias et al. 2014, Melser et al. 2015, Sorbara & Girardin 2015, Svenning & Johansen 2013). For an in-depth review of the subject, including the growing understanding of the role of selective autophagy in cancer, readers are referred to recent reviews (Khaminets et al. 2015a, Mancias & Kimmelman 2016).
3. AUTOPHAGY IN TUMOR SUPPRESSION
Autophagy was initially considered a tumor suppressive mechanism based on indirect evidence from oncogene and tumor suppressor gene alteration studies. Gain-of-function mutations or amplifications in PI3K, or AKT or PTEN loss or silencing, which all activate mTOR and, thereby, inhibit autophagy, are common oncogenic alterations, suggesting a potential importance of suppressing autophagy during tumor initiation (Kimmelman 2011, Maiuri et al. 2009).
The tumor suppressor p53 appears to have opposing roles in autophagy based on its subcellular localization (Tang et al. 2015b). Nuclear p53 has been proposed to activate autophagy via a number of transcriptional mechanisms. Indeed, a comprehensive high-throughput chromatin immunoprecipitation sequencing study revealed a large number of autophagy genes as direct p53 target genes and that autophagy assists in p53-dependent apoptosis and cancer suppression (Kenzelmann Broz et al. 2013). With the loss of functional p53 seen in many tumors, the expectation is that this would lead to a decrease in autophagy, which would be consistent with a role for autophagy as constraining tumor initiation. In contrast to the role of nuclear p53 in activating autophagy, cytoplasmic p53 can inhibit autophagy, mainly via protein–protein interactions with autophagic machinery (Tang et al. 2015b).
More direct evidence of a role for autophagy in suppressing tumor initiation comes from mouse genetic studies of autophagic machinery, including Atg7, Atg5, and Becn1, showing that when autophagy is impaired, there is an increase in tumor initiation (Qu et al. 2003, Takamura et al. 2011, Yue et al. 2003). Interestingly, tumors that develop in models in which autophagy is completely ablated are benign (Takamura et al. 2011). One exception is the studies on Becn1 heterozygous mice in which the authors found that tumors developed and, in many cases, were able to progress to malignant lesions. This may reflect the fact that these mice, although autophagy impaired, still were autophagy competent (Qu et al. 2003). Taken together, the data suggest that even though autophagy loss may predispose to tumor initiation, active autophagy also supports the progression to invasive cancers. Although the initial studies on BECN1 showed that many ovarian and breast cancers have a monoallelic loss of this gene, recent, large-scale, human tumor sequencing studies have suggested that this may be a passenger alteration, given the proximity to BRCA1 on chromosome 17q21 and the lack of any BECN1-only mutation or loss in cancers (Laddha et al. 2014, Lebovitz et al. 2015). However, a study of BECN1 messenger RNA expression patterns in breast cancers suggested an association between low BECN1 expression and poor prognosis in Her2, basal-like, and p53-mutant cancers, which may indicate an additional mechanism of downregulation in certain cancers (Tang et al. 2015a).
From a mechanistic standpoint, the inhibition of autophagy leads to excess ROS, increases in DNA damage, and impaired mitochondria, all potentially protumorigenic (Figure 2) (White 2015). Indeed, studies by the White lab (Karantza-Wadsworth et al. 2007, Mathew et al. 2007) and others have shown that the loss of autophagy leads to genomic instability and aneuploidy. Interestingly, beyond protumorigenic intrinsic effects, autophagy loss in vivo can also trigger tumor cell extrinsic effects, including a protumorigenic inflammatory microenvironment (Degenhardt et al. 2006). Studies on the selective autophagy receptor p62 (SQSTM1) and tumorigenesis have suggested a potential mechanistic link between tumor suppression and selective autophagy. In mouse models with defective autophagy, p62 ablation decreases tumorigenesis, suggesting that p62 accumulation upon autophagy loss can contribute to tumorigenesis (Duran et al. 2008, Inami et al. 2011). Indeed, p62 overexpression promotes oxidative stress and tumor growth (Mathew et al. 2009). Interestingly, amplification of chromosome 5q, where p62 resides, and thereby of p62 expression, is associated with the development of clear cell renal cell carcinoma (Li et al. 2013, Moscat & Diaz-Meco 2009).
Figure 2.
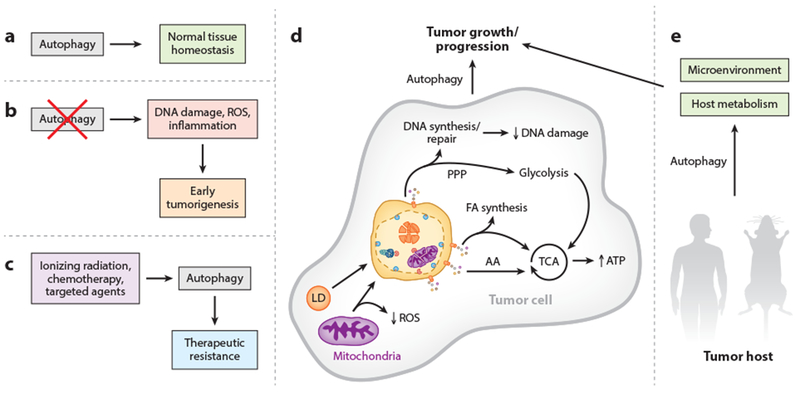
Role of autophagy in tumors and cancer metabolism. (a) In normal tissue, autophagy performs homeostatic functions, such as protein and organelle quality control. (b) If autophagy is suppressed in tissues, normal homeostasis is disrupted, leading to increased DNA damage (genomic instability and aneuploidy), reactive oxygen species (ROS), and inflammation. Together, these changes can promote tumor initiation and lead to early tumorigenesis. (c) Cancer-directed therapies, including ionizing radiation (IR), chemotherapy, and various targeted agents, can induce a cytoprotective autophagy that contributes to therapeutic resistance. (d) Activated autophagy can promote tumor growth and progression in established tumors by allowing tumor cells to keep up with their metabolic demand as well as regulate oxidative stress. Specifically, recycled breakdown products, such as amino acids (AA), can fuel the tricarboxylic acid (TCA) cycle [producing adenosine triphosphate (ATP)]; building blocks for fatty acid (FA) synthesis can also fuel the TCA cycle; and recycled nucleotides can be used for DNA synthesis and repair (contributing to reduced DNA damage), as well as for the nonoxidative arm of the pentose phosphate pathway (PPP) that supports glycolysis. Targeting damaged mitochondria for autophagic degradation (mitophagy) can also regulate ROS levels in the tumor. Overall, autophagy acts in multiple ways to support a protumorigenic phenotype in a cell-autonomous manner. (e) Autophagy also acts in a nonautonomous manner in the tumor host to support tumor growth and progression. Autophagy activation in nontumor cells, such as tumor stroma, can provide tumors with critical metabolic support. Furthermore, autophagy may have a role in producing an environment favorable for tumor progression.
Another potential mechanism by which autophagy acts as a tumor suppressor is via its requirement in cellular senescence, a program of permanent cell division arrest that can be induced in response to oncogenic stress to avoid malignant transformation (Pérez-Mancera et al. 2014). Recent reports have shown a potential role for selective autophagy in this process (Dou et al. 2015, Kang et al. 2015).
4. AUTOPHAGY IN TUMOR PROMOTION
Although autophagy can act as a suppressor of early tumorigenesis through a number of mechanisms (White 2015), work from many groups has also shown that autophagy can support tumor growth in multiple tumor types (Guo et al. 2013b, Kimmelman 2011, White 2015), as well as promote resistance to a variety of therapies (Amaravadi et al. 2007). This apparent dual role of autophagy can be explained, in part, because the same elements that promote tumorigenesis at initial stages (ROS, inflammation, DNA damage) can be deleterious at later stages (Imlay & Linn 1988, Poillet-Perez et al. 2015). Indeed, under stress conditions, such as hypoxia or nutrient deprivation, autophagy is a prosurvival mechanism that eliminates unfolded proteins and provides substrates for adenosine triphosphate (ATP) production by rapidly degrading endogenous substrates (Figure 2) (Kuma et al. 2004, Singh & Cuervo 2011). Therefore, in a tumor’s hypoxic regions, autophagy is usually elevated and promotes cell survival (Degenhardt et al. 2006).
In addition, autophagy is also highly activated across a wide variety of cancer types (Lazova et al. 2012), including KRAS- and BRAF-driven tumors (Guo et al. 2011, Lock et al. 2011, Yang et al. 2011). The latter rely on autophagy even under basal conditions for the proper functioning of organelles and to meet their metabolic demands. In fact, autophagy has been shown to be important for RAS transformation. Breast epithelial cells transformed with KRASV12 upregulated expression of various ATG genes through a ROS/JNK-dependent mechanism, leading to the increased formation of autophagosomes (Kim et al. 2011b). Autophagy induction alone was not sufficient to promote transformation, but it was required for KRAS-induced tumorigenesis (Kim et al. 2011b). Furthermore, mouse embryonic fibroblasts isolated from a Fip200 (Atg17 ortholog)-null model transformed with HRASV12 showed reduced proliferation (Wei et al. 2011). In agreement with this concept, autophagy is required to support adhesion-independent transformation by mutant RAS (Kim et al. 2011b, Lock et al. 2011).
Consistent with the requirement for autophagy in KRAS tumors, our laboratory has described an increase in basal autophagy in human PDAC, a tumor that nearly universally possesses activating KRAS mutations (Biankin et al. 2012, Jones et al. 2008). Indeed, autophagy inhibition by RNA interference or chloroquine (CQ) treatment [an inhibitor of lysosomal acidification and autophagosomal degradation (Rubinsztein et al. 2012)] decreased growth and colony formation in vitro, as well as tumor growth in vivo (Yang et al. 2011). Similar results have been obtained in the White laboratory (Guo et al. 2011), in a study in which RAS transformation induced an increase in basal autophagy while autophagy inhibition impaired cell viability in starvation conditions as well as tumor growth in vivo (Guo et al. 2011).
Although there is significant evidence that autophagy is required for the tumorigenic growth of multiple cancer types, a recent study concluded that it is dispensable in KRAS-mutant tumor cell lines, and it also questioned whether the antitumor effects of CQ and its derivative, hydroxychloroquine (HCQ), are due to autophagy inhibition (Eng et al. 2016). There are several reasons for the apparent discrepancy between this and a large number of prior studies. One issue is that although autophagy inhibition has been shown to be inhibitory in multiple KRAS-mutant tumors, the in vitro effects in short-term, two-dimensional (2D) growth assays in complete media are typically cytostatic and more modest than those in vivo. Most in vitro effects are seen predominantly in tumorigenesis assays, such as soft agar and low-density clonogenic assays, since these assays are themselves stresses, similar to in vivo tumor growth, and autophagy is a critical stress response. Another potential issue involves the use of gene editing to delete autophagy genes (Eng et al. 2016). Although this approach provides a true knockout phenotype, it can be susceptible to selective pressures and the generation of resistant clones. Such studies may be informative for understanding the resistance that may ultimately develop to autophagy inhibitors, but they do not exclude the initial requirement for autophagy in those cell lines. Indeed, autophagy gene knockout studies in PDAC mouse models have demonstrated that although tumor formation is decreased, those tumors that do manage to form no longer require autophagy (Yang et al. 2014). Perhaps most importantly, some of the most robust tumor responses to autophagy loss have been seen in autochthonous models with homotypic tumor-stromal interactions and an intact immune system (discussed below) (Guo et al. 2013a, Karsli-Uzunbas et al. 2014, Rao et al. 2014), suggesting additional non-cell-autonomous factors for which autophagy is critical, which cannot be assessed in cell culture and standard xenograft studies. Ongoing and future studies will explore these possibilities, including the role of the immune system and other features of the tumor microenvironment.
In regard to the effects of CQ and HCQ, it is well accepted that they inhibit autophagy. Although these drugs do inhibit the lysosome and can affect other lysosomal pathways, studies have shown that at certain dose ranges, the antitumor effects of HCQ are likely to occur through autophagy inhibition (Amaravadi et al. 2007). Despite the possible clinical activity of HCQ (discussed below), the therapeutic validation of autophagy inhibition will benefit from the development of more potent and specific inhibitors.
4.1. Autophagy Regulates Energy Homeostasis and Cell Metabolism
Highly proliferating tumor cells require lipids, carbohydrates, amino acids, and nucleotides as substrates for biosynthesis and energy production. Autophagy can generate all of these metabolic intermediates to support the increased metabolic demand of proliferating tumors (Figure 2). Glycogen can be hydrolyzed to carbohydrates that can feed glycolysis, and protein degradation provides amino acids that feed the tricarboxylic acid (TCA) cycle at different points or that can be used for protein synthesis. Nucleotides can be degraded to obtain ribose-phosphate, which can either be converted to glycolytic intermediates in the nonoxidative pentose phosphate pathway to generate ATP or be used anabolically for DNA replication and repair (Rabinowitz & White 2010).
Autophagy can also target substrates selectively (Mancias & Kimmelman 2016, Mizushima & Komatsu 2011), in some cases with important metabolic consequences. Mitophagy selectively degrades defective mitochondria to prevent oxidative stress (Mizushima & Komatsu 2011) and to maintain mitochondrial metabolic processes, such as fatty acid oxidation (Guo et al. 2013a). Lipid requirements are elevated in tumor cells, either for ATP production or membrane synthesis. Lipid stores or lipid droplets can be degraded by autophagy in a selective process known as lipophagy (Kaur & Debnath 2015), releasing free fatty acids that support fatty acid β-oxidation and the TCA cycle (Singh et al. 2009). In fact, autophagy blockade by liver-specific Atg7 knockout leads to the accumulation of lipid droplets even in the absence of nutrient deprivation, showing that lipophagy is an essential mechanism for cell metabolism (Singh et al. 2009).
These studies strongly suggest a role for autophagy in tumor metabolism and that modulation, either genetically or pharmacologically, would impact tumor growth. Indeed, Debnath and colleagues (Lock et al. 2011) showed that autophagy inhibition decreased anchorage-independent growth, proliferation, and glucose metabolism in Ras-transformed mouse embryonic fibroblasts. Similar results regarding decreased glycolysis have been observed after deletion of Fip200 in a conditional model of mammary tumors (Wei et al. 2011). One of the most important metabolic effects observed throughout many studies is that autophagy inhibition decreases mitochondrial respiration and ATP production by reducing protein turnover and the supply of intermediates to the TCA cycle (Guo et al. 2011, 2013a; Rao et al. 2014; Strohecker et al. 2013; Yang et al. 2011, 2014). In fact, KRAS-driven tumors increase autophagic flux and lysosomal degradation of extracellular scavenged proteins by macropinocytosis to maintain intracellular pools of amino acids (Perera et al. 2015). The role of autophagy in fatty acid β-oxidation seems to be more heterogeneous. Although autophagy is important for fatty acid catabolism in a mouse model of KrasG12D non-small-cell lung carcinoma (NSCLC) (Guo et al. 2013a), this effect is not observed in BrafV600E-driven lung tumors (Strohecker et al. 2013), suggesting differential fatty acid catabolism in lung cancer depending on oncogene activation.
In summary, autophagy facilitates tumor growth by multiple mechanisms, including providing intermediates that support oxidative metabolism or anabolic pathways. Further studies are necessary to identify whether and how the selectivity of autophagic cargo is programmed in a cancer-specific manner to support tumor metabolism.
4.2. Autophagy and Therapeutic Resistance
Autophagy activation in response to internal and external stressors is a well-known mechanism for cell survival. Accordingly, autophagy activation has been shown to be a mediator of therapeutic resistance in a variety of situations, including genotoxic and metabolic stresses, as well as those that inhibit proliferation and replication. Many studies have shown an induction of autophagy in response to cytotoxic chemotherapeutics (e.g., cisplatin, doxorubicin, temozolomide, etoposide), including an early study by Amaravadi et al. (2007) in which alkylating agents were shown to induce autophagy. Notably, autophagy inhibition synergized with alkylating agents, showing that autophagy inhibition can be a means for overcoming autophagy-induced therapeutic resistance (Ding et al. 2011, Kanzawa et al. 2004, Zhang et al. 2012). Ionizing radiation induces autophagy in a wide array of cancer cell lines (as well as in normal nontumor cell lines), and autophagy inhibition can increase radiosensitization in a subset of these cell lines (Apel et al. 2008, Ito et al. 2005). However, there are some conflicting data in the literature showing that in some cases radiosensitization is seen with autophagy activation (Ondrej et al. 2016). Autophagy has also been shown to cause therapeutic resistance to targeted agents such as histone deacetylase inhibitors (Carew et al. 2007), AKT inhibition (Degtyarev et al. 2008), and tyrosine kinase inhibitors, such as imatinib (Ertmer et al. 2007), to name a few.
In some instances, the molecular mechanisms of autophagy activation in response to the cancer-directed therapies discussed above have been determined; however, further work is necessary to understand whether there are other context-specific activation pathways. Furthermore, it is unclear in most instances whether the autophagy that is activated in response to cancer-directed therapies is more targeted to specific substrates or to handle specific aspects of stress. It is important to note that despite the many situations in which autophagy acts a resistance mechanism, there are circumstances whereby autophagy inhibition can decrease the efficacy of a particular therapy (Levy & Thorburn 2011). Therefore, therapeutic combinations should be carefully vetted, in particular by using robust, genetically engineered mouse models of cancer.
4.3. Autophagy in Genetically Engineered Mouse Models of Cancer
The data discussed above support the protumorigenic role of autophagy in multiple tumor types. To more thoroughly evaluate the role of autophagy in cancer, genetically engineered mouse models (GEMMs) of a variety of cancers are being used to identify the mechanisms by which autophagy contributes to tumor growth. In general, GEMMs have many advantages over xenograft tumor models or 2D cancer cell line experiments. These models allow the study of spontaneous tumor growth in situ in the corresponding tissue of origin and with the appropriate tumor-microenvironment interactions. Importantly, given the role of autophagy in modulating the immune system (discussed below), GEMMs allow for the study of tumors in an immune-competent host. Because of the context and stage-dependent role of autophagy, highly representative models are indispensable for understanding the potential clinical use of autophagy inhibitors. To that end, several laboratories have developed mouse models to address important issues, such as the effects of autophagy inhibition in premalignant or malignant lesions and the consequences of inhibiting autophagy in either a cancer cell–specific or systemic manner (Table 1).
Table 1.
Genetically engineered mouse models for studying autophagy deficiency in cancer
| Cancer type | Genotype | Autophagy gene deleted | Initiation | Progression or tumor growth | Metabolic consequences | Reference |
|---|---|---|---|---|---|---|
| Pancreas | Pdx1Cre; lslKrasG12D/+; Trp53flox/+ | Atg5 | Increased | Decreased | Decreased OCR | Yang et al. 2014 |
| Pancreas | Pdx1Cre; lslKrasG12D/+ |
Atg5 Atg7 |
Increased | Decreased | ND | Rosenfeldt et al. 2013 |
| Pancreas | Pdx1Cre; lslKrasG12D/+; Trp53flox/flox |
Atg5 Atg7 |
Increased | Increased | Increased glycolysis and PPP | Rosenfeldt et al. 2013 |
| Lung | lslKrasG12D/+ | Atg7 | ND | Decreased | ND | Guo et al. 2013a |
| Lung | lslKrasG12D/+; Trp53flox/flox | Atg7 | ND | Decreased | Decreased FAO; increased glycolysis | Guo et al. 2013a |
| Lung | lslKrasG12D | Atg5 | Increased | Decreased | Decreased OCR | Rao et al. 2014 |
| Lung | lslKrasG12D; Trp53flox/flox | Atg5 | No difference | No difference | ND | Rao et al. 2014 |
| Lung | BrafV600E; Tp53flox/flox | Atg7 | Increased | Decreased | Decreased OCR | Strohecker et al. 2013 |
| Lung | fsfKrasG12D; Tp53frt/frt | Atg7 | No difference | Decreased | ND | Karsli-Uzunbas et al. 2014 |
| Breast | MMTV-PyMT | FIP200 | Decreased | Decreased | Decreased glycolysis | Wei et al. 2011 |
| Breast | Palb2flox/flox; Wap-cre | Becn1 | ND | Decreased | ND | Huo et al. 2013 |
| Breast | Palb2flox/flox; Trp53flox/flox; Wap-cre | Becn1 | ND | No difference | ND | Huo et al. 2013 |
| Melanoma | TgTyr-cre/ERT2/+; Lsl-BrafV600E/+; Ptenflox/+ | Atg7 | Decreased | Decreased | ND | Xie et al. 2015 |
| Prostate | Nkx3.1CreERT2/+; Ptenflox/flox | Atg7 | No difference | Decreased | ND | Santanam et al. 2016 |
| Colon | VilCre-ERT2 Apcflox/+ | Atg7 | Decreased | Decreased | ND | Lévy et al. 2015 |
Abbreviations: Atg, autophagy-related; Becn, Beclin; FAO, fatty acid oxidation; flox, conditional allele using the Cre-Lox system; frt, conditional allele using FLP-FRT system; fsf, frt-stop-frt; lsl, lox-stop-lox; ND, not determined; OCR, oxygen consumption rate; PPP, pentose phosphate pathway.
In a KrasG12D GEMM of NSCLC, tumor-specific Atg7 deletion, regardless of p53 status, decreased proliferation and led to the formation of oncocytomas, a benign tumor type that accumulates dysfunctional mitochondria (Guo et al. 2013a). Atg 7−/−, p53−/− tumor-derived cell lines (TDCLs) accumulated lipids and decreased mitochondrial respiration, making them sensitive to nutrient starvation due to defects in fatty acid β-oxidation and decreased production of substrates for the TCA cycle (Guo et al. 2013a). Interestingly, a similar study using Atg5-null lung tumors showed that p53 deletion impacted the response to autophagy loss (Rao et al. 2014), which may indicate a distinct role for Atg5 versus Atg7 or may be due to technical reasons, such as mouse strain differences.
To study another Kras-driven cancer, our group used a conditional Atg5 knockout in the context of a PDAC GEMM [Kras mutation and p53 loss of heterozygosity (LOH)] (Yang et al. 2014). Autophagy loss decreased PDAC formation, thereby prolonging survival. Consistent with an initial role in tumor suppression, the Atg5 homozygous deletion increased the number of premalignant pancreatic intraepithelial neoplasia, but significantly impaired the progression to invasive PDAC. The tumors that did form in the Atg5-null mice showed impaired proliferation in vivo, and in TDCLs, there was increased cell death and DNA damage. Interestingly, a second study using a similar model confirmed that autophagy loss prevented tumor progression in the KrasG12D PDAC model (Rosenfeldt et al. 2013). However, when a p53 homozygous deletion was incorporated, the results were paradoxical, with decreased survival. Differences in the mouse models explain these apparently contradictory results, given that p53 homozygous deletion in the pancreas during embryogenesis is distinct from heterozygous deletion and subsequent p53 loss via LOH (analogous to the situation in human tumors). Indeed, CQ treatment in patient-derived pancreatic cancer xenografts impaired tumor growth independently of p53 status (Yang et al. 2014). Furthermore, CQ treatment or RNA interference-mediated Atg5 or Atg7 depletion decreased colony formation and oxidative phosphorylation in a panel of mouse PDAC cell lines with varying p53 statuses, consistent with results from the p53 heterozygous GEMM. Thus, in the physiological situation in which p53 is lost via LOH, autophagy is required for PDAC progression and growth. The results from mice with homozygous p53 embryonic deletion likely indicate an important biological role for p53 in directing an intact autophagy program, as has been shown previously (Kenzelmann Broz et al. 2013).
Because KrasG12D-driven models appear to depend on autophagy, an interesting question was whether the same was true for tumors with constitutive activation of Braf, a downstream effector of Kras. BRAF mutations have been described in lung adenocarcinoma, melanoma, ovarian cancer, and colorectal cancer (Davies et al. 2002). A study in a BrafV600E-driven lung cancer GEMM with either p53 intact or co-deleted showed that Atg7 deletion extended survival (Strohecker et al. 2013). The decreased tumor growth was a consequence of a metabolic crisis resulting from limited nutrient supply to the TCA cycle. Indeed glutamine, a major anaplerotic fuel source for cancer cells in culture, was able to rescue starvation-induced death in Atg7-null TDCLs (Strohecker et al. 2013). In contrast to KrasG12D-induced tumors, autophagy ablation did not lead to an accumulation of lipids or induce inflammation (Guo et al. 2013a), suggesting differential regulation of mitochondrial metabolism according to the driver oncogene. Similarly, in an Atg7-deficient melanoma GEMM driven by BrafV600E with or without Pten deletion, autophagy loss also induced the accumulation of defective mitochondria, oxidative stress, and DNA damage as well as increased survival (Xie et al. 2015). In addition to Ras- and Raf-driven tumors, autophagy has been shown to have a role in a multitude of tumor types in a variety of GEMMs. For instance, in a polyomavirus middle T antigen (PyMT) model of breast cancer, autophagy inhibition by conditional Fip200 (Atg17 ortholog) deletion suppressed tumor initiation, progression, and metastasis (Wei et al. 2011). In another model of hereditary breast cancer driven by Palb2 deletion, autophagy inhibition by single allelic loss of Becn1 in the mouse mammary gland decreased tumorigenesis in a p53-dependent manner (Huo et al. 2013). Additionally, in mouse models of prostate cancer (driven by Pten loss) (Santanam et al. 2016) and colon cancer (Apc+/−) (Lévy et al. 2015), genetic inhibition of autophagy showed significant antitumor responses.
Interestingly, the mechanisms by which autophagy contributes to tumorigenicity might be different according to tumor type. Previous studies in lung cancer models have shown that autophagy inhibition attenuated tumor growth by inducing the accumulation of dysfunctional mitochondria (Guo et al. 2013a). However, in the prostate cancer model mentioned above, Atg7-deficient tumors accumulated swollen ER, which is indicative of ER stress, suggesting a critical role for autophagy in these tumors for eliminating the ER and regulating the accumulation of unfolded proteins (Santanam et al. 2016). Similar results have been seen in a colon cancer model with Atg5 deficiency (Sakitani et al. 2015).
The use of GEMMs is helping to clarify the temporal and tissue-dependent role of autophagy. These models show how the modulation of autophagy has significant cell-autonomous effects in tumor metabolism and regulation of the DNA damage response. Importantly, these effects can vary among tumor types and even for the same tumor type, depending on the activation of different oncogenes.
4.4. Nontumor Cell-Autonomous Effects of Autophagy
Because any autophagy inhibitor that will be used in patients will impact autophagy in the entire patient, it is essential to differentiate between cell-autonomous and non-cell-autonomous effects. Indeed, many of the aforementioned studies have analyzed the role of autophagy from only the tumor cell perspective. However, recent data have begun to address this question genetically, and the data suggest that the impact of systemic autophagy inhibition may be more profound than just the inhibition of autophagy in tumor cells.
To assess the impact of systemic autophagy loss in an adult mouse and to model autophagy inhibitor treatment in a patient, Karsli-Uzunbas et al. (2014) ablated Atg7 systemically in adult mice. Remarkably, Atg7−/− mice survived for more than two months without active autophagy. Ultimately, the majority of the mice succumbed to neurodegeneration (Karsli-Uzunbas et al. 2014), a known sequelae of autophagy loss. The authors then used this Atg7 mouse model in combination with a dual recombinase system (Cre-Lox and FLP-Frt) to generate fully formed lung tumors (KrasG12D, p53-null), after which acute ablation of Atg7 systemically was induced. This model showed dramatic reductions in tumor size. This study illustrates several key points. First, it demonstrates that tumors rely on autophagy more than the host initially, indicating that there may be a therapeutic index for autophagy inhibition in the clinic. Clearly, potential central nervous system effects will have to be accounted for in any therapeutic strategy of long-term autophagy inhibition, either through intermittent dosing strategies or by restricting central nervous system penetration. Second, because the antitumor response was more profound when autophagy was deleted in the whole host (tumor and mouse) than in prior studies when it was deleted only in the tumor, this indicates that there are significant host factors that contribute to the antitumor effects. Importantly, despite the fact that autophagy may be important for various aspects of the immune response (Dengjel et al. 2005, Hubbard et al. 2010, Jacquel et al. 2012, Kondylis et al. 2013, Lee et al. 2010, Pua et al. 2007, Ushio et al. 2011, Willinger & Flavell 2012), these data indicate that systemic autophagy inhibition does not compromise and, in fact, enhances the antitumor response.
The tumor stroma is a complex system composed of the extracellular matrix, cells [immune cells, fibroblasts, myofibroblasts, and cancer-associated fibroblasts (CAFs)], cytokines, and blood and lymph vessels (Kalluri & Zeisberg 2006) that support tumor proliferation, progression, and metastasis (Maes et al. 2013). Although little is known about the specific mechanisms that mediate communication between the stroma and the epithelial compartment, recent work has described a model in which tumor-mediated inactivation of p62 in fibroblasts led to activation of CAFs. This induced a metabolic reprogramming through mTORC1/c-MYC inactivation that ultimately led to the production of interleukin (IL)-6, an inflammatory cytokine that enhances the proliferation and invasion of tumor cells (Valencia et al. 2014). These results suggest a link between the activation of autophagy in the stromal compartment and inflammation that would enhance tumor development and, conversely, suggest that autophagy inhibition in the stroma may provide an antitumor effect. Work from our lab has also demonstrated the importance of autophagy in the stromal compartment, as the inhibition of autophagy specifically in CAFs decreased PDAC tumor growth in an orthotopic transplant model. Moreover, this protumorigenic role of autophagy in the stroma is due to an autophagy-dependent metabolic cross-talk. In this case, CAFs degrade proteins via autophagy, which leads to the secretion of amino acids that can be taken up by the tumor cells to fuel various metabolic pathways (Sousa et al. 2016).
Another mechanism by which autophagy may promote tumor growth, in a non-cell-autonomous manner, is by contributing to the secretion of proteins that are not secreted by conventional pathways (Ponpuak et al. 2015). In one example, autophagy-dependent secretion can mediate the release of IL-6 in the tumor microenvironment of HRAS-transformed cells, contributing to RAS-driven invasion (Lock et al. 2014). Further work is necessary to elucidate the mechanisms that differentiate degradative from secretory autophagy and the role of autophagy-dependent secretion in other cancers.
From a therapeutic perspective, autophagy inhibition in the tumor cell may modulate the immune response, thereby regulating cancer progression. Previous work from the White laboratory (Degenhardt et al. 2006) has shown that when apoptosis and autophagy are inhibited, necrosis induces an inflammatory response characterized by NF-κB and IL-6 secretion and macrophage infiltration. These results are in line with other studies in which autophagy inhibition by Atg7 deletion in NSCLC precipitated an inflammatory response (Guo et al. 2013a, Karsli-Uzunbas et al. 2014). Consistent with these findings, the antitumor effects of autophagy inhibition on both Apc+/− colon GEMMs (Lévy et al. 2015) and the PyMT breast cancer model (Wei et al. 2011) have been shown to depend, at least in part, on intact T cell responses.
In fact, CD8+ T cell depletion accelerated mammary tumor initiation in the Fip200-null mice, suggesting that autophagy contributes to increased tumorigenesis by suppressing the antitumor immune response (Wei et al. 2011). Similar results have been obtained in an Apc+/−, Atg7−/− model of colon cancer, which showed increased secretion of IL-12 and increased infiltration of CD45+ and CD11c+ cells, as well as T regulatory and CD8 interferon (IFN)-γ T cells. Strikingly, autophagy-deficient mice had more bacterial burden, and antibiotics limited the antitumor response, reinforcing the antitumoral role of the gut microbiota when autophagy is inhibited. Further work needs to be performed to elucidate the mechanisms by which autophagy can regulate the IFN pathway and other antitumoral immune responses. In this sense, a recent paper (Mathew et al. 2014) has shown that starvation-induced autophagy can selectively degrade proteins in the RIG-I pathway, which are involved in inflammatory processes, thus inhibiting the innate immune response. In a clinical context, it will be important not only to evaluate the effects on the tumor-induced immune response but also to determine the consequences of systemic autophagy ablation treatment on the immune system, given the known role of autophagy in various immune cell types (Michaud et al. 2011).
5. AUTOPHAGY AS A THERAPEUTIC TARGET
Because autophagy can support tumor growth and preclinical evidence has demonstrated the role of autophagy inhibition as a promising therapeutic strategy, there are now numerous ongoing clinical trials (http://www.cancer.gov/clinicaltrials) assessing the efficacy of the lysosomal inhibitor HCQ. Like CQ, HCQ is a weak basic tertiary amine that can accumulate in the acidic lysosome where it is protonated, thereby inhibiting diffusion out of the lysosome. This results in an increase in the pH of the lysosome, which inhibits lysosomal function (Homewood et al. 1972) and autophagy in the process. Early studies have refined the appropriate doses of lysosomal inhibitors and shown potential efficacy with regard to tumor response. Although most of these trials are ongoing, there have been encouraging results from several Phase I and Phase II trials (Rebecca & Amaravadi 2016, Wang et al. 2016). The majority of the trials have used a combination of HCQ and other antineoplastic regimens, including chemotherapy, targeted therapy, and radiation therapy, given the multiple studies showing that autophagy is activated as a survival response to antineoplastic therapy (discussed above) (Kimmelman 2011). Examples of completed studies include a Phase I trial combining HCQ with bortezomib (a proteasome inhibitor) in relapsed or refractory multiple myeloma in which an improved effect was noted in comparison with historical use of bortezomib alone (Vogl et al. 2014). A trial with temozolomide and HCQ for patients primarily with metastatic melanoma showed a 41% partial response or stable disease (Rangwala et al. 2014). However, a Phase II trial of HCQ, temozolomide, and radiation in glioblastoma patients showed a median survival of 15.6 months, which was not significantly improved compared with historical controls (Rosenfeld et al. 2014, Stupp et al. 2005). Notably, the mean tolerated dose of HCQ in this trial was 600 mg/day and at that dose autophagy inhibition was not consistently achieved. Larger studies are necessary to determine the utility of HCQ as an anticancer therapy, and several are ongoing.
Based on work from our laboratory and others, clinical trials using HCQ as part of a PDAC therapeutic regimen have been initiated. Although HCQ as monotherapy in a Phase II trial of PDAC patients with metastatic disease who had progressed through multiple lines of therapy showed no objective responses, this may have been a result of the fact that these patients were a heavily pretreated population and did not remain on HCQ for a long period and, thus, may have not achieved therapeutic doses (Wolpin et al. 2014). Alternatively, it may reflect the need for combination therapy in this highly treatment-refractory disease. More recent studies of HCQ-mediated autophagy inhibition in PDAC have been promising, including a Phase I and II study of preoperative HCQ and gemcitabine therapy that showed CA19-9 tumor marker response and improvement in overall survival compared with historical controls (Boone et al. 2015). Based on these data, a randomized Phase II trial has opened, combining HCQ with gemcitabine and nab-paclitaxel (nanoparticle albumin-bound paclitaxel) in the neoadjuvant setting.
There are many unanswered questions with regard to the use of autophagy inhibition as a cancer therapeutic. Understanding which patients will respond to autophagy inhibition a priori is an ongoing challenge (Mancias & Kimmelman 2011). Whereas GEMM studies suggest a reliance of autophagy in Ras-driven cancers, this bears further testing, as preclinical data suggest that this might not be a sufficient biomarker for selecting patients (Morgan et al. 2014). Interestingly, other tumor-driver mutations may predict responsiveness to autophagy inhibition, including the BRAFV600E mutation. Indeed, a pediatric patient with a brainstem ganglioglioma harboring a V600E mutation had a significant and sustained response to a combination of CQ and RAF inhibition with vemurafenib (Levy et al. 2014). More generally, it is possible that tumors that have a high basal level of autophagy may respond best to autophagy inhibition. Unfortunately, our methods for assessing autophagy levels in vivo or the pharmacodynamic response to autophagy inhibition are limited to monitoring LC3-II levels and LC3 puncta or using electron micrographs to detect autophagosomes. Therefore, better biomarkers and methods require further development (Kimmelman 2011, Mancias & Kimmelman 2011). Although CQ and HCQ are effective inhibitors of autophagy in vitro, it is unclear whether the doses used in clinical trials effectively inhibit autophagy in vivo. Another issue with HCQ is that the pharmacokinetics are unfavorable, with long periods of time required for adequate micromolar dose levels to be reached (Munster et al. 2002, Tett et al. 1993). Therefore, there is an intensive effort to identify not only new analogs of CQ, such as Lys05, but also drugs targeting other aspects of the autophagic pathway (Solomon et al. 2010). Although many of the efforts have sought to target the canonical autophagic pathway, this may miss the noncanonical pathways, such as LC3-associated phagocytosis (Kim & Overholtzer 2013), that cancer cells may depend upon. Both canonical and noncanonical autophagic pathways, as well as macropinocytosis, an extracellular scavenging mechanism used by some cancers to support metabolism (Commisso et al. 2013), converge on the lysosome; therefore, it may be that the lysosome is the best target. To that end, Lys05 is being tested preclinically, and additional antimalarial analogs are being actively studied, such as VATG-027, which disrupts lysosomal processes (Goodall et al. 2014).
There are multiple additional components of the autophagic process that are potentially targets for therapeutic intervention. Given the success with targeting kinases, multiple pharmaceutical companies have designed specific inhibitors of VPS34 (Figure 1) (Dowdle et al. 2014). Many of these are highly specific and avoid cross-reactivity with others of the class I and class II PI3Ks; however, there remain concerns with regard to altering endosomal trafficking, given the role of VPS34 in multiple trafficking events (Rebecca & Amaravadi 2016, Wang et al. 2016). BECN1 is a binding partner of VPS34 and has an important regulatory role in the PI(3)KCIII complex (Figure 1) and, therefore, has been identified as a target for autophagy inhibition. Spautin-1 has been shown to stimulate degradation of the BECN1–VPS34 complex by inhibiting two ubiquitin-specific proteases that regulate the stability of the complex. Spautin-1 has been shown in preclinical studies to enhance cancer cell death in the setting of nutrient deprivation when autophagy would generally act as a survival mechanism. Spautin-1 has also been shown to have other activities, including roles in activating nucleotide excision repair, highlighting the need for more specific inhibitors of BECN1–VPS34 as a means of autophagy inhibition (Liu et al. 2011).
Because ULK1–2 is an integral component of the autophagy initiation machinery, it is an obvious target for autophagy inhibitors (Figure 1). To date, groups have attempted to directly inhibit ULK1–2 kinase activity through various small molecule inhibitors, including MRT68921, MRT67307, and SBI-0206965. Off-target inhibitory effects on other kinases may limit the utility of these compounds for autophagy-specific inhibition (Egan et al. 2015, Petherick et al. 2015). Additionally, ULK1-independent autophagy has been described (Cheong et al. 2011). Initial efforts toward developing inhibitors against ATG4B (Figure 1) are in the preclinical phase, but they show promise with regard to inhibiting ATG8 processing (Akin et al. 2014). Likewise, targeting the downstream ubiquitin-like conjugation machinery, such as the ATG7 enzyme (Figure 1), may represent a viable therapeutic strategy.
Whichever approach is taken to inhibit autophagy, the balance between potency and toxicity must be considered, as autophagy clearly has a key role in normal tissue homeostasis. Novel inhibitors and combination therapies should be critically evaluated in GEMMs to assess both the efficacy and effects on normal tissues.
6. FUTURE PERSPECTIVES
The role of autophagy in cancer is complex, with the data supporting its role in constraining cancer initiation and, later, in a protumorigenic process. The tumor-promoting roles of autophagy support tumor growth by providing necessary nutrients and managing ROS. Furthermore, autophagy can support therapeutic resistance to cytotoxic chemotherapy, molecularly targeted agents, and radiotherapy. There are many questions that are the focus of ongoing work in the field of autophagy in cancer. Most fundamentally, the molecular events that tumor cells employ to switch on higher basal levels of autophagy have only begun to be elucidated (Perera et al. 2015, Wong et al. 2015). Also unclear at this time is whether there are particular cargos that are selectively degraded in tumors to support growth. Some proteomic studies have begun to address this issue (Mancias et al. 2014, Mathew et al. 2014). Given the complexity of the tumor-microenvironment interaction, further in vivo studies are required to understand how the microenvironment may modulate the dependence of tumors on autophagy. Although there have been advances in using organotypic cultures, these studies will most likely have to be performed in vivo and will require advanced mouse models with genetic manipulation of autophagy activity. These studies will also improve our understanding of the effects of autophagy inhibition in the tumor and microenvironment, and inform our understanding of the relative contribution of the tumor cell-autonomous versus nonautonomous effects of autophagy inhibition on tumor efficacy studies. A great deal of preclinical data support autophagy inhibition as an anticancer strategy. Much work is required to understand the subset of tumors that would benefit most from autophagy inhibition. In part, this will depend on developing more robust biomarkers of the basal autophagy level in tumors. These biomarkers will also be useful in ongoing clinical trials of autophagy inhibition as a cancer therapeutic.
ACKNOWLEDGMENTS
The authors apologize for the omission of any primary references. This work was supported by National Institutes of Health grant GM095567, National Cancer Institute grants R01CA15749 and R01CA188048, and American Cancer Society Research Scholar Grant RSG-13-298-01-TBG to A.C.K. J.D.M. is supported by a KL2/Catalyst Medical Research Investigator Training award (TR001100) and a Burroughs Wellcome Fund Career Award for Medical Scientists.
Glossary
- Atg
autophagy related
- ROS
reactive oxygen species
- PDAC
pancreatic ductal adenocarcinoma
- ATP
adenosine triphosphate
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- CQ
chloroquine
- HCQ
hydroxychloroquine
- TCA
tricarboxylic acid
- NSCLC
non-small-cell lung carcinoma
- GEMM
genetically engineered mouse model
- TDCL
tumor-derived cell line
- LOH
loss of heterozygosity
- CAF
cancer-associated fibroblast
- IFN
interferon
Footnotes
DISCLOSURE STATEMENT
J.D.M and A.C.K. have grant support to study the role of autophagy in pancreatic cancer.
LITERATURE CITED
- Akin D, Wang SK, Habibzadegah-Tari P, Law B, Ostrov D, et al. 2014. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 10:2021–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, et al. 2007. Autophagy inhibition enhances therapy-induced apoptosis in aMyc-induced model of lymphoma. J. Clin. Investig. 117:326–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. 2008. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 68:1485–94 [DOI] [PubMed] [Google Scholar]
- Bago R, Malik N, Munson MJ, Prescott AR, Davies P, et al. 2014. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J. 463:413–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW. 2010. Network organization of the human autophagy system. Nature 466:68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras M-C, Muthuswamy LB, et al. 2012. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491:399–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, et al. 2015. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine patients with pancreatic adenocar-cinoma. Ann. Surg. Oncol. 22:4402–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, et al. 2007. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 110:313–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. 2011. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. PNAS 108:11121–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. 2013. Autophagy in human health and disease. N. Engl. J. Med. 368:651–62 [DOI] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, et al. 2013. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497:633–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Wong E. 2014. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24:92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, et al. 2002. Mutations of the BRAF gene in human cancer. Nature 417:949–54 [DOI] [PubMed] [Google Scholar]
- De Duve C, Wattiaux R. 1966. Functions of lysosomes. Annu. Rev. Physiol. 28:435–92 [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, et al. 2006. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10:51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtyarev M, De Mazière A, Orr C, Lin J, Lee BB, et al. 2008. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 183:101–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, et al. 2005. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. PNAS 102:7922–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZB, Hui B, Shi YH, Zhou J, Peng YF, et al. 2011. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin. Cancer Res. 17:6229–38 [DOI] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan J-A, et al. 2015. Autophagy mediates degradation of nuclear lamina. Nature 527:105–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, et al. 2014. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 16:1069–79 [DOI] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, et al. 2008. The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell 13:343–54 [DOI] [PubMed] [Google Scholar]
- Egan DF, Chun MGH, Vamos M, Zou H, Rong J, et al. 2015. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 59:285–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, et al. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, et al. 2016. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. PNAS 113:182–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, et al. 2007. The anticancer drug imatinib induces cellular autophagy. Leukemia 21:936–42 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Levine B, Kroemer G. 2014. Metabolic control of autophagy. Cell 159:1263–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall ML, Wang T, Martin KR, Kortus MG, Kauffman AL, et al. 2014. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy 10:1120–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, et al. 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25:460–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, et al. 2013a. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 27:1447–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Xia B, White E. 2013b. Autophagy-mediated tumor promotion. Cell 155:1216–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–74 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. 2012. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13:251–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homewood CA, Warhurst DC, Peters W, Baggaley VC. 1972. Lysosomes, pH and the anti-malarial action of chloroquine. Nature 235:50–52 [DOI] [PubMed] [Google Scholar]
- Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. 2010. Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunol. 185:7349–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, et al. 2013. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. CancerDiscov. 3:894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA, Linn S. 1988. DNA damage and oxygen radical toxicity. Science 240:1302–9 [DOI] [PubMed] [Google Scholar]
- Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, et al. 2011. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 193:275–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Daido S, Kanzawa T, Kondo S, Kondo Y. 2005. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 26:1401–10 [PubMed] [Google Scholar]
- Jacquel A, Obba S, Boyer L, Dufies M, Robert G, et al. 2012. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 119:4527–31 [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, et al. 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321:1801–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. 2010MTOR regulation of autophagy. FEBS Lett. 584:1287–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBOJ. 19:5720–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. 2006. Fibroblasts in cancer. Nat. Rev. Cancer 6:392–401 [DOI] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, et al. 2015The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349:aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. 2004. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 11:448–57 [DOI] [PubMed] [Google Scholar]
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, et al. 2007. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 21:1621–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, et al. 2014. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4:915–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J, Debnath J. 2015. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16:461–72 [DOI] [PubMed] [Google Scholar]
- Kenific CM, Debnath J. 2015. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 25:37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Mello SS, Bieging KT, Jiang D, Dusek RL, et al. 2013. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27:1016–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I. 2015a. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 26:6–16 [DOI] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, et al. 2015b. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522:354–58 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan K-L. 2011a. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13:132–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, et al. 2011b. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 286:12924–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SE, Overholtzer M. 2013. Autophagy proteins regulate cell engulfment mechanisms that participate in cancer. Semin. Cancer Biol. 23:329–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC. 2011. The dynamic nature of autophagy in cancer. Genes Dev. 25:1999–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Schulman BA. 2014. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 21:336–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondylis V, Van Nispen Tot Pannerden HE, Van Dijk S, Ten Broeke T, Wubbolts R, et al. 2013. Endosome-mediated autophagy: an unconventional MIIC-driven autophagic pathway operational in dendritic cells. Autophagy 9:861–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, et al. 2004. The role of autophagy during the early neonatal starvation period. Nature 432:1032–36 [DOI] [PubMed] [Google Scholar]
- Laddha SV, Ganesan S, Chan CS, White E. 2014. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol. Cancer Res. 12:485–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. 2012. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 18:370–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, et al. 2015. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11:1668–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, et al. 2010. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32:227–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévy J, Cacheux W, Bara MA, L’Hermitte A, Lepage P, et al. 2015. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 17:1062–73 [DOI] [PubMed] [Google Scholar]
- Levy JMM, Thompson JC, Griesinger AM, Amani V, Donson AM, et al. 2014. Autophagy inhibition improves chemosensitivity in BRAFV600E brain tumors. Cancer Discov. 4:773–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JMM, Thorburn A. 2011. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol. Ther. 131:130–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shen C, Nakamura E, Ando K, Signoretti S, et al. 2013. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell 24:738–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WW, Li J, Bao JK. 2012. Microautophagy: lesser-known self-eating. Cell. Mol. Life Sci. 69:1125–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xia H, Kim M, Xu L, Li Y, et al. 2011Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147:223–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. 2014. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 4:466–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, et al. 2011. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 22:165–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes H, Rubio N, Garg AD, Agostinis P. 2013. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 19:428–46 [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, et al. 2009. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 16:87–93 [DOI] [PubMed] [Google Scholar]
- Mancias JD, Kimmelman AC. 2011. Targeting autophagy addiction in cancer. Oncotarget 2:1302–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Kimmelman AC. 2016. Mechanisms of selective autophagy in normal physiology and cancer. J. Mol. Biol. 428:1659–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. 2014. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509:105–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, et al. 2009. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:1062–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, White E. 2014. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol. Cell 55:916–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, et al. 2007. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 21:1367–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melser S, Lavie J, Bénard G. 2015. Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta 1853:2812–21 [DOI] [PubMed] [Google Scholar]
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, et al. 2011. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334:1573–77 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell 147:728–41 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, et al. 1998. A protein conjugation system essential for autophagy. Nature 395:395–98 [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Gamez G, Menke C, Hernandez A, Thorburn J, et al. 2014. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy 10:1814–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT. 2009. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137:1001–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster T, Gibbs JP, Shen D, Baethge BA, Botstein GR, et al. 2002. Hydroxychloroquine concentration-response relationships in patients with rheumatoid arthritis. Arthritis Rheum. 46:1460–69 [DOI] [PubMed] [Google Scholar]
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, et al. 2009. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461:654–58 [DOI] [PubMed] [Google Scholar]
- Noda NN, Inagaki F. 2015. Mechanisms of autophagy. Annu. Rev. Biophys. 44:101–22 [DOI] [PubMed] [Google Scholar]
- Ondrej M, Cechakova L, Durisova K, Pejchal J, Tichy A. 2016. To live or let die: unclear task of autophagy in the radiosensitization battle. Radiother. Oncol. 119:265–75 [DOI] [PubMed] [Google Scholar]
- Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, et al. 2014. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol. Cell 53:471–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier B, El-Ahmad Y, Filoche-Rommé B, Dureuil C, Fassy F, et al. 2014. Discovery of (2S)-8-[(3R)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)-3,4-dihydro-2H-pyrimido[1,2-a] pyrimidin-6-one: a novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J. Med. Chem. 58:376–400 [DOI] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, et al. 2015. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 524:361–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Mancera PA, Young ARJ, Narita M. 2014. Inside and out: the activities of senescence in cancer. Nat. Rev. Cancer 14:547–58 [DOI] [PubMed] [Google Scholar]
- Petherick KJ, Conway OJL, Mpamhanga C, Osborne SA, Kamal A, et al. 2015. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 290:11376–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. 2015. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 4:184–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V. 2015. Secretory autophagy. Curr. Opin. Cell Biol. 35:106–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He Y-W. 2007. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 204:25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, et al. 2003. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 112:1809–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. 2010. Autophagy and metabolism. Science 330:1344–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, et al. 2014. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 10:1391–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, et al. 2014. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 5:3056. [DOI] [PubMed] [Google Scholar]
- Rebecca VW, Amaravadi RK. 2016. Emerging strategies to effectively target autophagy in cancer. Oncogene 35:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, et al. 2014. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 10:1013–19 [DOI] [PubMed] [Google Scholar]
- Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, et al. 2014. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10:1359–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, et al. 2013. P53 status determines the role of autophagy in pancreatic tumour development. Nature 504:296–300 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. 2012. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 11:709–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakitani K, Hirata Y, Hikiba Y, Hayakawa Y, Ihara S, et al. 2015. Inhibition of autophagy exerts anti-colon cancer effects via apoptosis induced by p53 activation and ER stress. BMC Cancer 15:795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, Dipaola RS. 2016. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 30:399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Elazar Z. 2007. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17:422–27 [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. 2007. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBOJ. 26:1749–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Cuervo AM. 2011. Autophagy in the cellular energetic balance. Cell Metab. 13:495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, et al. 2009. Autophagy regulates lipid metabolism. Nature 458:1131–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobodkin MR, Elazar Z. 2013. The Atg8 family: multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 55:51–64 [DOI] [PubMed] [Google Scholar]
- Solomon VR, Hu C, Lee H. 2010. Design and synthesis of chloroquine analogs with anti-breast cancer property. Eur. J. Med. Chem. 45:3916–23 [DOI] [PubMed] [Google Scholar]
- Sorbara MT, Girardin SE. 2015. Emerging themes in bacterial autophagy. Curr. Opin. Microbiol. 23:163–70 [DOI] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, et al. 2016. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. In press. doi: 10.1038/nature19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, et al. 2013. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 3:1272–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason W, van den Bent MJ, Weller M, Fisher BM, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352:987–96 [DOI] [PubMed] [Google Scholar]
- Svenning S, Johansen T. 2013. Selective autophagy. Essays Biochem. 55:79–92 [DOI] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, et al. 2011. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25:795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Sebti S, Titone R, Zhou Y, Isidoro C, et al. 2015a. Decreased BECN1 mRNA expression in human breast cancer is associated with estrogen receptor-negative subtypes and poor prognosis. EBioMedicine 2:255–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Di J, Cao H, Bai J, Zheng J. 2015b. p53-mediated autophagic regulation: a prospective strategy for cancer therapy. Cancer Lett. 363:101–7 [DOI] [PubMed] [Google Scholar]
- Tett SE, Day RO, Cutler DJ. 1993. Concentration-effect relationship of hydroxychloroquine in rheumatoid arthritis—a cross sectional study. J. Rheumatol. 20:1874–79 [PubMed] [Google Scholar]
- Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, et al. 2011. Crucial role for autophagy in degranulation of mast cells. J. Allergy Clin. Immunol. 127:1267–76 [DOI] [PubMed] [Google Scholar]
- Valencia T, Kim J, Abu-Baker S, Moscat-Pardos J, Ahn C, et al. 2014. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 26:121–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl DT, Stadtmauer EA, Tan K-S, Heitjan DF, Davis LE, et al. 2014. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 10:1380–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Hu Q, Shen H-M. 2016. Pharmacological inhibitors of autophagy as novel cancer therapeutic agents. Pharmacol. Res. 105:164–75 [DOI] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. 2011. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 25:1510–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. 2015. The role for autophagy in cancer. J. Clin. Investig. 125:42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willinger T, Flavell RA. 2012. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. PNAS 109:8670–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, et al. 2014. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 19:637–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P-M, Feng Y, Wang J, Shi R, Jiang X. 2015. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 6:8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Koh JY, Price S, White E, Mehnert JM. 2015. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 5:410–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, et al. 2014. Autophagyis critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 4:905–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, et al. 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25:717–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. 2010. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12:814–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. 2003. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. PNAS 100:15077–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cheng Y, Ren X, Zhang L, Yap KL, et al. 2012. NAC1 modulates sensitivity of ovarian cancer cells to cisplatin by altering the HMGB1-mediated autophagic response. Oncogene 31:1055–64 [DOI] [PMC free article] [PubMed] [Google Scholar]