

Abstract
The immunosuppressive microenvironment is one of the major factors promoting the growth of glioblastoma multiforme (GBM). Infiltration of CD4+CD25+Foxp3+ regulatory T cells (Tregs) into the tumor microenvironment plays a significant role in the suppression of the antitumor immunity and portends a dismal prognosis for patients. Glioma-mediated secretion of chemo-attractant C-C motif ligand 2 and 22 (CCL2/22) has previously been shown by our group to promote Treg migration in-vitro. In this study, we show that a local implantation of platelet rich fibrin patch (PRF-P) into the brain of GL-261 glioma-bearing mice prolonged the survival of affected animals by 42.85% (p=0.0011). Analysis performed on brain tumor tissue harvested from PRF-P-treated mice revealed a specific decrease in intra-tumoral lymphocytes with a preferential depletion of immunosuppressive Tregs. Importantly, co-culture of GL261 or chemo-attractants (CCL2/22) with PRF-P abrogated Treg migration. Pharmacological blockade of the CCL2/22 interaction with their receptors potentiated the inhibitory effect of PRF-P on Tregs recruitment in culture. Moreover, our findings revealed the soluble CD40 ligand (sCD40L) as a major Treg inhibitory player produced by activated platelets entrapped within the fibrin matrix of the PRF-P. Blockade of sCD40L restored the migratory capacity of Tregs, emphasizing the role of PRF-P in preventing the Treg migration to glioma tissue. Our findings highlight autologous PRF-P as a personalized, Treg-selective suppression platform that can potentially supplement and enhance the efficacy of glioma therapies.
Keywords: glioblastoma multiforme, immunosuppressive microenvironment, platelet rich fibrin patch, regulatory T cells and glioma immune-therapies
INTRODUCTION
The highly aggressive and devastating nature of glioblastoma multiforme (GBM) is reflected in short patient survival that ranges from 12 to 20 months despite surgical management, radiation, and chemotherapy [1,2]. One of the major factors contributing to the aggressiveness of these tumors is the suppression of the local immune responses within the glioma microenvironment.
Gliomas are known to recruit immune-regulatory cells such as Tregs, which promote immunosuppressive functions within the cancer’s microenvironment together with the tumor-associated macrophages/microglia (TAMs) [3–5]. The recruitment of these immunosuppressive cells is mediated by the increased presence of chemokine motif C-C ligand 2 and 22 (CCL2 and CCL22) chemo-attractants [6,7]. This accumulation of Tregs around the GBM microenvironment negatively impacts the efficacy of immune therapies including dendritic cell vaccines and immune checkpoint inhibitors [8]. Thus, the effective and safe approaches selectively unmasking the glioma protective shield created by Treg-mediated immune suppression could weaken the tumor and potentiate the effect of these and other therapies [9]. The successful development of Treg targeted therapy can bring current efforts to the next level and prospects a promising pathway leading towards complete eradication of glioma [8]. Although not well investigated, the role of platelets in the modulation of anti-tumor proinflammatory processes can also induce anti-tumor immunity [10]. These cells fine-tune leucocyte effector function, extravasation, differentiation, and cytokine release [4,11]. Intriguingly, many immunomodulatory molecules, such as sCD40L secreted by platelets, have been shown to inhibit the migration of CD40 expressing Tregs [12]. The immunomodulatory properties of CD40 make this molecule particularly attractive in immunotherapeutic discoveries for solid tumors, such as glioma [13,14]. Currently, an agonistic anti-CD40 antibody is being evaluated in phase 1 clinical trials in a variety of cancers in the context of immune stimulation (NCT02376699). Therefore, sCD40L holds high therapeutic potential as it inhibits Treg recruitment and boosts anti-tumor immunity [13,14].
In this study, we generated PRF as patch (PRF-P) to evaluate its effect on glioma. This PRF-P treatment extended survival in a murine model of glioma. Our in vitro analysis shows that PRF-P treatment induces selective apoptosis of glioma but not normal cells. Furthermore, PRF-P can negate the immunosuppressive microenvironment of GBM by blocking the recruitment of Tregs. This blockade is partially mediated by suppressing the expression of Treg migratory signal molecules (CCL2/CCL22) from glioma cells. Importantly, sCD40L secreted by PRF-P play major role on blocking the Treg recruitment.
MATERIALS AND METHODS
Cell culture
The GL-261 glioma cell lines cultured in DMEM, 10% FBS (Hyclone) with penicillin/streptomycin (Invitrogen). For in-vitro assays of proliferation, dye uptake, and cell death, GL-261were plated at density of 5×104 per well in a 24 well plate for all assays. Primary glia cultures were prepared from the cortices of postnatal day 2 C57BL/6 pups [6,15]. Cells were maintained in DMEM/F12 (Gibco) medium supplemented with 1X non-essential amino acids (NEAA, Gibco), 10U/mL of penicillin, 10 μg/mL of streptomycin (Gibco), and 5 mL GlutaMax (Gibco), and plated on poli-L-lysine coated dishes (Sigma). Fourteen days after isolation from (postnatal day) PND 3, mixed cortical glia were used for the co-culture with PRF-P co-cultures).
Isolation and culture of the regulatory T cells
Splenocytes were harvested into single cell suspension from C57BL/6-Foxp3-GFP. The single cell suspension was then lysed for two minutes at RT with ACK lysis buffer. Pre-enrichment of T-cells was performed using the EasySep Pan T-cell isolation kit following the manufacturer’s protocol including CD8 biotin for depletion (Biolegend, San Diego, CA). Cells were then sorted using a BD FACS Aria II cell sorter (Becton Dickinson, Franklin Lakes, NJ) using CD4-APC and GFP+ expression and expanded with Dynabeads T-cell (Invitrogen, Waltham, MA) expander beads per manufacturer’s instructions. Expanded Tregs were used for migration assays as described below.
Flow Cytometry
For flow cytometry analysis of infiltrative myeloid cells, C57/Bl6 were sacrificed 10 days after tumor implantation and 5 days after L-PRF implantation. Single cell suspension of peripheral organs was achieved by homogenizing tissue through a 70μm strainer before antibody staining. Isolation of lymphocytes and myeloid cells was performed homogenizing the tumor and patch, followed by a centrifugation of the tumors using 30%/70% discontinuous Percoll gradient centrifugation at 1000xg for 30 minutes at RT. The interface was collected, and all samples were then stained in 96 well V-bottom plates (Corning, Corning, NY). Cells were washed in PBS and stained with fixable viability dye Efluor 780 (Fisher, NJ). Cells were washed and stained in 2% FBS/PBS, incubated with anti-CD16/32 blocking antibody (Biolegend, San Diego, CA) 1:100 before staining with fluorescence conjugated antibodies. The fluorescence conjugated antibodies used were as follows: anti-CD4, anti-CD45 AmCyan, anti-CD8 BV605, anti-CD11b Pacific Blue, anti-CD11c APC, anti-CD45 PE-Cy7, and anti-CD25 Alexa-Fluor 700, and anti-Foxp3 PE (Fisher, Hampton, NH). All antibodies were purchased from Biolegend except when noted. Flow cytometry analysis was performed using the BD Fortessa (Becton Dickinson, Franklin Lakes, NJ).
The generation of PRF-P
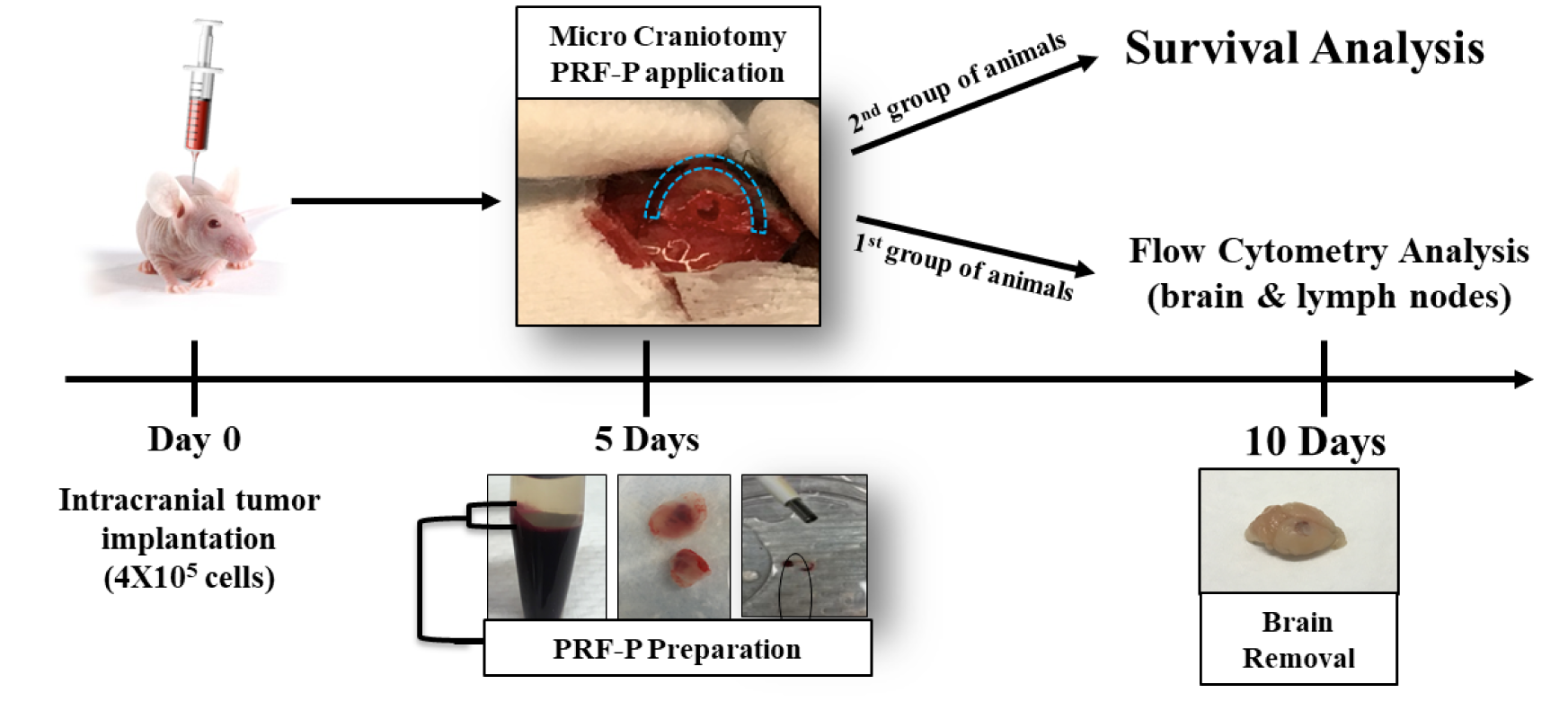
The PRF has been prepared from the blood drawn from C57BL/6 (WT) donor mice five days after GL 261 malignant glioma cell line implantation. The protocol for the preparation of our PRF clots was adapted from previously described technique for Advanced Platelet Rich Fibrin (A-PRF) [16]. Blood samples were taken during the surgery, prior to the craniotomy or prior to performed in vitro assays. 1.5 ml tubes without anti-coagulant were used and were immediately centrifuged at 1,500 rpm for 14 minutes. Obtained PRF clots were placed in sterile petri dishes and compressed to achieve 2 mm thickness. PRF patches were cut using 3 mm in diameter punch biopsy needle. Freshly generated PRF patches (PRF-P) were place over the implanted tumor site in the direct contact with the formed brain tumor achieved by craniotomy and further secured with preserved bone flap.
Migration assays
Tregs were cultured overnight in 5% reduced serum RPMI before being plated at a density of 2.5×105 Tregs/well on the top chamber of 5μm trans well inserts (Corning, Corning, NY). Depending on the group, the bottom wells were seeded with 5×104 GL261 overnight or left empty and co-cultured with or without the addition of PRF-P for 12 hours before introducing top chamber with Tregs (Supp. Fig. 1). To further verify suppressive effects of PRF-P on the ligands for Treg recruitment, 5% FBS RPMI was supplemented with blockades for CC chemokine receptor 4 (CCR4) (R&D Systems, Inc, MN), and C-X-C chemokine receptor type 4 (CXCR4) (R&D Systems, Inc, MN) or anti-sCD40L antibodies (R&D Systems, Inc, MN). For all experimental settings Tregs were allowed to migrate for either 6 hours or overnight before percent migration was assessed. Cells that remained in the top chamber were manually counted to validate migration.
Mice and treatments
C57BL/6 (WT) and C57BL/6-Foxp3-IRES-GFP mice were obtained and bred from Jackson Laboratories, and housed in Northwestern University animal facility at the Center for Comparative Medicine (CCM). GL-261, a malignant glioma cell line that is syngeneic to C57BL/6 mice was administered via intracranial injection using a stereotactic apparatus at a concentration of 4×105 GL-261 cells in 2.5μL PBS to all experimental groups at 6–8 weeks of age as described previously [17]. Depending on the group, craniotomy with bone flap preservation followed by PRF-P application or craniotomy with bone flap preservation with no PRF-P application has been performed (control group) (Supp Fig 2). Group of blood donor mice implanted with 4×105 GL-261 cells in 2.5μL PBS had been used to generate PRF patches. Animal survival was conducted following endpoint protocols outlined in our approved animal protocols.
Statistical analysis.
All statistical analyses were performed using Graphpad Prism 4 (GraphPad Software Inc., San Diego CA). Numerical data was reported as Mean ± SEM. Student’s t test was used for two group comparisons. ANOVA with Tukey’s post hoc test was used for comparisons between multiple groups. Kaplan-Meier survival curve was generated, and log rank test was utilized to compare survival distributions. All reported p values were considered to be statistically significant at * p<0.05, **p<0.01, and ***p<0.001.
RESULTS
Application of PRF-P prolongs the survival of glioma-bearing mice.
GBM induces immunosuppression creating a favorable microenvironment for tumor growth [18,19]. Since PRF-P holds cells and molecules capable of efficiently fine-tuning the immunological milieu [11], we sought to determine if locally applied PRF-P influences on the growth of GBM when applied 5 days after tumor establishment (Supp. Fig. 2). We observed that glioma-bearing animals treated with PRF-P survived significantly longer over the non-treated control mice (p≤0.001). The median survival of glioma-bearing mice treated with patch was improved by 43% (Fig. 1 A). These findings suggest that local application of PRF-P suppresses the GBM progression. In order to investigate if this survival benefit was a direct effect of the PRF-P on the growth of glioma cells, we co-cultured GL261 with PRF-P. We performed a cell viability analysis by staining with crystal violet and found that the viability of GL261 cells was significantly lower after 5 days of exposure to PRF-treatment (Fig. 1 C) while there was no change in the cell death in normal mixed cortical glia cells (Fig. 1D). This specific glioma cell death was due to the modulations of apoptosis related genes as we observed a transcriptional increase for pro-apoptotic death receptor 5 (DR5) and reduction of the anti-apoptotic gene, B-cell lymphoma (BCL2) (Fig. 1E). Therefore, it is plausible to conclude that the decreased cell number was at least in part due to the induction of apoptosis in glioma cells by PRF-P.
Figure 1. PRF-P has anti-glioma activity in mice bearing the GL261 tumors.

4.0×105 GL261 glioma cells were administrated intra-cranially into C57BL/6 mice. Five days later, animals were treated with a PRF-P. a. PRF-P treatment significantly extended the survival of C57BL/6 animals from 14 to 20 days (n≥6). b. PRF-P reduced the viability of the glioma but not normal glial cells after five days of in vitro co-culture (n=4). B) The mRNA of proteins involved in apoptosis, DR5 and BCL2, were deregulated upon the treatment with the PRF-P in vitro (n=4).
PRF-P does not directly contribute to cellular composition of the tumor microenvironment.
It has been also shown that PRF-P is a source of cells and soluble factors capable of modulating the immune landscape [16,11]. Therefore, we asked if the suppression of glioma growth was due to the activity of cellular infiltrates from PRF-P. In order to determine this, we generated the PRF-P from CD45.1 congenic mice and implanted it into CD45.2 glioma-bearing mice. There was a minimal CD45.1 cellular infiltration to the brain parenchyma (Fig. 2 A, B). The majority of CD45+ cells were that of CD45.2+, hence originating from the host animal. Flow cytometry analysis of PRF-P-treated animals did not show any significant differences in myeloid infiltrates or microglia (Fig. 2 C-E). Additionally, there was minimal infiltration present of the CD45.1+CD3+ cells (Fig. 2F, G). These results suggested that cellular infiltrates were entrapped in the patch and not directly interacting with glioma cells. Therefore, soluble factors present within the patch could be responsible for the observed enhanced survival of PRF-P treated glioma-bearing animals.
Figure 2. PRF-P treatment does not contribute to the glioma cellular environment.

a. Sample of the density plot representing brain cellular environment of glioma-bearing mice with and without PRF-P treatment derived from the blood of C57BL/6 congenic CD45.1 mice. b. Quantification of the total CD45.1+ cellularity in brains of glioma-bearing mice (n=5). c. Example of flow cytometry density plot showing gating strategy employed in quantification of: d. myeloid and e. microglia (n=5) immunological compartments. f. and g. Gating strategy used for the analysis of the CD3+ cells of the host and PRF-P origin in brains of glioma-bearing animals. h. Quantification of brain T cell types (CD4 and CD8) derived from the transplanted PRF-P (n=5).
PRF-P treatment causes selective aberration of Treg around GBM microenvironment.
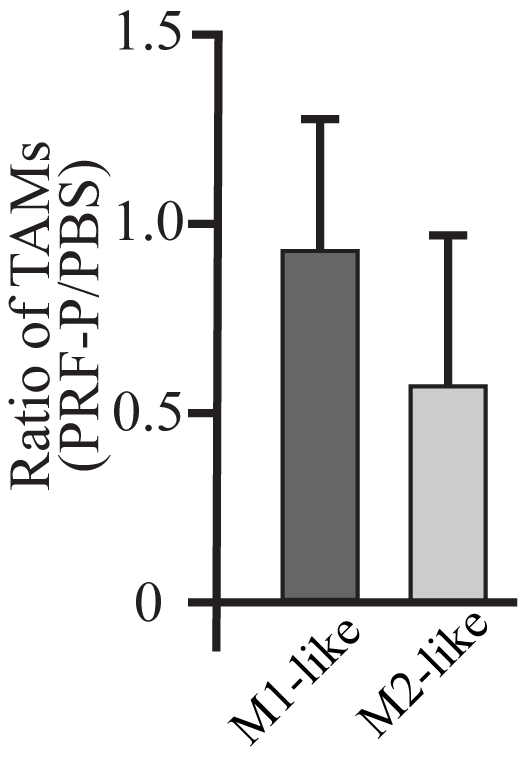
GBM mediated immunosuppression selectively induces accumulation of regulatory T cells and inhibits the infiltrations of pro-inflammatory T cells or M1 polarized macrophages/microglia to evade immune recognition [6,7]. Since PRF-P contains immunomodulatory molecules, we next investigated if PRF-P treatment promotes survival of mice through changing the compositions of immune compartment within the GBM-microenvironment. We detected a specific reduction in the CD45hiCD11b- negative cell compartment, mainly represented by host T lymphocytes (Fig. 2C, F, G). Although there was a reduction of CD8+T cells, the most dramatic was a 5-fold decreased in Treg infiltration into the brain of PRF-P treated animals (Fig. 3A). Importantly, ratios of CD8/Treg and conventional CD4/Treg were significantly increased in PRF-P treated group (Fig. 3A), indicating an increase in anti-tumor T cell mediated immunity. Additionally, the number of Foxp3+ CD4+ Tregs was decreased in the deep cervical lymph nodes, but not in the superficial lymph nodes (Fig 3 B, C). Similarly, an increased in the CD4/Treg ratio was observed in the deep, but not the superficial cervical lymph nodes (Fig 3 B, C). Additionally, we could observe that PRF-P treatment skewed the type of tumor associated macrophages from M2 to M1 (Sup Fig 3). Therefore, the specific reduction of Tregs in the brain and deep cervical lymph nodes but not in the superficial lymph nodes suggests that PRF-P treatment either inhibits the recruitment of these immunosuppressive cells to the tumor sites or causes the death of Tregs.
Figure 3. Anti-glioma immunity after PRF-P treatment.

Analyses of host CD4+, CD8+, and Treg subsets were performed at five days from PRF-P treatment in animals bearing GL261 glioma. Lower numbers of CD8 and Tregs were observed in the a. brain, Tregs only in the b. deep draining lymph nodes (dLN), but not in the c. superficial dLN (n=5). An increased anti-glioma immunity was observed as the ratios of CD4/Tregs and CD8/Tregs was increased in the a. brains and b. deep dLNs of PRF-P treated glioma-bearing animals (n=5).
PRF-P reduces the migration of regulatory T cells toward GL261 glioma cells.
In order to describe the reason behind the observed reduction in the number of Tregs, we measured the migration capacity and cell death in Tregs by co-culturing glioma cells and Tregs, with or without PRF-P (Fig. 4 A). We found that there was a significant reduction in the amount of migrated Tregs when PRF-P was present in the glioma-Treg co-culture within the first 6 hours of the assay (52.2±2.7% vs. 39.4±7.7%) (Fig. 4 B). The reduced number of migrated Tregs was sustained over the 12 h co-cultured period with glioma cells, Tregs, and PRF-P and was 39.4±7.7% and 36.9±3.9% at 6 h. and 12 h., respectively (Fig. 4 B). On the contrary, the number of migrated Tregs increased significantly by 21.6% from 52.2±2.7% at 6 h. to 73.85±3.2% for the 12 h. co-cultured of glioma and Tregs without PRF-P (Fig. 4 B). Next, we investigated if the reduced number of Tregs in the presence of the PRF-P is due to the induction of Treg death. We found that there is no difference in the survival rate of Tregs when cultured in the presence or absence of PRF-P (Fig. 4 C). Our data demonstrates that PRF-P does not affect the viability of Tregs but specifically inhibits the migration of Tregs toward glioma likely by modulating the release of soluble molecules.
Figure 4. PRF-P reduces the migration of regulatory T cells to GL261cells in vitro.

a. Sample histogram of FACS sorted CD4+Foxp3-GFP+ cells used for the in vitro migration assays. b. PRF-P treatment significantly reduced the migration of the Tregs to GL261 after 6 and 12 hours of co-culture (n≥3) without affecting their c. viability (n≥3).
PRF-P treatment inhibits the recruitment of Tregs
It has been shown that glioma cells overexpress CCL2 and CCL22 to recruit immune-regulatory cells [1,7]. In order to determine if PRF-P affects the expression or activity of these molecule to inhibit the recruitment of Tregs, we first checked the mRNA levels of these ligands in glioma cells after co-culturing with PRF-P. Interestingly, the mRNA level of these chemokines produced by glioma cells was significantly increased when co-cultured with Tregs, showing a 3.4±0.6 fold increase in the production of CCL2 and 2.77±0.19 fold increase in the generation of CCL22 mRNAs. However, this increased expression of both mRNAs were diminished in the presence of PRF-P (Fig. 5 A, B), decreasing from 3.4±0.6 to 1±0. 2-fold change for CCL2 mRNA and 2.4±0.02 to 0.76±0.02-fold change for CCL22 mRNA. We the verified that PRF-P blocks the Treg recruiting effects of these chemokines already secreted by GL261 cells. Blockade of the CCR4 receptor with CCL22 resulted in 17.2% reduction of Treg migration. PRF-P alone was capable of blocking Treg recruitment by 23.2%. Upon combining the CCR4 blockade with PRF-P, the rate of the Treg recruitment was severely decreased by 49.6% (Fig. 5 C). These findings suggest that PRF-P regulates the activity of the CCL2/22 chemokines at the transcription level and protein secretion. Since the blockade of CCL2/22 chemotactic properties incompletely abrogated the Treg migration toward glioma, we aimed to determine if the soluble isoform of the CD40 ligand (L) could be yet another immunomodulatory molecule affecting the Treg migration toward glioma cells in the presence of the PRF-P. The soluble isoform of CD40 ligand (sCD40L) produced by PRF-P [20–22] has been shown to reduce the number of Tregs in the mouse model of atherosclerosis [23]. Therefore, we investigated if sCD40L produced by the PRF-P might suppress the Treg movement toward glioma. We co-cultured GL261 and PRF-P in the presence of the CD40L blocking antibodies to investigate if the blocking of CD40L can reverse the inhibitory effect of PRF-P on the Tregs migration. Anti-CD40L antibodies negated the effect of PRF-P on the Tregs migration, normalizing the amount of migrated Tregs (Fig. 5 D).
Figure 5. PRF-P induces microenvironmental changes reducing the Tregs migration toward GL261 cells.

The mRNA level for chemokine (C-C motif) ligand a. 2 and b. 22 (CCL2 and CCL22, respectively) was significantly reduced by the PRF-P treatment in GL261-Treg co-culture (n=3). c. PRF-P caused cessation of the Treg migration after pharmacological blockade of the CCL2/22 interaction with their receptors (n=3). d. Pharmacological inhibition of the soluble CD40 ligand (sCD40L) reversed the PRF-P induced reduction of the Tregs’ migration (n=3).
Therefore, PRF-P directly suppresses the Treg recruitment by modulating the expression of CCL2/22 chemokines by glioma cells and blocking the chemotactic activity of the already present CCL2/22. Furthermore, PRF-P releases the sCD40L to glioma environment effectively inhibiting the migration of regulatory T cells (Fig 6).
Figure 6. Schematic diagram of a potential mechanism how PRF-P affects on glioma cells.

DISCUSSION
In the present work, we show that local PRF-P treatment extends the survival of glioma-bearing mice by both directly inducing the apoptosis of glioma cells and preventing Treg infiltration into the brain of glioma-bearing mice. Mechanistically, PRF-P treatment attenuated the CCL2/22 and CD40-CD40L activity, which are key signaling pathways for Treg recruitment.
We mimicked the aggressiveness of human GBM in our in vivo experimental approach by using an animal model with the median survival of 15 days. Although the PRF-P treatment was applied in a fairly late stage of glioma development at the day 5 after tumor implantation, PRF-P treatment was able to reduce the aggressiveness of this model and prolonged the survival of animals by 43% (6 days). Therefore, our PRF-P treatment approach may offer an immunotherapeutic opportunity for GBM patients.
The fibrin matrix of the PRF-P acts as a scaffold for leucocytes and platelets containing healing, regenerative, and immune modulating molecules [13]. In our study, we observed that the cellularity of the brain in PRF-P treated mice was not different from that of un-treated mice, indicating that the cellular components of the PRF-P were successfully entrapped within the fibrin matrix. Therefore, the PRF-P induced its effects mainly through the secreted molecules produced de novo by cells in the PRF-P.
PRF-P specifically induced apoptosis of glioma cells but not normal cells in co-culture, suggesting that the events orchestrated by the application of the patch might lead to the death of glioma cells in vivo. Concomitantly, we observed an increase in anti-tumor immunity as the ratio of CD8/Treg and conventional CD4/Treg were significantly increased in PRF-P treated mice. Even though the population of the conventional effector and cytotoxic T cells were also adversely affected by the application of PRF-P. Specifically the quantity of Foxp3+ Tregs were significantly diminished. Interestingly, we observed a decrease in Foxp3+ CD4+ cells in the deep cervical lymph nodes but not in the superficial lymph nodes in the animals treated with PRF-P. This suggests the patch causes a decline in the migration of immunosuppressive cells to the tumor rather than just the survival of lymphocytes within the tumor. All together, these findings strongly support our hypothesis that PRF-P exerted its effect via secreted molecules, which reduced the migration of the regulatory T cells to the tumor site.
At the mechanistic level, our analysis showed that PRF-P treatment suppresses the expression of CCL2 and CCL22 from glioma cells. These chemokines are responsible for the recruitment of inhibitory immune cells to the glioma microenvironment [1]. Additionally, as shown in our migration analysis in vitro, PRF-P suppressed the recruiting activity of CCL2/22. However, the reduced expression and inhibition in the efficacy of these molecules only partially inhibited the recruitment of Tregs. Stronger blockade of Treg recruitment was observed with the CD40L blocking antibodies. This is consistent with previous work showing that sCD40L was capable of inhibiting the recruitment of Tregs in a mouse model of arteriosclerosis [12]. Together, our findings indicate that sCD40L secreted from PRF-P is the major factor that inhibits the Treg migration toward glioma along with the suppression of CCL2 and CCL22 expression.
In conclusion, PFR-P treatment suppresses glioma bi-directionally by inducing apoptosis of glioma cells and inhibiting Treg recruitment via suppressing T cell migratory signals (CCL2/CCL22 and CD40/CD40L). Our findings indicate that autologous PFR-P treatment could be a novel personalized immunotherapeutic platform as it can fine-tune the local immune responses in the GBM microenvironment leading to a specific eradication of cancerous cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We would also like to thank Irina Balyasnikova and Atique U. Ahmed for ther comments and suggestions. This work is supported by National Cancer Institute Outstanding Investigator Award from NIH/National Cancer Institute to M.S.L. (R35CA197725) and by a grant from NIH/National Cancer Institute to M.S.L. (R01 NS087990).
REFERENCES
- 1.Stupp R, Dietrich PY, Ostermann Kraljevic S, Pica A, Maillard I, Maeder P, Meuli R, Janzer R, Pizzolato G, Miralbell R, Porchet F, Regli L, de Tribolet N, Mirimanoff RO, Leyvraz S (2002) Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 20 (5):1375–1382. doi: 10.1200/jco.2002.20.5.1375 [DOI] [PubMed] [Google Scholar]
- 2.Pituch KC, Miska J, Krenciute G, Panek WK, Li G, Rodriguez-Cruz T, Wu M, Han Y, Lesniak MS, Gottschalk S, Balyasnikova IV (2018) Adoptive Transfer of IL13Rα2-Specific Chimeric Antigen Receptor T Cells Creates a Pro-inflammatory Environment in Glioblastoma. Mol Ther 26 (4):986–995. doi: 10.1016/j.ymthe.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panek WK, Kane JR, Young JS, Rashidi A, Kim JW, Kanojia D, Lesniak MS (2017) Hitting the nail on the head: combining oncolytic adenovirus-mediated virotherapy and immunomodulation for the treatment of glioma. Oncotarget 8 (51):89391–89405. doi: 10.18632/oncotarget.20810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, Xiao HL, Wang B, Yi L, Wang QL, Jiang XF, Yang L, Zhang P, Qian C, Cui YH, Zhang X, Bian XW (2012) Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. Journal of immunology (Baltimore, Md : 1950) 189 (1):444–453. doi: 10.4049/jimmunol.1103248 [DOI] [PubMed] [Google Scholar]
- 5.Crane CA, Ahn BJ, Han SJ, Parsa AT (2012) Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol 14 (5):584–595. doi: 10.1093/neuonc/nos014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, Kanojia D, Pituch KC, Qiao J, Pytel P, Han Y, Wu M, Zhang L, Horbinski CM, Ahmed AU, Lesniak MS (2016) CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer research 76 (19):5671–5682. doi: 10.1158/0008-5472.Can-16-0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB (2008) Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer immunology, immunotherapy : CII 57 (1):123–131. doi: 10.1007/s00262-007-0336-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, Han Y, Lesniak MS (2014) Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 20 (20):5290–5301. doi: 10.1158/1078-0432.Ccr-14-0514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grauer OM, Sutmuller RP, van Maren W, Jacobs JF, Bennink E, Toonen LW, Nierkens S, Adema GJ (2008) Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. International journal of cancer 122 (8):1794–1802. doi: 10.1002/ijc.23284 [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z (2017) In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nature Biomedical Engineering 1:0011. doi:10.1038/s41551-016-001110.1038/s41551-016-0011https://www.nature.com/articles/s41551-016-0011#supplementary-informationhttps://www.nature.com/articles/s41551-016-0011#supplementary-information [Google Scholar]
- 11.Kral JB, Schrottmaier WC, Salzmann M, Assinger A (2016) Platelet Interaction with Innate Immune Cells. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie 43 (2):78–88. doi: 10.1159/000444807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moura R, Tjwa M (2010) Platelets suppress Treg recruitment. Blood 116 (20):4035–4037. doi: 10.1182/blood-2010-09-303396 [DOI] [PubMed] [Google Scholar]
- 13.Chonan M, Saito R, Shoji T, Shibahara I, Kanamori M, Sonoda Y, Watanabe M, Kikuchi T, Ishii N, Tominaga T (2015) CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol 17 (11):1453–1462. doi: 10.1093/neuonc/nov090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shoji T, Saito R, Chonan M, Shibahara I, Sato A, Kanamori M, Sonoda Y, Kondo T, Ishii N, Tominaga T (2016) Local convection-enhanced delivery of an anti-CD40 agonistic monoclonal antibody induces antitumor effects in mouse glioma models. Neuro Oncol 18 (8):1120–1128. doi: 10.1093/neuonc/now023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moyano AL, Pituch K, Li G, van Breemen R, Mansson JE, Givogri MI (2013) Levels of plasma sulfatides C18 : 0 and C24 : 1 correlate with disease status in relapsing-remitting multiple sclerosis. Journal of neurochemistry 127 (5):600–604. doi: 10.1111/jnc.12341 [DOI] [PubMed] [Google Scholar]
- 16.Ghanaati S, Booms P, Orlowska A, Kubesch A, Lorenz J, Rutkowski J, Landes C, Sader R, Kirkpatrick C, Choukroun J (2014) Advanced platelet-rich fibrin: a new concept for cell-based tissue engineering by means of inflammatory cells. The Journal of oral implantology 40 (6):679–689. doi: 10.1563/aaid-joi-D-14-00138 [DOI] [PubMed] [Google Scholar]
- 17.Miska J, Rashidi A, Chang AL, Muroski ME, Han Y, Zhang L, Lesniak MS (2016) Anti-GITR therapy promotes immunity against malignant glioma in a murine model. Cancer Immunol Immunother 65 (12):1555–1567. doi: 10.1007/s00262-016-1912-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JW, Kane JR, Panek WK, Young JS, Rashidi A, Yu D, Kanojia D, Hasan T, Miska J, Gomez-Lim MA, Ulasov IV, Balyasnikova IV, Ahmed AU, Wainwright DA, Lesniak MS (2018) A Dendritic Cell-Targeted Adenoviral Vector Facilitates Adaptive Immune Response Against Human Glioma Antigen (CMV-IE) and Prolongs Survival in a Human Glioma Tumor Model. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. doi: 10.1007/s13311-018-0650-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JW, Miska J, Young JS, Rashidi A, Kane JR, Panek WK, Kanojia D, Han Y, Balyasnikova IV, Lesniak MS (2017) A Comparative Study of Replication-Incompetent and -Competent Adenoviral Therapy-Mediated Immune Response in a Murine Glioma Model. Molecular therapy oncolytics 5:97–104. doi: 10.1016/j.omto.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phipps RP, Kaufman J, Blumberg N (2001) Platelet derived CD154 (CD40 ligand) and febrile responses to transfusion. Lancet (London, England) 357 (9273):2023–2024 [DOI] [PubMed] [Google Scholar]
- 21.Blumberg N, Gettings KF, Turner C, Heal JM, Phipps RP (2006) An association of soluble CD40 ligand (CD154) with adverse reactions to platelet transfusions. Transfusion 46 (10):1813–1821. doi: 10.1111/j.1537-2995.2006.00979.x [DOI] [PubMed] [Google Scholar]
- 22.Blumberg N, Spinelli SL, Francis CW, Taubman MB, Phipps RP (2009) The platelet as an immune cell-CD40 ligand and transfusion immunomodulation. Immunologic research 45 (2–3):251–260. doi: 10.1007/s12026-009-8106-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winkels H, Meiler S, Lievens D, Engel D, Spitz C, Bürger C, Beckers L, Dandl A, Reim S, Ahmadsei M, Hartwig H, Holdt LM, Hristov M, Megens RTA, Schmitt MM, Biessen EA, Borst J, Faussner A, Weber C, Lutgens E, Gerdes N (2017) CD27 co-stimulation increases the abundance of regulatory T cells and reduces atherosclerosis in hyperlipidaemic mice. Eur Heart J 38 (48):3590–3599. doi: 10.1093/eurheartj/ehx517 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.