

Abstract
Adenosine is a well-characterized endogenous anticonvulsant and seizure terminator of the brain. Through a combination of adenosine receptor-dependent and -independent mechanisms, adenosine affects seizure generation (ictogenesis), as well as the development of epilepsy and its progression (epileptogenesis). Maladaptive changes in adenosine metabolism, in particular increased expression of the astroglial enzyme adenosine kinase (ADK), play a major role in epileptogenesis. Increased expression of ADK has dual roles in both reducing the inhibitory tone of adenosine in the brain, which consequently reduces the threshold for seizure generation, and also driving an increased flux of methyl-groups through the transmethylation pathway, thereby increasing global DNA methylation. Through these mechanisms, adenosine is uniquely positioned to link metabolism with epigenetic outcome. Therapeutic adenosine augmentation therefore not only holds promise for the suppression of seizures in epilepsy, but moreover the prevention of epilepsy and its progression overall. This review will focus on adenosine-related mechanisms implicated in ictogenesis and epileptogenesis and will discuss therapeutic opportunities and challenges.
Keywords: Seizures, Epileptogenesis, Adenosine kinase, Epigenetics, DNA methylation, Disease modification
1. Introduction
Epilepsy is the third most common neurological disorder, affecting about 75 million people worldwide. Despite several decades of investigation and the introduction of more than 20 novel anti-seizure drugs (ASDs) in recent years, almost a third of all epilepsies are refractory against conventional forms of drug treatment [1, 2]. Current ASDs have been developed to suppress neuronal hyperexcitability by either decreasing excitation or enhancing inhibition however, without an emphasis on pathogenic mechanisms involved in the generation and progression of epilepsy (epileptogenesis). Pharmacoresistance to modern ASDs has called for a shift in focus from seizure control towards therapies that may intervene with epileptogenesis and ultimately prevent the development of epilepsy and its progression [3, 4]. This has called for attention to antiepileptogenic drug (AEG) development, with a focus on interfering with endogenous mechanisms that turn a healthy brain into an epileptic brain [3, 4]. The final goal of AEG development is to intervene with mechanisms of the brain that are intricately involved in the development and progression of epilepsy along with its comorbidities. This review will primarily focus on adenosine’s role in epilepsy and its development. Adenosine is a well-characterized endogenous seizure suppressor and seizure terminator of the brain [5, 6], which acts via presynaptic inhibition and stabilization of the postsynaptic membrane potential. The discovery that adenosine not only acts as an adenosine receptor agonist, but also as a regulator of methylation reactions, including DNA methylation [7, 8], has led to the utilization of therapeutic adenosine augmentation as an epigenetic therapy capable of the prevention of epilepsy and its progression [8]. Therapeutic benefits of adenosine augmentation have been demonstrated in well established rodent models, such as kindling and post status epilepticus (SE) models, which have established face value to model clinical temporal lobe epilepsy. Although promising in therapeutic applications for experimental epilepsy in rodents, therapeutic adenosine augmentation for the treatment of epilepsy has not yet been translated into the clinic.
2. Epilepsy and its development
2.1. Seizures
Seizures are a characteristic pathological hallmark of epilepsy. Whereas seizures per se can be triggered by a variety of stimuli, the condition epilepsy is defined by the occurrence of recurrent, non-provoked seizures. Clinically, seizures can range from brief periods of altered consciousness (e.g. staring, changes in sense of smell, sound or vision) to convulsive seizures, which can have tonic and/or clonic components. Seizures are due to synchronized neuronal hyperexcitability, caused by excessive excitation (glutamate) and/or deficient inhibition (GABA). Experimentally, seizures can be caused by excitotoxic agents, e.g. compounds that enhance excitation (e.g. kainic acid) or reduce inhibition (e.g. pentylentetrazole). Normally, seizures are associated with a rise in adenosine [5], which acts as a seizure terminator [6]. Therefore, seizures are normally self-terminating. When endogenous seizure control mechanisms fail, status epilepticus results, which can cause major injury to the brain and which can trigger subsequent epileptogenesis. A clinical example highlighting the importance of adenosine-related control mechanisms is theophylline-induced status epilepticus in susceptible individuals, which can develop into a life- threatening condition [9, 10]. Because theophylline is an adenosine receptor antagonist, those findings show that seizures are normally kept under control by endogenous adenosine acting on adenosine receptors. This example, as well as experimental data in rodents, shows a critical role of adenosine A1 receptors in preventing seizure spread [11].
2.2. Epileptogenesis
Epileptogenesis is the process that turns a healthy brain into an epileptic brain and includes mechanisms that drive the development of epilepsy and its progression. Whereas different types of injury to the brain can cause epilepsy, certain commonalities in the epileptogenic process have been identified [12]. Briefly, insults to the brain, which may include traumatic brain injury, status epilepticus, febrile seizures, or infection, trigger a cascade of events including inflammatory processes, microglial and astroglial activation, as well as epigenetic changes [12]. During a so-called seizure-free ‘latent period’ the brain undergoes structural, biochemical, and epigenetic remodeling until the first spontaneous seizures (i.e. the ‘diagnosis’ of epilepsy) occur. The latency period between the epilepsy triggering primary insult and the onset of spontaneous recurrent seizures can range between months and years in humans and between days and weeks in rodents. Epileptogenesis can be a continuous process leading to the progression of epilepsy and its severity. Progression of epilepsy is associated with the development of comorbidities (e.g. depression, sleep disorders, cognitive impairment, or psychosis) and increased risk for sudden unexpected death in epilepsy (SUDEP) [13–16]. Because of the progressive nature of many forms of epilepsy, preventative therapy and the development of AEGs is an urgent unmet clinical need.
2.3. The role of adenosine kinase in epileptogenesis
Adenosine kinase (ADK) is a ribokinase, which adds a phosphate group to adenosine and forms AMP [17]. In the adult brain, ADK is predominantly expressed in astrocytes [18] and is the major metabolic clearance route for adenosine. Several neurological conditions are associated with inflammatory processes and the development of astrogliosis, characterized by astroglial cell proliferation and hypertrophy [19]. A range of animal models has demonstrated that overexpression of ADK appears to be a general response to astroglial activation [14, 20, 21]. Patients with mesial temporal lobe epilepsy have significantly overexpressed ADK [22, 23]. This has demonstrated a tight link between ADK expression levels and seizure susceptibility [17]. Importantly, overexpression of ADK has been linked to the epileptogenic process itself [24, 25]. Experimental data indicate two distinct phases of epileptogenesis, an acute immediate phase followed by chronic changes implicated in the progression and sustenance of epileptogenesis.
Phase 1:
acute, trigger: The first phase is caused by insults to the brain ranging from traumatic brain injury, seizures, infection, or a stroke leading to an immediate surge in adenosine associated with the transient downregulation of ADK expression within the first few hours post injury [24, 26]. Interestingly, ADK acts both as a sensor for the energy state of a cell and as a switch, whereby an energy crisis, as caused by an injury or a seizure, leads to increases in ADP (derived from rapid ATP breakdown), which in turn leads to aggregation and inactivation of ADK, thus boosting the resulting adenosine surge [17]. This adenosine surge has been linked to hypomethylation of DNA [8] and it can be postulated that an injury-induced adenosine surge can exacerbate epileptogenesis through epigenetic mechanisms where the consequential hypomethylation of DNA may stimulate transcription and expression of epileptogenesis promoting genes. Future studies are necessary to establish genes modulated by an injury-induced adenosine surge in order to grow our understanding of the pathogenic mechanisms involved in epileptogenesis.
Phase 2:
epilepsy progression: Taking place during the ‘latent period’ of epileptogenesis, inflammatory responses are triggered within days that prompt reactive microgliosis and astrogliosis [27–30]. As previously mentioned, astroglial activation influences a robust increase in ADK expression and consequential adenosine deficiency. It has been demonstrated that seizures derive from areas of astrogliosis with an overexpression of ADK [31], that onset of seizure activity during epileptogenesis temporally coincides with the first signs of astrogliosis and overexpressed ADK [32], that transgenic overexpression of ADK by itself is capable to trigger electrographic seizures [25, 32], and that overexpression of ADK is associated with hypermethylation of DNA [8]. Whereas therapeutic adenosine augmentation rescues normal DNA methylation levels and prevents epilepsy progression long-term [8], we propose that increased ADK expression and increased DNA methylation status are two interrelated features that propagate a vicious cycle driving the progression and perpetuation of the epileptic state. For this reason, dysregulation of ADK plays a major role in the transition from a normal brain into an epileptic brain (Figure 1).
Figure 1.
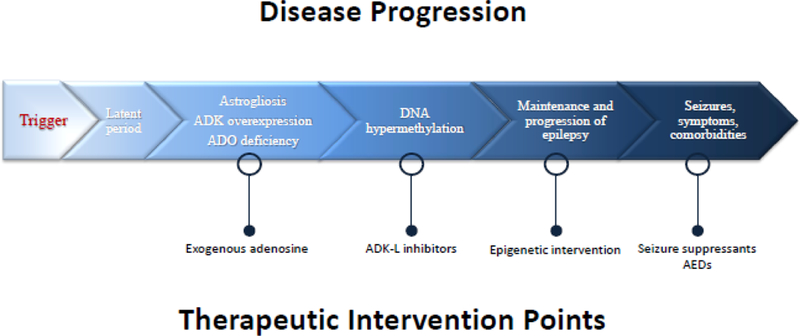
A model of epileptic pathology progression with strategic therapeutic intervention points. As the disease state of epilepsy develops, maladaptive changes to ADK expression and adenosine tone build in series to a progressively more severe outcome. In this review, many therapeutic strategies are discussed; select opportunities for such interventions are shown at their appropriate time points here.
2.4. The epigenetics of epilepsy
Epigenetic modifications are acquired changes to the genome and its organization, which include chemical modifications such as methylation or acetylation to DNA or histones, and the expression of micro RNAs. Acquired epigenetic changes are both widespread and common, and have, for example, been shown to be interrelated with rapid and lasting effects of stress [33]. Epigenetic modifications can be ingrained quickly and trigger functional effects within minutes, can diminish again hours later, or result in lasting epigenetic changes [34–36]. One major finding is that epigenetic changes can persist long after initiation and therefore determine long-term functional modifications in the brain [33].
2.4.1. DNA methylation
The methylation hypothesis of epileptogenesis suggests that ictal activity itself is sufficient to trigger epigenetic chromatin modifications and stimulate epilepsy development [4]. This hypothesis is based on findings that increased DNA methyltransferase (DNMT) activity is associated with human temporal lobe epilepsy (TLE) [37]. In line with those findings, increased methylation in the reelin gene promoter was found to be associated with human TLE and granule cell dispersion, whereas differential DNA methylation at 2573 different loci was associated with chronic epilepsy in the rat pilocarpine model [38, 39]. Independent research work has shown significant methylation changes in 321 gene loci as a result of status epilepticus, and accordingly, differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human TLE [34, 40]. DNA methylation impacts the function of DNA by activating or repressing the transcriptional activity of a gene [41]. DNA methylation relies on the donation of a methyl group from S-adenosylmethionine (SAM), a step that is facilitated by DNMTs (Figure 2A). The product of methyl group transfer to DNA is S-adenosylhomocysteine (SAH), which is then cleaved into adenosine and homocysteine by SAH hydrolase. The equilibrium of this reaction, and therefore the flux of methyl groups through the methylation pathway, is determined by the local adenosine concentration in the microenvironment of the cell nucleus. Because ADK removes adenosine, it drives DNA methylation. Overexpression of ADK, as determined in chronic epilepsy [42], will therefore promote global DNA hypermethylation in the brain (Figure 2B). While maladaptive changes in DNA methylation have been associated with chronic epilepsy and its development, therapeutic adenosine augmentation is a potential methylation intervention strategy with antiepileptogenic potential, as will be discussed in more detail below [8].
Figure 2.
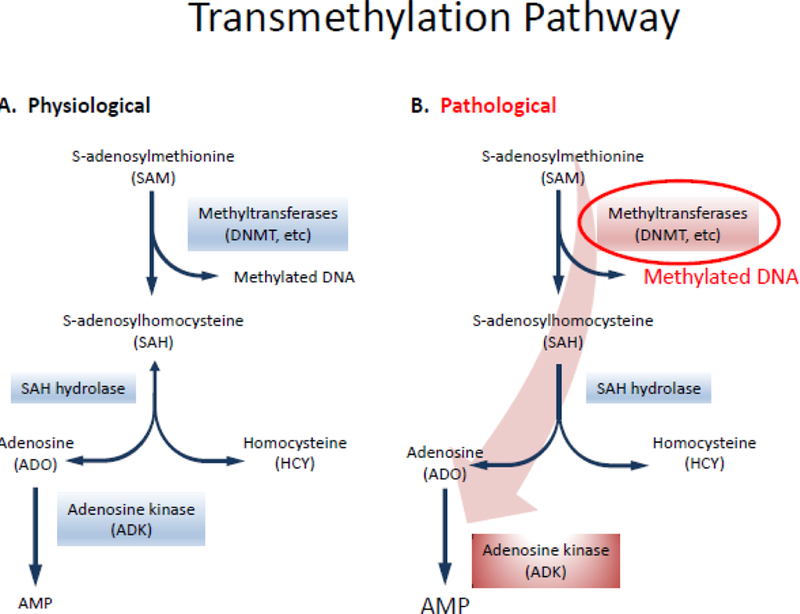
The epigenetic role of adenosine: a comparison of the normal physiological (A) and epileptic(B) states in the transmethylation pathway. Due to clearance of adenosine by overexpression of ADK in the pathological state, the shifted equilibrium (indicated by the large red arrow) drives this pathway to hypermethylation, a hallmark of epileptogenesis.
2.4.2. Histone modification
Histone modifications include methylation and acetylation and, similar to DNA methylation changes, have been identified in human TLE as well as in rodent models of TLE [35, 43–45]. In line with altered histone methylation and acetylation markers, histone modifying enzymes, such as histone deacetylases (HDACs), are dysregulated during epileptogenesis and in the epileptic state [46, 47]. Histones are methylated on their lysine or arginine residues by histone methyltransferases (HMT). Generally these modifications are located at the amino-terminal tail of histones and present two modes of action: chromatin condensation and recruitment of histone-binding proteins that modify the transcription process [33]. These processes can expose DNA stretches containing particular genes for extended or shortened periods of time, essentially switching genes on or off and causing repressive or active translational changes in protein production. Because the nuclear isoform of ADK (ADK-L) regulates DNA methylation, it is highly likely that histone methylation is regulated through a similar mechanism. This is an exciting perspective that warrants further studies.
3. Adenosine receptor dependent effects of adenosine
As mentioned above, adenosine is an endogenous anticonvulsant and seizure terminator of the brain. In addition, it has been shown that maladaptive changes in adenosine receptor signaling contribute to the pathophysiology of epilepsy. Neuronal excitability in the brain is modulated by the activation of G-protein coupled adenosine receptors (A1, A2A, A2B, and A3). Neuronal excitability therefore depends on the equilibrium of receptor effects, expression levels in different brain regions, and their availability for receptor activation. A shift in the ratio of inhibitory A1R towards stimulatory A2ARs directly influences neuronal excitability. In general, the epileptic state is marked by decreased A1R signaling and/or enhanced A2AR signaling. It is currently unknown whether modifications in adenosine receptor expression are cause or consequence of epilepsy.
3.1. Adenosine A1R
Research has shown that a reduction in A1R density and a deficiency of endogenous adenosine-based seizure control mechanisms occur in the rat kindling model of epilepsy, indicating a failure of endogenous seizure control mechanisms [48]. Receptor knockout designs have demonstrated that mice without A1R have spontaneous electrographic seizures [49] and develop fatal status epilepticus after intrahippocampal injection of kainic acid or after a traumatic brain injury [11, 50]. These experiments exemplify that A1R activation is necessary for the suppression of seizures and their spread. Desensitization of A1Rs, but normal expression levels, have been described in rodents after status epilepticus [51]; upregulation of A1R, however, has been described as a response to spontaneous seizures induced by electrical stimulation [52]. Variants in the A1R gene have been associated with the progression of posttraumatic seizures after brain injury, indicating that a failure in A1R signaling could potentially be connected to epileptogenesis [53]. These findings suggest that disruption of A1R signaling is dynamically linked to the pathophysiology of epilepsy.
3.2. Adenosine A2AR
A reduction in the synaptic expression of A2A receptors can mitigate synaptotoxic consequences of the synaptic pool of adenosine [54–56]. This function is directly linked to the neuronal release of adenosine or its precursor ATP, which leads to neuronal hyperexcitability, which is likely dependent on increased synaptic A2A receptor activation. This aggravation of synaptotoxicity could further the degeneration of normal circuitry and contribute to the exacerbation of epilepsy. Genetic changes in the A2AR gene have been linked with acute encephalopathy and seizures in children, indicating that dysregulation of A2ARs may play a role in seizure progression and excitotoxic brain injury [57]. In line with a possible pro-convulsive role of A2ARs, A2AR knockout mice were partially resistant to limbic seizures, further suggesting that promotion of the epileptic state is a consequence of increased A2AR activation (Figure 3).
Figure 3.
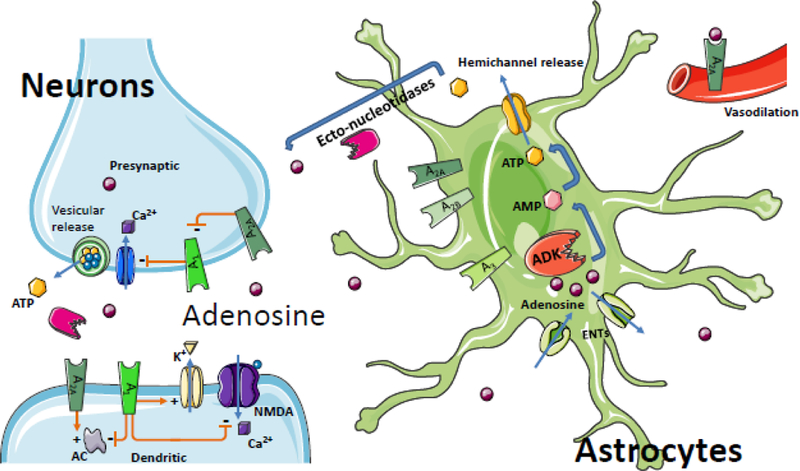
Adenosine flux between neurons and astrocytes in the brain. Astrocytes serve as a sink for synaptic levels of adenosine. Equilibrative nucleoside transporters (ENTs) normally equilibrate extra- and intracellular adenosine levels; however conditions of increased intracellular ADK drive the influx of adenosine into the cell yielding reduced extracellular levels of adenosine and – consequentially – reduced adenosine receptor activation; ATP, the major source of adenosine, can be released from neurons and astrocytes via vesicles or directly through hemichannels from astrocytes. Adenosine can then be generated through the activity of ectonucleotidases to complete the balance of the ATP / adenosine conversion cycle. The physiological functions of adenosine receptors are more extensively covered in other articles of this Special Issue; briefly, the interactions of select adenosine receptors and metabolite transporters are shown here. Presynaptically, A2A receptors antagonize the inhibitory effect of the A1 receptor on inhibiting calcium (Ca2+) import, which ultimately modulates vesicular release of ATP and glutamate. Postsynaptically, the A1 receptor inhibits calcium import of the N-methyl-D- aspartate receptor (NMDA), increases the conductance of potassium (K+), and inhibits adenylate cyclase (AC); A2A receptors have an opposing stimulation of dendritic AC. A2A receptors also play a role in vasodilation of blood vessels in the brain and periphery. Because the implications of the adenosine system in oligodendrocytes and microglia for epilepsy are largely unknown, those cell-types have been omitted to enhance clarity.
3.3. Adenosine A2BR and A3R
Based on their low expression levels in the brain and lower affinities for adenosine, A2BRs and A3Rs do not appear to play a direct role in epilepsy. However tissue from human resected epileptic tissue transplanted into Xenopus oocytes showed that selective A2BR and A3R antagonists decreased the run-down of GABA currents. Thus, it has been suggested that cortical adenosine A2B and A3 receptors alter the stability of GABAA receptors and thus potentially calibrate neuronal excitability [58].
4. Adenosine receptor independent effects of adenosine
In addition to adenosine receptor dependent effects, adenosine itself acts as a biochemical metabolite and plays a major role in the transmethylation pathway, which includes DNA methylation [8, 59, 60]. Because adenosine is a product of transmethylation, ADK and, in particular, its nuclear isoform ADK-L, assumes a unique role as regulator of DNA methylation.
4.1. Transmethylation pathway
DNA methylation relies on the donation of a methyl group from S-adenosylmethionine (SAM) which is facilitated by DNMT enzymes that yield S-adenosylhomocysteine (SAH) [8]. SAH is then converted into adenosine and homocysteine by SAH hydrolase. The direction of this pathway is contingent on the equilibrium of the SAH hydrolase reaction towards SAH formation [61]; thus, the reaction will only continue when adenosine and homocysteine are continually removed [7, 61]. Since adenosine is a necessary end product of SAM-dependent transmethylation reactions, ADK drives the flux of methylation through this pathway by removing adenosine; therefore, increased ADK expression directly promotes hypermethylated DNA [8]. On the contrary, reduced ADK expression or increased adenosine, along with increased homocysteine, shifts the equilibrium of the methylation pathway in reverse, favoring the formation of SAH. This significant shift caused by SAH hydrolase towards SAH is known to block DNA methyltransferase activity through means of product inhibition [8]. This block corresponds to a decrease in methyltransferase activity and promotes a global reduction in DNA methylation levels, which is of therapeutic value in the intervention of epileptogenesis [8].
These effects have been demonstrated by intracerebroventricular injections of the two end products adenosine and homocysteine; this intervention revealed significantly decreased global methylation levels in the hippocampus of rats at two distinct time points [8]. In contrast, the augmentation of SAM (the main methyl donor for the transmethylation pathway) led to hypermethylation in the same setting [8].
In line with these findings, transgenic mice with reduced ADK expression in the brain had hypomethylated DNA. Likewise, the systemic administration of an ADK inhibitor led to a reduction of DNA methylation status in the brain, an effect sustained in the presence of caffeine, indicating that adenosine-induced hypomethylation of DNA is independent of adenosine receptors [8]. Finally, BHK cells engineered to either lack ADK or to exclusively express only one of the two isoforms of ADK were used to establish a direct mechanistic link between ADK expression and DNA methylation. Cells lacking ADK were hypomethylated, whereas cells overexpressing the long nuclear isoform (ADK-L) were hypermethylated, with cells expressing the short cytoplasmic isoform (ADK-S) having an intermediate phenotype [8]. Those data show that increased expression of ADK (as observed during epileptogenesis) drives increased DNA methylation.
4.2. Epigenetic modification
Due to the combination of increased expression of ADK being associated with epileptogenesis [8, 24], increased ADK driving increased DNA methylation [8], and maladaptive changes in DNA methylation having already been linked with epilepsy development [39, 62], it follows that increases in ADK expression likely play a causative role in epileptogenesis. This hypothesis was directly tested in a kainic acid induced rat model of progressive epilepsy. After the emergence of the first spontaneous seizures, a transient dose of adenosine was given by the intraventricular implantation of silk-based adenosine releasing polymers. The polymers were designed to release adenosine for a restricted time span of only 10 days. During active adenosine release, the seizures were almost completely suppressed. However, after the cessation of adenosine release, the adenosine treated rats still did not progress in the severity of their epilepsy as quantified by the number of daily seizures, whereas all control animals progressed with epilepsy development [8]. This disease modifying effect was long-lasting (at least 3 months) and associated with a lasting normalization of global DNA methylation levels in the adenosine treated animals, while DNA in the hippocampus of control animals remained hypermethylated. In line with a sustained suppression of epileptogenesis, mossy fiber sprouting within the epileptogenic hippocampus of the adenosine treated rats was also blocked. These data support a critical role of maladaptive DNA hypermethylation in the epileptogenic process and demonstrate that a transient dose of adenosine can effectively block this process and prevent epileptogenesis. Adenosine as such may be save to deliver in a clinical scenario, because intrathecal adenosine up to 2 mg was considered to be safe in healthy human volunteers [87, 88].
5. Adenosine augmentation therapies
Experimental and clinical findings outlined above present a neurochemical rationale for using therapeutic adenosine augmentation in the treatment of seizures and the associated comorbidities of epilepsy. Several strategies have been assessed, which collectively demonstrate a potent antiictogenic and antiepileptogenic potential of adenosine augmentation therapies. Whereas the transient injury- or seizure-induced adenosine surge is an endogenous protective mechanism of the brain, excessive adenosine in the brain can lead to postictal brain shutdown and thereby might contribute to sudden unexpected death in epilepsy (SUDEP) [17]. It is important to note that adenosine augmentation therapies are not designed to further boos the acute adenosine response, but rather to restore normal adenosine equilibrium in conditions of acquired adenosine deficiency.
5.1. Pharmacology
Adenosine augmentation therapies capitalize on the anticonvulsant and neuroprotectant therapeutic properties of endogenous adenosine in the brain with the potential to not only suppress seizures, but more importantly, prevent epileptogenesis [63]. ADK inhibitors have demonstrated to be powerful therapeutic agents to enhance the tissue tone of endogenous adenosine [17]. These inhibitors can mediate an endogenous adenosine response through the disruption of adenosine metabolic clearance; for example, ADK inhibitors are uniquely suited to potentiate a seizure-induced adenosine increase in a site- and event-specific manner [64–67]. Since ADK overexpression is a pathological hallmark in epileptogenic brain regions [22, 24, 42, 68] and because overexpression of ADK has been linked to seizure generation [31, 42, 69, 70], the rationale for the therapeutic use of ADK inhibitors in epilepsy is strongly supported. Notably, the global ADK inhibitor 5-ITU has demonstrated robust efficacy in seizure suppression in a mouse model of pharmacoresistant temporal lope epilepsy [24], implying that the potential value of ADK inhibitors may prove to be superior to conventional antiepileptic agents. In addition, multiple experiments have suggested that the high-affinity, low-capacity enzyme ADK, rather than the low-affinity, high-capacity enzyme adenosine deaminase (ADA), is the major adenosine metabolizing enzyme in the brain. Specifically, ADK inhibition, but not ADA disruption, enhanced endogenous adenosine and limited neuronal activity in hippocampal sections [71]; the genetic knockout of ADK has also shown superior adenosine release in comparison to cells with a genetic knockout of ADA [72]. Along with those findings, the ADK inhibitors 5’-amino-5’-deoxyadenosine or 5- ITU, but not the ADA inhibitor 2’-deoxycoformycin, were able to suppress bicuculline-induced seizures in rats, further supporting the anti-seizure effect of ADK inhibition as superior to ADA inhibition [73]. Unfortunately, permanent or long-term therapeutic adenosine augmentation by systemic global ADK inhibition may not be a viable therapeutic intervention due to liver toxicity [7], as well as cognitive and sedative side effects [17].
However, since the recent discovery of ADK protein existing in two isoforms, derived through splicing and alternative promoter use, which exert either cell-autonomous (nuclear ADK-L) or non-cell-autonomous (cytoplasmic ADK-S) functions [8], the selective modulation of ADK isoforms has gained increased interest due to their ability to specifically target dynamic adenosine expression. The nuclear expression of ADK is seen in cell types that manage plastic properties into adulthood, such as astrocytes or granular cells in the dentate gyrus; however, terminally differentiated cells, like the majority of neurons, lack ADK-L expression [17]. Understanding the mechanisms associated with isoform expression could be of further clinical value in the regulation of adenosine homeostasis through specific and focal manipulation. Neurological disorders could be dynamically ADK isoform specific but further studies are needed in order to finely delineate Adk expression post injury. Localized therapeutic approaches that could potentially target specific ADK-isoforms may be better suited to refine the therapeutic potential of adenosine and avoid the adverse side effects associated with conventional non-specific global ADK inhibitors [74].
5.2. Cell Based Therapy
A distinct approach for the local augmentation of adenosine is the transplantation of cells engineered to release adenosine. This can be accomplished by removing the Adk gene in cultured cells to induce therapeutic adenosine delivery followed by focally transplanting the adenosine-releasing cells into a brain to specifically exploit locally enriched levels of adenosine [17]. One way to perform this adenosine augmentation therapy is with engineered BHK (baby hamster kidney) cells where the Adk gene has been deleted via a combination of mutagenesis and selection for the ADK deficiency. As mentioned, disruption of the Adk gene induced more cellular adenosine release than disrupting the adenosine deaminase gene [72]. These ADK-deficient BHK cells therapeutically delivered about 40ng adenosine per 10^5 cells per day. When encased into semipermeable polymer membranes and focally transplanted into the brain ventricles of kindled epileptic rats, the implants mediated robust suppression of seizures in an A1R dependent manner [72]. The strength of seizure suppression was limited to two weeks however, due to the finite longevity of the encapsulated cells. In order to create a more adaptable cell-based process for seizure regulation, both alleles of the Adk gene were disrupted in mouse embryonic stem (ES) cells via homologous recombination producing Adk−/− ES cells [75]. Once differentiated into neural precursor cells and implanted into the infrahippocampal fissure of rats, the adenosine releasing cells elicited profound antiepileptogenic effects in the kindling model [49]. In addition, when identically grafted into mice 24 h post status epilepticus, the adenosine releasing cells prevented the progression of epilepsy in a post-status model of epileptogenesis as well [42]. Recipients of the Adk−/− cells demonstrated reduced astrogliosis, the prevention of endogenous ADK overexpression, and completely eliminated seizure activity; this contrasts with the recipients of wild-type cells, which developed epileptogenic histopathology and spontaneous seizures at a rate of 4 seizures per hour [42]. To better suit clinical translation of engineered human adenosine-releasing stem cells, human mesenchymal stem cells were infected with a lentivirus designed to express a micro RNA targeted against Adk. This strategy reduced ADK expression to 20% of standard levels and generated the release of approximately 1 ng adenosine per 10^5 cells per hour [76]. Acute seizure-induced cytotoxicity was alleviated when adenosine-releasing cells were transplanted into the infrahippocampal fissure of mice [76] with partial antiepileptogenic activity [77]. This partial therapeutic influence is likely due to a 40-fold decreased rate of adenosine release from those cells, in contrast with the engineered ES cells that globally lacked any Adk expression. Collectively, cell-based adenosine augmentation has emerged as a means to locally augment adenosine signaling within the brain as a potent therapy yielding benefits of neuroprotection and seizure suppression, in addition to antiepileptogenic effects.
5.3. Gene Therapy
An alternative strategy to achieve a more specific and cell-type selective adenosine enrichment is gene therapy. In contrast to conventional adenosine augmentation therapies that supply a systemic or locally implanted delivery, the expression of a transgene can yield a far more refined target; in this case, the attenuation of the pathological over- expression of the endogenous Adk gene in epilepsy. In line with this rationale, the overexpression of ADK in hippocampal or cortical astrocytes via viral vectors has been shown to be sufficient to trigger and induce seizures [70, 78]. It is worth noting that even modest ADK increases of 25% are adequate to provoke seizures in both rodent [24, 42] and human [22] temporal lobe epilepsy. This supports the rationale to therapeutically decrease the expression of the endogenous Adk gene in epileptic brains to physiological levels.
One method for attaining this reduction is through the use of antisense approaches [79] designed to knock down expression of the Adk gene. One successful study implemented an adeno-associated virus (AAV) vector which was designed to express an Adk cDNA in the antisense orientation under an astrocytic promoter [80]. It was shown in a transgenic model of spontaneous recurrent seizures (global overexpression of ADK) that an intrahippocampal injection of this antisense AAV produced considerable reduction in seizure frequency; experimental animals treated with an ADK knockdown vector only experienced an average of 0.6 +/− 0.6 seizures/h in the ipsilateral hippocampus, in contrast to 5.8 +/− 0.5 seizures/h in the contralateral control side [70]. This study suggested that a modest knockdown of <5% ADK in these mice was sufficient to suppress seizure activity [70]. Similarly, a viral vector expressing a miRNA directed against ADK was shown to be neuroprotective in an ex vivo gene therapy approach [81]. These results help establish a viral strategy as a feasible approach by targeting ADK in a localized manner (hippocampus) and to a specific cell type (astrocyte) with the goal to exploit the anticonvulsive effects of endogenous adenosine. Three potential gene therapy strategies are worth further exploration.
As an alternative to the use of a cDNA expressed in antisense orientation, seizure suppression can be induced via a miRNA based knockdown of ADK. This method would have the benefits of combining a permanent intervention with a more simplistic construct design (large target genes may be incompatible with the restrictive size limits of AAV capsids). The principle behind using miRNA to silence the gene expression of ADK would provide an incentive for further translational work. This treatment paradigm has the potential to reveal miRNA’s efficacy in restoring a physiological state to epileptic subjects by downregulating pathological ADK expression, which may provide alternative therapeutic options for those who suffer from epilepsy. If limited clinically to an epileptogenic focus in patients scheduled for epilepsy surgery, this might eventually be a ‘reversible gene therapy’ if there are no benefits and/or adverse events occur.
A more refined gene therapy approach aimed for enhanced potency is through the use of a direct gene editing tool, such as the CRISPR-Cas9 system. This approach would enable the selective elimination of either ADK-L (nuclear) or ADK-S (cytoplasmic) expression. Preliminary results have indicated that the two isoforms of ADK act on separate pools of adenosine [17]. The ADK-S isoform acts on the cytoplasmic pool of adenosine, which determines extracellular levels of adenosine and adenosine receptor activation, and ultimately contributes to the regulation of neuronal excitability. On the other hand, the ADK-L isoform, confined to the nucleus [17], is directly linked to the methylation status of the DNA [8]. Excessive DNA methylation is detrimental and aggravates the progression of epilepsy in response to an overabundance of ADK-L. In order to capitalize on the antiepileptogenic effect of ADK-L inhibition, the focus would be to selectively delete ADK-L via viral delivery of a tailored CRISPR-Cas9 based gene editing protein.
5.4. Strategies for antiepileptogenesis
It has been suggested that DNA methylation inhibitors have therapeutic value in the intervention of epilepsy by restoring non-pathological epigenetic homeostasis [83]. Conventional antineoplastic DNA methylation inhibitors are incorporated into the DNA, and thus indirectly block DNA methylation [84]; however, this treatment is not considered to be suitable for the treatment of epilepsy due to risks of cell death and mutagenesis. In order to avoid adverse events related to antineoplastic DNA methylation inhibitors, focal adenosine therapy may prove to be a safer epigenetically acting alternative.
There are several potential approaches to studying the antiepileptogenic effects of adenosine and how they coincide with epigenetic mechanisms. For instance, transgenic mice with a genetic reduction of forebrain ADK expression have been shown to be resistant to the development of epilepsy, even when a pro-epileptogenic stimulus was coupled with a transient blockade of A1R [42]. In another study, adenosine-releasing stem cells were also demonstrated to attenuate astrogliosis, restrain ADK expression, and mitigate the progression of spontaneous ictal activity after triggering epileptogenesis [42]. One particular method, however, involving transient adenosine exposure via silk polymers, has yielded promising results that warrant further discussion.
This unique approach towards focally delivering therapeutic adenosine has been achieved via bilateral silk-based biodegradable polymer implants into the brain ventricles, designed to deliver pre-defined target doses of adenosine for a restricted time span [8]. This focal and temporal approach has been used in two separate models where adenosine-releasing silk methods have proven to be effective strategies in not only demonstrating the utility of anti- seizure, but also anti-epileptogenic, adenosine-based therapies.
First, to assess the translational antiepileptogenic role of adenosine-releasing silk-based polymers, a more clinically relevant model of epilepsy was used. After the emergence of the first spontaneous recurrent seizures (i.e. ‘diagnosis of epilepsy) nine weeks after triggering TLE via systemic injection of KA in rats adenosine-releasing polymers designed to release adenosine for only 10 days were implanted into the lateral brain ventricles. The intraventricular implants of adenosine-releasing silk were determined to block DNMT activity and to restore DNA methylation levels during active adenosine release; in contrast, the control-implant recipients’ DNA status remained hypermethylated in line with their epileptic phenotype. This substantial therapeutic change persisted for at least three weeks after depletion of adenosine-release from the polymers, indicating a transient and localized dose of adenosine can exert long-lasting influence on maladaptive DNA methylation changes in epilepsy [8].
In support of this finding, further investigation went into this intervention in order to assess if adenosine-releasing silk could potentially offer anti-epileptogenic properties. Similarly, rats at nine weeks post systemic KA administration received adenosine polymer implants with a ten day release duration. Epilepsy progression was then monitored for a total of three months, where the adenosine treatment groups showed almost complete suppression of all seizures during the first week after implantation [8]. Notably, reduced seizure activity was maintained well beyond the ten day lifespan of adenosine released from the polymer. Even more impressively, a 75% reduction of seizure activity was observed for at least twelve weeks after polymer implantation, whereas seizures in control animals increased in number and intensity, and three animals died due to excessive seizures [8].
In order to substantiate the focal therapeutic value of adenosine and provide an independent outcome measure for the anti-epileptogenic performance of silk-based adenosine delivery, the degree of mossy fiber sprouting was investigated. Since mossy fiber sprouting is indicative of epileptogenesis and the formation of new recurrent excitatory circuits in the dentate gyrus [82], epileptic rats nine weeks post systemic KA treatment were compared to naïve rats and notable differences were observed in axon spread [8]. At this nine week time point, adenosine-silk implants were administered and compared with controls twelve weeks later. In contrast, animals exposed to transient adenosine-releasing silk lacked progressive changes in mossy fiber sprouting, while the sham- or empty polymer-treated controls proceeded to express increased sprouting of mossy fibers corresponding to the detrimental expansion of mossy fiber synaptic terminal growth [8].
Together these results demonstrate (i) that a transient local dose of adenosine can prevent the progression of epilepsy, and (ii) that increased DNA methylation (normalized by transient adenosine therapy) is a contributing factor of epilepsy development and progression.
6. Conclusions and outlook
Neurons are connected into vast sensory networks controlled by the homeostatic milieu of their environment, in particular through glial cell regulation. As we try to unravel the associated comorbidities of complex neurological syndromes, it becomes clear that these comorbid conditions that emerge a newly recognized spectrum disorders are best explained by the disruption of network homeostasis. The disruption of network homeostasis involves simultaneous and universal dysregulation of several molecular pathways, which consequently become pervasive in neurodegeneration in the sense that ‘seizures beget seizures’. This old concept holds genuine truth and can be best described by the self-reinforcing interplay of several homeostatic systems that become increasingly dysfunctional following disease onset. Conventional antiepileptic drugs with narrow action targets are unlikely to impact network homeostasis or to exert preventative value in the progressive maladaptive changes associated with epileptogenesis. Novel therapeutic interventions based on adenosine, epigenetic mechanisms, or dietary and lifestyle intervention might hold promise to influence global homeostasis on the network level. Thus adenosine augmentation (Figure 4) has become a forerunner of therapeutic value in suppressing ictal activity [42, 49], preventing disease exacerbation and epileptogenesis [8, 42], averting psychosis, and enhancing cognition [85] without any known adverse side effects when delivered locally or transiently. The antipsychotic and pro-cognitive response in mice [85] implies that therapeutic adenosine augmentation might couple anticonvulsant with pro-cognitive effects. In preclinical toxicity investigation of intrathecal adenosine in canines, no adverse side effects were noted with chronic adenosine administration for 26 days [86]. Similarly, intrathecal adenosine was conducted in humans in escalating doses ranging up to 2 mg without any negative side effects [87, 88]. This is of substantial value because safety and feasibility must be paramount, and already several adenosine therapies are actively in pre-clinical development. It is within the realm of possibilities that adenosine augmentation will be introduced into the clinic as an ameliorative homeostatic network therapy within the next decade. This novel class of therapy will have developmental challenges to limit adenosine’s action to identifiable target areas and cells; the prospect of cell specificity in gene vectors could offer a promising strategy to shape adenosine homeostasis in a localized and cell-type selective way.
Figure 4.
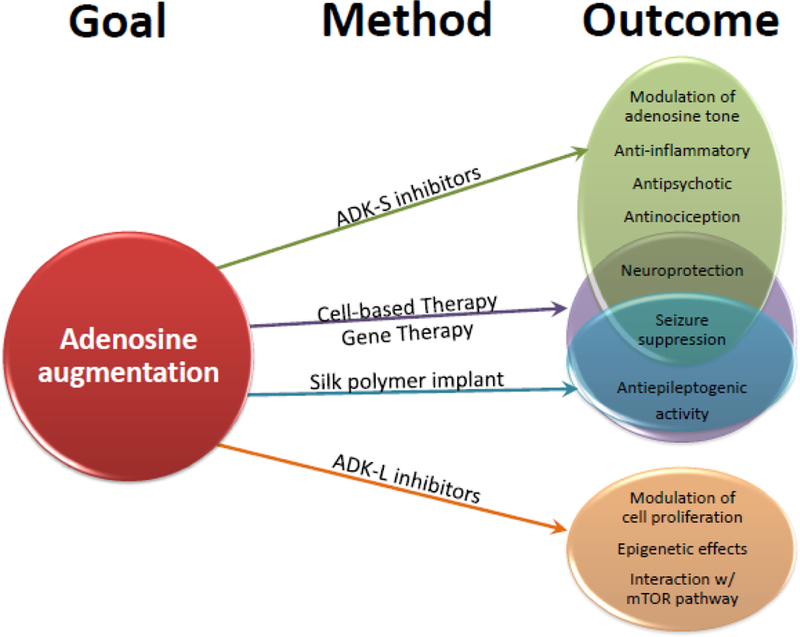
Summary of key findings in adenosine augmentation methods. Many of the potential outcomes of adenosine augmentation are seen to overlap in ameliorating the symptoms and comorbidities associated with adenosine deficiency and epilepsy. As new therapeutic strategies emerge, it is useful to consider the interplay and novel effects of these different methods.
Highlights:
Adenosine is an endogenous anticonvulsant and seizure terminator.
Adenosine exerts potent anti-seizure effects through adenosine A1 receptor activation.
Adenosine has novel adenosine receptor independent effects as regulator of DNA methylation.
Adenosine has a novel role for disease modification and epilepsy prevention.
Therapeutic adenosine augmentation has potential to stop seizures and to prevent epilepsy.
Acknowledgements:
We wish to thank Citizens United for Research in Epilepsy (CURE) and the NIH (NS088024, NS065957) for prior funding of our work on adenosine in the epilepsies. We thank Bradley Hansen for his assistance in editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None
REFERENCES
- 1.Bialer M, et al. , Progress report on new antiepileptic drugs: A summary of the Thirteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIII). Epilepsia, 2017. 58(2): p. 181–221. [DOI] [PubMed] [Google Scholar]
- 2.Younus I and Reddy DS, A resurging boom in new drugs for epilepsy and brain disorders. Expert Rev Clin Pharmacol, 2018. 11(1): p. 27–45. [DOI] [PubMed] [Google Scholar]
- 3.Younus I and Reddy DS, Epigenetic interventions for epileptogenesis: A new frontier for curing epilepsy. Pharmacol Ther, 2017. 177: p. 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobow K, et al. , Finding a better drug for epilepsy: antiepileptogenesis targets. Epilepsia, 201253(11): p. 1868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.During MJ and Spencer DD, Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol, 1992. 32(5): p. 618–24. [DOI] [PubMed] [Google Scholar]
- 6.Lado FA and Moshe SL, How do seizures stop? Epilepsia, 2008. 49(10): p. 1651–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boison D, et al. , Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci USA, 2002. 99(10): p. 6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams-Karnesky RL, et al. , Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Inv, 2013. 123(8): p. 3552–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukuda M, et al. , Adenosine A1 receptor blockage mediates theophylline-associated seizures. Epilepsia, 2010. 51(3): p. 483–7. [DOI] [PubMed] [Google Scholar]
- 10.Korematsu S, et al. , Theophylline-associated seizures and their clinical characterizations. Pediatr Int, 2008. 50(1): p. 95–8. [DOI] [PubMed] [Google Scholar]
- 11.Fedele DE, et al. , Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol, 2006. 200(1): p. 184–190. [DOI] [PubMed] [Google Scholar]
- 12.Klein P, et al. , Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia, 2018. 59(1): p. 37–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazarati AM, Lewis ML, and Pittman QJ, Neurobehavioral comorbidities of epilepsy: Role of inflammation. Epilepsia, 2017. 58 Suppl 3: p. 48–56. [DOI] [PubMed] [Google Scholar]
- 14.Boison D and Aronica E, Comorbidities in Neurology: Is adenosine the common link? Neuropharmacology, 2015. 97: p. 18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shankar R, et al. , Sudden unexpected death in epilepsy (SUDEP): what every neurologist should know. Epileptic Disord, 2017. 19(1): p. 1–9. [DOI] [PubMed] [Google Scholar]
- 16.Young C, et al. , Does intellectual disability increase sudden unexpected death in epilepsy (SUDEP) risk? Seizure, 2015. 25: p. 112–6. [DOI] [PubMed] [Google Scholar]
- 17.Boison D, Adenosine kinase: exploitation for therapeutic gain. Pharmacological Reviews, 201365: p. 906–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Studer FE, et al. , Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience, 2006. 142(1): p. 125–37. [DOI] [PubMed] [Google Scholar]
- 19.Pekny M and Nilsson M, Astrocyte activation and reactive gliosis. Glia, 2005. 50(4): p. 427–434. [DOI] [PubMed] [Google Scholar]
- 20.Boison D and Steinhauser C, Epilepsy and astrocyte energy metabolism. Glia, 2018. 66(6): p.1235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinhauser C and Boison D, Epilepsy: crucial role for astrocytes. Glia, 2012. 60(8): p. 1191. [DOI] [PubMed] [Google Scholar]
- 22.Aronica E, et al. , Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia, 2011. 52(9): p. 1645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masino SA, et al. , Purines and neuronal excitability: links to the ketogenic diet. Epilepsy Res, 2012. 100(3): p. 229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gouder N, et al. , Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci, 2004. 24(3): p. 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li T, et al. , Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Inv, 2008. 118(2): p. 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pignataro G, et al. , Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J Cereb Blood Flow Metab, 2008. 28: p. 17–23. [DOI] [PubMed] [Google Scholar]
- 27.Devinsky O, et al. , Glia and epilepsy: excitability and inflammation. Trends Neurosci, 201336(3): p. 174–84. [DOI] [PubMed] [Google Scholar]
- 28.Nabbout R, et al. , Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol, 2011. 10(1): p. 99–108. [DOI] [PubMed] [Google Scholar]
- 29.Vezzani A, et al. , The role of inflammation in epilepsy. Nat Rev Neurol, 2011. 7(1): p. 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vezzani A and Friedman A, Brain inflammation as a biomarker in epilepsy. Biomark Med, 20115(5): p. 607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li T, et al. , Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: a first step in epileptogenesis? Glia, 2012. 60: p. 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li T, et al. , Adenosine dysfunction in astrogliosis: cause for seizure generation? Neuron Glia Biology, 2007. 3: p. 353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michalak Z, et al. , Spatio-temporally restricted blood-brain barrier disruption after intra- amygdala kainic acid-induced status epilepticus in mice. Epilepsy Res, 2013. 103(2–3): p. 167–79. [DOI] [PubMed] [Google Scholar]
- 34.Miller-Delaney SF, et al. , Differential DNA methylation patterns define status epilepticus and epileptic tolerance. J Neurosci, 2012. 32(5): p. 1577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowe SL, et al. , Phosphorylation of histone H2A.X as an early marker of neuronal endangerment following seizures in the adult rat brain. J Neurosci, 2011. 31(21): p. 7648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang HY, et al. , Histone deacetylase inhibition mediates urocortin-induced antiproliferation and neuronal differentiation in neural stem cells. Stem Cells, 2012. 30(12): p. 2760–73. [DOI] [PubMed] [Google Scholar]
- 37.Zhu Q, et al. , Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy. J Mol Neurosci, 2012. 46(2): p. 420–6. [DOI] [PubMed] [Google Scholar]
- 38.Kobow K, et al. , Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J Neuropathol Exp Neurol, 2009. 68(4): p. 356–64. [DOI] [PubMed] [Google Scholar]
- 39.Kobow K, et al. , Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol, 2013. 126(5): p. 741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller-Delaney SF, et al. , Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain, 2015. 138(Pt 3): p. 616–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang CY, et al. , Growth stimulating effect on queen bee larvae of histone deacetylase inhibitors. J Agric Food Chem, 2012. 60(24): p. 6139–49. [DOI] [PubMed] [Google Scholar]
- 42.Huang D, et al. , A novel series of l-2-benzyloxycarbonylamino-8-(2-pyridyl)-disulfidyloctanoic acid derivatives as histone deacetylase inhibitors: design, synthesis and molecular modeling study. Eur J Med Chem, 2012. 52: p. 111–22. [DOI] [PubMed] [Google Scholar]
- 43.Sng JC, Taniura H, and Yoneda Y, Histone modifications in kainate-induced status epilepticus. Eur J Neurosci, 2006. 23(5): p. 1269–82. [DOI] [PubMed] [Google Scholar]
- 44.Taniura H, Sng JC, and Yoneda Y, Histone modifications in status epilepticus induced by kainate. Histol Histopathol, 2006. 21(7): p. 785–91. [DOI] [PubMed] [Google Scholar]
- 45.Tsankova NM, Kumar A, and Nestler EJ, Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci, 2004. 24(24): p. 5603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Y, et al. , Increased expression of histone deacetylases 2 in temporal lobe epilepsy: a study of epileptic patients and rat models. Synapse, 2012. 66(2): p. 151–9. [DOI] [PubMed] [Google Scholar]
- 47.Jagirdar R, et al. , Expression of class II histone deacetylases in two mouse models of temporal lobe epilepsy. J Neurochem, 2016. 136(4): p. 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rebola N, et al. , Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. Eur J Neurosci, 2003. 18(4): p. 820–8. [DOI] [PubMed] [Google Scholar]
- 49.Li T, et al. , Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain, 2007. 130(Pt 5): p. 1276–88. [DOI] [PubMed] [Google Scholar]
- 50.Kochanek PM, et al. , Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab, 2006. 26: p. 565–575. [DOI] [PubMed] [Google Scholar]
- 51.Knoflach F, et al. , Pharmacological modulation of the diazepam-insensitive recombinant gamma-aminobutyric acidA receptors alpha 4 beta 2 gamma 2 and alpha 6 beta 2 gamma 2. Mol Pharmacol, 1996. 50(5): p. 1253–61. [PubMed] [Google Scholar]
- 52.Hargus NJ, et al. , Enhanced actions of adenosine in medial entorhinal cortex layer II stellate neurons in temporal lobe epilepsy are mediated via A(1)-receptor activation. Epilepsia, 2012. 53(1): p. 168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner AK, et al. , Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res, 2010. 90(3): p. 259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matos M, et al. , Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J Alzheimers Dis, 2012. 31(3): p. 555–67. [DOI] [PubMed] [Google Scholar]
- 55.Popoli P, et al. , Modulation of glutamate release and excitotoxicity by adenosine A2A receptors. Neurology, 2003. 61(11 Suppl 6): p. S69–71. [DOI] [PubMed] [Google Scholar]
- 56.Silva CG, et al. , Blockade of adenosine A(2A) receptors prevents staurosporine-induced apoptosis of rat hippocampal neurons. Neurobiol Dis, 2007. 27(2): p. 182–189. [DOI] [PubMed] [Google Scholar]
- 57.Shinohara M, et al. , ADORA2A polymorphism predisposes children to encephalopathy with febrile status epilepticus. Neurology, 2013. 80(17): p. 1571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roseti C, et al. , Adenosine receptor antagonists alter the stability of human epileptic GABAA receptors. Proc Natl Acad Sci U S A, 2008. 105(39): p. 15118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boison D, et al. , Seizure suppression by adenosine-releasing cells is independent of seizure frequency. Epilepsia, 2002. 43(8): p. 788–96. [DOI] [PubMed] [Google Scholar]
- 60.Mato JM, Martinez-Chantar ML, and Lu SC, Methionine metabolism and liver disease. Annu Rev Nutr, 2008. 28: p. 273–93. [DOI] [PubMed] [Google Scholar]
- 61.Kredich NM and Martin DV Jr., Role of S-adenosylhomocysteine in adenosinemediated toxicity in cultured mouse T lymphoma cells. Cell, 1977. 12(4): p. 931–8. [DOI] [PubMed] [Google Scholar]
- 62.Kobow K and Blumcke I, The emerging role of DNA methylation in epileptogenesis. Epilepsia, 2012. 53 Suppl 9: p. 11–20. [DOI] [PubMed] [Google Scholar]
- 63.Boison D, Adenosine augmentation therapies (AATs) for epilepsy: prospect of cell and gene therapies. Epilepsy Res, 2009. 85(2): p. 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kowaluk EA, Bhagwat SS, and Jarvis MF, Adenosine kinase inhibitors. Curr Pharm Des, 19984(5): p. 403–16. [PubMed] [Google Scholar]
- 65.Kowaluk EA and Jarvis MF, Therapeutic potential of adenosine kinase inhibitors. Expert Opin Investig Drugs, 2000. 9(3): p. 551–64. [DOI] [PubMed] [Google Scholar]
- 66.McGaraughty S, Cowart M, and Jarvis MF, Recent developments in the discovery of novel adenosine kinase inhibitors: mechanism of action and therapeutic potential. CNS Drug Rev, 2001. 7(4): p. 415–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGaraughty S, et al. , Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors. Curr Top Med Chem, 2005. 5(1): p. 43–58. [DOI] [PubMed] [Google Scholar]
- 68.Boison D, Adenosine dysfunction in epilepsy. Glia, 2012. 60(8): p. 1234–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Etherington LA, et al. , Astrocytic adenosine kinase regulates basal synaptic adenosine levels and seizure activity but not activity-dependent adenosine release in the hippocampus. Neuropharmacology, 2009. 56(2): p. 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Theofilas P, et al. , Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia, 2011. 52(3): p. 589–601; PMCID: PMC3075862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pak MA, et al. , Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacol, 1994. 33: p. 1049–1053. [DOI] [PubMed] [Google Scholar]
- 72.Huber A, et al. , Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc. Natl. Acad. Sci. USA, 2001. 98(13): p. 7611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang G, Franklin PH, and Murray TF, Manipulation of endogenous adenosine in the rat prepiriform cortex modulates seizure susceptibility. J Pharmacol Exp Ther, 1993. 264(3): p. 1415–24. [PubMed] [Google Scholar]
- 74.Boison D, Adenosinergic signaling in epilepsy. Neuropharmacology, 2015. [DOI] [PMC free article] [PubMed]
- 75.Fedele DE, et al. , Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett, 2004. 370(2–3): p. 160–165. [DOI] [PubMed] [Google Scholar]
- 76.Ren G, et al. , Lentiviral RNAi-induced downregulation of adenosine kinase in human mesenchymal stem cell grafts: a novel perspective for seizure control. Exp Neurol, 2007. 208: p. 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li T, et al. , Human mesenchymal stem cell grafts engineered to release adenosine reduce chronic seizures in a mouse model of CA3-selective epileptogenesis. Epilepsy Res, 2009. 84: p. 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen HY, et al. , Overexpression of adenosine kinase in cortical astrocytes and focal neocortical epilepsy in mice. J Neurosurg, 2014. 120(3): p. 628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boison D, Inhibitory RNA in epilepsy: Research tool and therapeutic perspectives. Epilepsia, 2010. 51(9): p. 1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee Y, et al. , GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia, 2008. 56(5): p. 481–93. [DOI] [PubMed] [Google Scholar]
- 81.Ren G and Boison D, Engineering human mesenchymal stem cells to release adenosine using miRNA technology. Methods Mol Biol, 2010. 650: p. 225–40. [DOI] [PubMed] [Google Scholar]
- 82.Dudek FE and Sutula TP, Epileptogenesis in the dentate gyrus: a critical perspective. Prog Brain Res, 2007. 163: p. 755–73. [DOI] [PubMed] [Google Scholar]
- 83.Kobow K and Blumcke I, The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia, 2011. 52 Suppl 4: p. 15–9. [DOI] [PubMed] [Google Scholar]
- 84.Christman JK, 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene, 2002. 21(35): p. 5483–95. [DOI] [PubMed] [Google Scholar]
- 85.Shen HY, et al. , Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia. J Clin Invest, 2012. 122(7): p. 2567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chiari A, et al. , Preclinical toxicity screening of intrathecal adenosine in rats and dogs. Anesthesiology, 1999. 91(3): p. 824–32. [DOI] [PubMed] [Google Scholar]
- 87.Eisenach JC, Hood DD, and Curry R, Preliminary efficacy assessment of intrathecal injection of an American formulation of adenosine in humans. Anesthesiology, 2002. 96(1): p. 29–34. [DOI] [PubMed] [Google Scholar]
- 88.Eisenach JC, Hood DD, and Curry R, Phase I safety assessment of intrathecal injection of an American formulation of adenosine in humans. Anesthesiology, 2002. 96(1): p. 24–8. [DOI] [PubMed] [Google Scholar]