

ABSTRACT
Hypoxia, a common condition of the tumor microenvironment, induces changes in the proteome of cancer cells, mainly via HIF-1, a transcription factor conformed by a constitutively expressed β-subunit and an oxygen-regulated α-subunit. In hypoxia, HIF-1α stabilizes, forms the heterodimeric complex with HIF-1β, and binds to Hypoxia Response Elements (HRE), activating gene expression to promote metabolic adaptation, cell invasion and metastasis. Furthermore, the focal adhesion kinase, FAK, is activated in hypoxia, promoting cell migration by mechanisms that remain unclear. In this context, integrins, which are glycoproteins required for cell migration, are possibly involved in hypoxia-induced FAK activation. Evidence suggests that cancer cells have an altered glycosylation metabolism, mostly by the expression of glycosyltransferases, however the relevance of glycosylation is poorly explored in the context of hypoxia. Here, we discuss the role of hypoxia in cancer, and its effects on protein glycosylation, with emphasis on integrins and cell migration.
KEYWORDS: Hypoxia, glycosylation, cell migration, integrins
Introduction
Cancer is the second most common cause of death worldwide, with an estimated of 171,2 deaths per 100,000 people per year, based on the 2008–2012 period, which is projected to continue increasing [1] . For this reason, it becomes important to study the molecular mechanisms involved in the development of tumors, in order to find new therapeutic targets. Altered glycosylation in human cancer cells has been acknowledged decades ago, and recent studies have increased our understanding about the role of protein glycosylation in tumor progression [2]. It has been suggested that the wide variety of changes in the glycoproteome depends on the origin and malignity of tumor cells, which could be due to transcriptional regulation of glycosyltransferases (GTs), the responsible enzymes for glycosylation [3]. Although, most studies have been focused on understanding the mechanisms by which glycosylation is altered in cancer cells, the effects of conditions in the tumor microenvironment and particularly hypoxia in the glycoproteome, is poorly explored.
Hypoxia is a common condition of the tumor microenvironment, which is associated with poor patient prognosis, enhanced tumor cell migration, invasion, and metastasis [4–6]. In this review, we will discuss the current state of the art about the relevance of hypoxia on protein glycosylation, and its consequences on tumor cell migration.
Hypoxia in cancer
Low oxygen concentration or hypoxia is a potent microenvironmental factor observed in solid tumors that promotes metastasis [7], which is also associated with resistance to radiation therapy [8] and chemotherapy [9] . Normal oxygen concentration ranges from 19,7% to 3,8%, depending on a particular tissue; however, in solid tumors, oxygen concentration values are close to 0,3% to 2,1% depending on the tumor type [10,11]. In fact, the general consensus defines normoxia as the oxygen concentration commonly used in cell cultures (19,95%), whereas hypoxia is considered as any oxygen concentrations below physiologic normoxia (generally 1% oxygen) [10,11].
Cell adaptation to hypoxia involves the expression of a group of genes, many of which are transcriptionally regulated by the hypoxia-inducible factor 1 (HIF-1), which can induce changes in the proteome of tumor cells [12]. HIF-1, is a heterodimeric transcription factor consisting of a constitutively expressed β-subunit and an oxygen-regulated α-subunit. In normoxia, HIF-1α is hydroxylated on P402 and P564, which allows its interaction with the von Hippel-Lindau (VHL) E3 ubiquitin ligase, followed by its rapid polyubiquitynation and proteasomal degradation [13]. Alternatively, in hypoxia, HIF-1α is no longer degraded, allowing to its stabilization, and in a complex with HIF-1β, it binds to Hypoxia Response Element sequences (HRE; 5′[AG]CGTG-3′) localized in the promoter of HIF-1 target genes, to initiate gene expression [13]. More than 60 putative direct HIF-1 target genes have been identified, which could have a function in the development of the tumor phenotype [14]. Several groups, including our laboratory, have established that tumor cell migration and invasion are stimulated by hypoxia [15–20], and it has been suggested that hypoxia activates these phenotypes in tumor cells via multiple mechanisms, for example, through the direct and indirect regulation of the epithelial to mesenchymal transition (EMT) transcription factors Snail, Slug, Twist, and Zeb1 [21], or by promoting the invasion of surrounding tissues via upregulation and secretion of proteolytic enzymes, such as matrix metalloproteinases (MMPs), cathepsins, lysyl oxidases, and prolyl-4-hydroxylses (P4H) [22,23]. Another interesting mechanism whereby hypoxia increases cell migration is via activation of the Focal Adhesion Kinase (FAK) [15–17,24]. Our laboratory recently showed that hypoxia increases the phosphorylating activation of FAK (phosphorylation on Y397) in epithelial tumor cells, leading to sustained migration, invasion, and ECM remodeling [17]. Although the mechanisms underlying FAK activation by hypoxia are poorly understood, one possibility is that FAK is activated downstream of integrin receptors, which is supported by evidence showing that hypoxia induces the expression of integrins in several tumor cell models [25–28]. Interestingly, integrins and other cell surface proteins involved in cell migration, and known to be upregulated in hypoxia, are highly glycosylated [29,30]. Nevertheless, the role of hypoxia on protein glycosylation remains poorly understood.
Glycosylation in cancer
Glycosylation is one of the most complex post-translational modifications, because it varies according to the expression of glycosylating enzymes, namely glycosyltransferases (GTs) [30,31]. GTs expression and their glycosylating activity depend on the cellular environment and the physio-pathological states, generating structural diversity on each glycoprotein repertoire [31]. The complete set of glycoproteins encoded by the genome is known as the cell glycoproteome, which can be structurally different between cells [31]. Changes in glycosylation, especially sialylation and fucosylation of glycans, are hallmarks of cancer cells, linked to an increase in oncogenic potential and invasiveness [2]. Alterations in glycan expression may be due to under or over-expression of GTs (Table 1). For instance, N-acetylglucosaminyltransferase V (GnT-V), sialyltransferases ST3Gal-I, ST6Gal1 and ST6GalNAc, and fucosyltransferases FUT1, FUT2, FUT3, FUT4 and FUT8 are often overexpressed in tumor cells (Table 1) [2,32]. Upregulation of these GTs leads to the expression of common tumor-cell epitopes such as sialyl-Lewisx and sialyl-Lewisa (sLex/sLea), Thomsen-nouvelle antigen (Tn), and sialyl-Tn (sTn), commonly used as tumor markers [33]. Additionally, it has been shown that GTs subcellular distribution is important for glycosylation. For instance, overexpression of ST6GalNAc-I, an enzyme that participates in sTn biosynthesis, leads to its accumulation throughout all Golgi cisternae, disrupting glycosylation by prematurely adding sialic acid to form the STn antigen [34]. Also, pH-dependent mislocalization of ST3 in Golgi, was correlated with impaired α(2,3)-sialylation of N-glycans [35].
Table 1.
Regulation of glycosyltransferases in cancer. Glycosyltransferases upregulated or downregulated on cancer.
| Enzyme | Regulation | |
|---|---|---|
| Sialyltransferases | ST3GAL1 ST3GAL3, ST3GAL4, ST3GAL6, ST6GAL1, ST6GALNAC1, ST6GALNAC2 ST8SIA1 | Up-regulated |
| ST3GAL3, ST3GAL 4 ST3GAL6, ST6GALNAC | Down-regulated | |
| Fucosyltransferases | FUT1, FUT2, FUT3, FUT4, FUT5, FUT6 FUT7 FUT8 FUT9, FUT10 FUT11 |
Up-regulated |
| FUT3, FUT4, FUT9, FUT10 | Down-regulated | |
| Others | - Glucosaminyl (N-acetyl) transferase (GNT1) - N-Acetylgalactosaminyltransferase 14 (GALNT14) - β-1,4-galactosyltransferase 1–2-3–5 (B4GALT) - Mannosyl (β1,4-)-glycoprotein β-1,4-N-acetylglucosaminyltransferas (MGAT3) - Mannosyl (alpha-1,6-)-glycoprotein β −1,6-N-acetyl-glucosaminyltransferase (MGAT5) |
Up-regulated |
| - Mannosyl (β1,4-)-glycoprotein β-1,4-N-acetylglucosaminyltransferas (MGAT3) | Down-regulated |
Glycosylation as a marker of malignancy
Increased branching of glycans and GTs expression have been associated with an increased malignancy in different tumor cells. Specifically, in breast cancer cells, upregulation of sialyltransferases, fucosyltransferases and N-acetylglucosaminyltransferases is associated with increased malignancy [36–39] metastasis and poor patient prognosis [40–42]. In colon cancer, augmented expression of mucins (heavily glycosylated proteins) and sialylated Tn and Lex antigens in tissues, is associated with increased metastasis [43], whereas increased fucosylation is associated with increased cellular adhesion [44]. Similar observations have been made in other tumors cells, such as liver, ovarian and melanoma cancer, among others, highlighting the importance of glycosylation in tumor malignancy, however, it remains poorly understood if microenvironmental conditions, such as hypoxia, could affect differentially protein glycosylation of malignant tumors cells [2].
Glycosylation in hypoxia
Growing evidence suggest that hypoxia affects the expression of GTs in tumor cells, including human colon cancer cell lines SW480, C1 and Colo20, where a 7-day exposure to hypoxia was shown to increase the transcription of genes encoding for FUT7, ST3Gal-I and UDP-galactose transporter-1 (UGT1) [45]. Similarly, hypoxia promotes the transcription of FUT7, FUT1, GCNT2 and GCNT3 in immortalized prostate RWPE1 cells [46], although importantly, it remains unclear if HIF-1α is directly involved in such responses. The only evidence indicating that HIF-1α regulates GT expression was obtained by siRNA-based approaches, where HIF1α downregulation was followed by increased levels of mRNA encoding for FUT1 and FUT2, which was correlated with increased binding of the lectin UEA-1 and the antibody anti-LeY to the surface of HIFα-depleted cells [47]. However, whether HIF1α can bind to the promoter of GTs, allowing the transcription of GTs, and the putative effects of hypoxia on GTs protein levels and subcellular localization, remain largely unexplored. Consequently, the effects on protein glycosylation remain poorly understood.
Alternatively, protein glycosylation depends on both, monosaccharide synthesis and transport from cytosol to the ER and Golgi being central [48].UDP-GlcNAc, an intermediate necessary for N and O-linked glycosylation, is synthesized in the hexosamine pathway, from monosaccharide precursors, such as glucose [49]. Consequently, alterations in this biosynthetic pathway or in previous transport of precursors affect the glycosylation pattern. In this respect, changes in levels of the different transporters and enzymes involved in their synthesis, have been reported during hypoxia [50]. Specifically, hypoxia has been shown to increase the expression of glucose transporters GLUT1 and GLUT3, as well as the enzyme hexokinase-2 (HKII) in a variety of cell lines [50]. Moreover, hypoxia promotes the expression of the enzyme pyruvate dehydrogenase kinase 1 (PDK1) in human Burkitt’s lymphoma cell line P493-6, in a time-dependent manner [51]. Additionally, that study reported that HIF-1 directly regulates the expression of PDK1 by chromatin immunoprecipitation assays [51]. Alternatively, a Hypoxia Response Element (HRE) was found in the promoter of glycogen synthase 1 (GYS1), as shown in skeletal muscle myotube cells C2CL2, where HIF-1α binding allows the expression of GYS1, resulting in an increased activity and the accumulation of glycogen [52]. Other studies showed that HIF-1α controls glycogen levels by inducing the expression of PPP1R3C (also known as ‘protein targeting to glycogen’ (PTG)), via binding to a functional HRE sequence [53]. Also, mRNA levels of Glutamine-fructose-6-phosphate (GFPT1) transaminase increases during hypoxia, which correlates with an increased enzyme activity [54]. Intriguingly, despite Glycosylated Phosphofructokinase 1 (PFK1) levels are increased during hypoxia, PFK1 activity was shown to decrease [55].
Collectively, all these data suggest that hypoxia has a relevant role in protein glycosylation, either directly or indirectly, leading to altered functional activities. In this respect, it will be critical assessing the role of glycosylation-induced by hypoxia on proteins relevant to the migratory, invasive, and metastatic characteristics in tumor cells. This is particularly relevant, because several proteins involved in tumor cell migration and invasion are heavily glycosylated, those including integrins, metalloproteinases, and cadherins, amongst others [56,57]. Most importantly, several proteins from this group, specially integrins, are known to be deregulated in hypoxia [23,25]. Hence, the putative role of hypoxia on integrin glycosylation, is just beginning to be explored, and thus initial studies in the field will be discussed in the upcoming paragraphs.
Integrin activation and trafficking
Integrins are a family of large transmembrane cell surface receptors composed of 18 α and 8 β subunits, which can form 24 different heterodimers and interact with different extracellular matrix ligands such as laminin, fibronectin and collagen, among others. Integrins regulate diverse cellular processes including cell survival, motility, and proliferation, and their contribution to cell migration and invasion is one of their most studied functions in tumor cell biology [58,59]. The function of integrins is controlled by activation and inactivation mechanisms of the receptor complex. In their inactive state, integrins are in a low-affinity closed conformation (bent) (Figure 1) [60]. Upon extracellular ligand binding (outside-in signaling), integrins undergo conformational changes, first to an intermediate affinity conformation (closed), and then to a high-affinity open conformation (extended), allowing to separation of the tail domain and subsequent interaction with cytoplasmic regulators (Figure 1) [60]. Alternatively, integrin signaling can be triggered by intracellular activators (inside-out signaling), such as talin or kindlin, which bind to the β tail, leading to conformational changes that increase the affinity to extracellular ligands [60]. After these initial events, integrin-containing adhesion complexes (referred to as focal adhesions, FAs) grow in size, and mature to form macromolecular complexes, which contain more than hundreds of different adhesion proteins, including FAK [60,61].
Figure 1.
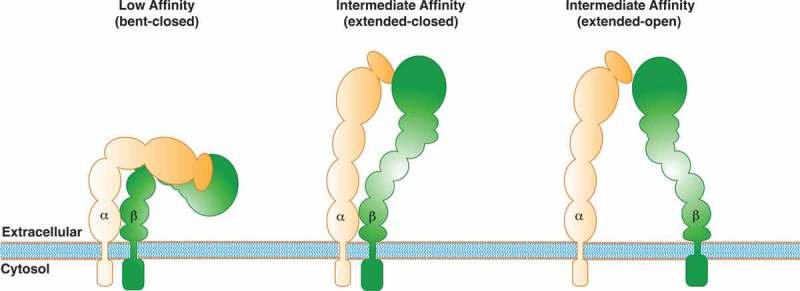
Schematic model of β1-integrin activation. The model shows different conformations of integrins, along with their different affinities: the bent conformation (low-affinity), and the two extended head-piece conformations (intermediate-affinity and high-affinity).
As for other cell surface receptors, integrin signaling is also regulated by the delivery of newly synthesized proteins to the cell surface, as well as their endocytosis from the plasma membrane, and their recycling or degradative trafficking routes [61–63]. It has been shown that inactive and active (ligand-bound) conformations of integrin heterodimers can be internalized via macropinocytosis [64], clathrin-dependent endocytosis [65] and clathrin-independent endocytosis, which includes caveolae-mediated endocytosis [66] and clathrin-independent carriers (CLICs) [67]. Intriguingly, regardless the endocytic mechanism, a subset of regulators of integrin trafficking has been extensively studied, namely a group of Rab small GTPases [61]. For instance, active and inactive β1-integrin are endocytosed through the clathrin and dynamin routes, allowing its co-localization with Rab5 and Rab21 in early endosomes, as shown in PC-3, MDA-MB-231 and NCI- H460 cells [68]. Also, in endothelial cells, the Ras and Rab Interactor 2 (RIN2) localizes to adhesion complexes and plays a role in the endocytosis of active β1-integrin [69]. On the other hand, endocytosed integrins traffic en route to late endosomes/lysosomes, or recycle back to the plasma membrane via recycling endosomes or alternative routes [61]. In this regard, active β1-integrin was shown to recycle via Rab11-dependent, long recycling loop, whereas the inactive conformation of this integrin is rapidly recycled in an Rab4-dependent manner [70]. Importantly, integrins have a long half-life (18 hours) when compared to the rates of their own recycling (30 minutes approximately, depending on the pathway), and several reports have demonstrated that non-recycled integrins are targeted for lysosomal degradation instead [61,70]. However, lysosomal targeting does not necessarily result in integrin degradation, because integrins can even recycle back to the plasma membrane from these compartments [68,70].
Alternatively, glycosylation has been proposed as a relevant post-translational mechanism contributing to integrin regulation [27–29]. Despite altered glycosylation has been proposed as a hallmark of cancer cells, and integrins are recognized as highly glycosylated proteins, only a few studies have addressed the effects of glycosylation on integrin expression, trafficking, localization and biological function. In this respect, current knowledge is rather limiting. The role of integrins in cancer cells is one of their most studied functions, because of their relevance in cell migration and invasion (reviewed in [71,72]). In this regard, a wide variety of integrins contribute to tumor progression and integrins known to be overexpressed in cancer include α6β4, α6β1, αvβ5, α2β1 and α3β1 [59]. Also, it has been suggested that several additional integrins are crucial for tumor angiogenesis, such as αVβ3, α1β1, α2β1, α4β1, α5β1, α6β1, α9β1 and α6β4 [59]. Noteworthy, several of these studies have pointed out to the relevance of β1-integrin in cancer (reviewed in [73]), and inhibition of this specific integrin with blocking antibodies, significantly impaired tumor growth in vitro and in vivo, which was also paralleled with antiangiogenic effects [74,75]. These compelling data highlight the central role of β1-integrin in tumor progression.
Effects of glycosylation on integrin function
Protein glycosylation is central to different cellular functions, because it affects protein folding, trafficking, subcellular localization, and degradation [76]. In this regard, accumulating evidence suggest that integrins possess over 20 potential N-glycosylation sites (Figure 2 shows a schematic summary of potential glycosylation sites, obtained from the Uniprot data base), and that N-glycosylation is crucial for integrin function [27,29,30]. Specifically, β1,6 branching of N-linked oligosaccharides on β1-integrin is important for both function and structure, because it is required for β1 clustering and interaction with the ECM [77]. Also, hyposialylation (i.e. specific changes in glycosylation branching size) of β1-integrin increases its binding to fibronectin and cell adhesion [78]. Moreover, overexpression of GnT-III blocked α5β1-mediated cell spreading, migration and FAK phosphorylation [79], suggesting that modification of the N-glycan branch size affects integrin downstream signaling and function. In this work, the authors focused on the effects of the overexpression of GnT-III on α2, α3 and α5, which substantially increased its reactivity to lectin E4-PHA in transfected cells.
Figure 2.
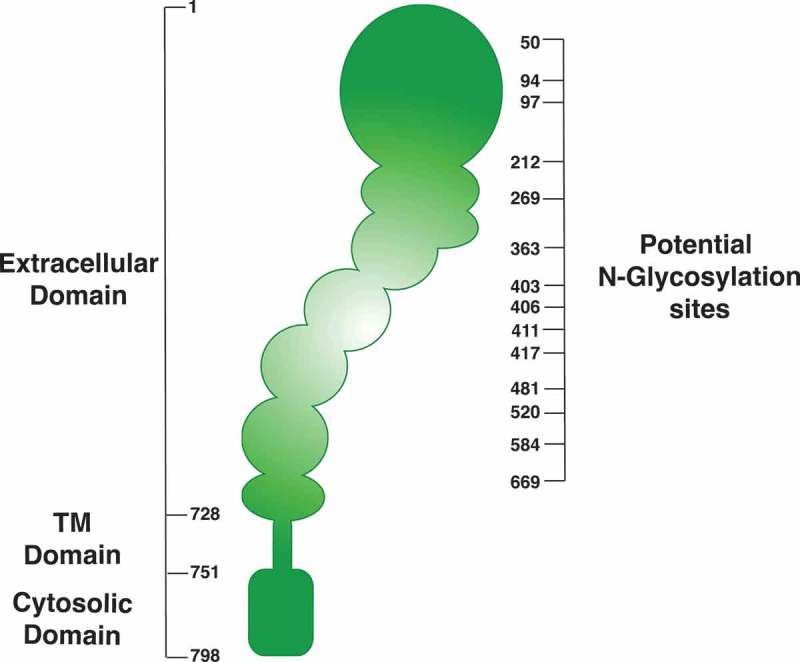
Schematic diagram of potential N-glycosylation sites of β1-integrin. Data was obtained using the Uniprot data base. http://www.uniprot.org/uniprot/P05556.
Finally, it has been shown that complete ablation of putative N-glycosylation sites in β1-integrin, by treatment with N-glycosidase F and tunicamycin, decreases integrin cell surface level and binding to fibronectin, respectively [80], whereas the ablation of glycosylation sites by mutagenesis leads to decreases in the cell surface levels of α5 [81] and β1 [82,83] and sustained FAK activity [81–83]. However, it remains unclear how specific changes in the glycosylation sites and their branching size could affect integrin subcellular localization, and most importantly, their regulation in the context of hypoxia, remains largely unexplored.
Effects of hypoxia on integrins
Hypoxia has been shown to enhance the expression of integrins, including β1-integrin in fibroblasts and keratinocytes [26,84], αvβ4 in breast carcinoma cells [28], β3 and β5 integrins in endothelial cells [85], αvβ3 in melanoma cells [24], β2 in leukocytes [86] and the α5 subunit in colon cancer cells [45]. Also, in human mesenchymal stem cells, hypoxia increased RNA expression of various integrins (α1, α3, α6, α11, αv, β1, β3) [87]. However, the underlying mechanisms involved in such regulations by hypoxia remain to be assessed. Specifically, it has been shown that HIF-1 and HIF-2 bind to the promoter of α5-integrin under hypoxic conditions [88], and that HIF-1 binds to the promoter of β1-integrin [26], however, whether hypoxia alters post-translational events leading to integrin expression and function, such as their trafficking and degradation, is not clear. The only available study that connects integrin glycosylation with hypoxia, showed that N-glycosylation of α3-integrin subunit is upregulated in A431 squamous carcinoma cells during hypoxia, leading to increased integrin surface levels, and enhanced cell invasion [89]. In the same work, the authors found an increase in overall β1-integrin glycosylation in hypoxia, however, they did not determine if hypoxia-dependent glycosylation on β1-integrin affects its subcellular localization or function [89]. Most importantly, the nature of the glycosylation sites, patterns, and branching sizes, were not explored. Collectively these pieces of evidences suggest that integrin glycosylation is a key event accounting for hypoxia dependent cell migration; however, determining the molecular mechanisms and precise residues undergoing glycosylation is a demanding issue.
Conclusions
Hypoxia is one of the most important factors within the tumor microenvironment as it is a determining factor in the prognosis of patients. However, to date, the mechanisms by which hypoxia controls tumor progression are not yet fully understood. Nevertheless, growing evidence support the roles that hypoxia and HIFs play in the control of tumor cell migration, invasion and metastasis. Despite that several proteins controlled by hypoxia and HIFs have been determined, the role of glycosylation and the expression of GTs in the biology of cancer in the context of hypoxia have been poorly addressed. Here, we spotlight the recent literature that shows the importance of protein glycosylation under the context of hypoxia, specifically in key proteins associated to cell migration and invasion. In this context, our focus was on integrins, since this family of cellular surface proteins offers many possibilities for regulation in the context of glycosylation, such as their own trafficking and subcellular localization, as well as their stability, activity status and downstream signaling (Figure 3). Despite this interesting approach, limited studies have shown the regulation of glycosylation of integrins in hypoxia and its role in the induction of cellular processes such as tumor cell migration and invasion. Even so, this hypothesis unfolds an interesting window for future design of therapeutic strategies that address cellular glycosylation regulated by hypoxia within the tumor context, specifically metastasis, that seek to control the cancer spread and improve the prognosis of patients.
Figure 3.
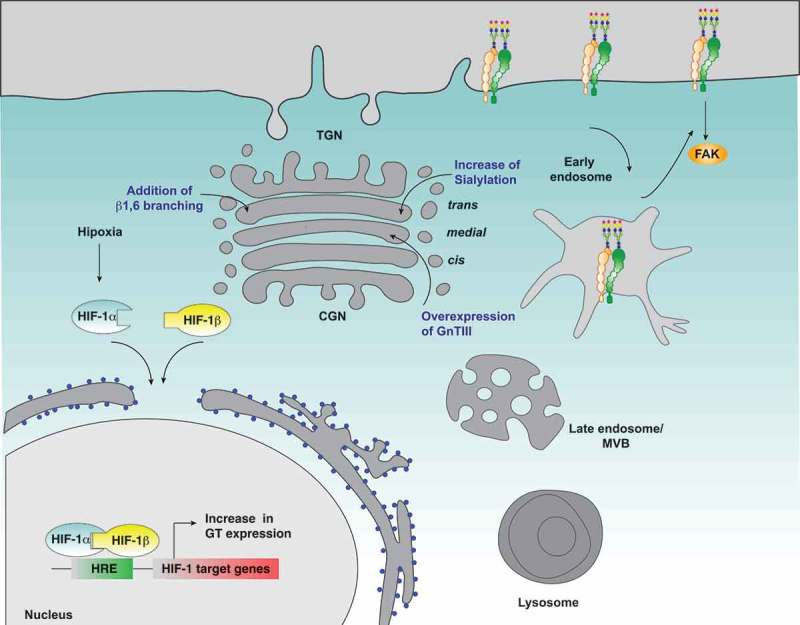
Possible effect of hypoxia on β1-integrin glycosylation and cell migration. Hypoxia induces the expression of glycosyltransferases in a HIF1α dependent manner, which could lead to changes in the glycosylation profile of β1-integrin, and thus affecting its subcellular distribution and function, its interaction with extracellular ligands, FAK signaling and cell migration. In blue letters are indicated the main modifications on β1-integrin glycosylation that affect its function and the Golgi compartments where they occur.
Funding Statement
This work was supported by the Fondo Nacional de Desarrollo Científico y Tecnológico, FONDECYT [1180495];
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Group USCSW United States cancer statistics: 1999–2012 incidence and mortality Available from: https://nccd.cdc.gov/uscs/cancersrankedbystate.aspx2015.
- [2].Vajaria BN, Patel PS.. Glycosylation: a hallmark of cancer? Glycoconj J. 2017. April;34(2):147–156. 10.1007/s10719-016-9755-2. PubMed PMID: 27975160. [DOI] [PubMed] [Google Scholar]
- [3].Comelli EM, Head SR, Gilmartin T, et al. A focused microarray approach to functional glycomics: transcriptional regulation of the glycome. Glycobiology. 2006. February;16(2):117–131. PubMed PMID: 16237199. [DOI] [PubMed] [Google Scholar]
- [4].Vaupel P. Hypoxia and aggressive tumor phenotype: implications for therapy and prognosis. Oncologist. 2008;13 Suppl 3:21–26. . PubMed PMID: 18458121. [DOI] [PubMed] [Google Scholar]
- [5].Muz B, De La Puente P, Azab F, et al. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl). 2015;3:83–92. . PubMed PMID: 27774485; PubMed Central PMCID: PMCPMC5045092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chang J, Erler J. Hypoxia-mediated metastasis. Adv Exp Med Biol. 2014;772:55–81. . PubMed PMID: 24272354. [DOI] [PubMed] [Google Scholar]
- [7].Rankin EB, Nam JM, Giaccia AJ. Hypoxia: signaling the metastatic cascade. Trends Cancer. 2016. June;2(6):295–304. .PubMed PMID: 28741527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rockwell S, Dobrucki IT, Kim EY, et al. Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med. 2009. May;9(4):442–458. PubMed PMID: 19519402; PubMed Central PMCID: PMCPMC2752413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shannon AM, Bouchier-Hayes DJ, Condron CM, et al. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev. 2003. August;29(4):297–307. PubMed PMID: 12927570. [DOI] [PubMed] [Google Scholar]
- [10].Carreau A, El Hafny-Rahbi B, Matejuk A, et al. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011. June;15(6):1239–1253. PubMed PMID: 21251211; PubMed Central PMCID: PMCPMC4373326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Byrne MB, Leslie MT, Gaskins HR, et al. Methods to study the tumor microenvironment under controlled oxygen conditions. Trends Biotechnol. 2014. November;32(11):556–563. PubMed PMID: 25282035; PubMed Central PMCID: PMCPMC4254115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. 2014. Jan-Feb;49(1):1–15. .PubMed PMID: 24099156; PubMed Central PMCID: PMCPMC4342852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Masoud GN, Li W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015. September;5(5):378–389. .PubMed PMID: 26579469; PubMed Central PMCID: PMCPMC4629436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003. October;3(10):721–732. .PubMed PMID: 13130303. [DOI] [PubMed] [Google Scholar]
- [15].Hu X, Wei L, Taylor TM, et al. Hypoxic preconditioning enhances bone marrow mesenchymal stem cell migration via Kv2.1 channel and FAK activation. Am J Physiol Cell Physiol. 2011. August;301(2):C362–72. PubMed PMID: 21562308; PubMed Central PMCID: PMCPMC3154562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee SH, Lee YJ, Han HJ. Role of hypoxia-induced fibronectin-integrin beta1 expression in embryonic stem cell proliferation and migration: involvement of PI3K/Akt and FAK. J Cell Physiol. 2011. February;226(2):484–493. .PubMed PMID: 20677223. [DOI] [PubMed] [Google Scholar]
- [17].Silva P, Mendoza P, Rivas S, et al. Hypoxia promotes Rab5 activation, leading to tumor cell migration, invasion and metastasis. Oncotarget. 2016. May 17;7(20):29548–29562. PubMed PMID: 27121131; PubMed Central PMCID: PMCPMC5045416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Meininger CJ, Schelling ME, Granger HJ. Adenosine and hypoxia stimulate proliferation and migration of endothelial cells. Am J Physiol. 1988. September;255(3 Pt 2):H554–62. .PubMed PMID: 3414822. [DOI] [PubMed] [Google Scholar]
- [19].Djagaeva I, Doronkin S. Hypoxia response pathway in border cell migration. Cell Adh Migr. 2010. Jul-Sep;4(3):391–395. PubMed PMID: 20523117; PubMed Central PMCID: PMCPMC2958615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Leong HS, Chambers AF. Hypoxia promotes tumor cell motility via RhoA and ROCK1 signaling pathways. Proc Natl Acad Sci U S A. 2014. January 21;111(3):887–888. . PubMed PMID: 24398519; PubMed Central PMCID: PMCPMC3903196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jiang J, Tang YL, Liang XH. EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther. 2011. April 15;11(8):714–723. PubMed PMID: 21389772. [DOI] [PubMed] [Google Scholar]
- [22].Fahling M, Perlewitz A, Doller A, et al. Regulation of collagen prolyl 4-hydroxylase and matrix metalloproteinases in fibrosarcoma cells by hypoxia. Comp Biochem Physiol C Toxicol Pharmacol. 2004. October;139(1–3):119–126. PubMed PMID: 15556073. [DOI] [PubMed] [Google Scholar]
- [23].Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014. June;14(6):430–439. .PubMed PMID: 24827502; PubMed Central PMCID: PMCPMC4283800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Corley KM, Taylor CJ, Lilly B. Hypoxia-inducible factor 1alpha modulates adhesion, migration, and FAK phosphorylation in vascular smooth muscle cells. J Cell Biochem. 2005. December 1;96(5):971–985. . PubMed PMID: 16149050. [DOI] [PubMed] [Google Scholar]
- [25].Cowden Dahl KD, Robertson SE, Weaver VM, et al. Hypoxia-inducible factor regulates alphavbeta3 integrin cell surface expression. Mol Biol Cell. 2005. April;16(4):1901–1912. PubMed PMID: 15689487; PubMed Central PMCID: PMCPMC1073670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Keely S, Glover LE, MacManus CF, et al. Selective induction of integrin beta1 by hypoxia-inducible factor: implications for wound healing. FASEB J. 2009. May;23(5):1338–1346. PubMed PMID: 19103643; PubMed Central PMCID: PMCPMC2669428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Janik ME, Litynska A, Vereecken P. Cell migration-the role of integrin glycosylation. Biochim Biophys Acta. 2010. June;1800(6):545–555. .PubMed PMID: 20332015. [DOI] [PubMed] [Google Scholar]
- [28].Yoon SO, Shin S, Mercurio AM. Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 2005. April 1;65(7):2761–2769. . PubMed PMID: 15805276. [DOI] [PubMed] [Google Scholar]
- [29].Gu J, Taniguchi N. Regulation of integrin functions by N-glycans. Glycoconj J. 2004;21(1–2):9–15. . PubMed PMID: 15467392. [DOI] [PubMed] [Google Scholar]
- [30].Bellis SL. Variant glycosylation: an underappreciated regulatory mechanism for beta1 integrins. Biochim Biophys Acta. 2004. May 27;1663(1–2):52–60. . PubMed PMID: 15157607. [DOI] [PubMed] [Google Scholar]
- [31].Kolarich D, Jensen PH, Altmann F, et al. Determination of site-specific glycan heterogeneity on glycoproteins. Nat Protoc. 2012. June 7;7(7):1285–1298. PubMed PMID: 22678432. [DOI] [PubMed] [Google Scholar]
- [32].Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. 2015;10:473–510. . PubMed PMID: 25621663; PubMed Central PMCID: PMCPMC4396820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Adamczyk B, Tharmalingam T, Rudd PM. Glycans as cancer biomarkers. Biochim Biophys Acta. 2012. September;1820(9):1347–1353. .PubMed PMID: 22178561. [DOI] [PubMed] [Google Scholar]
- [34].Sewell R, Backstrom M, Dalziel M, et al. The ST6GalNAc-I sialyltransferase localizes throughout the Golgi and is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J Biol Chem. 2006. February 10;281(6):3586–3594. PubMed PMID: 16319059. [DOI] [PubMed] [Google Scholar]
- [35].Rivinoja A, Hassinen A, Kokkonen N, et al. Elevated Golgi pH impairs terminal N-glycosylation by inducing mislocalization of Golgi glycosyltransferases. J Cell Physiol. 2009. July;220(1):144–154. PubMed PMID: 19277980. [DOI] [PubMed] [Google Scholar]
- [36].Potapenko IO, Haakensen VD, Luders T, et al. Glycan gene expression signatures in normal and malignant breast tissue; possible role in diagnosis and progression. Mol Oncol. 2010. April;4(2):98–118. PubMed PMID: 20060370; PubMed Central PMCID: PMCPMC5527897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Burchell J, Poulsom R, Hanby A, et al. An alpha2,3 sialyltransferase (ST3Gal I) is elevated in primary breast carcinomas. Glycobiology. 1999. December;9(12):1307–1311. PubMed PMID: 10561455. [DOI] [PubMed] [Google Scholar]
- [38].Wu C, Guo X, Wang W, et al. N-Acetylgalactosaminyltransferase-14 as a potential biomarker for breast cancer by immunohistochemistry. BMC Cancer. 2010. April 1;(10):123 . PubMed PMID: 20356418; PubMed Central PMCID: PMCPMC2873381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Recchi MA, Hebbar M, Hornez L, et al. Multiplex reverse transcription polymerase chain reaction assessment of sialyltransferase expression in human breast cancer. Cancer Res. 1998. September 15;58(18):4066–4070. PubMed PMID: 9751611. [PubMed] [Google Scholar]
- [40].Handerson T, Camp R, Harigopal M, et al. Beta1,6-branched oligosaccharides are increased in lymph node metastases and predict poor outcome in breast carcinoma. Clin Cancer Res. 2005. April 15;11(8):2969–2973. PubMed PMID: 15837749. [DOI] [PubMed] [Google Scholar]
- [41].Madjd Z, Parsons T, Watson NF, et al. High expression of Lewis y/b antigens is associated with decreased survival in lymph node negative breast carcinomas. Breast Cancer Res. 2005;7(5):R780–7. . PubMed PMID: 16168124; PubMed Central PMCID: PMCPMC1242157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nakagoe T, Fukushima K, Itoyanagi N, et al. Expression of ABH/Lewis-related antigens as prognostic factors in patients with breast cancer. J Cancer Res Clin Oncol. 2002. May;128(5):257–264. PubMed PMID: 12029441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bresalier RS. Intestinal mucin and colorectal cancer: it’s not just goo. Gastroenterology. 2002. August;123(2):648–649. PubMed PMID: 12145823. [DOI] [PubMed] [Google Scholar]
- [44].Osumi D, Takahashi M, Miyoshi E, et al. Core fucosylation of E-cadherin enhances cell-cell adhesion in human colon carcinoma WiDr cells. Cancer Sci. 2009. May;100(5):888–895. PubMed PMID: 19302290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Koike T, Kimura N, Miyazaki K, et al. Hypoxia induces adhesion molecules on cancer cells: A missing link between Warburg effect and induction of selectin-ligand carbohydrates. Proc Natl Acad Sci U S A. 2004. May 25;101(21):8132–8137. PubMed PMID: 15141079; PubMed Central PMCID: PMCPMC419569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nonaka M, Fukuda MN, Gao C, et al. Determination of carbohydrate structure recognized by prostate-specific F77 monoclonal antibody through expression analysis of glycosyltransferase genes. J Biol Chem. 2014. June 6;289(23):16478–16486. PubMed PMID: 24753248; PubMed Central PMCID: PMCPMC4047414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Belo AI, Van Vliet SJ, Maus A, et al. Hypoxia inducible factor 1alpha down regulates cell surface expression of alpha1,2-fucosylated glycans in human pancreatic adenocarcinoma cells. FEBS Lett. 2015. August 19;589(18):2359–2366. PubMed PMID: 26232512. [DOI] [PubMed] [Google Scholar]
- [48].Varki A. Factors controlling the glycosylation potential of the Golgi apparatus. Trends Cell Biol. 1998. January;8(1):34–40. PubMed PMID: 9695806. [DOI] [PubMed] [Google Scholar]
- [49].Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta. 2010. February;1800(2):80–95. .PubMed PMID: 19647043; PubMed Central PMCID: PMCPMC2815088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Natsuizaka M, Ozasa M, Darmanin S, et al. Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp Cell Res. 2007. September 10;313(15):3337–3348. PubMed PMID: 17651733. [DOI] [PubMed] [Google Scholar]
- [51].Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006. March;3(3):177–185. PubMed PMID: 16517405. [DOI] [PubMed] [Google Scholar]
- [52].Pescador N, Villar D, Cifuentes D, et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS One. 2010. March 12;5(3):e9644 PubMed PMID: 20300197; PubMed Central PMCID: PMCPMC2837373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shen GM, Zhang FL, Liu XL, et al. Hypoxia-inducible factor 1-mediated regulation of PPP1R3C promotes glycogen accumulation in human MCF-7 cells under hypoxia. FEBS Lett. 2010. October 22;584(20):4366–4372. PubMed PMID: 20888814. [DOI] [PubMed] [Google Scholar]
- [54].Manzari B, Kudlow JE, Fardin P, et al. Induction of macrophage glutamine: fructose-6-phosphate amidotransferase expression by hypoxia and by picolinic acid. Int J Immunopathol Pharmacol. 2007. Jan-Mar;20(1):47–58. PubMed PMID: 17346427. [DOI] [PubMed] [Google Scholar]
- [55].Yi W, Clark PM, Mason DE, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012. August 24;337(6097):975–980. PubMed PMID: 22923583; PubMed Central PMCID: PMCPMC3534962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhao Y, Sato Y, Isaji T, et al. Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J. 2008. May;275(9):1939–1948. PubMed PMID: 18384383. [DOI] [PubMed] [Google Scholar]
- [57].Boon L, Ugarte-Berzal E, Vandooren J, et al. Glycosylation of matrix metalloproteases and tissue inhibitors: present state, challenges and opportunities. Biochem J. 2016. June 1;473(11):1471–1482. PubMed PMID: 27234584; PubMed Central PMCID: PMCPMC4888457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol. 2011. March 1;3(3). DOI: 10.1101/cshperspect.a004994 PubMed PMID: 21421922; PubMed Central PMCID: PMCPMC3039929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010. January;10(1):9–22. .PubMed PMID: 20029421; PubMed Central PMCID: PMCPMC4383089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010. April;11(4):288–300. .PubMed PMID: 20308986; PubMed Central PMCID: PMCPMC3929966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Margadant C, Monsuur HN, Norman JC, et al. Mechanisms of integrin activation and trafficking. Curr Opin Cell Biol. 2011. October;23(5):607–614. PubMed PMID: 21924601. [DOI] [PubMed] [Google Scholar]
- [62].Wiley HS, Burke PM Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001. January;2(1):12–18. PubMed PMID: 11208164. [DOI] [PubMed] [Google Scholar]
- [63].Hanyaloglu AC, Von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. . PubMed PMID: 18184106. [DOI] [PubMed] [Google Scholar]
- [64].Gu Z, Noss EH, Hsu VW, et al. Integrins traffic rapidly via circular dorsal ruffles and macropinocytosis during stimulated cell migration. J Cell Biol. 2011. April 4;193(1):61–70. PubMed PMID: 21464228; PubMed Central PMCID: PMCPMC3082178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ezratty EJ, Bertaux C, Marcantonio EE, et al. Clathrin mediates integrin endocytosis for focal adhesion disassembly in migrating cells. J Cell Biol. 2009. November 30;187(5):733–747. PubMed PMID: 19951918; PubMed Central PMCID: PMCPMC2806590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wickstrom SA, Alitalo K, Keski-Oja J. Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res. 2002. October 1;62(19):5580–5589. PubMed PMID: 12359771. [PubMed] [Google Scholar]
- [67].Dozynkiewicz MA, Jamieson NB, Macpherson I, et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev Cell. 2012. January 17;22(1):131–145. PubMed PMID: 22197222; PubMed Central PMCID: PMCPMC3507630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Arjonen A, Alanko J, Veltel S, et al. Distinct recycling of active and inactive beta1 integrins. Traffic. 2012. April;13(4):610–625. PubMed PMID: 22222055; PubMed Central PMCID: PMCPMC3531618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sandri C, Caccavari F, Valdembri D, et al. The R-Ras/RIN2/Rab5 complex controls endothelial cell adhesion and morphogenesis via active integrin endocytosis and Rac signaling. Cell Res. 2012. October;22(10):1479–1501. PubMed PMID: 22825554; PubMed Central PMCID: PMCPMC3463263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bridgewater RE, Norman JC, Caswell PT. Integrin trafficking at a glance. J Cell Sci. 2012. August 15;125(Pt 16):3695–3701. . PubMed PMID: 23027580; PubMed Central PMCID: PMCPMC3462077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011. September 1;3(9):a005074 . PubMed PMID: 21885598; PubMed Central PMCID: PMCPMC3181029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Paul NR, Jacquemet G, Caswell PT. Endocytic trafficking of integrins in cell migration. Curr Biol. 2015. November 16;25(22):R1092–105. . PubMed PMID: 26583903. [DOI] [PubMed] [Google Scholar]
- [73].Hamidi H, Pietila M, Ivaska J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br J Cancer. 2016. October 25;115(9):1017–1023. . PubMed PMID: 27685444; PubMed Central PMCID: PMCPMC5117799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Park CC, Zhang H, Pallavicini M, et al. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006. February 1;66(3):1526–1535. PubMed PMID: 16452209; PubMed Central PMCID: PMCPMC2933188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bhaskar V, Zhang D, Fox M, et al. A function blocking anti-mouse integrin alpha5beta1 antibody inhibits angiogenesis and impedes tumor growth in vivo. J Transl Med. 2007. November 27;(5):61 . PubMed PMID: 18042290; PubMed Central PMCID: PMCPMC2235829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Varki A, Lowe JB., et al. Biological roles of glycans In: Varki A, Cummings RD, editors. Essentials of glycobiology. (NY): Cold Spring Harbor; ; 2009. [PubMed] [Google Scholar]
- [77].Guo HB, Lee I, Kamar M, et al. Aberrant N-glycosylation of beta1 integrin causes reduced alpha5beta1 integrin clustering and stimulates cell migration. Cancer Res. 2002. December 1;62(23):6837–6845. PubMed PMID: 12460896. [PubMed] [Google Scholar]
- [78].Semel AC, Seales EC, Singhal A, et al. Hyposialylation of integrins stimulates the activity of myeloid fibronectin receptors. J Biol Chem. 2002. September 6;277(36):32830–32836. PubMed PMID: 12091385. [DOI] [PubMed] [Google Scholar]
- [79].Isaji T, Gu J, Nishiuchi R, et al. Introduction of bisecting GlcNAc into integrin alpha5beta1 reduces ligand binding and down-regulates cell adhesion and cell migration. J Biol Chem. 2004. May 7;279(19):19747–19754. PubMed PMID: 14998999. [DOI] [PubMed] [Google Scholar]
- [80].Zheng M, Fang H, Hakomori S. Functional role of N-glycosylation in alpha 5 beta 1 integrin receptor. De-N-glycosylation induces dissociation or altered association of alpha 5 and beta 1 subunits and concomitant loss of fibronectin binding activity. J Biol Chem. 1994. April 22;269(16):12325–12331. PubMed PMID: 7512965. [PubMed] [Google Scholar]
- [81].Hang Q, Isaji T, Hou S, et al. A key regulator of cell adhesion: identification and characterization of important N-glycosylation sites on integrin alpha5 for cell migration. Mol Cell Biol. 2017. May 1;37(9). DOI: 10.1128/MCB.00558-16 PubMed PMID: 28167607; PubMed Central PMCID: PMCPMC5394273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Isaji T, Sato Y, Fukuda T, et al. N-glycosylation of the I-like domain of beta1 integrin is essential for beta1 integrin expression and biological function: identification of the minimal N-glycosylation requirement for alpha5beta1. J Biol Chem. 2009. May 1;284(18):12207–12216. PubMed PMID: 19261610; PubMed Central PMCID: PMCPMC2673289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hou S, Hang Q, Isaji T, et al. Importance of membrane-proximal N-glycosylation on integrin beta1 in its activation and complex formation. FASEB J. 2016. December;30(12):4120–4131. PubMed PMID: 27565712. [DOI] [PubMed] [Google Scholar]
- [84].Tang D, Yan T, Zhang J, et al. Notch1 signaling contributes to hypoxia-induced high expression of integrin beta1 in keratinocyte migration. Sci Rep. 2017. March 7;(7):43926 . PubMed PMID: 28266574; PubMed Central PMCID: PMCPMC5339698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Walton HL, Corjay MH, Mohamed SN, et al. Hypoxia induces differential expression of the integrin receptors alpha(vbeta3) and alpha(vbeta5) in cultured human endothelial cells. J Cell Biochem. 2000. June 12;78(4):674–680. PubMed PMID: 10861864. [DOI] [PubMed] [Google Scholar]
- [86].Kong T, Eltzschig HK, Karhausen J, et al. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci U S A. 2004. July 13;101(28):10440–10445. PubMed PMID: 15235127; PubMed Central PMCID: PMCPMC478589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Saller MM, Prall WC, Docheva D, et al. Increased stemness and migration of human mesenchymal stem cells in hypoxia is associated with altered integrin expression. Biochem Biophys Res Commun. 2012. June 29;423(2):379–385. PubMed PMID: 22664105. [DOI] [PubMed] [Google Scholar]
- [88].Ju JA, Godet I, Ye IC, et al. Hypoxia selectively enhances integrin alpha5beta1 receptor expression in breast cancer to promote metastasis. Mol Cancer Res. 2017. June;15(6):723–734. PubMed PMID: 28213554; PubMed Central PMCID: PMCPMC5510543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ren Y, Hao P, Law SK, et al. Hypoxia-induced changes to integrin alpha 3 glycosylation facilitate invasion in epidermoid carcinoma cell line A431. Mol Cell Proteomics. 2014. November;13(11):3126–3137. PubMed PMID: 25078904; PubMed Central PMCID: PMCPMC4223496. [DOI] [PMC free article] [PubMed] [Google Scholar]