

Abstract
Biomolecules have many properties that make them promising for intracellular therapeutic applications, but delivery remains a key challenge because large biomolecules cannot easily enter the cytosol. Furthermore, quantification of total intracellular versus cytosolic concentrations remains demanding, and the determination of delivery efficiency is thus not straightforward. In this review, we discuss strategies for delivering biomolecules into the cytosol and briefly summarize the mechanisms of uptake for these systems. We then describe commonly used methods to measure total cellular uptake and, more selectively, cytosolic localization, and discuss the major advantages and drawbacks of each method. We critically evaluate methods of measuring “cell penetration” that do not adequately distinguish total cellular uptake and cytosolic localization, which often lead to inaccurate interpretations of a molecule’s cytosolic localization. Finally, we summarize the properties and components of each method, including the main caveats of each, to allow for informed decisions about method selection for specific applications. When applied correctly and interpreted carefully, methods for quantifying cytosolic localization offer valuable insight into the bioactivity of biomolecules and potentially the prospects for their eventual development into therapeutics.
Graphical Abstract
INTRODUCTION
The majority of successful biotherapeutics target extracellular receptors because efficient intracellular delivery is a challenge. Small, sufficiently hydrophobic molecules can cross the plasma membrane without hindrance, but most biological macromolecules, which are much larger and more polar, cannot. Intracellular delivery of macromolecules has been the subject of intense research for many years, ever since the observation of specialized translocation domains that permit cellular transduction. The allure of using biomolecules such as proteins, peptides, and nucleic acids as therapies has increased interest in intracellular delivery strategies. Here, we discuss these advances with a critical eye toward methods used to quantitate intracellular delivery.
At the outset of any discussion of cell penetration, it is essential to clarify the concepts of “inside” and “outside” the cell. A eukaryotic cell is surrounded by a plasma membrane, which routinely engulfs extracellular molecules in a process called endocytosis.1,2 Importantly, after endocytosis, the macromolecule is still topologically “outside” the cell, as it remains trapped in a vesicle and separated from the cytosol by a plasma membrane. Yet the great majority of physiologically important cellular processes occur in the cytosol or in the nucleus, which is topologically connected to the cytosol by the nuclear pores.
Several intracellular delivery strategies have been developed based on naturally occurring translocation domains or other physical principles. However, not all of them have been scrutinized regarding the fraction of the cargo that actually reaches the cytosol. Most of the delivery strategies described in this review rely on the active uptake of cargo molecules by the cell via endocytosis. Mammalian cells are capable of a number of different endocytosis mechanisms, and their engagement depends on the molecule that is taken up.1,2 Larger particles and volumes of the extracellular fluid are generally taken up via phagocytosis and macropinocytosis. These two mechanisms operate by the engulfment of extracellular material by the plasma membrane via remodeling of actin filaments and further processing into endosomal compartments. Smaller particles, however, can also be taken up via pathways that depend on coat proteins, specifically clathrin or caveolin. Upon initiation of endocytosis, clathrin or caveolin coat the bending membrane until the particles are fully engulfed, while further factors, such as dynamin, separate the vesicle from the membrane. Upon uncoating, endocytosed vesicles are processed into early endosomes, and the particles inside are further sorted: for degradation into lysosomes or else into other cellular compartments such as the trans-Golgi via retrograde transport. Clathrin- and caveolin-independent pathways have also been described, which can be also dependent or independent of dynamin.1,2
One major issue intrinsic to internalization via endocytosis is the entrapment of cargoes within endosomes. Another issue is that upon acidification of these endosomes and further fusion with lysosomal compartments, trapped molecules are degraded.3 For molecules with cytosolic targets, however, biological activity requires the molecule not just to be taken up by the cell, but to efficiently escape the endosome and access the cytosol. Accessing the cytosol can be accomplished by different mechanisms, which are further detailed in subsequent sections. Some molecules have the intrinsic property of forming pores or using existing protein pores and can thus enter the cytosol from endolysosomal compartments without any impact on the endosome itself. A very different “escape mechanism” relies on the rupture of endosomal membranes. Some delivery systems, as detailed below, can effectively buffer the acidification of the endosome. The high water and ion influx then leads to the rupture of endosomal membranes, allowing the diffusion of the molecule-of-interest into the cytosol.
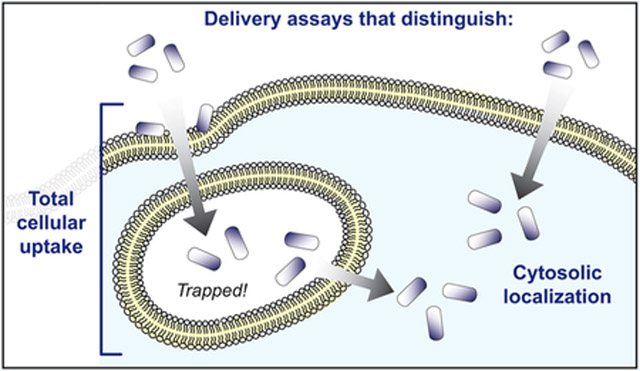
A critical question for research on cell-penetrating biomolecules is therefore to distinguish with confidence molecules that have been taken up by the cell (total cellular uptake) from molecules that have reached the cytosol (cytosolic localization). These are very different measurements of “intracellular” localization or “cell penetration”, and we avoid these latter terms because of their imprecise nature. Measuring total cellular uptake refers to all material within the cell, including endosomal and lysosomal compartments and, sometimes, material adhered to the plasma membrane. Measuring cytosolic localization refers only to material in the cytosol, which is typically the material that will confer a biological effect. Distinguishing between total cellular uptake and cytosolic localization is therefore extremely important in the screening and development of biologically active macromolecules. Recent work has highlighted the concept of translocation efficiency, which can be measured as the ratio of cytosolic localization to total cellular uptake. If both total cellular uptake and cytosolic localization of a cargo are measured, these quantities provide not only a contrast between these two measurements, but a quantitative measure of endosomal escape efficiency for a specific molecule and its translocation mechanism.
In this review, we first describe various translocation strategies with a focus on their endosomal escape mechanisms. We then discuss several assays currently used to measure “cell penetration” in detail and discuss their ability to differentiate between total cellular uptake and cytosolic localization. Finally, we also review some procedures and their associated artifacts that have led to false interpretations of delivered cargo molecules in the past, and thus should no longer be used.
DELIVERY METHODS AND THEIR MODES OF ACTION
Over the past few decades several different protein translocation methods have been developed. In the next paragraphs we describe the most commonly used delivery methods, which are also summarized in Figure 1.
Figure 1.

Common methods for the intracellular delivery of biomolecules. Cargo molecules can be fused to positively charged molecules, fused to bacterial protein toxins, or packed in nanoparticles or virus-like particles for cellular uptake and eventual endosomal escape. Physical methods and liposomes can directly deliver cargo molecules to the cytosol.
Physical Methods.
Some of the oldest and most direct methods to facilitate intracellular delivery are mechanical methods, in which the membrane is physically disrupted during incubation of the cargo molecule. Various physical methods such as electroporation, microinjection, sonoporation, or mechanical deformation were recently reviewed quite extensively.4 Physical methods for intracellular delivery of macromolecules bypass endocytosis and allow for direct access of exogenous molecules into the cytosol, but they can be highly damaging. Also, most are poorly translatable to live organisms.
Cell-Penetrating Peptides (CPPs).
Cell-penetrating peptides (CPPs), first developed about 30 years ago,5 have drawn a great deal of attention due to their potential for nucleic acid and protein delivery.6–8 Tat and penetratin were the first CPPs to be reported, derived from the natural proteins HIV Tat (“trans-activator of transcription”) and Antennapedia homeoprotein, respectively.9–12 Many synthetic CPPs have since been designed, mostly linear peptide sequences up to 40 amino acids long. CPPs have received this name by their observed property to “penetrate” membranes and translocate themselves and an attached cargo to the cytosol of cells, yet their efficacy and mechanism depend on many factors and are often not clear.3 Positively charged functional groups, particularly guanidinium groups, are essential for total cellular uptake for most CPPs, and this property is also related to cytosolic localization.13–23 However, amphipathic, hydrophobic, or even anionic CPPs have also been described.24,25
Although there has been quite an effort to clarify the mechanism(s) by which CPPs translocate to the inside of a cell, it remains elusive how exactly a particular peptide or peptide–cargo complex crosses the membrane.3One proposed mechanism is the interaction between the positively charged peptide and negatively charged cell surface proteoglycans. Thus, the CPP electrostatically interacts with the membrane itself or its glycoprotein components and gets internalized via endocytosis, without a clearly defined mechanism for a subsequent endosomal escape.3 Besides rupture of the endosomal membrane (see below), other proposed mechanisms for CPPs are direct penetration, pore formation, micelle formation, membrane thinning, receptor-specific uptake, transporter-mediated uptake, as described previously.2–37 Various mechanistic investigations such as ATP depletion or low-temperature incubation have been conducted, with contradictory results regarding whether a separate uptake mechanism is relevant other than endocytosis.18 For instance, it has been shown that direct translocation occurs only upon a certain threshold concentration, but it remains elusive how these high concentrations may affect membrane integrity of cellular and endosomal membranes, as we will discuss below.38,39 Data also suggest that, at least in some cases, uptake is dependent on the peptide-to-cell ratio rather than on the peptide concentration.40
Upon endocytosis, one major problem of CPP-mediated delivery is endosomal entrapment.41–44 It is generally unclear whether escape from endosomes relies on a specific mechanism, and it likely depends on the physicochemical characteristics of the CPP and attached cargo, its concentration, and/or the cell line and culture conditions used for the assay.45 Because of the diverse properties of CPPs, a universal uptake mechanism that applies to all molecules will be unlikely.22,46 However, a few key studies have provided a simple framework for understanding the critical parameters for possible cell penetration mechanisms. Among others Fuchs et al.34 demonstrated in 2004 that leakage from endocytic vesicles can occur, dependent on high concentrations of CPP.34,35 In this mechanism, the CPP induces internalization via endocytosis and then leads to rupture of the endosomal membrane, finally “penetrating” into the cytosol.36,37 We discuss below the potential role of massive hydration and subsequent rupture of the endosomes. These high concentrations thus might lead to toxic effects that need to be tested for each peptide cargo complex.15
Since cytosolic localization is not as easily achieved as initially assumed, various efforts have been undertaken to improve delivery efficiencies of CPPs. More recently, machine-learning based predictions of cellular uptake as well as the simulation of improved cargo delivery have been explored to reduce screening efforts for suitable peptide–cargo combinations.16,23,47–50 Inherent in these efforts is the assumption that the predicted molecular structure can actually enhance endosomal escape, beyond the hydration and rupture effect. It would be extremely crucial to train such algorithms by reliable data on how much cargo has reached the cytosol, compared to how much has remained in endosomes. The importance of distinguishing these data emphasizes the need for a careful approach to data interpretation that is the focus of the present review. Enhancing endosomal escape and therefore increasing cytosolic concentrations are challenges that remain under investigation.51
As will be discussed further below, CPPs are part of a larger group of “cell-penetrating” cationic molecules, including supercharged proteins, naturally occurring cationic proteins (such as most DNA-binding proteins), cationic nanoparticles, cationic detergents, and auxiliary substances such as chitosan and polyethylenimine.
Zinc Fingers and Other DNA-Binding Proteins.
Zinc-finger domains and other DNA-binding proteins usually have a high net-positive charge as their natural role is to bind negatively charged DNA. Zinc-finger proteins have up to 20 positive charges for a typical protein with six zinc-finger domains. Similarly to CPPs, their positive charges interact with the cell surface. It is therefore not unreasonable to treat them in one group together with CPPs and supercharged proteins (see next section), and positively charged nanoparticles.
Genome engineering applications have utilized zinc-finger domains by taking advantage of their reported self-delivering properties.52 Recently, the delivery of proteins and enzymes as cargoes fused to zinc-finger domains has also been reported.53 Proteins genetically fused to zinc-finger domains appear to enter mammalian cells via macropinocytosis and, to a limited extent, through caveolin-dependent endocytosis.53 This process is dose-dependent, and delivery efficiencies varied with the number of zinc-finger domains, with an optimum of two or three domains. This limit appeared to be due to overall protein stability.53 It is not clear how proteins escape the endosome upon macropinocytosis, but the endosomal escape efficiency varied from 12–80%, depending on the assay used.53,54 Further studies are clearly needed to elucidate their detailed translocation mechanisms and assess their potential as transporters.
Supercharged Proteins.
In 2010 another class of protein-based delivery vehicle was described that maximizes the most common feature of successful CPPs and DNA-binding proteins: high positive charge. “Supercharged” GFP containing varying numbers of surface-exposed charges has been reported to potently and rapidly deliver proteins in vitro and in vivo.55—57 It was also employed as an effective method to enhance siRNA and DNA transfection.58 Other naturally occurring human proteins with high positive net charges have also been tested for their delivery potential.59
Supercharged proteins seem to utilize uptake mechanisms similar to those of CPPs. This may not be surprising, since the more efficient transfection agents typically have a high positive net charge as well as a minimum proportion of hydrophobic surface area. The positively charged proteins do not passively translocate through the membrane, but rather are endocytosed upon binding to sulfated proteoglycans on the cell surface. This induces endocytosis pathways dependent on clathrin and dynamin, leading to uptake into endosomes. Translocation efficiency is strongly dependent on the net positive charge and can be designed to be pH dependent.60,61 Supercharged proteins can also have higher net charges than short CPPs, but when molecules of similar net charge are compared, supercharged proteins show higher total cellular uptake. The mechanism of this enhanced uptake remains elusive.59 It has been proposed that proteoglycan cross-linking is the major driver for macropinocytosis and that the larger surface area is advantageous over CPP-mediated endocytosis.60 Resistance to proteolysis may also contribute to this effect.
It has been observed that uptake of supercharged GFP is characterized by punctate endosomal staining in peripheral parts of a cell. Some authors have proposed that these endosomal compartments potentially fail to undergo further transport or processing into lysosomal compartments, thus giving more time for endosomal escape of trapped cargo.60 This hypothesis would view endosomal escape as a competing pathway with lysosomal degradation, with the role of endosomal rupture to be established.36,37 Clearly, further studies are required to elucidate the exact mechanism of endosomal escape of supercharged proteins.
Nanoparticles and Liposomes.
Nanoparticles represent a diverse group of carriers designed to deliver small molecules, nucleic acids, peptides, and proteins to the cytosol. Nanocarriers have been used to deliver several proteins, most commonly GFP but also other proteins like CRISPR/Cas9 designed for future therapeutic applications, as reviewed by Yu et al.62–65
Various synthetic and naturally occurring polymers, lipids, polysaccharides, and proteins have been used as carriers for nanoparticle-based delivery.66–70 Many parameters must be considered when designing nanoparticles to encapsulate, deliver, and release cargo, including size, aggregation rate, surface characteristics, efficiency of encapsulation, and drug release kinetics.66,67 The most common drawbacks of these carriers are their high cytotoxicity, low stability, variable efficacy, and various issues associated with biocompatibility and biodegradability. These drawbacks vary widely among different nanocarriers and thus must be tested carefully for each individual carrier and cargo.67,71,72 The most prominent synthetic polymers used as nanocarriers for protein delivery are copolymers consisting of polylactic-co-glycolic acid (PLGA), N-(2-hydroxypropyl) methacrylamide, or polyvinylpyrrolidone.70 Several proteins, such as albumin, have been cross-linked to form protein-based nanoparticles. These carriers represent promising alternatives to synthetic nanoparticles due to reduced toxicity and reduced affinity to off-target tissues.66,67,72
The proposed uptake mechanism for nonliposomal nanocarriers depends on the nature of the carrier. Interaction with the cellular membrane leads to, among others, different types of endocytosis, phagocytosis, macropinocytosis, or direct membrane fusion. Several mechanisms have been proposed for endosomal escape; however, various nanocarrier–protein complexes have been shown to remain trapped in the endosome and undergo lysosomal degradation, leading to low cytosolic levels of cargo molecules.72
As with other delivery methods, endosomal escape of nanoparticles is a complicated and diverse process that can involve several different mechanisms.73,74 Some cytosolic delivery methods take advantage of the “proton sponge” effect to mediate endosomal escape. Membrane pumps create a proton gradient within the late endosome, leading to acidification, a process stabilized by a carrier with buffering capabilities. Negatively charged ions and water molecules flood into the endosome from the cytosol to offset the proton gradient. This high influx of ions results in endosomal rupture and release of trapped cargo. Cationic polymers often have considerable pH-buffering capacity, enhancing endosomal escape through the same mechanism. However, as previously mentioned for CPP-mediated delivery, high net positive charge may lead to cellular toxicity. To decrease overall toxicity, pH-sensitive nanocarriers have been designed, such as carriers based on calcium phosphate. They have a neutral charge in circulation and reveal their buffering or membrane disruption capabilities only in the acidified endosome.74 Nanocarriers that are membrane-disruptive are believed to have a high endosomal escape efficiency.74
Nanoparticles based on liposomes can be treated as a separate class of carriers as they can effectively fuse with the cellular or endosomal membrane, thereby delivering their encapsulated cargo to the cytosolic space. Liposomes with CPPs on their surface have further shown enhanced cellular uptake and endosomal escape.75–77
Additional nanocarrier-based delivery systems have been developed with different proposed mechanisms. Hydrophobic polyampholytes have been proposed to interact with and destabilize the endosomal membranes leading to cargo release to the cytosol.72 Another method, a freeze concentration protocol, was able to result in highly internalized complexes; however, a clear understanding of the uptake mechanism and endosomal escape remains elusive, and this particular method is only suitable for in vitro delivery.71
Virus-like Particles.
Compared to other delivery methods, virus-like particles (VLPs) are still in their infancy. VLPs have been generated both from enveloped and nonenveloped viruses. The nonenveloped ones are tightly packed and ordered assemblies of viral coat proteins, but they are devoid of viral DNA or RNA. Because of these characteristics, VLPs have been used extensively as vaccine carriers, since the high immunogenicity of these particles is ideal for vaccine applications.78–80 VLPs from many different virus types have been used as protein delivery platforms.81–84 Cargo proteins are either fused to the surface of the VLP or encapsulated within the VLP during their self-assembly. Through attaching receptor-targeting molecules to surface proteins, VLPs can also be made cell-specific.81
Although natural viruses possess the ability to transduce cells and deliver nucleic acids to the cytosol, usually with subsequent transport to the nucleus, the exact mechanism by which a cargo molecule reaches the cytosol from a VLP remains elusive. There are some data to support a model in which, upon receptor-mediated endocytosis, the VLPs escape the endosomes by their natural endosomal escape mechanism. This function would have to be provided by one of the viral coat proteins.85
Bacterial Toxins.
Some bacteria naturally secrete protein toxins with a component that can translocate a toxic protein to the cytosol of host cells. These toxins have evolved a highly specific translocation mechanism to deliver to the cytosol an enzyme that fatally damages the cell. The best studied bacterial translocation complexes are those associated with anthrax toxin (Bacillus anthracis), diphtheria toxin (Corynebacterium diphtheriae), and exotoxin A (Pseudomonas aeruginosa).86,87 These bacterial protein toxins are of the AB type and usually consist of three major domains, an A-component with an enzymatic/toxic domain, as well as a B component with a cell-binding domain and a translocation domain.
Because of their modular structure, domains can be engineered for altered cell specificity and customized cargo, making bacterial protein toxin transport systems an adaptable and potentially general approach for protein delivery.88–92 For instance, more than 30 cargo proteins have been tested in anthrax-mediated delivery, as reviewed previously.86 A prerequisite for engineering bacterial toxins is a thorough understanding of the natural translocation mechanisms, which are not yet fully understood and have also been reviewed elsewhere.87,93 Here we provide a brief overview of these mechanisms, specifically focusing on AB toxins that have been engineered with altered cell specificity and alternative cargoes.
For effective, cell-specific targeting, the natural binding domain of AB toxins must either be completely replaced by a retargeting molecule,94 or mutated to prevent wild-type receptor binding and additionally fused to a binding molecule for cell-specific interaction.95 Since the receptor-binding domains are loosely associated with the translocation domain, they can be exchanged in a modular fashion. Successful retargeting of various toxins has been shown for tumor-associated receptors like EpCAM and Her2.96–98 Upon receptor binding, proteolytic activation of some toxins occurs at the cell surface. Wild-type protective antigen (PA), the translocation domain of anthrax toxin, is proteolytically activated by furin; however, this cleavage site can be altered to a substrate for proteases that are highly overexpressed in various cancer types.99,100 The toxin complexes get internalized via receptor-mediated endocytosis. For a number of toxins, it is not yet fully understood if this process is clathrin-dependent or -independent.101 In the early endosomes, proteolytic activation of some toxins occurs at this stage by cleaving off the A component.87
Once inside endosomes, further processing differs depending on the toxin. Two distinct endosomal escape mechanisms have been described that ensure cytosolic delivery and prevent endosomal degradation. The first, used by Shiga toxin and Pseudomonas exotoxin A (ETA), relies on the cellular translocation machinery via the trans-Golgi network and the endoplasmic reticulum (ER). Shiga toxin traffics through the Golgi apparatus directly from early endosomes,101 whereas ETA requires a pH shift as occurs in late endosomes before undergoing a similar retrograde transport. Both translocate their cargo via the Golgi to the ER and eventually to the cytosol. Cargoes that undergo retrograde transport rely on the unfolding and refolding machinery of the host cell, and it is unclear how efficiently alternative cargoes get processed. The rate-limiting step, however, is the efficiency by which proteins get translocated from the endosome to the trans-Golgi network, with a proposed efficiency of 5–10%.93
The other endosomal escape mechanism, used by anthrax toxin, diphtheria toxin, Clostridium botulinum C2 toxin and others, directly translocates cargo proteins across the endosomal membrane. Diphtheria toxin and anthrax toxin might require a pH shift to late endosomes, depending on the receptor to which they are bound.102 Direct translocation from these late endosomes then occurs by insertion of a pore, formed by a domain of its B component, into the endosomal membrane. Because of the lower pH of the late endosome, the bound cargo molecules unfold and translocate to the cytosol through this pore.90,103 Direct translocation via toxin-encoded membrane pores is independent of any cellular machinery but requires the cargo to have a certain surface charge to pass through the selective pore and the ability to unfold prior to translocation and to refold after translocation. The delivery of alternative cargoes with bacterial protein toxins was observed to have a wide range of efficiencies, depending on the energy needed to unfold the protein.96 It has been shown that the native toxins must refold to become catalytically active and effectively kill the host cells,104 and this also applies for alternative cargoes.
ASSAYS FOR TOTAL CELLULAR UPTAKE
From CPPs to supercharged proteins to VLPs, a key aspect of research into cell-penetrating molecules is the assay used to measure cellular internalization. As defined above, it is prudent to distinguish clearly between assays that measure total cellular uptake, which may include endosomally trapped material, and assays that specifically measure cytosolic localization. In this section and in Table 1, we describe the development of assays that measure total cellular uptake. Notably, while measurements of total cellular uptake remain valuable (for instance, in calculating endosomal release efficiency), we emphasize that these assays do not measure cytosolic localization. Assays that do measure cytosolic localization are described in the next section. The results of assays that measure total cellular uptake and those assays that measure cytosolic localization are two different measurements, and these do not necessarily correlate with each other. Early on, these two measurements were routinely conflated, but recent evidence has demonstrated the critical importance of distinguishing them because, in general, only cytosolically present molecules can elicit a biologic effect (see section below entitled Artifacts and Misinterpretations in Measurements of Total Cellular Uptake and Cytosolic Localization).
Table 1.
Summary of Methods for Measuring the Total Cellular Uptake of Biomolecules
| method | tag | detection method | throughputa | caveatsb | refs |
|---|---|---|---|---|---|
| model membranes | none | UV or LC-MS | high | PAS | 109 |
| monolayer | none | LC-MS | high | PAS | 121, 125 |
| PARA | |||||
| HPLC | QUANT | ||||
| analysis of cell lysate | varies | MS | medium | LABEL | 129, 134, 138, 139, 141, 155 |
| Western blot | DEG | ||||
| LABEL | |||||
| fluorescence | dye | fluorescence microscopy/flow cytometry | medium/high | DEG | 18, 142 |
| QUANT | |||||
| SICM/SECM | none | electrochemistry | low | INST | 154 |
Throughput: Low throughput refers to methods that require multiple readings per cell, processing large bulk quantities of cells, or preparation of cell lysates; medium throughput refers to methods that require one reading per cell, such as simple or automated fluorescence microscopy; high throughput refers to methods that require one reading per cell but can process many cells rapidly, such as flow cytometry.
Caveats: Important caveats and limitations for each assay are noted as follows: measures passive penetration of artificial phospholipid bilayers, not necessarily of cells (PAS); signal can be derived from processes other than cellular uptake, such as paracellular transport or vesicular transcellular transport (PARA); quantitation requires careful calibration (QUANT); requires a tagged or labeled molecule (LABEL); potential artifacts due to degradation (DEG); sophisticated instrumentation, algorithms, and/or data processing required (INST).
Assays That Measure Passive Translocation Across Membranes and Cell Monolayers.
For decades, medicinal chemists have been developing and refining assays for predicting oral bioavailability, typically of small molecules. These assays do not measure cytosolic localization directly, but their results have generally correlated well with cytosolic localization of typical small-molecule drugs.105-107 Some assays commonly used in medicinal chemistry employ synthetic model membranes, while others use cell monolayers. Despite being developed to predict gut absorption of small molecules, these assays have also been used to characterize biological macromolecules, especially to identify those which may enter cells via a passive penetration mechanism.
Synthetic Model Membranes.
Before 1998, measuring translocation across synthetic model membranes typically involved chromatography with immobilized artificial membrane (IAM) columns.108 To address the low-throughput nature of IAM chromatography, Kansy et al.109 developed the parallel artificial membrane permeability assay (PAMPA). PAMPA measures diffusion of a molecule-of-interest across an artificial lipid bilayer. Originally, UV spectroscopy was used to measure the flux of molecules across the artificial membrane,109 but modern implementations commonly use quantitative LC-MS.107,110 Variations of PAMPA have recently been reviewed elsewhere.111 As an alternative to PAMPA, a few groups reported similar assays for passive penetration of synthetic membrane vesicles.112,113 For instance, synthetic vesicles were used to monitor the internalization of dye-labeled molecules-of-interest by quantitating the flux across the membrane by HPLC.112,113 Similar work was performed with synthetic giant unilamellar vesicles that have enclosed inner vesicles containing a membrane-impermeant fluorescent dye.114 This assay measures membrane permeabilization because the giant vesicle will only become homogeneously fluorescent if a peptide transiently permeabilizes the membrane.114 Synthetic multilamellar vesicles can also measure the extent of membrane translocation of a dye-labeled cargo by fluorescence microscopy.115 The same setup, but with a fluorescence polarization readout, was used to determine if fluorescein-labeled cyclic peptides bound extensively to membrane phospholipids of small unilamellar vesicles.116
Artificial membrane assays are excellent assays to measure the behavior of molecules with respect to the phospholipid bilayer. When it comes to being proxies for cell penetration of live cells, there are several important limitations. These assays measure only passive penetration of the lipid composition studied and cannot take into account any effects due to protein binding outside or inside the cell, any effects of cellular internalization pathways such as endocytosis, and most importantly, endosomal escape mechanisms. Despite this caveat, the results obtained from these types of assays have proven to correlate well with cytosolic localization mostly for small molecules, but also for a subset of CPPs, some intrinsically cell-penetrant peptides, and some CPP-cargo complexes.107,111,117,118 Since the assay by itself does not measure cytosolic localization, any hypothesis derived from these assays needs to be verified by true cytosolic localization assays (section below entitled Assays that Quantitate Cytosolic Localization).
Monolayer Assays.
Two assays are commonly used in medicinal chemistry to detect translocation across a cell monolayer. The first uses human colon adenocarcinoma (Caco-2) cells, which spontaneously grow in monolayers and differentiate into polarized enterocytes, and therefore mimic many of the properties of enterocytes in the small intestine.119,120 Caco-2 monolayers are grown on a transwell permeable support to allow quantitation of the translocation of molecules-of-interest across the monolayer.121,122 Originally, a liquid scintillation counter was used to monitor the translocation of radioactive molecules that had reached the basal side of the Caco-2 monolayer,122 but LC-MS is the current detection method of choice.123,124 The second assay, inspired by the Caco-2 monolayer system, uses Madin-Darby canine kidney (MDCK) cells.125 MDCK cells proliferate more quickly than Caco-2 cells and have more stable expression of membrane transporters, making them an attractive alternative, but both assays are still commonly used.126,127
Caco-2 and MDCK assays are commonly used as a proxy measure for small-molecule cell permeability with the goal of predicting oral bioavailability. Cell monolayer assays can be high-throughput, and their results typically correlate well with gut permeation in vivo.120,128 However, it is important to note that they monitor both transcellular transport (which depends on total cellular uptake and, potentially, cytosolic localization and the release on the distal side) and paracellular transport (in which cargo passes through junctions between cells and does not enter these cells).120,122,129 Paracellular transport would not be expected to be related to cytosolic localization, and transcellular transport of macromolecules may be vesicular, meaning that the traversing molecule may never enter the cytosol.
Overall, synthetic model membranes remain a useful tool for predicting potential cytosolic localization for molecules-of-interest, but only for a subset of molecules expected to enter cells passively. Yet, neither total cellular uptake nor cytosolic localization of a molecule can be measured directly using these assays.
Assays That Measure the Amount of Molecule in a Cell Lysate.
One approach for measuring total cellular uptake is to quantitate the amount of molecule-of-interest in a cell lysate, as opposed to detecting molecules in intact cells. The general approach for these cell lysate assays is to incubate cells in culture with a molecule-of-interest, lyse the cells, and quantitate the amount of molecule present within the cell lysate (typically analyzing only the soluble fraction of the lysate). Quantitation can be accomplished in several ways, most notably HPLC, mass spectrometry, peptide nucleic acid (PNA) hybridization, and quantitative Western blotting (Figure 2). Typically, stringent washing is necessary to try to remove material from the exterior of the cell prior to lysis, in order to quantify only internalized material, and it is not always easy to verify that all external material was truly removed.
Figure 2.

Assays that measure the amount of molecule in a cell lysate. The cells are incubated with the molecule or cargo being investigated, followed by cell lysis. The efficient removal (or their exclusion from the assay) of molecules still bound to the surface is a critical step. Subcellular fractionation is an optional step that can be used to isolate specific subcellular compartments, requiring additional controls for excluding postlysis redistribution. The cell lysate (or fraction of the cell lysate) can then be used in several different assays to quantitate internalized molecule, including HPLC, mass spectrometry, and quantitative Western blotting.
Shift in HPLC Retention Time after Chemical Labeling.
One of the earliest cell penetration assays relied on chemically labeling extracellular and surface-bound molecules, in a manner that did not label internalized molecules. HPLC was then used to separate the unmodified form of the molecule-of-interest to detect total cellular uptake. For this purpose, Oehlke et al.129 incubated cells in culture with a nonlabeled peptide of interest and then washed cells and treated with a diazotized 2-nitroaniline, which reacted with extracellular and surface-exposed peptide. The cell lysates were then analyzed by reverse-phase HPLC. Material that eluted at the normal retention time was interpreted as having been internalized by the cells, while modified peptides showed a shift in retention time; the relative amounts of modified and unmodified peptide were used to make conclusions on the degree of internalization.129,130
Mass Spectrometry.
Matrix-assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI-TOF-MS) has most commonly been used as a label-free method to quantitate the total internalization and degradation of peptides in cell lysates and conditioned cell culture media. To ensure reliable quantitation, a deuterated form of the molecule-of-interest was used as an internal calibration standard.131–133 To reduce the background signal from the lysate, Chassaing and co-workers134,135 used a biotinylated molecule to enable a preanalysis purification step. Specifically, cultured cells were incubated with a biotin-labeled molecule, washed, and lysed, and then the lysate was spiked with a known concentration of deuterated, biotinylated molecule as an internal standard. Biotinylated molecules were purified with streptavidin-coated magnetic beads, and the mass spectrometry peak ratio of nondeuterated to deuterated molecules allowed for the quantification of the internalized molecule-of-interest.134-137 More recently, a label-free variation was developed that used size-exclusion chromatography as a purification step, followed by reverse-phase HPLC.138
Mass spectrometry is very sensitive, allowing the measurement of small amounts of internalized cargo. One drawback to mass spectrometry-based detection methods is the need for an internal standard for accurate quantitation. Most commonly, a deuterated form of each molecule must be prepared as the standard, which places limits on throughput with respect to the number of different molecules that can be analyzed in parallel.
PNA Hybridization Assays.
Peptide nucleic acid (PNA) hybridization assays are used to quantitate the amount of nucleic acid (often, therapeutic RNA) in a sample. PNA hybridization has been used routinely to monitor total cellular uptake of nucleic acids for cells in culture and accumulation in tissues in vivo. To detect an RNA of interest, cell lysates are incubated with a complementary, dye-labeled PNA either in solution or with the PNA immobilized as a capture probe. The RNA that is captured through hybridization to the PNA is subsequently quantitated by HPLC.139,140
This detection method is highly specific, and it also allows for detection of internalization in ex vivo tissue samples just as easily as in cell culture.
Quantitative Western Blotting.
While less common, quantitative Western blotting can also be used to measure the amount of a cargo in a cell lysate. Quantitation is achieved by normalizing band intensities to those of loading controls of known concentration.141 This method is limited to the analysis of molecules that have an epitope for a detection antibody, such as proteins or peptides carrying an affinity tag.
Assays That Measure the Amount of Molecule in Intact Cells.
Measuring the amount of cargo in a cell lysate has the advantage of producing a bulk signal for a large number of cells. This can lead to increased sensitivity, especially for molecules that have small degrees of total cellular uptake. By contrast, many assays have been developed that measure internalization of the molecule-of-interest at the level of individual, intact cells. These assays can have decreased sensitivity, but allow for more direct measurement of total cellular uptake. In this section, we discuss assays that measure total cellular uptake in intact cells without quantitative measurement of cytosolic localization. These assays integrate signal from material localized in any compartment, including those trapped in endosomes and lysosomes. Just as for assays using cell lysates, these assays require efficient removal or exclusion from the assay of molecules still bound to the surface.
Fluorescence-Based Methods.
The most common methods used to monitor the cell penetration of biomolecules involve conjugating a fluorescent dye to the molecule-of-interest, then observing or quantitating the localization of the dye. The extent of internalization is most often measured by fluorescence microscopy or flow cytometry.
Fluorescence microscopy is only qualitative with respect to the amount of fluorophore delivered and the relative localization of the fluorescence throughout the cell. A common phenomenon observed is punctate staining due to endosomal entrapment. However, if the microscope cannot resolve individual puncta, and/or if it cannot eliminate out-of-plane fluorescence, then diffuse fluorescence can be erroneously interpreted as cytosolic localization. Even confocal fluorescence microscopy can have difficulties distinguishing dye-labeled molecule in the cytosol from small aggregates, or molecules that are trapped inside small endosomes or lysosomes.24,142–144 Careful high-content microscopy, custom-tailored algorithms and colocalization with lysosomal/endosomal markers can increase the reliability and reproducibility of analyzing images from fluorescence microscopy, but not necessarily the quantitation and definitive discernment of cytosolic material.145 Fluorescence microscopy, even confocal microscopy, is thus very challenging for quantitating total cellular uptake and can be unreliable for determining cytosolic localization.
An appealing alternative for measuring the degree of association of dye-labeled cargo with cultured cells is flow cytometry. Flow cytometry is quantitative and very high-throughput. However, flow cytometry cannot provide even qualitative data on the subcellular localization of a dye-labeled cargo. Thus, it cannot distinguish endosomally trapped material from cytosolic material. Furthermore, signals can include fluorescence from dye-labeled molecules associated with the cell membrane.18 To address this issue, washing steps with an enzymatic digest or trypan blue have been used to reduce or somewhat quench fluorescence of dye-labeled peptides at the outer membrane.146 Additionally, trypsin and/or heparin treatment can be used to eliminate excess signal from noninternalized molecules at the cell membrane, though the completeness of this step must be verified.146,147
Often, a combination of fluorescence microscopy and flow cytometry has been used for qualitative assessment of subcellular localization and quantitative measurement of total cellular uptake, respectively. This approach can distinguish highly penetrant molecules from poorly penetrant molecules, but may not be suited for careful comparisons among subtly different molecules. Another limitation is that all fluorescence-based assays require a bulky, hydrophobic dye to be attached to the molecule-of-interest. As with all chemical tags, attachment of a fluorescent dye can alter the solubility, cellular uptake, and endosomal escape of the tagged molecule.148–150 If the molecule-of-interest can be degraded in the culture medium or in the cell (particularly relevant to lysosomally trapped material), the dye may become cleaved from the cargo, leading to staining of the cytosol and misinterpretation of the results. One approach that could sidestep some of these drawbacks is to label the molecule in situ after incubation of the molecule-of-interest with the cells, but high background and low sensitivity have prevented this approach from being widely adopted.151
Overall, only carefully interpreted data from microscopy and flow cytometry have generally correlated with results from mass spectrometry and other cell penetration assays that measure total cellular uptake,146,152,153 but none of these methods are suitable for measuring cytosolic localization.
Ion Conductance Microscopy and Electrochemical Microscopy.
In 2017, Unwin and co-workers154 used scanning ion conductance microscopy (SICM) and scanning electrochemical microscopy (SECM) in tandem to monitor the internalization of biomolecules across specific, localized patches of the cell membrane. This assay involved a two-barrel probe system in proximity to a single cell. One compartment of the probe contained the molecule-of-interest, and the other housed a carbon electrode that measured the local concentration and flux of molecules as they were delivered across the plasma membrane.154 This method measures not only the amount of uptake, but also the rate of uptake, at several localized areas of a single cell. Yet, the method cannot distinguish the subcellular localization of the molecule-of-interest.
ASSAYS THAT QUANTITATE CYTOSOLIC LOCALIZATION
Methods that monitor the total cellular uptake of biomolecules were at one time widely accepted and trusted as measurements of “cell penetration”. It was common 10 years ago to assume that these measurements were reflective of a molecule’s cytosolic localization. As the fields of peptide, protein, and nucleic acid delivery grew, increasing evidence surfaced that common methodology was not adequate, as it did not report on cytosolic localization, but rather total cellular uptake, which includes membrane-associated and endosomally trapped material.
While these assays were standard several years ago and still offer useful information on total cellular uptake, most investigators today are careful to avoid conflating these two measurements. As described in this section and in Table 2, several methods have been developed to measure the degree to which a molecule-of-interest reaches the cytosol, distinct from total cellular uptake. As a whole, cytosolic localization assays have largely surpassed total internalization assays as the methods of choice for detecting cellular penetration.
Table 2.
Summary of Methods for Monitoring the Cytosolic Localization of Biomolecules
| method | tag | detection method | throughputa | caveatsb | refs |
|---|---|---|---|---|---|
| environment-sensitive fluorescence | dye | fluorescence microscopy/flow cytometry | high | LABEL | 160, 163 |
| DEG | |||||
| NONCYT | |||||
| fluorescence correlation spectroscopy (FCS) | dye | fluorescence microscopy | low | LABEL | 168, 169 |
| DEG | |||||
| INST | |||||
| splice-correcting | single-stranded complementary RNA | fluorescence or luminescence spectroscopy | high | LABEL | 171, 172 |
| DEG | |||||
| CELL | |||||
| AMPL | |||||
| dexamethasone reporter genes | dexamethasone | fluorescence or luminescence spectroscopy | high | LABEL | 54, 175 |
| DEG | |||||
| CELL | |||||
| AMPL | |||||
| split protein complementation | peptide | fluorescence or luminescence spectroscopy | high | LABEL | 183,186-188 |
| DEG | |||||
| QUANT | |||||
| CELL | |||||
| farnesylation | CaaX tetrapeptide | SDS-PAGE | medium | LABEL | 197 |
| DEG | |||||
| deubiquitination | dye and ubiquitin | SDS-PAGE and fluorimaging | medium | LABEL | 198 |
| DEG | |||||
| glucocorticoid-induced eGFP translocation (GIGT) | dexamethasone | fluorescence microscopy | medium | LABEL | 54 |
| DEG | |||||
| INST | |||||
| CELL | |||||
| calcein release | none or calcein-AM | fluorescence microscopy/flow cytometry | high | LABEL | 209, 210 |
| DEG | |||||
| fluorogenic probe and enzyme pair | dye and additional functional group | fluorescence microscopy/flow cytometry | high | LABEL | 200, 228 |
| DEG | |||||
| QUANT | |||||
| CELL | |||||
| biotin ligase assay (BirA) | Avi tag | quantitative Western blot | medium | LABEL | 229 |
| DEG | |||||
| CELL | |||||
| chloroalkane penetration assay (CAPA) | chloroalkane | flow cytometry | high | LABEL | 220 |
| DEG | |||||
| CELL |
Throughput: Low throughput refers to methods that require multiple readings per cell, processing large bulk quantities of cells, or preparation of cell lysates; medium throughput refers to methods that require one reading per cell, such as simple or automated fluorescence microscopy; high throughput refers to methods that require one reading per cell but can process many cells rapidly, such as flow cytometry.
Caveats: Important caveats and limitations for each assay are noted as follows: quantitation requires careful calibration (QUANT); requires a tagged or labeled molecule (LABEL); potential artifacts due to degradation (DEG); sophisticated instrumentation, algorithms, and/or data processing required (INST); transfected or stable cell line required (CELL); signal is amplified, and thus is not proportional to amount of molecule translocated (AMPL); noncytosolic material may contribute to assay signal (NONCYT).
Assays That Separate the Cytosol Using Ultracentrifugation.
As mentioned above, one of the major drawbacks of analyzing cell lysates is that the lysate is a mixture of all cell-associated material, including material from the cytosol, organelles, vesicles, and plasma membrane. To isolate the cytosolic fraction from membrane-enclosed compartments, a lysate can be physically separated by high-speed ultracentrifugation. Subcellular fractionation is a common technique in cell biology, where Western blotting is used to confirm fractionation of different compartments and to compare the levels of endogenous biomolecules in those compartments.156–158 Applied to any of the lysate-based cell penetration assays described above, fractionation potentially allows independent quantitation of the amount of molecule-of-interest localized in each cellular compartment. However, subcellular fractionation is technically demanding, particularly for separation of small endosomes from cytosolic material. It can also be difficult to verify complete separation of each compartment, particularly if the cargo is small enough to diffuse through membranes permeabilized in the lysis or fractionation process.159
Assays That Distinguish Cytosolic Fluorescence.
Some of the first assays designed to selectively measure the cytosolic access of a molecule-of-interest exploited the unique chemical environment of the cytosol. In 2001, Langel and co-workers160 labeled molecules-of-interest with a fluorescence quencher and further conjugated these to a fluorophore-containing peptide via a disulfide bond. Treatment of cultured cells with these disulfide-conjugated, self-quenched molecules would result in a fluorescence signal only if the disulfide was broken, which was assumed to only happen in the reducing environment of the cytosol (Figure 3a). However, one drawback of this assay is the possibility of disulfide reduction in the endosome or even at the cell membrane, making it harder to distinguish between different cellular compartments.161,162 This approach required a large label, but it did allow monitoring of the kinetics of accumulation, albeit not the subcellular localization.
Figure 3.

Assays that distinguish cytosolic fluorescence from fluorescence in endosomes or other compartments. (a) If a molecule-of-interest or CPP cargo is labeled with a fluorophore (magenta), and also linked to a fluorescence quencher (blue) via a disulfide bond, then cytosolic localization can be inferred from dequenching of the fluorophore following reduction of the disulfide bond. (b) The pH-sensitive dye naphthofluorescein has low fluorescence in acidic environments such as endosomes and higher fluorescence in the cytosol where pH is close to 7. (c) Fluorescence correlation spectroscopy uses a diffusion model to quantitate absolute concentrations of a fluorescent dye within a focal volume chosen to exclude endosomes and other subcellular compartments. Caveats for each of these methods are summarized in Table 2.
Another fluorescence-based method takes advantage of the pH difference between the cytosol and the endolysosomal compartment by conjugating a pH-sensitive dye, naphthofluorescein, to the molecule-of-interest (Figure 3b).163 Naphthofluorescein is nearly completely protonated and exhibits no fluorescence when trapped in the endosome or lysosome, where the pH is between 5 and 6, but becomes fluorescent when it escapes the endosome and enters the cytosol.163,164 Naphthofluorescein fluorescence thus reports on any cellular compartments with neutral pH, so careful additional analysis is needed to distinguish between cytosolic staining and other neutral, membrane-enclosed compartments. Notably, by comparing the ratio of fluorescence from internalized rhodamine-labeled molecules to that of internalized naphthofluorescein-labeled molecules, the relative efficiencies of endosomal escape for different molecules-of-interest could be estimated.35,163,165–167
While these assays were designed to report exclusively on cytosolic localization, they required additional independent information, and they lacked absolute quantitation. Recently, a new fluorescence-based method was reported that provided absolute quantitation of fluorophore concentration in the cytosol (Figure 3c).146,168,169 This method uses fluorescence correlation spectroscopy (FCS) to measure the diffusion of a dye within a small, defined focal volume of the cell over time, as calculated using a modified three-dimensional diffusion model.146,168,169 For FCS measurements to report exclusively on the cytosol, focal volumes must be manually selected to exclude areas likely to contain endosomes or other membranes or compartments. This judgment requires both excellent spatial resolution and familiarity with the morphology of the particular cell line under study. FCS correlated well with related measurements using fluorescence microscopy and flow cytometry, but with the ability to exclude material trapped in endosomes or bound to membranes.153,168 FCS was later combined with independent FACS measurements to compare both total cellular uptake and cytosolically localized material.169 A dual-color variation of FCS, fluorescence cross-correlation spectroscopy (FCCS), was developed to study the dynamic colocalization of two molecules labeled with orthogonal fluorophores.170 Provided that the chosen cytosolic region does not inadvertently contain other compartments, a major advantage of FCS and FCCS is that they provide an absolute measurement of cytosolic concentration, while most other assays provide only a measurement of relative concentration.168 While FCS relies on manual identification of nonendosomal locations that may be difficult to cross-validate with other methods, the published data correlate well to other measures of cytosolic localization.
There are several important drawbacks for all cell penetration assays that measure localization of a dye-labeled cargo. Foremost among these are the potential effects of the dye itself. The dye changes the physicochemical properties of the molecule-of-interest (or CPP cargo), often in ways that can affect uptake and endosomal escape. Thus, results with a dye-labeled molecule may not be representative of the cytosolic localization of the molecule alone. In addition, if the molecule can be degraded in the culture medium or in the endolysosomal compartment, this can lead to release of the dye and diffusion to cellular compartments which are not accessible to the molecule-of-interest being investigated. These drawbacks are characteristic of any assay that requires covalent labeling of the molecule-of-interest. Ultimately, the potential effects of the dye and of cargo degradation must be estimated and minimized using carefully designed control experiments.
Assays that Measure Expression of a Reporter Protein.
Several research groups have produced cell lines that report on cytosolic localization of an exogenously added molecule-of-interest by turning on expression of a reporter protein. An early example of this approach is the luciferase splice-correction assay (Figure 4a), used to monitor the cytosolic localization of a molecule-of-interest labeled with a short, splice-altering RNA.171,172 The assay used a cell line that incorporated a luciferase gene interrupted by a mutated β-globin intron. Upon delivery to the cytosol, the RNA label spliced out the intron and allowed for the expression of active luciferase, which was quantified using luciferin as a substrate.171,172
Figure 4.

Assays that measure expression of a reporter protein. (a) In the splice-correcting assay, molecules-of-interest are labeled with a nucleic acid sequence that is complementary to an interrupted luciferase mRNA transcript. When the molecule reaches the cytosol, the nucleic acid label corrects the aberrantly spliced transcript, resulting in luciferase expression that can be detected using standard luminescence methods. (b) Molecule-of-interest labeled with dexamethasone can bind to the cytosolic glucocorticoid receptor. This leads to translocation to the nucleus and expression of a reporter gene (luciferase or GFP). (c) A molecule-of-interest labeled with a GFP-derived peptide can reconstitute GFP upon cytosolic localization by complementing a genetically expressed, larger fragment. Caveats for each of these methods are summarized in Table 2.
In 2004, Dowdy and co-workers173 developed a similar approach using Cre recombinase.173,174 Cells expressed a reporter gene for eGFP or luciferase, but the reporter gene was disrupted by a loxP site. Reconstitution of the active, expressible reporter gene was achieved by delivery of active Cre to the nucleus. For instance, Tat-Cre fusion proteins were incubated with mouse T cells in culture, and delivery of Cre was measured by eGFP expression using fluorescence microscopy and flow cytometry.173 This method reports unambiguously on the delivery of Cre to the nucleus, but only a single turnover of one Cre molecule is needed for regenerating the active gene, after which the signal is amplified by transcription and translation. Thus, while very sensitive, the output of this assay is unlikely to scale linearly with the amount of Cre delivered and may overestimate delivery efficiency for methods with poor endosomal escape efficiency.
In 2005, Kodadek and colleagues175 reported a different approach to measuring cytosolic delivery using expressed reporters (Figure 4b). This assay used cells transfected with plasmids encoding luciferase downstream from a Gal4 promoter and a hybrid transcription factor made up of Gal4 and the glucocorticoid receptor (GR).175 Dexamethasone, a GR ligand, was covalently attached to the molecule-of-interest. When the dexamethasone-tagged molecule reached the cytosol, binding of dexamethasone to GR induced translocation of the transcription factor to the nucleus and thus expression of the reporter. An increase in the luciferase signal was correlated to increased extent of cytosolic localization, allowing the comparison of hundreds of peptides and peptoids in a high-throughput format.175–181 Schepartz and co-workers54 reported improved versions of this assay that used a modified GR that binds much more specifically to dexamethasone, as well as eGFP instead of luciferase to provide a linear, stoichiometric readout. A direct fusion of GR to eGFP was similarly used to monitor cytosolic localization based on quantitating the degree of nuclear localization of the GR-eGFP conjugate.54
Each of these dexamethasone-based assays is high-throughput, sensitive, and cytosol-specific. Because the signal obtained from transcription-dependent assays is amplified, they allow for greater sensitivity for comparing molecules with low levels of cytosolic localization. However, transcriptional amplification also means that the signal is not linearly correlated with the extent of penetration; the assay that monitors the extent of nuclear localization of GR-–eGFP relies on a nonamplified signal and may better provide quantitative comparisons among cell-penetrant molecules. Even with these methods, it is difficult to obtain absolute quantitation.
Another method commonly used to measure cell penetration of biomolecules can be grouped as protein complementation assays (Figure 4c), including reporters such as split-GFP,182 split-luciferase,183 and split-ubiquitin.184 GFP can be separated into two fragments, with a smaller peptide containing only 15 amino acids and the larger fragment being nonfluorescent. When combined, the two fragments complement to a functional, fluorescent GFP.182,185 To assay cytosolic localization, the smaller fragment was conjugated to molecules-of-interest, and the larger fragment was transiently or stably expressed in the cytosol.186 Once the molecule enters the cytosol, the peptide bound the incomplete, larger GFP fragment, and the resulting fluorescent signal was quantified by flow cytometry.186 Many variations of this strategy have been reported with different reporters and tags, often investigating both cytosolic localization and disruption of protein–protein interactions in live cells.51,187–196 These assays, while reporting exclusively on cytosolic access, are still an indirect measure of cytosolic localization because they depend on the extent to which the molecule accesses the cytosol, but also its binding to its unique cellular target.
Tag-based reporter assays share some of the same issues as dye-dependent assays, namely, that the chemical tag may alter cargo properties and that degradation of the cargo in lysosomes may release the tag, leading to artifacts that falsely indicate efficient cytosolic localization. For all reporter assays, a stable cell line would ideally be produced, since throughput and reproducibility can be limited by the need to transfect cells with reporter plasmids prior to each run of the assay, as transfection efficiencies will vary. Finally, as noted above, any signal amplification will increase sensitivity, but will also make the signal nonlinear with respect to the amount of cargo in the cytosol. Overall, these drawbacks are relatively mild, and reporter-based assays continue to see robust use, especially for specific and high-value drug targets for which cell line construction is worth the investment.
Assays That Measure Direct Interactions with Cytosolic Enzymes.
More recently, several groups have devised assays that rely on cytosolic enzyme activity to generate a quantitative signal. The general principle of these assays is to make the readout dependent on a cytosolically localized enzyme, thus reporting exclusively on cytosolic localization of the cargo. An early example of this strategy was the farnesylation penetration assay (Figure 5a) developed by Falnes et al.197 The delivered protein was engineered with a C-terminal CaaX motif, a tetrapeptide that is a well-characterized farnesylation substrate. Following addition of the CaaX-labeled protein to the cell, the extent of protein farnesylation was analyzed by SDS-PAGE and correlated to cytosolic localization.197 The molecule-of-interest is given a membrane association, however, by the farnesyl anchor.
Figure 5.

Assays that measure direct interactions with cytosolic enzymes. (a) The farnesylation assay involves the transfer of a farnesyl group to a CaaX motif appended to the molecule-of-interest. The extent of farnesylation can be monitored by SDS-PAGE or Western blot. (b) In the deubiquitination assay, a dye-labeled ubiquitin is released from the molecule-of-interest by cytosolic deubiquitinases. This change is also measured by SDS-PAGE or Western blot. (c) Using a molecule-of-interest tagged with a diglycosylated fluorescein, cytosolic localization is quantitated based on extent of galactopyranoside cleavage as measured by fluorescent signal. This signal can be detected by flow cytometry since it can only originate from the cytosol. (d) When it enters the cytosol, molecule-of-interest is biotinylated by cytosolic bacterial biotin ligase. The extent of biotinylation can be measured using a quantitative Western blot. (e) Chloroalkane-labeled molecule-of-interest irreversibly labels HaloTag protein expressed in the cytosol. The relative amount of unreacted HaloTag is measured by chasing with a chloroalkane dye and measured by flow cytometry. Caveats for each of these methods are summarized in Table 2.
The farnesylation assay inspired a ubiquitin-based assay (Figure 5b), which monitored a protein’s access to the cytosol using ubiquitin (Ub) cleavage. After cytosolic delivery of the Ub-protein fusion, ubiquitin-specific C-terminal proteases recognized and cleaved Ub.198,199 The Ub-protein fusion was fluorescently labeled, so the extent of cleavage could be monitored by SDS-PAGE and Western blot.198 Notably, using Ub as a tag is prohibitively large for most small molecules, nucleic acids, and peptides, and so this system is most practical for quantitating protein penetration.
Several enzyme-activated fluorogenic probes exist that can assay cytosolic localization (Figure 5c); these have been extensively reviewed previously.200 One common example requires labeling a cargo with fluorescein-di-β-D-galactopyrano-side (FDG, a di-O-glycosylated derivative of fluorescein).201–203 In this assay, the two sugars are cleaved off by β-galactosidase expressed in the cytosol of transfected cells, resulting in the unmasking of fluorescein. The dye is then localized and quantified by fluorescence microscopy and flow cytometry.201,202,204 A similar system was reported that uses an acetoxymethyl ester of coumarin-cephalosporin-fluorescein (CCF2-AM), which is a FRET-based substrate probe developed specifically for Escherichia coli TEM-β-lactamase.203,205–207 CCF2-AM exhibits green fluorescence, but its emission wavelength switches to blue upon cleavage by cytosolic β-lactamase,208 which was monitored by fluorescence microscopy and flow cytometry.209‘210 The cytosolic calcein release assay’ originally developed to measure cell viability’ involves either covalently labeling the cargo with calcein-AM, or coencapsulating the cargo with an acetoxymethyl ester version of calcein (calcein-AM) within a liposome to investigate whether the cargo disrupts membrane integrity.209,211–213 When the nonfluorescent calcein-AM reaches the cytosol, it is cleaved by cytosolic esterases, releasing fluorescent calcein which can be measured by microscopy or flow cytometry. A similar assay using luminescence instead of fluorescence for more sensitive detection involves conjugation of the molecule to luciferin with a disulfide bond. The release of luciferin from the molecule-of-interest in the reducing environment of the cytosol allows for detection via exogenously expressed luciferase in the cytosol.214 This approach reports on the cytosolic location of the molecule, with the limitation that any luciferin cleaved upon degradation of the molecule-of-interest would also passively diffuse to the cytosol.
Instead of cleaving a functional group from the labeled molecule; the biotin ligase assay (Figure 5d) introduced by Pluckthun and co-workers96 relies on biotinylation of the cargo by a cytosolically expressed biotin ligase (E. coli BirA).96,215 This requires fusing the cargo to an avi-tag, which is a 15-residue substrate for BirA216–218 but not a substrate of eukaryotic biotin ligases. Importantly, the assay can distinguish between total cellular uptake and cytosolic localization. Total internalization can be measured by a second tag fused to the cargo, while cytosolic localization is independently measured by blotting for biotinylated protein.96,219 This provides a means of calculating the relative efficiency of endosomal escape, as well as the absolute amount of cytosolic and total molecule-of-interest when Western blots are compared to calibration curves with known standards. The Western blots also test for the integrity of the molecule but limit sample throughput.
A conceptually similar assay, but with a smaller tag, was reported in 2017 by Kritzer and co-workers.220,221 The chloroalkane penetration assay (CAPA) (Figure 5e) uses a cell line that expresses cytosolic HaloTag protein. HaloTag is a modified bacterial haloalkane dehalogenase that covalently labels itself with a short chloroalkane, and it does so with fast kinetics and high specificity.222 Cells expressing cytosolic HaloTag protein were incubated with a chloroalkane-conjugated molecule-of-interest. Molecules that reached the cytosol reacted with HaloTag and blocked its active site. Subsequent incubation with chloroalkane-labeled dye resulted in labeling of all unreacted HaloTag, providing a signal that was inversely proportional to the extent of cytosolic localization of the molecule-of-interest.220,221 CAPA signal was quantified by flow cytometry, allowing quantitation in a 96-well plate format. One downside of this assay is that the signal is inversely proportional to the amount of internalized material, limiting sensitivity for detecting small amounts of material localized to the cytosol.
Cell penetration assays that rely on enzymes are versatile and compartment-specific, and can be very high-throughput. However, assays that do not use native enzymes, such as the biotin ligase assay and CAPA, must use transiently transfected or, preferably, stably transfected cell lines. These assays are also tag-based, and thus careful controls are required to minimize perturbations by the tag and/or limit the extent of signal that might be due to degradation of the cargo.
Assays That Measure Cell Death Following Delivery of Toxic Cargo.
Some reports have used a direct phenotypic readout to measure delivery of a cargo that was toxic to cells, or induced apoptosis.223,224 Zahaf et al.225 measured effective translocation of the catalytic part of ADP-ribosyltransferase TccC3 by anthrax toxin by evaluating the viability of two cancer lines. Quantification has also been accomplished by detecting markers of apoptosis.226,227 Importantly, analyzing the delivery efficiency with a biological readout must be performed very carefully and with a thorough understanding of the delivery mechanism. Potential effects on cellular integrity by individual components of the delivery system and/or stress induced by the delivery process itself must be excluded to ensure that the biologic effects are solely due to the cytosolic or nuclear presence of the cargo. Finally, there are many factors other than cytosolic localization of the cargo that might alter the health of cultured cells. As a result, assays that measure cell death as a proxy for cytosolic localization can be less specific, and thus less reliable, than other options.
ARTIFACTS AND MISINTERPRETATIONS IN MEASUREMENTS OF TOTAL CELLULAR UPTAKE AND CYTOSOLIC LOCALIZATION
Maximal cytosolic delivery remains the goal of most delivery systems. Clearly, assays are needed that can unambiguously differentiate between total cellular uptake, which includes material trapped in endosomes, and cytosolic localization. Unfortunately, some assays are employed in a way that leads to artifacts and results in misinterpretation. These are most disruptive to the field when they overestimate the efficiency of a delivery mechanism. In this section, we highlight some common pitfalls and how to avoid them.
Artifacts and Misinterpretations of Fluorescence Microscopy.
Microscopy is a versatile tool to qualitatively observe the intracellular location of delivered cargo molecules. Many reports have claimed the cytosolic localization of a cargo protein with a preferred delivery method based solely on microscopy using a dye-labeled cargo. In the final analysis, this method requires absolute differentiation of punctate staining from diffuse staining, distinguishing endosomally trapped material from cytosolic material. Both nuclear exclusion and nuclear enrichment can be misinterpreted and actually instead indicate that the cargo has not reached the cytosol. Nuclear exclusion can indicate endosomal localization (particularly for cargoes small enough that they should have traversed the nuclear pores if they had accessed the cytosol). Similarly, at low resolution, apparent nuclear enrichment can also instead indicate endosomal localization, since membrane-enclosed vesicles can appear nuclear or perinuclear.
Because the desired outcome is typically to achieve some degree of cytosolic delivery, there is an inherent temptation to interpret diffuse staining as cytosolic localization of a dye-labeled molecule. However, diffuse staining in fluorescence micrographs must be interpreted cautiously, as it can have multiple origins. Something as simple as poor resolution may give the impression of homogeneous staining, but even higher-resolution microscopy can misinterpret out-of-frame fluorescence or very small puncta as diffuse, cytosolic staining. Conversely, only the highest confocal microscopy standards, combined with the most meticulous safeguarding against permeabilization artifacts in preparing and treating microscopy samples, can protect against an erroneous conclusion of delivery to the cytosol.
Prior to the mid-2000s, cells were often fixed prior to observation by fluorescence microscopy, in part to improve resolution. These fixation conditions ranged from mild (4% paraformaldehyde) to harsh (methanol).88,230–234 A historically relevant example is the discussion of fixation artifacts in the measurement of the delivery efficacy of VP22, a type I herpes simplex virus tegument protein.235 Results suggested that a greater degree of internalization was observed with methanol fixation as compared to paraformaldehyde fixation. The authors rationalized this observation by proposing that methanol fixation was able to “concentrate”, “refold”, or “dequench” internalized GFP–VP22 fusion proteins, as compared to the milder fixation by PFA, which was proposed to quench GFP signals.235 Since that time, it has become clear that the appearance and distribution of intracellular fluorescence is dependent on the fixation method for translocated proteins and oligonucleotides.236‘237 Lundberg et al.238 further showed the apparent delivery of VP22 before and after fixation with methanol and demonstrated that highly positively charged molecules locate to plasma membranes before fixation and, upon fixation relocate to the nucleus, interact with negatively charged DNA. The same behavior was shown with histone H1, which was found in the nucleus of cells only after fixation.238
In 2003 the uptake mechanism of TAT and polyarginine (Arg)9 was evaluated with respect to fixation conditions. Very different location patterns for translocated CPP were observed in fixed and nonfixed samples.18 Cells fixed with formaldehyde showed nuclear staining and some cytosolic distribution, but in unfixed cells, the CPPs colocalized mainly with endosomal marker. The authors concluded that the highly charged CPP was bound to the endosomal membrane, and the membrane was disrupted upon fixation. Thus, cytosolic distribution of the peptide observed following fixation was an artifact of the fixation process. This was further underlined by Fischer et al. in 2007,239 who concluded that membrane integrity is lost upon fixation, and the delivered peptides redistribute and associate with negatively charged DNA.
On the basis of these studies, cells should never be fixed prior to assessing the cytosolic localization of a molecule. While modern assay development avoids fixation altogether, care must be taken in interpreting the literature, particularly older reports that have examined cytosolic or nuclear staining after fixation. Only live cell imaging should be used to examine subcellular localization of a cargo or molecule-of-interest.240,241 To minimize errors of interpretation, confocal microscopy should be used to eliminate out-of-frame fluorescence, with a resolution high enough to unequivocally differentiate punctate staining from true diffuse, cytosolic staining. Explaining detection after fixation by concentration effects or removal of interfering or quenching components can nowadays not be justified. In general, the authors must rigorously exclude any processes, additives, or conditions that may compromise membrane integrity or demonstrate experimentally that these conditions do not permeabilize membranes.
Extremely High Concentrations of Cargo Molecules.
Another common experimental condition that may contribute to misinterpretation is extremely high concentrations (10 μM, and even 50 μM or above have been reported) of molecules-of-interest.14,43,242–244 Because of the low efficiency of many delivery methods, it is common for investigators to increase molecule concentrations until an internalized or cytosolic fraction can be detected. Instead of taking low fluorescent signals as a sign of inefficient delivery to the cytosol, such low signals have also been interpreted as being caused by quenching by other molecules, motivating some investigators to use concentrations as high as 50 μM. For more efficient delivery systems, this should be unnecessary—for instance, Verdurmen et al.219 showed that cytosolic localization can be detected at low nanomolar concentrations, with outside concentrations in the nanomolar range, when appropriate methods of detection and efficient translocation systems are used.
Delivery mediated by CPPs and supercharged proteins can show evidence of endosomal escape, typically at or above a certain threshold concentration,36 partly as an effect of endosomal membrane rupture of highly packed endosomes. An attractive model for this effect is that, due to the high net positive charge of CPPs and supercharged proteins, a water influx occurs into the endosomes and leads to the rupture of endosomal membranes, allowing cytosolic diffusion of delivered cargo.37 The higher the concentration of positively charged molecules, the faster and more efficient is this process.36,37 To avoid endosomal membrane rupture as a possible mechanism for endosomal leakage, concentrations below 10 μM should ideally be used; if needed, a more sensitive assay should be used to detect the presence of cargo molecules at lower concentrations.
Another problem with increasing concentrations until some degree of penetration is observed is the likelihood of aggregation or insolubility at high concentrations. Even at more reasonable concentrations, molecules of interest can become insoluble, forming aggregates that may or may not be visible as cloudiness or turbidity. Insolubility is often exacerbated by the addition of large, hydrophobic tags. Molecules that are soluble in buffer may also be poorly soluble in culture medium, and this insolubility should not be ignored since cultured cells require growth medium to minimize stress. For all cell penetration experiments, the possibility of molecule insolubility or aggregation should be eliminated by careful preparation of solutions, including filtering and/or centrifugation, and inspection of treated cells using microscopy to check for the presence of aggregates. These practices should be routine to ensure material remains soluble under assay conditions.
If authors intend to show an energy-independent uptake mechanism, delivery experiments are often performed at 4 °C and compared to those at 37 °C. Many authors propose that an alternative, energy-independent uptake mechanism must be occurring if the molecule-of-interest is internalized at 4 °C. Yet, some reports investigated uptake at 4 °C by incubation at concentrations up to 500 μM.245–247 At such high concentrations, molecules sticking to the cell surface or residual presence of cargo protein after washes can give rise to a signal falsely interpreted as intracellular delivered protein.75 The interpretation of observed intracellular cargo might thus become questionable and especially so when combined with fixation.43,246 Careful processing of cells, avoiding sudden temperature changes or similar stress-inducing procedures before or after the incubation with cargo protein, is also crucial to prevent stress-induced rupture and apparent cytosolic delivery.28,59,63,248 Sudden changes in physiological conditions can potentially alter membrane integrity and/or intracellular organization and should therefore be avoided.
Medium Components and Cellular Integrity.
Translocation efficiencies have been studied for delivery of specific cargoes in serum-containing medium, serum-free medium and PBS.28,59,61,242,246,249 Intracellular accumulation of cargo molecules increases upon incubation with serum-free medium or PBS for some CPPs, compared to accumulation in cells treated with serum-containing medium.242,250 The underlying mechanism is the interaction between the positively charged CPP and negatively charged serum components that inhibit cellular interaction of the CPP.
If cell-based assays are not performed in growth medium, cellular viability and membrane integrity should be evaluated with great care, especially when other additives are used.248 Dimethyl sulfoxide (DMSO) for instance; has been widely used for cryopreservation of cells and tissue, as a DNA transfection enhancer, as well as an enhancer in topical pharmaceutical formulations.251–254 Studies suggest that high concentrations of DMSO lead to membrane loosening, pore formation, and even the collapse of the lipid bilayer.255 While some reports have investigated the effects of high concentrations of DMSO,246,256,257 if cellular membrane integrity cannot be ensured, it is not possible to evaluate the effectiveness of a delivery mechanism. Even low (1% or less) concentrations of DMSO can lead to phenotypic changes in cultured cells. Because of significant safety considerations DMSO would also not be useable in vivo.
Therefore, ideally a delivery system would be tested in the standard growth medium of the cell line used, with no other additives.
Clinical Applications of Presented Delivery Mechanisms.
Clinical or in vivo applications of most described delivery mechanisms, notably those relying on positive charges, remain very challenging due to poor pharmacokinetics, nonspecific interactions with serum components and other molecules, lack of cell specificity, endosomal entrapment, and poor efficiency for cytoplasmic delivery of cargo molecules.27,258–260
In a recent review, Bernkop-Schnürch261 pointed out that a common issue for the charge-based delivery system is that during in vivo applications they pass through a gauntlet of polyanions—mucus glycoproteins, proteoglycans, and serum proteins—on the way to their target tissue or organ. He termed this the “polycation dilemma”. If efficient translocation in vitro can only be obtained in the absence of serum, then such a delivery mechanism would be very challenging to develop for any in vivo applications. To solve the “polycation dilemma”, more advanced constructs have been developed, such as cleavable CPPs with an anionic counterpart.261 However, it is currently unclear whether they can be developed for in vivo use.
As stated above, several delivery systems have been shown to work effectively only above a high threshold concentration. The higher the concentration needed for cytosolic delivery, the lower the efficiency of cellular uptake and endosomal escape. If one only sees cytosolic localization at very high concentrations, one should ask whether the translocation mechanism is truly relevant for potential in vivo applications, or whether it will be limited to in vitro applications.46 For many biomolecules, it remains questionable whether high micromolar concentrations can be delivered to tissues of interest, and if a cytosolic localization mechanism that requires such a high concentration could be used in vivo without severe side effects.
Another issue of most delivery systems is the potential for immunogenic effects. VLPs, for instance, have been used as vaccines due to their high immunogenicity. This makes it uncertain whether they would be viable vectors for protein delivery in vivo.81
CONCLUSIONS AND PERSPECTIVES
The toolbox for measuring “cell penetration” is continuously being expanded. We advocate that the consideration of “cell penetration” should be replaced by an emphasis on distinguishing total cellular uptake and cytosolic localization of a cargo. This emphasis acknowledges the reality that a majority of internalized material typically remains trapped in endosomes or in lysosomes and that many desirable biological effects require efficient delivery into the cytosol or nucleus. There is no universal assay for quickly and quantitatively measuring the uptake and cytosolic localization of any cargo, and each method discussed above offers unique advantages and disadvantages. As with all reductionist approaches to biology, making measurements using multiple different assays will be the most reliable way to make firm conclusions about the cytosolic localization of a specific molecule-of-interest or delivery method.
Intracellular biotherapeutics offer great promise, providing distinct solutions to fundamental problems that hamper small molecules. If efficiently delivered to the cytosol, protein drugs would expand the “druggable” proteome far beyond the small subset of intracellular targets that possess a binding pocket for small molecules. RNA or DNA therapeutics, in turn’ would be able to modulate many steps that are inaccessible by modulating protein targets. Gene editing complexes would ultimately allow the fundamental reprogramming of a cell’s function. All of these modalities depend on cytosolic or nuclear delivery’ but many hurdles remain. Today’ the overwhelming majority of delivery strategies have not demonstrated any cell specificity, and some show toxicity. It remains unclear how to widen the therapeutic window if such biomolecules not only interact with most tissues, but also with many serum components. Future research will have to address cell specificity and tissue targeting in a major way.
Upon reading some of the literature of the last 25 years, one might erroneously conclude that delivery to the cytosol is very straightforward. If that were true, there would already be numerous FDA-approved drugs that are based on biomolecules delivered to the cytosol. The degree of difficulty should not dull the excitement surrounding the promise of cytosolically active biotherapeutics, but further progress will rely on quantitative and accurate assessments of biomolecule delivery to the cytosol. The field is not served by misinterpretations, such as inferring cytosolic localization when only total cellular uptake has been measured. Widespread success of therapeutic biomolecules with cytosolic targets will only become a reality if data are generated using robust experiments that unambiguously measure cytosolic localization.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Mayor S, and Pagano RE (2007) Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol 8, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kaksonen M, and Roux A (2018) Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol 19, 313–326. [DOI] [PubMed] [Google Scholar]
- (3).Pei D, and Buyanova M (2019) Overcoming Endosomal Entrapment in Drug Delivery. Bioconjugate Chem 30, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Stewart MP, Langer R, and Jensen KF (2018) Intracellular delivery by membrane disruption: Mechanisms, strategies, and concepts. Chem. Rev 118, 7409–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bechara C, and Sagan S (2013) Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett 587, 1693–1702. [DOI] [PubMed] [Google Scholar]
- (6).McClorey G, and Banerjee S (2018) Cell-penetrating peptides to enhance delivery of oligonucleotide-based therapeutics. Biomedicines 6, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kebebe D, Liu Y, Wu Y, Vilakhamxay M, Liu Z, and Li J (2018) Tumor-targeting delivery of herb-based drugs with cell-penetrating/tumor-targeting peptide-modified nanocarriers. Int. J. Nanomed 13, 1425–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lindgren M, and Langel Ü (2011) Classes and prediction of cell-penetrating peptides In Cell-Penetrating Peptides (Langel Ü, Ed.) Vol. 683, pp 3–19, Humana Press. [DOI] [PubMed] [Google Scholar]
- (9).Frankel AD, and Pabo CO (1988) Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55, 1189–1193. [DOI] [PubMed] [Google Scholar]
- (10).Vivès E, Brodin P, and Lebleu B (1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem 272, 16010–16017. [DOI] [PubMed] [Google Scholar]
- (11).Dupont E, Prochiantz A, and Joliot A (2011) Penetratin story: An overview In Cell-Penetrating Peptides (Langel Ü, Ed.) pp 21–29, Vol. 683, Humana Press. [DOI] [PubMed] [Google Scholar]
- (12).Joliot A, Pernelle C, Deagostini-Bazin H, and Prochiantz A (1991) Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. U. S. A 88, 1864–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, and Sugiura Y (2001) Arginine-rich Peptides: an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem 276, 5836–5840. [DOI] [PubMed] [Google Scholar]
- (14).Shirazi A, Mozaffari S, Sherpa R, Tiwari R, and Parang K (2018) Efficient intracellular delivery of cell-impermeable cargo molecules by peptides containing tryptophan and histidine. Molecules 23, 1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ramaker K, Henkel M, Krause T, Röckendorf N, and Frey A (2018) Cell penetrating peptides: a comparative transport analysis for 474 sequence motifs. Drug Delivery 25, 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Manavalan B, Subramaniyam S, Shin TH, Kim MO, and Lee G (2018) Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res 17, 2715–2726. [DOI] [PubMed] [Google Scholar]
- (17).Favaro M.T. de P., Serna N, Sánchez-García L, Cubarsi R, Roldáan M, Sánchez-Chardi A, Unzueta U, Mangues R, Ferrer-Miralles N, Azzoni AR, et al. (2018) Switching cell penetrating and CXCR4-binding activities of nanoscale-organized arginine-rich peptides. Nanomedicine 14, 1777–1786. [DOI] [PubMed] [Google Scholar]
- (18).Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, and Lebleu B (2003) Cell-penetrating Peptides: A reevaluation of the mechanism of cellular uptake. J. Biol. Chem 278, 585–590. [DOI] [PubMed] [Google Scholar]
- (19).Pescina S, Sala M, Padula C, Scala MC, Spensiero A, Belletti S, Gatti R, Novellino E, Campiglia P, Santi P, et al. (2016) Design and synthesis of new cell penetrating peptides: Diffusion and distribution inside the cornea. Mol. Pharmaceutics 13, 3876–3883. [DOI] [PubMed] [Google Scholar]
- (20).Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, and Rothbard JB (2000) The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. U. S. A 97, 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kobayashi H, Misawa T, Oba M, Hirata N, Kanda Y, Tanaka M, Matsuno K, and Demizu Y (2018) Structural development of cell-penetrating peptides containing cationic proline derivatives. Chem. Pharm. Bull. 66, 575–580. [DOI] [PubMed] [Google Scholar]
- (22).Kumar V, Agrawal P, Kumar R, Bhalla S, Usmani SS, Varshney GC, and Raghava GPS (2018) Prediction of cell-penetrating potential of modified peptides containing natural and chemically modified residues. Front. Microbiol 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wolfe JM, Fadzen CM, Choo Z-N, Holden RL, Yao M, Hanson GJ, and Pentelute BL (2018) Machine learning to predict cell-penetrating peptides for antisense delivery. ACS Cent. Sci 4, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Peraro L, and Kritzer JA (2018) Emerging methods and design principles for cell-penetrant peptides. Angew. Chem., Int. Ed 57, 11868–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Quartararo JS, Eshelman MR, Peraro L, Yu H, Baleja JD, Lin Y-S, and Kritzer JA (2014) A bicyclic peptide scaffold promotes phosphotyrosine mimicry and cellular uptake. Bioorg. Med. Chem 22, 6387–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Toyama K, Nomura W, Kobayakawa T, and Tamamura H (2018) Delivery of a proapoptotic peptide to EGFR-positive cancer cells by a cyclic peptide mimicking the dimerization arm structure of EGFR. Bioconjugate Chem 29, 2050–2057. [DOI] [PubMed] [Google Scholar]
- (27).Koren E, and Torchilin VP (2012) Cell-penetrating peptides: breaking through to the other side. Trends Mol. Med 18, 385–393. [DOI] [PubMed] [Google Scholar]
- (28).Ichimizu S, Watanabe H, Maeda H, Hamasaki K, Nakamura Y, Chuang VTG, Kinoshita R, Nishida K, Tanaka R, Enoki Y, et al. (2018) Design and tuning of a cell-penetrating albumin derivative as a versatile nanovehicle for intracellular drug delivery. J. Controlled Release 277, 23–34. [DOI] [PubMed] [Google Scholar]
- (29).Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, and Prochiantz A (1996) Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J. Biol. Chem 271, 18188–18193. [DOI] [PubMed] [Google Scholar]
- (30).Matsuzaki K, Yoneyama S, Murase O, and Miyajima K (1996) Transbilayer transport of ions and lipids coupled with Mastoparan X translocation. Biochemistry 35, 8450–8456. [DOI] [PubMed] [Google Scholar]
- (31).Pouny Y, Rapaport D, Mor A, Nicolas P, and Shai Y (1992) Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry 31, 12416–12423. [DOI] [PubMed] [Google Scholar]
- (32).Ferreira APA, and Boucrot E (2018) Mechanisms of carrier formation during clathrin-independent endocytosis. Trends Cell Biol 28, 188–200. [DOI] [PubMed] [Google Scholar]
- (33).Jones AT (2007) Macropinocytosis: searching for an endocytic identity and role in the uptake of cell penetrating peptides. J. Cell. Mol. Med 11, 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fuchs SM, and Raines RT (2004) Pathway for polyarginine entry into mammalian cells. Biochemistry 43, 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, and Pei D (2016) Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Vermeulen LMP, Brans T, Samal SK, Dubruel P, Demeester J, De Smedt SC, Remaut K, and Braeckmans K (2018) Endosomal size and membrane leakiness influence proton sponge-based rupture of endosomal vesicles. ACS Nano 12, 2332–2345. [DOI] [PubMed] [Google Scholar]
- (37).Danielsen EM, and Hansen GH (2018) Impact of cell-penetrating peptides (CPPs) melittin and Hiv-1 Tat on the enterocyte brush border using a mucosal explant system. Biochim. Biophys. Ada, Biomembr 1860, 1589–1599. [DOI] [PubMed] [Google Scholar]
- (38).Palm-Apergi C, Lönn P, and Dowdy SF (2012) Do cell-penetrating peptides actually “penetrate” cellular membranes? Mol. Ther 20, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hirose H, Takeuchi T, Osakada H, Pujals S, Katayama S, Nakase I, Kobayashi S, Haraguchi T, and Futaki S (2012) Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol. Ther 20, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hällbrink M, Oehlke J, Papsdorf G, and Bienert M (2004) Uptake of cell-penetrating peptides is dependent on peptide-to-cell ratio rather than on peptide concentration. Biochim. Biophys. Acta, Biomembr 1667, 222–228. [DOI] [PubMed] [Google Scholar]
- (41).Lee Y-J, Johnson G, Peltier GC, and Pellois J-P (2011) A HA2-Fusion tag limits the endosomal release of its protein cargo despite causing endosomal lysis. Biochim. Biophys. Acta, Gen. Subj 1810, 752–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Allen J, Brock D, Kondow-McConaghy H, and Pellois J-P (2018) Efficient delivery of macromolecules into human cells by improving the endosomal escape activity of cell-penetrating peptides: Lessons learned from dfTAT and its analogs. Biomolecules 8, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Maiolo JR, Ottinger EA, and Ferrer M (2004) Specific redistribution of cell-penetrating peptides from endosomes to the cytoplasm and nucleus upon laser illumination. J. Am. Chem. Soc 126, 15376–15377. [DOI] [PubMed] [Google Scholar]
- (44).Lee Y-J, Johnson G, and Pellois J-P (2010) Modeling of the endosomolytic activity of HA2-TAT peptides with red blood cells and ghosts. Biochemistry 49, 7854–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pazo M, Juanes M, Lostalé-Seijo I, and Montenegro J (2018) Oligoalanine helical callipers for cell penetration. Chem. Commun 54, 6919–6922. [DOI] [PubMed] [Google Scholar]
- (46).Guidotti G, Brambilla L, and Rossi D (2017) Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci 38, 406–424. [DOI] [PubMed] [Google Scholar]
- (47).Gautam A, Chaudhary K, Kumar R, Sharma A, Kapoor P, Tyagi A, Open source drug discovery consortium, and Raghava GPS (2013) In silico approaches for designing highly effective cell penetrating peptides. J. Transl. Med 11, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sanders WS, Johnston CI, Bridges SM, Burgess SC, and Willeford KO (2011) Prediction of cell penetrating peptides by support vector machines. PLoS Comput. Biol 7, e1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tang H, Su Z-D, Wei H-H, Chen W, and Lin H (2016) Prediction of cell-penetrating peptides with feature selection techniques. Biochem. Biophys. Res. Commun 477, 150–154. [DOI] [PubMed] [Google Scholar]
- (50).Pandey P, Patel V, George NV, and Mallajosyula SS (2018) KELM-CPPpred: Kernel extreme learning machine based prediction model for cell-penetrating peptides. J. Proteome Res 17, 3214–3222. [DOI] [PubMed] [Google Scholar]
- (51).Kauffman WB, Guha S, and Wimley WC (2018) Synthetic molecular evolution of hybrid cell penetrating peptides. Nat. Commun 9, 2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Gaj T, Guo J, Kato Y, Sirk SJ, and Barbas CF (2012) Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat. Methods 9, 805–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Gaj T, Liu J, Anderson KE, Sirk SJ, and Barbas CF (2014) Protein delivery using cys2 - his2 zinc-finger domains. ACS Chem. Biol 9, 1662–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Holub JM, LaRochelle JR, Appelbaum JS, and Schepartz A (2013) Improved assays for determining the cytosolic access of peptides, proteins, and their mimetics. Biochemistry 52, 9036–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cronican JJ, Thompson DB, Beier KT, McNaughton BR, Cepko CL, and Liu DR (2010) Potent delivery of functional proteins into mammalian cells in vitro and in vivo using a supercharged protein. ACS Chem. Biol 5, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Motevalli F, Bolhassani A, Hesami S, and Shahbazi S (2018) Supercharged green fluorescent protein delivers HPV16E7 DNA and protein into mammalian cells in vitro and in vivo. Immunol. Lett 194, 29–39. [DOI] [PubMed] [Google Scholar]
- (57).Fuchs SM, and Raines RT (2007) Arginine grafting to endow cell permeability. ACS Chem. Biol 2, 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).McNaughton BR, Cronican JJ, Thompson DB, and Liu DR (2009) Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. Proc. Natl. Acad. Sci. U. S. A 106, 6111–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Cronican JJ, Beier KT, Davis TN, Tseng J-C, Li W, Thompson DB, Shih AF, May EM, Cepko CL, Kung AL, et al. (2011) A class of human proteins that deliver functional proteins into mammalian cells in vitro and in vivo. Chem. Biol 18, 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Thompson DB, Villaseñor R, Dorr BM, Zerial M, and Liu DR (2012) Cellular uptake mechanisms and endosomal trafficking of supercharged proteins. Chem. Biol 19, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Hu S, Chen X, Lei C, Tang R, Kang W, Deng H, Huang Y, Nie Z, and Yao S (2018) Charge designable and tunable GFP as a target pH-responsive carrier for intracellular functional protein delivery and tracing. Chem. Commun 54, 7806–7809. [DOI] [PubMed] [Google Scholar]
- (62).Yu M, Wu J, Shi J, and Farokhzad OC (2016) Nanotechnology for protein delivery: Overview and perspectives. J. Controlled Release 240, 24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tang R, Kim CS, Solfiell DJ, Rana S, Mout R, Velázquez-Delgado EM, Chompoosor A, Jeong Y, Yan B, Zhu Z-J, et al. (2013) Direct delivery of functional proteins and enzymes to the cytosol using nanoparticle-stabilized nanocapsules. ACS Nano 7, 6667–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Zhang Z, Shen W, Ling J, Yan Y, Hu J, and Cheng Y (2018) The fluorination effect of fluoroamphiphiles in cytosolic protein delivery. Nat. Commun 9, 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Mout R, Ray M, Yesilbag Tonga G, Lee Y-W, Tay T, Sasaki K, and Rotello VM (2017) Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano 11, 2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Sharma AR, Kundu SK, Nam J-S, Sharma G, Priya Doss CG, Lee S-S, and Chakraborty C (2014) Next generation delivery system for proteins and genes of therapeutic purpose: Why and how? BioMed Res. Int 2014, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Lohcharoenkal W, Wang L, Chen YC, and Rojanasakul Y (2014) Protein nanoparticles as drug delivery carriers for cancer therapy. BioMed Res. Int 2014, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Agrahari V, Agrahari V, and Mitra AK (2016) Nanocarrier fabrication and macromolecule drug delivery: Challenges and opportunities. Ther. Delivery 7, 257–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Fu A, Tang R, Hardie J, Farkas ME, and Rotello VM (2014) Promises and pitfalls of intracellular delivery of proteins. Bioconjugate Chem 25, 1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Mukherjee B, Karmakar S, Hossain C, and Bhattacharya S (2014) Peptides, proteins and peptide/protein-polymer conjugates as drug delivery system. Protein Pept. Lett 21, 1121–1128. [DOI] [PubMed] [Google Scholar]
- (71).Ahmed S, Hayashi F, Nagashima T, and Matsumura K (2014) Protein cytoplasmic delivery using polyampholyte nanoparticles and freeze concentration. Biomaterials 35, 6508–6518. [DOI] [PubMed] [Google Scholar]
- (72).Ahmed S, Fujita S, and Matsumura K (2016) Enhanced protein internalization and efficient endosomal escape using polyampholyte-modified liposomes and freeze concentration. Nano scale 8, 15888–15901. [DOI] [PubMed] [Google Scholar]
- (73).Mundra V, and Mahato RI (2014) Design of nanocarriers for efficient cellular uptake and endosomal release of small molecule and nucleic acid drugs: learning from virus. Front. Chem. Sci. Eng 8, 387–404. [Google Scholar]
- (74).Ma D (2014) Enhancing endosomal escape for nanoparticle mediated siRNA delivery. Nanoscale 6, 6415–6425. [DOI] [PubMed] [Google Scholar]
- (75).Iwasa A, Akita H, Khalil I, Kogure K, Futaki S, and Harashima H (2006) Cellular uptake and subsequent intracellular trafficking of R8-liposomes introduced at low temperature. Biochim. Biophys. Acta, Biomembr 1758, 713–720. [DOI] [PubMed] [Google Scholar]
- (76).Li X-X, Chen J, Shen J-M, Zhuang R, Zhang S-Q, Zhu Z-Y, and Ma J-B (2018) pH-Sensitive nanoparticles as smart carriers for selective intracellular drug delivery to tumor. Int. J. Pharm 545, 274–285. [DOI] [PubMed] [Google Scholar]
- (77).Li H, Tsui T, and Ma W (2015) Intracellular delivery of molecular cargo using cell-penetrating peptides and the combination strategies. Int. J. Mol. Sci 16, 19518–19536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Chroboczek J, Szurgot I, and Szolajska E (2014) Virus-like particles as vaccine. Acta Biochim. Polym 61, 531–539. [PubMed] [Google Scholar]
- (79).Grgacic EVL, and Anderson DA (2006) Virus-like particles: Passport to immune recognition. Methods 40, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Al-Barwani F, Donaldson B, Pelham SJ, Young SL, and Ward VK (2014) Antigen delivery by virus-like particles for immunotherapeutic vaccination. Ther. Delivery 5, 1223–1240. [DOI] [PubMed] [Google Scholar]
- (81).Zdanowicz M, and Chroboczek J (2016) Virus-like particles as drug delivery vectors. Acta Biochim. Polym 63, 469–473. [DOI] [PubMed] [Google Scholar]
- (82).Muratori C, Bona R, and Federico M (2010) Lentivirus-based virus-like particles as a new protein delivery tool In Lentivirus Gene Engineering Protocols (Federico M, Ed.) Vol. 614, pp 111–124, Humana Press. [DOI] [PubMed] [Google Scholar]
- (83).Voelkel C, Galla M, Maetzig T, Warlich E, Kuehle J, Zychlinski D, Bode J, Cantz T, Schambach A, and Baum C (2010) Protein transduction from retroviral Gag precursors. Proc. Natl. Acad. Sci. U. S. A 107, 7805–7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Miersch S, and Sidhu SS (2016) Intracellular targeting with engineered proteins. F1000Research 5, 1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Kaczmarczyk SJ, Sitaraman K, Young HA, Hughes SH, and Chatterjee DK (2011) Protein delivery using engineered viruslike particles. Proc. Natl. Acad. Sci. U. S. A 108, 16998–17003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Rabideau AE, and Pentelute BL (2016) Delivery of nonnative cargo into mammalian cells using anthrax lethal toxin. ACS Chem. Biol 11, 1490–1501. [DOI] [PubMed] [Google Scholar]
- (87).Beilhartz GL, Sugiman-Marangos SN, and Melnyk RA (2017) Repurposing bacterial toxins for intracellular delivery of therapeutic proteins. Biochem. Pharmacol 142, 13–20. [DOI] [PubMed] [Google Scholar]
- (88).Ryou J-H, Sohn Y-K, Hwang D-E, and Kim H-S (2015) Shiga-like toxin-based high-efficiency and receptor-specific intracellular delivery system for a protein. Biochem. Biophys. Res. Commun 464, 1282–1289. [DOI] [PubMed] [Google Scholar]
- (89).Clemons NC, Bannai Y, Haywood EE, Xu Y, Buschbach JD, Ho M, and Wilson BA (2018) Cytosolic delivery of multidomain cargos by the N terminus of Pasteurella multocida toxin. Infect. Immun 86, e00248–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Meusch D, Gatsogiannis C, Efremov RG, Lang AE, Hofnagel O, Vetter IR, Aktories K, and Raunser S (2014) Mechanism of Tc toxin action revealed in molecular detail. Nature 508, 61–65. [DOI] [PubMed] [Google Scholar]
- (91).Beer L-A, Tatge H, Schneider C, Ruschig M, Hust M, Barton J, Thiemann S, Fühner V, Russo G, and Gerhard R (2018) The binary toxin CDT of clostridium difficile as a tool for intracellular delivery of bacterial glucosyltransferase domains. Toxins 10, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Webb R (2018) Engineering of botulinum neurotoxins for biomedical applications. Toxins 10, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Sandvig K, and van Deurs B (2005) Delivery into cells: lessons learned from plant and bacterial toxins. Gene Ther 12, 865–872. [DOI] [PubMed] [Google Scholar]
- (94).Zielinski R, Lyakhov I, Jacobs A, Chertov O, Kramer-Marek G, Francella N, Stephen A, Fisher R, Blumenthal R, and Capala J (2009) Affitoxin—A novel recombinant, HER2-specific, anticancer agent for targeted therapy of HER2-positive tumors. J. Immunother 32, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Mechaly A, McCluskey AJ, and Collier RJ (2012) Changing the receptor specificity of anthrax toxin. mBio 3, e00088–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Verdurmen WPR, Luginbühl M, Honegger A, and Plückthun A (2015) Efficient cell-specific uptake of binding proteins into the cytoplasm through engineered modular transport systems. J. Controlled Release 200, 13–22. [DOI] [PubMed] [Google Scholar]
- (97).McCluskey AJ, Olive AJ, Starnbach MN, and Collier RJ (2013) Targeting HER2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol 7, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Zhang L, Zhao J, Wang T, Yu C-J, Jia L-T, Duan Y-Y, Yao L-B, Chen S-Y, and Yang A-G (2008) HER2-targeting recombinant protein with truncated Pseudomonas Exotoxin: A translocation domain efficiently kills breast cancer cells. Cancer Biol. Ther 7, 1226–1231. [DOI] [PubMed] [Google Scholar]
- (99).Liu S, Bugge TH, and Leppla SH (2001) Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J. Biol. Chem 276, 17976–17984. [DOI] [PubMed] [Google Scholar]
- (100).Liu S, Netzel-Arnett S, Birkedal-Hansen H, and Leppla SH (2000) Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res 60, 6061–6067. [PubMed] [Google Scholar]
- (101).Sandvig K, and van Deurs B (2002) Membrane traffic exploited by protein toxins. Annu. Rev. Cell Dev. Biol 18, 1–24. [DOI] [PubMed] [Google Scholar]
- (102).Rainey GJA, Wigelsworth DJ, Ryan PL, Scobie HM, Collier RJ, and Young JAT (2005) Receptor-specific requirements for anthrax toxin delivery into cells. Proc. Natl. Acad. Sci. U. S. A 102, 13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).J Lord JM, Smith DC, and Roberts LM (1999) Toxin entry: how bacterial proteins get into mammalian cells. Cell. Microbiol 1 85–91. [DOI] [PubMed] [Google Scholar]
- (104).Arora N, Klimpel KR, Singh Y, and Leppla SH (1992) Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J. Biol. Chem 267, 15542–15548. [PubMed] [Google Scholar]
- (105).Giordanetto F, and Kihlberg J (2014) Macrocyclic drugs and clinical candidates: What can medicinal chemists learn from their properties? J. Med. Chem 57, 278–295. [DOI] [PubMed] [Google Scholar]
- (106).Over B, Matsson P, Tyrchan C, Artursson P, Doak BC, Foley MA, Hilgendorf C, Johnston SE, Lee MD, Lewis RJ, et al. (2016) Structural and conformational determinants of macrocycle cell permeability. Nat. Chem. Biol 12, 1065–1074. [DOI] [PubMed] [Google Scholar]
- (107).Pye CR, Hewitt WM, Schwochert J, Haddad TD, Townsend CE, Etienne L, Lao Y, Limberakis C, Furukawa A, Mathiowetz AM, et al. (2017) Nonclassical size dependence of permeation defines bounds for passive adsorption of large drug molecules. J. Med. Chem 60, 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Yang CY, Cai SJ, Liu H, and Pidgeon C (1997) Immobilized Artificial Membranes — screens for drug membrane interactions. Adv. Drug Delivery Rev 23, 229–256. [Google Scholar]
- (109).Kansy M, Senner F, and Gubernator K (1998) Physicochemical high throughput screening: Parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- (110).Hewitt WM, Leung SSF, Pye CR, Ponkey AR, Bednarek M, Jacobson MP, and Lokey RS (2015) Cell-permeable cyclic peptides from synthetic libraries inspired by natural products. J. Am. Chem. Soc 137, 715–721. [DOI] [PubMed] [Google Scholar]
- (111).Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, et al. (2008) Improving oral bioavailability of peptides by multiple N-methylation: Somatostatin analogues. Angew. Chem. Int. Ed 47, 2595–2599. [DOI] [PubMed] [Google Scholar]
- (112).Marks JR, Placone J, Hristova K, and Wimley WC (2011) Spontaneous membrane-translocating peptides by orthogonal high-throughput screening. J. Am. Chem. Soc 133, 8995–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Fuselier T, and Wimley WC (2017) Spontaneous membrane translocating peptides: The role of leucine-arginine consensus motifs. Biophys. J. 113, 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Wheaten SA, Ablan FDO, Spaller BL, Trieu JM, and Almeida PF (2013) Translocation of cationic amphipathic peptides across the membranes of pure phospholipid giant vesicles. J. Am. Chem. Soc 135, 16517–16525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Qian Z, Liu T, Liu Y-Y, Briesewitz R, Barrios AM, Jhiang SM, and Pei D (2013) Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem. Biol 8, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Qian Z, LaRochelle JR, Jiang B, Lian W, Hard RL, Selner NG, Luechapanichkul R, Barrios AM, and Pei D (2014) Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry 53, 4034–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Onfelt B, Nedvetzki S, Benninger RKP, Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MAA, French PMW, and Davis DM (2006) Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol 177, 8476–8483. [DOI] [PubMed] [Google Scholar]
- (118).Räder AFB, Reichart F, Weinmäller M, and Kessler H (2018) Improving oral bioavailability of cyclic peptides by N-methylation. Bioorg. Med. Chem 26, 2766–2773. [DOI] [PubMed] [Google Scholar]
- (119).Wilson G, Hassan IF, Dix CJ, Williamson I, Shah R, Mackay M, and Artursson P (1990) Transport and permeability properties of human Caco-2 cells: An in vitro model of the intestinal epithelial cell barrier. J. Controlled Release 11, 25–40. [Google Scholar]
- (120).Balimane PV, and Chong S (2005) Cell culture-based models for intestinal permeability: A critique. Drug Discovery Today 10, 335–343. [DOI] [PubMed] [Google Scholar]
- (121).Hilgers AR, Conradi RA, and Burton PS (1990) Caco-2 monolayers as a model for drug transport across the intestinal mucosa. Pharm. Res 7, 902–910. [DOI] [PubMed] [Google Scholar]
- (122).Artursson P, and Karlsson J (1991) Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun 175, 880–885. [DOI] [PubMed] [Google Scholar]
- (123).Wong AK, Ross BP, Chan Y-N, Artursson P, Lazorova L, Jones A, and Toth I (2002) Determination of transport in the Caco-2 cell assay of compounds varying in lipophilicity using LC—MS: Enhanced transport of Leu-enkephalin analogues. Eur. J. Pharm. Sci 16, 113–118. [DOI] [PubMed] [Google Scholar]
- (124).Stevenson C (1999) Use of Caco-2 cells and LC/MS/MS to screen a peptide combinatorial library for permeable structures. Int. J. Pharm 177, 103–115. [DOI] [PubMed] [Google Scholar]
- (125).Cho MJ, Thompson DP, Cramer CT, Vidmar TJ, and Scieszka JF (1989) The Madin Darby canine kidney (MDCK) epithelial cell monolayer as a model cellular transport barrier. Pharm. Res 6, 71–77. [DOI] [PubMed] [Google Scholar]
- (126).Jin X, Luong T-L, Reese N, Gaona H, Collazo-Velez V, Vuong C, Potter B, Sousa JC, Olmeda R, Li Q, et al. (2014) Comparison of MDCK-MDR1 and Caco-2 cell based permeability assays for anti-malarial drug screening and drug investigations. J. Pharmacol. Toxicol. Methods 70, 188–194. [DOI] [PubMed] [Google Scholar]
- (127).Irvine JD, Takahashi L, Lockhart K, Cheong J, Tolan JW, Selick HE, and Grove JR (1999) MDCK (Madin-Darby Canine Kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci 88, 28–33. [DOI] [PubMed] [Google Scholar]
- (128).Artursson P, Palm K, and Luthman K (2001) Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Delivery Rev 46, 27–43. [DOI] [PubMed] [Google Scholar]
- (129).Oehlke J, Scheller A, Wiesner B, Krause E, Beyermann M, Klauschenz E, Melzig M, and Bienert M (1998) Cellular uptake of an α-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta, Biomembr 1414, 127–139. [DOI] [PubMed] [Google Scholar]
- (130).Bontenbal M, Sieuwerts AM, Peters HA, van Putten WLJ, Foekens JA, and Klijn JGM (1998) Uptake and distribution of doxorubicin in hormone-manipulated human breast cancer cells in vitro. Breast Cancer Res. Treat 51, 139–148. [DOI] [PubMed] [Google Scholar]
- (131).Gobom J, Kraeuter K-O, Persson R, Steen H, Roepstorff P, and Ekman R (2000) Detection and quantification of neurotensin in human brain tissue by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Chem 72, 3320–3326. [DOI] [PubMed] [Google Scholar]
- (132).Elmquist A, and Langel Ü (2003) In vitro uptake and stability study of pVEC and its all-D analog. Biol. Chem 384, 387–393. [DOI] [PubMed] [Google Scholar]
- (133).Fischer R, Köhler K, Fotin-Mleczek M, and Brock R (2004) A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J. Biol. Chem 279, 12625–12635. [DOI] [PubMed] [Google Scholar]
- (134).Burlina F, Sagan S, Bolbach G, and Chassaing G (2005) Quantification of the cellular uptake of cell-penetrating peptides by MALDI-TOF mass spectrometry. Angew. Chem., Int. Ed 44, 4244–4247. [DOI] [PubMed] [Google Scholar]
- (135).Burlina F, Sagan S, Bolbach G, and Chassaing G (2006) A direct approach to quantification of the cellular uptake of cell-penetrating peptides using MALDI-TOF mass spectrometry. Nat. Protoc 1, 200–205. [DOI] [PubMed] [Google Scholar]
- (136).Aubry S, Aussedat B, Delaroche D, Jiao C-Y, Bolbach G, Lavielle S, Chassaing G, Sagan S, and Burlina F (2010) MALDI-TOF mass spectrometry: A powerful tool to study the internalization of cell-penetrating peptides. Biochim. Biophys. Acta, Biomembr 1798, 2182–2189. [DOI] [PubMed] [Google Scholar]
- (137).Girault S, Chassaing G, Blais JC, Brunot A, and Bolbach G (1996) Coupling of MALDI-TOF mass analysis to the separation of biotinylated peptides by magnetic streptavidin beads. Anal. Chem 68, 2122–2126. [DOI] [PubMed] [Google Scholar]
- (138).Rakowska PD, Lamarre B, and Ryadnov MG (2014) Probing label-free intracellular quantification of free peptide by MALDI-ToF mass spectrometry. Methods 68, 331–337. [DOI] [PubMed] [Google Scholar]
- (139).Roehl I, Schuster M, and Seiffert S (2011) US 2011/0201006 A1. [Google Scholar]
- (140).Godinho BMDC, Gilbert JW, Haraszti RA, Coles AG, Biscans A, Roux L, Nikan M, Echeverria D, Hassler M, and Khvorova A (2017) Pharmacokinetic profiling of conjugated therapeutic oligonucleotides: A high-throughput method based upon serial blood microsampling coupled to peptide nucleic acid hybridization assay. Nucleic Acid Ther 27, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Taylor SC, Berkelman T, Yadav G, and Hammond M (2013) A defined methodology for reliable quantification of western blot data. Mol. Biotechnol 55, 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Jiao C-Y, Delaroche D, Burlina F, Alves ID, Chassaing G, and Sagan S (2009) Translocation and endocytosis for cell-penetrating peptide internalization. J. Biol. Chem 284, 33957–33965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Sanderson MJ, Smith I, Parker I, and Bootman MD (2014) Fluorescence microscopy. Cold Spring Harb. Protoc 2014, 1042–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Hell SW (2003) Toward fluorescence nanoscopy. Nat. Biotechnol 21, 1347–1355. [DOI] [PubMed] [Google Scholar]
- (145).Bird GH, Mazzola E, Opoku-Nsiah K, Lammert MA, Godes M, Neuberg DS, and Walensky LD (2016) Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol 12, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Illien F, Rodriguez N, Amoura M, Joliot A, Pallerla M, Cribier S, Burlina F, and Sagan S (2016) Quantitative fluorescence spectroscopy and flow cytometry analyses of cell-penetrating peptides internalization pathways: optimization, pitfalls, comparison with mass spectrometry quantification. Sci. Rep 6, 36938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Crim RL, Audet SA, Feldman SA, Mostowski HS, and Beeler JA (2007) Identification of linear heparin-binding peptides derived from human respiratory syncytial virus fusion glycoprotein that inhibit infectivity. J. Virol 81, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Jones AT, and Sayers EJ (2012) Cell entry of cell penetrating peptides: tales of tails wagging dogs. J. Controlled Release 161, 582–591. [DOI] [PubMed] [Google Scholar]
- (149).Birch D, Christensen MV, Staerk D, Franzyk H, and Nielsen HM (2017) Fluorophore labeling of a cell-penetrating peptide induces differential effects on its cellular distribution and affects cell viability. Biochim. Biophys. Acta, Biomembr 1859, 2483–2494. [DOI] [PubMed] [Google Scholar]
- (150).Puckett CA, and Barton JK (2009) Fluorescein redirects a ruthenium–octaarginine conjugate to the nucleus. J. Am. Chem. Soc 131, 8738–8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Ochocki JD, Mullen DG, Wattenberg EV, and Distefano MD (2011) Evaluation of a cell penetrating prenylated peptide lacking an intrinsic fluorophore via in situ click reaction. Bioorg. Med. Chem. Lett 21, 4998–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Shin M-K, Hyun Y-J, Lee JH, and Lim H-S (2018) Comparison of cell permeability of cyclic peptoids and linear peptoids. ACS Comb. Sci 20, 237–242. [DOI] [PubMed] [Google Scholar]
- (153).Quach K, LaRochelle J, Li XH, Rhoades E, and Schepartz A (2018) Unique arginine array improves cytosolic localization of hydrocarbon-stapled peptides. Bioorg. Med. Chem 26, 1197–1202. [DOI] [PubMed] [Google Scholar]
- 154.() Page A, Kang M, Armitstead A, Perry D, and Unwin PR (2017) Quantitative visualization of molecular delivery and uptake at living cells with self-referencing scanning ion conductance microscopy-scanning electrochemical microscopy. Anal. Chem 89, 3021–3028. [DOI] [PubMed] [Google Scholar]
- (155).Nikan M, Osborn MF, Coles AH, Godinho BM, Hall LM, Haraszti RA, Hassler MR, Echeverria D, Aronin N, and Khvorova A (2016) Docosahexaenoic acid conjugation enhances distribution and safety of siRNA upon local administration in mouse brain. Mol. Mol. Ther.-Nucleic Acids 5, e344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Lee YH, Tan HT, and Chung MCM (2010) Subcellular fractionation methods and strategies for proteomics. Proteomics 10, 3935–3956. [DOI] [PubMed] [Google Scholar]
- (157).Walker LR, Hussein HAM, and Akula SM (2016) Subcellular fractionation method to study endosomal trafficking of Kaposi’s sarcoma-associated herpesvirus. Cell Biosci 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).de Duve C, Pressman BC, Gianetto R, Wattiaux R, and Appelmans F (1955) Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Pasquali C, Fialka I, and Huber LA (1999) Subcellular fractionation, electromigration analysis and mapping of organelles. J. Chromatogr., Biomed. Appl 722, 89–102. [DOI] [PubMed] [Google Scholar]
- (160).Hällbrink M, Florén A, Elmquist A, Pooga M, Bartfai T, and Langel Ü (2001) Cargo delivery kinetics of cell-penetrating peptides. Biochim. Biophys. Acta, Biomembr 1515, 101–109. [DOI] [PubMed] [Google Scholar]
- (161).Yang J, Chen H, Vlahov IR, Cheng J-X, and Low PS (2006) Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc. Natl. Acad. Sci. U. S. A 103, 13872–13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Falnes PO, and Olsnes S (1995) Cell-mediated reduction and incomplete membrane translocation of diphtheria toxin mutants with internal disulfides in the A fragment. J. Biol. Chem 270, 20787–20793. [DOI] [PubMed] [Google Scholar]
- (163).Qian Z, Dougherty PG, and Pei D (2015) Monitoring the cytosolic entry of cell-penetrating peptides using a pH-sensitive fluorophore. Chem. Commun 51, 2162–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Murphy RF (1984) Endosome pH measured in single cells by dual fluorescence flow cytometry: rapid acidification of insulin to pH 6. J. Cell Biol 98, 1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Liao H, and Pei D (2017) Cell-permeable bicyclic peptidyl inhibitors against T-cell protein tyrosine phosphatase from a combinatorial library. Org. Biomol Chem 15, 9595–9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Stolle A-S, Norkowski S, Köorner B, Schmitz J, Lüken L, Frankenberg M, Rüter C, and Schmidt MA (2017) T3SS-independent uptake of the short-trip toxin-related recombinant NleC effector of enteropathogenic Escherichia coli leads to NF-κB p65 cleavage. Front. Cell. Infect. Microbiol 7, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Norkowski S, Körner B, Greune L, Stolle A-S, Lubos ML, Hardwidge PR, Schmidt MA, and Ruter C (2018) Bacterial LPX motif-harboring virulence factors constitute a species-spanning family of cell-penetrating effectors. Cell. Mol. Life Sci 75, 2273–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).LaRochelle JR, Cobb GB, Steinauer A, Rhoades E, and Schepartz A (2015) Fluorescence correlation spectroscopy reveals highly efficient cytosolic delivery of certain penta-Arg proteins and stapled peptides. J. Am. Chem. Soc 137, 2536–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Rezgui R, Blumer K, Yeoh-Tan G, Trexler AJ, and Magzoub M (2016) Precise quantification of cellular uptake of cell-penetrating peptides using fluorescence-activated cell sorting and fluorescence correlation spectroscopy. Biochim. Biophys. Acta, Biomembr 1858, 1499–1506. [DOI] [PubMed] [Google Scholar]
- (170).Vasconcelos L, Lehto T, Madani F, Radoi V, Hällbrink M, Vukojević V, and Langel Ü (2018) Simultaneous membrane interaction of amphipathic peptide monomers, self-aggregates and cargo complexes detected by fluorescence correlation spectroscopy. Biochim. Biophys. Acta, Biomembr 1860, 491–504. [DOI] [PubMed] [Google Scholar]
- (171).Kang S-H, Cho M-J, and Kole R (1998) Up-regulation of luciferase gene expression with antisense oligonucleotides: implications and applications in functional assay development. Biochemistry 37, 6235–6239. [DOI] [PubMed] [Google Scholar]
- (172).Dominski Z, and Kole R (1993) Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. U. S. A 90, 8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Wadia JS, Stan RV, and Dowdy SF (2004) Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med 10, 310–315. [DOI] [PubMed] [Google Scholar]
- (174).Sternberg N, and Hamilton D (1981) Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J. Mol. Biol 150, 467–486. [DOI] [PubMed] [Google Scholar]
- (175).Yu P, Liu B, and Kodadek T (2005) A high-throughput assay for assessing the cell permeability of combinatorial libraries. Nat. Biotechnol 23, 746–751. [DOI] [PubMed] [Google Scholar]
- (176).Yu P, Liu B, and Kodadek T (2007) A convenient, high-throughput assay for measuring the relative cell permeability of synthetic compounds. Nat. Protoc 2, 23–30. [DOI] [PubMed] [Google Scholar]
- (177).Tan NC, Yu P, Kwon Y-U, and Kodadek T (2008) High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorg. Med. Chem 16, 5853–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Kwon Y-U, and Kodadek T (2007) Quantitative evaluation of the relative cell permeability of peptoids and peptides. J. Am. Chem. Soc 129, 1508–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Liu B, and Kodadek T (2009) Investigation of the relative cellular permeability of DNA-binding pyrrole–imidazole polyamides. J. Med. Chem 52, 4604–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Cho S, Choi J, Kim A, Lee Y, and Kwon Y-U (2010) Efficient solid-phase synthesis of a series of cyclic and linear peptoid-dexamethasone conjugates for the cell permeability studies. J. Comb. Chem 12, 321–326. [DOI] [PubMed] [Google Scholar]
- (181).Alluri P, Liu B, Yu P, Xiao X, and Kodadek T (2006) Isolation and characterization of coactivator-binding peptoids from a combinatorial library. Mol. BioSyst 2, 568–579. [DOI] [PubMed] [Google Scholar]
- (182).Cabantous S, Terwilliger TC, and Waldo GS (2005) Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol 23, 102–107. [DOI] [PubMed] [Google Scholar]
- (183).Kato N, and Jones J (2010) The split luciferase complementation assay In Plant Developmental Biology (Hennig L, and Köhler C, Eds.) Vol. 655, pp 359–376, Humana Press. [DOI] [PubMed] [Google Scholar]
- (184).Johnsson N, and Varshavsky A (1994) Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U. S. A 91, 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Cabantous S, and Waldo GS (2006) In vivo and in vitro protein solubility assays using split GFP. Nat. Methods 3, 845–854. [DOI] [PubMed] [Google Scholar]
- (186).Milech N, Longville BA, Cunningham PT, Scobie MN, Bogdawa HM, Winslow S, Anastasas M, Connor T, Ong F, Stone SR, et al. (2016) GFP-complementation assay to detect functional CPP and protein delivery into living cells. Sci. Rep 5, 18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Lönn P, Kacsinta AD, Cui X-S, Hamil AS, Kaulich M, Gogoi K, and Dowdy SF (2016) Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci. Rep 6, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Kim J-S, Choi D-K, Park S-W, Shin S-M, Bae J, Kim D-M, Yoo H, and Kim Y-S (2015) Quantitative assessment of cellular uptake and cytosolic access of antibody in living cells by an enhanced split GFP complementation assay. Biochem. Biophys. Res. Commun 467, 771–777. [DOI] [PubMed] [Google Scholar]
- (189).Zhang P, Monteiro da Silva G, Deatherage C, Burd C, and DiMaio D (2018) Cell-penetrating peptide mediates intracellular membrane passage of human papillomavirus L2 protein to trigger retrograde trafficking. Cell 174, 1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Hoffmann K, Milech N, Juraja SM, Cunningham PT, Stone SR, Francis RW, Anastasas M, Hall CM, Heinrich T, Bogdawa HM, et al. (2018) A platform for discovery of functional cell-penetrating peptides for efficient multi-cargo intracellular delivery. Sci. Rep 8, 12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Feng S, Sekine S, Pessino V, Li H, Leonetti MD, and Huang B (2017) Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun 8, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Schmidt S, Adjobo-Hermans MJW, Kohze R, Enderle T, Brock R, and Milletti F (2017) Identification of short hydrophobic cell-penetrating peptides for cytosolic peptide delivery by rational design. Bioconjugate Chem 28, 382–389. [DOI] [PubMed] [Google Scholar]
- (193).Li Y-C, Rodewald LW, Hoppmann C, Wong ET, Lebreton S, Safar P, Patek M, Wang L, Wertman KF, and Wahl M (2014) A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time. Cell Rep 9, 1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Herce HD, Deng W, Helma J, Leonhardt H, and Cardoso MC (2013) Visualization and targeted disruption of protein interactions in living cells. Nat. Commun 4, 2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Michnick SW, Ear PH, Manderson EN, Remy I, and Stefan E (2007) Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discovery 6, 569–582. [DOI] [PubMed] [Google Scholar]
- (196).Schmidt S, Adjobo-Hermans MJW, Wallbrecher R, Verdurmen WPR, Bovée-Geurts PHM, van Oostrum J, Milletti F, Enderle T, and Brock R (2015) Detecting cytosolic peptide delivery with the GFP complementation assay in the low micromolar range. Angew. Chem., Int. Ed 54, 15105–15108. [DOI] [PubMed] [Google Scholar]
- (197).Falnes P, Wiedlocha A, Rapak A, and Olsnes S (1995) Farnesylation of CaaX-tagged diphtheria toxin A-fragment as a measure of transfer to the cytosol. Biochemistry 34, 11152–11159. [DOI] [PubMed] [Google Scholar]
- (198).Loison F, Nizard P, Sourisseau T, Le Goff P, Debure L, Le Drean Y, and Michel D (2005) A ubiquitin-based assay for the cytosolic uptake of protein transduction domains. Mol. Ther 11, 205–214. [DOI] [PubMed] [Google Scholar]
- (199).Reyes-Turcu FE, Ventii KH, and Wilkinson KD (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem 78, 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Chyan W, and Raines RT (2018) Enzyme-activated fluorogenic probes for live-cell and in vivo imaging. ACS Chem. Biol 13, 1810–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Rotman B, Zderic JA, and Edelstein M (1963) Fluorogenic substrates for beta-D-galactosidases and phosphatases derived from flurescein (3,6-dihydroxyfluoran) and its monomethy-lether. Proc. Natl. Acad. Sci. U. S. A 50, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Plovins A, Alvarez AM, Ibañez M, Molina M, and Nombela C (1994) Use of fluorescein-di-beta-D-galactopyranoside (FDG) and C12-FDG as substrates for beta-galactosidase detection by flow cytometry in animal, bacterial, and yeast cells. Appl Environ. Microbiol 60, 4638–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Campbell RE (2004) Realization of β-lactamase as a versatile fluorogenic reporter. Trends Biotechnol 22, 208–211. [DOI] [PubMed] [Google Scholar]
- (204).Nolan GP, Fiering S, Nicolas JF, and Herzenberg LA (1988) Fluorescence-activated cell analysis and sorting of viable mammalian cells based on beta-D-galactosidase activity after transduction of Escherichia coli lacZ. Proc. Natl. Acad. Sci. U. S. A 85, 2603–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Zlokarnik G, Negulescu P, Knapp T, Mere L, Burres N, Feng L, Whitney M, Roemer K, and Tsien R (1998) Quantitation of transcription and clonal selection of single living cells with β-lactamase as reporter. Science 279, 84–88. [DOI] [PubMed] [Google Scholar]
- (206).Hobson JP, Liu S, Rønø B, Leppla SH, and Bugge TH (2006) Imaging specific cell-surface proteolytic activity in single living cells. Nat. Methods 3, 259–261. [DOI] [PubMed] [Google Scholar]
- (207).Hu H, and Leppla SH (2009) Anthrax toxin uptake by primary immune cells as determined with a lethal factor-β-lactamase fusion protein. PLoS One 4, e7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Stone S, Heinrich T, Juraja S, Satiaputra J, Hall C, Anastasas M, Mills A, Chamberlain C, Winslow S, Priebatsch K, et al. (2018) β-lactamase tools for establishing cell internalization and cytosolic delivery of cell penetrating peptides. Biomolecules 8, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Torchilin VP, and Weissig V (2007) Liposomes: A Practical Approach, Oxford University Press. [Google Scholar]
- (210).Lichtenfels R, Biddison WE, Schulz H, Vogt AB, and Martin R (1994) CARE-LASS (calcein-release-assay), an improved fluorescence-based test system to measure cytotoxic T lymphocyte activity. J. Immunol. Methods 172, 227–239. [DOI] [PubMed] [Google Scholar]
- (211).Jobin M-L, Blanchet M, Henry S, Chaignepain S, Manigand C, Castano S, Lecomte S, Burlina F, Sagan S, and Alves ID (2015) The role of tryptophans on the cellular uptake and membrane interaction of arginine-rich cell penetrating peptides. Biochim. Biophys. Acta, Biomembr 1848, 593–602. [DOI] [PubMed] [Google Scholar]
- (212).Walrant A, Matheron L, Cribier S, Chaignepain S, Jobin M-L, Sagan S, and Alves ID (2013) Direct translocation of cell-penetrating peptides in liposomes: A combined mass spectrometry quantification and fluorescence detection study. Anal. Biochem 438, 1–10. [DOI] [PubMed] [Google Scholar]
- (213).Drin G, Cottin S, Blanc E, Rees AR, and Temsamani J (2003) Studies on the internalization mechanism of cationic cell-penetrating peptides. J. Biol. Chem 278, 31192–31201. [DOI] [PubMed] [Google Scholar]
- (214).Jones LR, Goun EA, Shinde R, Rothbard JB, Contag CH, and Wender PA (2006) Releasable luciferin–transporter conjugates: Tools for the real-time analysis of cellular uptake and release. J. Am. Chem. Soc 128, 6526–6527. [DOI] [PubMed] [Google Scholar]
- (215).Beckett D, Kovaleva E, and Schatz PJ (1999) A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci 8, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Schatz PJ (1993) Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: A 13 residue consensus peptide specifies biotinylation in Escherichia coli. Nat. Biotechnol 11, 1138–1143. [DOI] [PubMed] [Google Scholar]
- (217).Fairhead M, and Howarth M (2015) Site-specific biotinylation of purified proteins using BirA In Site-Specific Protein Labeling (Gautier A, and Hinner MJ, Eds.) Vol. 1266, pp 171–184, Springer, New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Chen I, Howarth M, Lin W, and Ting AY (2005) Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2, 99–104. [DOI] [PubMed] [Google Scholar]
- (219).Verdurmen WPR, Mazlami M, and Pluckthun A (2017) A quantitative comparison of cytosolic delivery via different protein uptake systems. Sci. Rep 7, 13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Peraro L, Deprey KL, Moser MK, Zou Z, Ball HL, Levine B, and Kritzer JA (2018) Cell penetration profiling using the chloroalkane penetration assay. J. Am. Chem. Soc 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Peraro L, Zou Z, Makwana KM, Cummings AE, Ball HL, Yu H, Lin YS, Levine B, and Kritzer JA (2017) Diversity-oriented stapling yields intrinsically cell-penetrant inducers of autophagy. J. Am. Chem. Soc 139, 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008) HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- (223).McCluskey AJ, Olive AJ, Starnbach MN, and Collier RJ (2013) Targeting HER2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol 7, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Arora N, and Leppla SH (1994) Fusions of anthrax toxin lethal factor with shiga toxin and diphtheria toxin enzymatic domains are toxic to mammalian cells. Infect. Immun 62, 4955–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Zahaf N-I, Lang AE, Kaiser L, Fichter CD, Lassmann S, McCluskey A, Augspach A, Aktories K, and Schmidt G (2017) Targeted delivery of an ADP-ribosylating bacterial toxin into cancer cells. Sci. Rep 7, 41252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Zelphati O, Wang Y, Kitada S, Reed JC, Feigner PL, and Corbeil J (2001) Intracellular delivery of proteins with a new lipid-mediated delivery system. J. Biol. Chem 276, 35103–35110. [DOI] [PubMed] [Google Scholar]
- (227).Ruttekolk IR, Chakrabarti A, Richter M, Duchardt F, Glauner H, Verdurmen WPR, Rademann J, and Brock R (2011) Coupling to polymeric scaffolds stabilizes biofunctional peptides for intracellular applications. Mol. Pharmacol 79, 692–700. [DOI] [PubMed] [Google Scholar]
- (228).Chao T-Y, and Raines RT (2013) Fluorogenic label to quantify the cytosolic delivery of macromolecules. Mol. BioSyst 9, 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Verdurmen WPR, Mazlami M, and Pluckthun A (2017) A biotin ligase-based assay for the quantification of the cytosolic delivery of therapeutic proteins In Synthetic Antibodies (Tiller T, Ed.) Vol. 1575, pp 223–236, Springer, New York. [DOI] [PubMed] [Google Scholar]
- (230).Ryou J-H, Sohn Y-K, Kim D-G, Kyeong H-H, and Kim H-S (2018) Engineering and cytosolic delivery of a native regulatory protein and its variants for modulation of ERK2 signaling pathway. Biotechnol. Bioeng 115, 839–849. [DOI] [PubMed] [Google Scholar]
- (231).Gomarasca M, Martins TFC, Greune L, Hardwidge PR, Schmidt MA, and Ruter C (2017) Bacterium-derived cell-penetrating peptides deliver gentamicin to kill intracellular pathogens. Antimicrob. Agents Chemother 61, e02545–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Xue X, Huang J, and Wang H (2014) The study of the intercellular trafficking of the fusion proteins of herpes simplex virus protein VP22. PLoS One 9, e100840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Takeuchi T, and Futaki S (2016) Current understanding of direct translocation of arginine-rich cell-penetrating peptides and its internalization mechanisms. Chem. Pharm. Bull. 64, 1431–1437. [DOI] [PubMed] [Google Scholar]
- (234).Marschall AL, Zhang C, Frenzel A, Schirrmann T, Hust M, Perez F, and Dubel S (2014) Delivery of antibodies to the cytosol: Debunking the myths. mAbs 6, 943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Brewis N, Phelan A, Webb J, Drew J, Elliott G, and O’Hare P (2000) Evaluation of VP22 spread in tissue culture. J. Virol 74, 1051–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Pichon C, Monsigny M, and Roche A-C (1999) Intracellular localization of oligonucleotides: Influence of fixative protocols. Antisense Nucleic Acid Drug Dev 9, 89–93. [DOI] [PubMed] [Google Scholar]
- (237).Aints A, Dilber MS, and Smith CIE (1999) Intercellular spread of GFP-VP22. J. Gene Med 1, 275–279. [DOI] [PubMed] [Google Scholar]
- (238).Lundberg M, and Johansson M (2002) Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem. Biophys. Res. Commun 291, 367–371. [DOI] [PubMed] [Google Scholar]
- (239).Fischer PM (2007) Cellular uptake mechanisms and potential therapeutic utility of peptidic cell delivery vectors: Progress 2001–2006. Med. Res. Rev 27, 755–795. [DOI] [PubMed] [Google Scholar]
- (240).Lemken M-L, Wolf C, Wybranietz WA, Schmidt U, Smirnow I, Bühring H-J, Mack AF, Lauer UM, and Bitzer M (2007) Evidence for intercellular trafficking of VP22 in living cells. Mol. Ther 15, 310–319. [DOI] [PubMed] [Google Scholar]
- (241).Elliott G, and O’Hare P (1999) Intercellular trafficking of VP22-GFP fusion proteins. Gene Ther 6, 149–151. [DOI] [PubMed] [Google Scholar]
- (242).Kosuge M, Takeuchi T, Nakase I, Jones AT, and Futaki S(2008) Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjugate Chem 19, 656–664. [DOI] [PubMed] [Google Scholar]
- (243).Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, and Brock R (2007) A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 8, 848–866. [DOI] [PubMed] [Google Scholar]
- (244).Herbig ME, Weller K, Krauss U, Beck-Sickinger AG, Merkle HP, and Zerbe O (2005) Membrane surface-associated helices promote lipid interactions and cellular uptake of human calcitonin-derived cell penetrating peptides. Biophys. J. 89, 4056–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Wei Y, Li C, Zhang L, and Xu X (2014) Design of novel cell penetrating peptides for the delivery of trehalose into mammalian cells. Biochim. Biophys. Acta, Biomembr 1838, 1911–1920. [DOI] [PubMed] [Google Scholar]
- (246).Maiolo JR, Ferrer M, and Ottinger EA (2005) Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim. Biophys. Acta, Biomembr 1712, 161–172. [DOI] [PubMed] [Google Scholar]
- (247).Holowka EP, Sun VZ, Kamei DT, and Deming TJ (2007) Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nat. Mater 6, 52–57. [DOI] [PubMed] [Google Scholar]
- (248).Fretz MM, Penning NA, Al-Taei S, Futaki S, Takeuchi T, Nakase I, Storm G, and Jones AT (2007) Temperature-, concentration- and cholesterol-dependent translocation of L- and Docta-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem. J. 403, 335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Weill CO, Biri S, Adib A, and Erbacher P (2008) A practical approach for intracellular protein delivery. Cytotechnology 56, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Phan NN, Li C, and Alabi CA (2018) Intracellular delivery via noncharged sequence-defined cell-penetrating oligomers. Bioconjugate Chem 29, 2628–2635. [DOI] [PubMed] [Google Scholar]
- (251).Marren K (2011) Dimethyl sulfoxide: An effective penetration enhancer for topical administration of NSAIDs. Phys. Sportsmed 39, 75–82. [DOI] [PubMed] [Google Scholar]
- (252).Notman R, den Otter WK, Noro MG, Briels WJ, and Anwar J (2007) The permeability enhancing mechanism of DMSO in ceramide bilayers simulated by molecular dynamics. Biophys. J. 93, 2056–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Cervera L, Fuenmayor J, González-Domínguez I, Gutiérrez-Granados S, Segura MM, and Gòdia F (2015) Selection and optimization of transfection enhancer additives for increased virus-like particle production in HEK293 suspension cell cultures. Appl Microbiol. Biotechnol 99, 9935–9949. [DOI] [PubMed] [Google Scholar]
- (254).Kligman AM (1965) Topical pharmacology and toxicology of dimethyl sulfoxide—Part 1. JAMA J. Am. Med. Assoc 193, 796–804. [DOI] [PubMed] [Google Scholar]
- (255).de Ménorval M-A, Mir LM, Fernández ML, and Reigada R (2012) Effects of dimethyl sulfoxide in cholesterol-containing lipid membranes: A comparative study of experiments in silico and with cells. PLoS One 7, e41733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Wang H, Zhong C-Y, Wu J-F, Huang Y-B, and Liu C-B (2010) Enhancement of TAT cell membrane penetration efficiency by dimethyl sulfoxide. J. Controlled Release 143, 64–70. [DOI] [PubMed] [Google Scholar]
- (257).Kurth F, Dittrich PS, Walde P, and Seebach D (2018) Influence of the membrane dye R18 and of DMSO on cell penetration of guanidinium-rich peptides. Chem. Biodiversity 15, e1800302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Wagstaff KM, and Jans DA (2006) Protein transduction: cell penetrating peptides and their therapeutic applications. Curr. Med. Chem 13, 1371–1387. [DOI] [PubMed] [Google Scholar]
- (259).Rosenblum D, Joshi N, Tao W, Karp JM, and Peer D (2018) Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun 9, 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Araste F, Abnous K, Hashemi M, Taghdisi SM, Ramezani M, and Alibolandi M (2018) Peptide-based targeted therapeutics: Focus on cancer treatment. J. Controlled Release 292, 141–162. [DOI] [PubMed] [Google Scholar]
- (261).Bernkop-Schnürch A (2018) Strategies to overcome the polycation dilemma in drug delivery. Adv. Drug Delivery Rev 136–137, 62–72. [DOI] [PubMed] [Google Scholar]