

Abstract
Essential to development, primary cilia are microtubule-based cellular organelles that protrude from the surface of cells. Acting as cellular antenna, primary cilia play central roles in transducing or regulating several signaling pathways, including Sonic hedgehog (Shh) and Wnt signaling. Defects in primary cilia contribute to a group of syndromic disorders known as “ciliopathies” and can adversely affect development of the brain and other essential organs, including the kidneys, eyes, and liver. The molecular mechanisms of how defective primary cilia contribute to neurological defects, however, remain poorly understood. In this mini review, we summarize recent advances in understanding of the interactions between primary cilia and signaling pathways essential to cellular homeostasis and brain development.
Keywords: primary cilia, Wnt, MTOR, autophagy, ciliopathy, FMCD
Introduction
Primary cilia are microtubule-based cellular organelles that aid in sensing and transducing environmental signals during development (Goetz and Anderson, 2010): primary cilia transduce or regulate several signaling pathways, including Sonic hedgehog (Shh) and Wnt signaling (Corbit et al., 2005; Rohatgi et al., 2007; Gerdes and Katsanis, 2008; Goetz et al., 2009). This antenna-like structure was first observed using electron microscopy at the lumen of kidney tubules in 1898 and has since been intensively studied (Zimmermann, 1898). Emanating from the apical surface of basal body, the axoneme structure of primary cilium comprises a radial array of nine microtubule doublets lacking a central pair (9+0 structure) (Figure 1A; Davenport and Yoder, 2005). At the base of primary cilium, protein entry and exit are regulated via a transition fiber that anchors the axoneme to the ciliary membrane, compartmentalizing this distinct organelle from the cytosol while remaining continuous with the plasma membrane (Gherman et al., 2006; Marshall, 2008; Reiter et al., 2012).
FIGURE 1.

Primary cilia and signal transduction. (A) Structure of primary cilium. Microtubules extend from the centriole constituting the basal body and form the axoneme. The surrounding membrane is called the ciliary membrane, distinct from other membranes. Unlike motile cilium with a 9+2 structure, primary cilium has a 9+0 structure. Only ciliary proteins are allowed to access the cilium, and the transition zone performs the filtering function. (B) In the absence of Shh, Patched (Ptch) inhibits the translocation of Smoothened (Smo), a membrane G protein-coupled receptor (GPCR)-like protein. Suppressor of Fused (SUFU) in the tip of the cilium represses Gli, causing Gli to be present in an inactive form. In the presence of Shh, Shh binds to Ptch, making it no longer able to suppress Smo. Smo, relieved from suppression, translocates into the cilium and represses SUFU. Gli is then converted to its active form and translocates from the cilium to the nucleus and transcribes target genes. (C) Primary cilia in Wnt signaling. In the absence of Wnt ligand, the β-catenin destruction complex, comprising glycogen synthase kinase 3β (GSK3β), adenomatous polyposis coli (APC), and Axin, phosphorylates β-catenin, leading to its proteasomal degradation, which occurs near the basal body. Wnt signaling is activated by binding extracellular Wnt ligand to the membrane-bounded receptor family, frizzled and low-density lipoprotein receptor-related protein (LRP). Then, the β-catenin destruction complex is destabilized by anchoring Axin to the plasma membrane through Dishevelled (Dvl), which leads to stabilization and nuclear localization of β-catenin for transcriptional activation of target genes under T-cell factor/lymphoid enhancer-binding factor (TCF/LEF) promoters (van Amerongen and Nusse, 2009). Inversin, for which ciliary localization has been reported, mediates proteasomal degradation of Dvl to regulate Wnt signaling.
Leading to defective primary cilia, mutations in genes necessary for ciliogenesis and ciliary structure and function are known to cause a group of human genetic disorders described as “ciliopathies.” To date, 187 mutated genes in 35 known ciliopathies and 241 ciliopathy-associated genes essential to ciliary structure and function that could potentially be causative for ciliopathies have been documented (Reiter and Leroux, 2017). These ciliopathies affect the body’s essential organs, including the kidneys, eyes, brain, liver, and skeleton, during development and tend to share clinical phenotypes (Hildebrandt et al., 2011): for example, as primary cilia are critical to development of the central nervous system (CNS), many ciliopathies, such as Joubert syndrome (JBTS), Meckel syndrome (MKS), and orofaciodigital syndrome (OFD), commonly exhibit neurological defects of CNS malformation, intellectual disability, ataxia, and retina dystrophy (Valente et al., 2014). Here, we summarize our recent understanding of the interactions between primary cilia and several signaling pathways essential for cellular homeostasis and for brain development.
Signaling Pathways and Biological Processes Mediated by Primary Cilia
Shh Signaling
Sonic hedgehog signal transduction is accomplished through binding of Shh to the transmembrane receptor Patched (Ptch). In the absence of Shh, Ptch inhibits the ciliary localization of Smoothened (Smo), a membrane G protein-coupled receptor (GPCR)-like protein. When present, Shh binds to Ptch to alleviate the suppression of Smo, which, in turn, translocates to the primary cilium to repress Suppressor of Fused (SUFU) (Rohatgi et al., 2007; Tukachinsky et al., 2010; Sasai and Briscoe, 2012). This initiates the activation of Gli transcriptional factors (Figure 1B; Alcedo et al., 1996): Gli regulates the transcription of target genes that regulate the Shh signaling (e.g., Ptch1, Gli1), proliferation (e.g., cyclin D1, MYC), and apoptosis (e.g., Bcl-2) (Scales and de Sauvage, 2009). Shh, Ptch, Smo, and Gli are expressed in the developing and adult brain (Echelard et al., 1993; Ekker et al., 1995).
Sonic hedgehog signaling is critical to spatial patterning of the neuroepithelium, cellular identity in CNS, axonal guidance, wiring of the neural network, and neuronal activity. As Shh signaling-related proteins are located within primary cilium, Shh signaling is not properly achieved in mice with defective cilia, resulting in several defects in brain development such as defective neural patterning, cerebellar hypoplasia, and defective hippocampal neurogenesis (reviewed in Ferent and Traiffort, 2015). For example, mice with mutation in Kinesin family member 3A (Kif3a), which is important for ciliogenesis, show reduced Gli1 expression, thereby leading to defects in hippocampal neurogenesis and formation of neural stem cells (Han et al., 2008). Stumpy mutant mice exhibit defective ciliogenesis leading to prenatal hydrocephalus and severe polycystic kidney disease. The Stumpy gene, located on mouse chromosome 7, encodes B9 protein domain 2 (B9D2), which is localized with basal bodies along ciliary axonemes and appears to play a role in ciliogenesis in association with intraflagellar transport proteins (IFTs) (Town et al., 2008). Conditional Stumpy mutant mice (driven by Nestin-Cre) have been found to show abrogated Shh signaling and Gli processing in the hippocampus (Breunig et al., 2008).
Wnt Signaling
Wnt signaling is another developmental signaling pathway mediated by primary cilia (Figure 1C; May-Simera and Kelley, 2012). Wnt signaling can be divided into (1) β-catenin dependent (canonical) and (2) β-catenin independent (non-canonical) signaling (reviewed in Berbari et al., 2009; Oh and Katsanis, 2013). Several studies have shown that perturbation of ciliary genes aberrantly activates canonical Wnt signaling and disrupts non-canonical Wnt signaling (Lin et al., 2003; Cano et al., 2004; Ross et al., 2005; Simons et al., 2005; Gerdes et al., 2007; Corbit et al., 2008). In particular, Inversin, which is localized in cilium, mediates the proteasomal degradation of Dishevelled (Dvl) to regulate both canonical and non-canonical Wnt signaling (Simons et al., 2005). Perturbations in several ciliopathy-related genes have been found to elicit de-regulated canonical Wnt signaling in fibroblasts and in the forebrain (Willaredt et al., 2008; Abdelhamed et al., 2013; Wheway et al., 2013). Also, ciliary ablation in adult-born dentate granule cells in the hippocampus has been shown to aberrantly activate canonical Wnt signaling, concomitant with severe defects in dendritic refinement and synapse formation (Kumamoto et al., 2012). Additionally, researchers have recently demonstrated that defective neuronal ciliogenesis caused by hyperactivating mutation in MTOR elicits abnormal activation of canonical Wnt signaling and inactivation of non-canonical Wnt signaling, resulting in defective neuronal migration due to disrupted neuronal polarization (Park et al., 2018). Notwithstanding, several studies have reported that ciliary dysfunction does not affect Wnt signaling, as evidenced by constant activity of Wnt signaling reporter and expression of Wnt target genes (Huang and Schier, 2009; Ocbina et al., 2009). Interestingly, however, deregulated canonical Wnt signaling via deletion of adenomatous polyposis coli (APC) was found to cause a loss of primary cilia in association with radial progenitor malformation in the neocortex (Nakagawa et al., 2017). In these regards, the interaction between primary cilia and Wnt signaling may be context-dependent, and more thorough studies will be necessary to elucidate it.
MTOR Signaling
MTOR is a serine/threonine kinase essential for protein translation, lipid synthesis, and autophagy, and plays an essential role in neural differentiation, neuronal migration, and synaptic formation, all of which are crucial to brain development. Disruption of MTOR signaling has been documented in numerous pathological conditions, including cancer, neurological disorder, and metabolic disorder (Lipton and Sahin, 2014; Saxton and Sabatini, 2017) Polycystic kidney disease (PKD) patients with inherited mutations in ciliary genes show deregulation of the MTOR signaling pathway (Shillingford et al., 2006; Wahl et al., 2006). In particular, polycystin-1, which is recurrently mutated in PKD, has been shown to inhibit MTOR through interaction with TSC2, a component of the TSC complex that inhibits MTOR (Shillingford et al., 2006). Studies have also identified that, by bending stimulus via fluid flow, primary cilia downregulate AMPK-MTOR signaling, which, in turn, induces autophagy to control cell size via LKB1 localized at primary cilia in the kidneys (Boehlke et al., 2010; Orhon et al., 2016). Recently, research has demonstrated that, during brain development, mice with defective cilia present abnormal increases in MTOR signaling, leading to enlarged apical domains of radial glial cells (RGCs) and subsequent dilatation of brain ventricles (Foerster et al., 2017). Conversely, in TSC1- or TSC2-null fibroblasts, TSC1 and TSC2 have been found to positively regulate ciliogenesis without using the TSC-MTOR signaling axis (i.e., rapamycin-insensitive) (Hartman et al., 2009). Nevertheless, a recent independent study which also performed with TSC1- or TSC2-null fibroblasts reported different ciliary phenotypes marked by a longer ciliary length in TSC1-null cells and a shorter ciliary length in TSC2-null cells, and there was a discrepancy between the two cell types in that only the elongated ciliary phenotype in TSC1-null cells could be rescued by rapamycin treatment (Rosengren et al., 2018). In focal malformations of cortical developments (FMCDs), such as hemimegalencephaly (HME) and focal cortical dysplasia (FCD), which are highly associated with intractable epilepsy and autism-spectrum disorders, brain somatic activating mutations in MTOR eliciting blockage of autophagy have been described as disrupting neuronal ciliogenesis in brain tissues from FMCD patients (Wegiel et al., 2010; Lim and Crino, 2013; Park et al., 2018; Figure 2A,B). Given the complex reciprocal interaction between primary cilia and MTOR signaling, further mechanism studies are yet needed.
FIGURE 2.
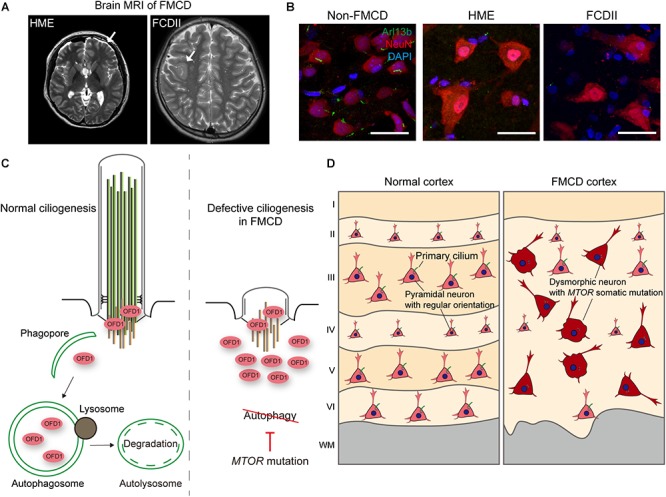
Defective ciliogenesis due to brain somatic mutations in MTOR accounts for cortical dyslamination in FMCDs. (A) Representative brain MRIs of patients with FMCDs, including hemimegalencephaly (HME) and focal cortical dysplasia (FCD) type II. Arrows indicate the affected region of the brain. Adapted from Park et al. (2018), with permission. (B) Immunostaining for Arl13b, a marker for primary cilia, and NeuN, a marker for neurons, with DAPI co-staining in brain tissue from FMCD patients. While primary cilium normally forms at each neuron in brain tissue from non-FMCD, FMCD patients with brain somatic mutations in MTOR exhibit defective neuronal ciliogenesis. Scale bars, 30 μm. Adapted from Park et al. (2018), with permission. (C) Autophagic degradation of the centriolar satellite pool of OFD1 induces primary ciliogenesis. However, brain somatic activating mutation in MTOR, which blocks autophagy, disrupts ciliogenesis in brain tissues with FMCDs. (D) Cortical dyslamination with ciliary defective dysmorphic neurons in the cerebral cortex of a FMCD patient. Somatic activating mutations in MTOR, causative for FMCDs, disrupt neuronal ciliogenesis through blockage of autophagy, resulting in cortical dyslamination.
Autophagy
Autophagy is a cellular degradative process by which a cell gains nutrients to maintain homeostasis (Mizushima, 2007). Two independent studies have provided evidence of links between ciliogenesis and autophagy: In serum-nutrient conditions, basal autophagy degrades the ciliary protein IFT20, while OFD1 protein at centriolar satellites inhibits ciliogenesis. Upon serum-withdrawal, the centriolar satellite pool of OFD1 is degraded by inducible autophagy, while IFT20 initiates ciliary trafficking, leading to ciliogenesis (Pampliega et al., 2013; Tang et al., 2013). Cilia-mediated Shh signaling has been found to assemble several proteins needed for autophagy at the periciliary region, thus activating Shh signaling induces autophagy flux. Interestingly, one study described defects in serum-withdrawal-induced autophagy and biogenesis of autophagosome among Ift20- or Ift88-compromised cells (Pampliega et al., 2013). In line with this, others have found that treatment of Shh to cultured-hippocampal neurons upregulates several autophagy-related genes and enhances autophagy (Petralia et al., 2013).
Meanwhile, dysregulated ciliogenesis in relation to defective autophagy has been described in several diseases. Previous studies have reported suppressed autophagic flux in ciliary dysfunctional PKD mouse models and impairment of autophagosome formation in cells derived from PKD patients (Belibi et al., 2011; Zhu et al., 2017). Interestingly, Hürthle cells found in thyroid cancer were found to show defective ciliogenesis, and this defect in Hürthle cells eliciting high basal autophagic flux was restored by autophagy inhibition (Lee et al., 2016). Dysregulated ciliogenesis has also been reported in Huntington’s disease (HD), for which defective autophagy has been well recognized (Keryer et al., 2011; Kaliszewski et al., 2015). Although Huntingtin, which is mutated in HD, is known to regulate autophagy, it also participates in trafficking pericentriolar material 1 (PCM1) to the centrosome. When Huntingtin is mutated, aberrant accumulation of PCM1 occurs, which, in turn, causes dysregulated ciliogenesis (Keryer et al., 2011; Rui et al., 2015). Further studies, however, are required to elucidate the direct interaction between ciliary abnormality and defective autophagy in HD. Finally, as stated in the previous section on MTOR signaling, defective autophagy and resultant disruption of ciliogenesis have been demonstrated in FMCDs (Park et al., 2018). Brain somatic activating mutations in MTOR, which are causative for FMCDs, block autophagy, resulting in defective neuronal ciliogenesis due to aberrant accumulation of OFD1 (Figure 2C; Park et al., 2018). Although defective autophagy has been implicated in many neurodevelopmental and neurodegenerative disorders, much more studies are needed to elucidate the pathophysiological role of primary cilia in these disorders (Levine and Kroemer, 2008; Lee et al., 2013; Marsh and Dragich, 2018).
Developmental Functions of Neuronal Cilia
Patterning and Morphogenesis of the Forebrain
One of the well-established developmental functions of primary cilia is to control forebrain patterning, which is severely defected in human ciliopathies (Vogel et al., 2012). Patterning in the neural tube of the CNS occurs through progressive subdivisions along the dorsal to ventral and the rostral to caudal axes (Dessaud et al., 2008). Cobblestone mice (Ift88cbs/cbs), which show severe disorganization of the telencephalon, have been found to exhibit an ambiguous dorsal-ventral forebrain boundary and dorsal telencephalic-diencephalic boundary, concomitant with increased levels of full-length Gli3 (Willaredt et al., 2008). Meanwhile, mice lacking primary cilia as a result of mutation in the ciliopathy gene Rpgrip1l exhibited defective phenotypes associated with MKS and JBTS. In these mice, a mislocalized olfactory bulb-like structure and ambiguous dorsal-ventral patterning were observed, both of which were rescued upon introduction of constitutively active repressor form of Gli3 (Besse et al., 2011). Other mice with defective cilia through loss of Ttc21b (encoding retrograde Ift139) have also been shown to display defects in dorsal–ventral patterning and rostral–caudal patterning. Researchers have reported that Ttc21b-null mice exhibit normal anterograde transport that permits entry of Shh signaling components into cilium but partially defective retrograde transport. Overactivation of GLI2 and GLI3A in these mice could accounts for activation of Shh signaling and abnormal patterning, suggesting a periciliary role for retrograde IFT in GLI3 processing and GLI2 activity (Huangfu and Anderson, 2005; May et al., 2005; Tran et al., 2008; Stottmann et al., 2009). Crossing Ttc21b-null mice with mice heterozygous for a null allele of Shh to reduce levels of Shh ligand was found to partially rescue their defective phenotypes (Tran et al., 2008; Stottmann et al., 2009). This paradoxical effect of retrograde IFT mutants on Shh signaling (i.e., activation of Shh signaling) was well reviewed by Pal and Mukhopadhyay (2015) and Bangs and Anderson (2017). In line with the importance of primary cilia in cortical patterning, mice with mutant Slb (encoding anterograde Ift172) that possess very short axonemes with no visible microtubules in their cilia have also been found to exhibit a global brain-patterning defect along the dorsal–ventral and rostral–caudal axes (Gorivodsky et al., 2009). Collectively, these studies suggest the importance of Gli3 processing, which could be mediated by primary cilia, in forebrain patterning.
The establishment of polarized radial glial scaffold formation during cortical development has been shown to be regulated by Arl13b, a small GTPase enriched in primary cilia (Higginbotham et al., 2013). Arl13b mutant mice (Arl13bhnn/hnn), which display defective cilia, surprisingly exhibit dramatic inversion of the apicobasal polarity of the radial glial scaffold, which, in turn, changes the location of the progenitor zone to the outer part of the cortex. This defect is also observed when Arl13b is removed just before the formation of radial glia from neuroepithelial cells using Foxg1-driven Cre. However, Arl13b removal after establishment of the radial glial scaffold using Nestin- or GFAP-driven Cre has not been found to generate this intriguing phenotype, suggesting that the role of Arl13b in polarization is temporally restricted to the period during which the radial glial scaffold develops (Higginbotham et al., 2013). This reversed apicobasal polarity seen in Arl13b mutant mice has not been reported in mice deficient of other ciliary genes. In line with this temporally restricted role of a ciliary gene during development, a recent study performed conditional knockout (KO) of Kif3a, Ift88, and Ttc21b in a series of specific spatiotemporal domains. Intriguingly, the observed neurological defects, including forebrain expansion, cortical malformations, impaired olfactory bulb development, and ventriculomegaly, were significantly different across four different Cre transgenic alleles (Foxg1-Cre, Emx1-Cre, Wnt1-Cre, and AP2-Cre), indicating that the roles of cilia in cortical development are discretely spatiotemporal. For example, the deletion of Ift88 at earlier stages of development, driven by Foxg1-Cre, was found to increase brain size; however, when deleted at later stages, driven by Emx1-Cre, there were no significant differences in brain size (Snedeker et al., 2017). Taken together, these experimental studies of perturbations of primary cilia in mice support the crucial role of primary cilia in forebrain development, which, when disrupted, lead to severe morphological malformations in the brain.
Neuronal Migration
During brain cortical development, newly born neurons follow one of two main migratory routes to reach their final destination, known as radial and tangential migration (Hatten, 1999). The role of primary cilia in radially migrating pyramidal neurons remains unclear. In a previous study, cerebral cortical lamination was not apparently affected in Arl13b-conditional KO mice driven by Nex-Cre or in Stumpy-, Kif3a-, and Ift88-conditional KO mice driven by Nestin-Cre (Arellano et al., 2012; Higginbotham et al., 2012; Tong et al., 2014). However, a recent in vivo systemic study revealed that genes linked to ciliopathies affect cortical development, including neuronal radial migration, neural progenitor development, neuronal differentiation, and early neuronal connectivity (Guo et al., 2015). In this study, knockdown of 17 ciliopathy genes in the developing neocortex resulted in delayed neuronal radial migration in association with transient multipolar stage, multipolar-to-bipolar transition, and glial-guided radial migration (Guo et al., 2015). Meanwhile, others have suggested that the defective phenotypes seen in ciliary conditional KO mice driven by Nestin-Cre, the expression of which is broad and is also seen in non-RGCs at later developmental stages, may be partly attributable to non-cell-autonomous effects (Foerster et al., 2017; Chen et al., 2018). Accordingly, the cell-autonomous function of primary cilia in neural stem cells at the late developmental stage was investigated via acute knockdown of ciliary genes (Kif3a or Ift88) in the developing cortex, which resulted in defective neuronal differentiation and migration; this was accompanied by delays in neural stem cell cycle progression and failures in interkinetic nuclear migration (Chen et al., 2018). In FMCDs, the molecular mechanism of how somatic mutations in MTOR lead to focal cortical dyslamination appears to be related to defective neuronal ciliogenesis by MTOR mutation, which affects the multipolar to bipolar transition essential for neuronal radial migration (Figure 2C,D; Park et al., 2018).
Interneurons originating from the medial ganglionic eminence migrate along a tangential path in the cortical plate, either at the marginal zone or intermediate zone. These neurons change their orientation from a tangential to radial path to colonize the cortical plate and to differentiate into GABAergic neurons (Guo and Anton, 2014). Recent studies have demonstrated that disruption of primary cilia at interneurons affects the interneuronal migratory process, suggesting the importance of primary cilia in neuronal migration (Baudoin et al., 2012; Higginbotham et al., 2012). In a previous study, mice with conditional KO of Arl13b genes driven by Dlx5/6-Cre showed disruption of interneuron placement at Arl13b-deleted cortices caused by defects in interneuron migration and branching. However, when the Arl13b gene was disrupted in post-migratory interneurons driven by PV-Cre, there was no significant difference in the morphology of dendritic arbors during neuronal differentiation. Such defective interneuron migration may be a potential pathophysiological mechanism underlying neurological defects in JBTS with Arl13b mutations (Higginbotham et al., 2012). Meanwhile, a recent study showed that disruption of cilia in interneurons using Nkx2.1-Cre to ablate Ift88 or Kif3a genes led to defective migration of interneurons, marked by an aberrant accumulation of interneurons at their first tangential path, not radial streams in the cortical plate, as well as abnormal positioning and density of interneurons in the postnatal cortex (Baudoin et al., 2012). Taken together, these studies emphasize that primary cilia in immature interneurons are crucial to interneuron migration in the developing cortex and the underlying mechanisms thereof (Baudoin et al., 2012). Nevertheless, the role of primary cilia in neuronal migration needs further study, especially in regards to which other cilia-mediated signaling pathways are involved. These future studies on neuronal migration should shed light on the pathophysiology of ciliopathies presenting with cognitive deficits, which may be attributable to defective neuronal migration and disrupted neural connectivity.
Cerebellum Development
Ataxia due to cerebellar malformation is one major symptom of ciliopathies, particularly JBTS. During development of the cerebellum, Purkinje cells secrete Shh, which regulates the proliferation of cerebellar neuronal precursor (Wechsler-Reya and Scott, 1999; Haldipur et al., 2012): Shh signaling mediated by primary cilia occurs in late embryonic stages of cerebellum development, during which Shh is required for expansion of the granule neuron precursor population, but not for the subsequent differentiation of these cells (Lewis et al., 2004). Primary cilia control cerebellar morphogenesis by promoting the expansion of the granule progenitor pool. Loss of either Ift88 or Kif3a causes severe cerebellar hypoplasia and inhibits the expansion of the granule cell progenitor population. Although Ift88 and Kif3a are not required for the specification and differentiation of cerebellar cell types, they affect granular cell progenitor proliferation. Researches have shown that the expression of Gli1 at the external granule cell layer in Kif3a mutant mice is lower than that in controls (Chizhikov et al., 2007; Spassky et al., 2008). These studies demonstrate that ablation of primary cilia impairs Shh signaling, which regulates the development of the cerebellum, particularly expansion of cerebellar progenitors. This mechanism could explain the hypoplasia of the cerebellum and cerebellar ataxia seen in patients with ciliopathies such as JBTS.
Learning and Memory
Patients with ciliopathies commonly present with intellectual disability (ID) as a neurological deficit. Although there are not enough studies to explain the mechanism underlying ID as a result of defects in primary cilia, several studies have shown that depletion of primary cilia leads to hippocampal-dependent learning and memory deficits (Breunig et al., 2008; Han et al., 2008; Amador-Arjona et al., 2011). Hippocampal neurogenesis plays a role in fear conditioning, recognition, spatial memory, and pattern separation (Cameron and Glover, 2015). Also, Shh signaling mediated by primary cilia controls the proliferation of neural progenitor cells (NPCs). As evidence thereof, Emx1-Cre-dependent Shh-conditional KO mice were found to show smaller dorsal telencephalons at E18.5 and decreased proliferation and increased apoptosis of neural stem cells (NSCs) and NPCs in the dorsal pallium (Komada et al., 2008). Meanwhile, Nestin-Cre- and hGFAP-Cre-dependent Smo KO mice exhibited smaller postnatal dentate gyruses and fewer proliferating cells than those in controls (Machold et al., 2003; Han et al., 2008). In Kif3a mutant and Ift88 mutant mice, neuronal proliferation in the subgranular zone (SGZ) and subventricular zone (SVZ) was decreased, and Gli1 expression was abolished (Han et al., 2008; Tong et al., 2014; Gazea et al., 2016) These demonstrate that Shh signaling is involved in the proliferation and survival of NSCs and NPCs. Hypomorphic Ift88-mutant mice, in which Ift88 mRNA and protein are reduced by 70–80%, showed disorganization of the midbrain dopominergic neuron (mDA) progenitors domain, and the number of mDA neurons was severly reduced (Gazea et al., 2016). Considering that primary cilia regulate neurogenesis by mediating Shh signaling, defects in learning and memory and ID might be caused by decreased neurogenesis due to aberrant Shh signaling. Expanding on the above research, additional studies are needed to determine how defective neuronal cilia lead to learning and memory impairment and ID.
Concluding Remarks
In this review, we briefly summarized the interaction between primary cilia and several signaling pathways, including Shh, Wnt, MTOR, and autophagy, all of which are essential to cellular homeostasis. Given that neurological disorders are typical symptoms of ciliopathies, the importance of primary cilia during brain development cannot be emphasized enough. At present, however, we are unable to fully understand the underlying mechanisms by which defective neuronal cilia contribute the pathogenesis of neurological defects in ciliopathies. Future studies of neuronal cilia in relation to signaling pathways may provide better understanding of the functions of neuronal cilia and may help to develop novel treatments for ciliopathies by targeting neuronal cilia-mediated signaling pathways. Compared with considerable studies on neuronal cilia, there is a lack of research on the role of primary cilia in glial cells, another major component of the brain. Glial cells, such astrocytes and oligodendrocytes, also possess a single primary cilium (Berbari et al., 2007; Bishop et al., 2007; Kasahara et al., 2014). Recently, studies reported defective ciliogenesis in glioblastoma (GBM), a high-grade astrocytic malignancy, and demonstrated the potential role of primary cilia on GBM progression (Moser et al., 2014; Hoang-Minh et al., 2016a,b; Loskutov et al., 2018). Further studies on the role of primary cilia in glial cells, as well as neurons, will advance our understanding of the role of primary cilia in brain development.
To help achieve a better understanding of human ciliopathies resulting from mutations in ciliary genes, CRISPR/Cas9 genome editing of identified mutations in human ciliopathies or 3D culture of brain organoids derived from pluripotent stem cells from ciliopathy patients can be used to generate more pathophysiologically relevant models for ciliopathy, thereby overcoming the gap between animal models and human subjects. Also, increasing studies on interactions between primary cilia and other signaling pathways may uncover a previously unknown role for cilia in the pathogenesis of human neurological diseases that are not considered classical ciliopathies, such as FMCD, brain cancer (medulloblastoma and GBM), and neurodegenerative diseases (Alzheimer’s disease and HD) (Sotthibundhu et al., 2009; Keryer et al., 2011; Eguether and Hahne, 2018). Future studies of these disorders may help us to further the molecular basis of human diseases characterized by defects in primary cilia.
Author Contributions
SP, HJ, and JL conceived the idea and wrote the manuscript.
Conflict of Interest Statement
JL is a co-founder of SoVarGen, Inc. that develops new diagnostics and therapeutics for brain disorders. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by grants from the Suh Kyungbae Foundation (to JL); Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (H15C3143 and H16C0415 to JL).
References
- Abdelhamed Z. A., Wheway G., Szymanska K., Natarajan S., Toomes C., Inglehearn C., et al. (2013). Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum. Mol. Genet. 22 1358–1372. 10.1093/hmg/dds546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcedo J., Ayzenzon M., Von Ohlen T., Noll M., Hooper J. E. (1996). The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 86 221–232. 10.1016/s0092-8674(00)80094-x [DOI] [PubMed] [Google Scholar]
- Amador-Arjona A., Elliott J., Miller A., Ginbey A., Pazour G. J., Enikolopov G., et al. (2011). Primary cilia regulate proliferation of amplifying progenitors in adult hippocampus: implications for learning and memory. J. Neurosci. 31 9933–9944. 10.1523/jneurosci.1062-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arellano J. I., Guadiana S. M., Breunig J. J., Rakic P., Sarkisian M. R. (2012). Development and distribution of neuronal cilia in mouse neocortex. J. Compar. Neurol. 520 848–873. 10.1002/cne.22793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs F., Anderson K. V. (2017). Primary cilia and mammalian hedgehog signaling. Cold Spring Harb. Perspect. Biol. 9:a028175. 10.1101/cshperspect.a028175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudoin J. P., Viou L., Launay P. S., Luccardini C., Espeso Gil S., Kiyasova V., et al. (2012). Tangentially migrating neurons assemble a primary cilium that promotes their reorientation to the cortical plate. Neuron 76 1108–1122. 10.1016/j.neuron.2012.10.027 [DOI] [PubMed] [Google Scholar]
- Belibi F., Zafar I., Ravichandran K., Segvic A. B., Jani A., Ljubanovic D. G., et al. (2011). Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol. Renal Physiol. 300 F1235–F1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari N. F., Bishop G. A., Askwith C. C., Lewis J. S., Mykytyn K. (2007). Hippocampal neurons possess primary cilia in culture. J. Neurosci. Res. 85 1095–1100. 10.1002/jnr.21209 [DOI] [PubMed] [Google Scholar]
- Berbari N. F., O’connor A. K., Haycraft C. J., Yoder B. K. (2009). The primary cilium as a complex signaling center. Curr. Biol. 19 R526–R535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse L., Neti M., Anselme I., Gerhardt C., Ruther U., Laclef C., et al. (2011). Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development 138 2079–2088. 10.1242/dev.059808 [DOI] [PubMed] [Google Scholar]
- Bishop G. A., Berbari N. F., Lewis J., Mykytyn K. (2007). Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J. Compar. Neurol. 505 562–571. 10.1002/cne.21510 [DOI] [PubMed] [Google Scholar]
- Boehlke C., Kotsis F., Patel V., Braeg S., Voelker H., Bredt S., et al. (2010). Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 12 1115–1122. 10.1038/ncb2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig J. J., Sarkisian M. R., Arellano J. I., Morozov Y. M., Ayoub A. E., Sojitra S., et al. (2008). Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc. Natl. Acad. Sci. U.S.A. 105 13127–13132. 10.1073/pnas.0804558105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron H. A., Glover L. R. (2015). Adult neurogenesis: beyond learning and memory. Annu. Rev. Psychol. 66 53–81. 10.1146/annurev-psych-010814-015006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano D. A., Murcia N. S., Pazour G. J., Hebrok M. (2004). Orpk mouse model of polycystic kidney disease reveals essential role of primary cilia in pancreatic tissue organization. Development 131 3457–3467. 10.1242/dev.01189 [DOI] [PubMed] [Google Scholar]
- Chen J. L., Chang C. H., Tsai J. W. (2018). Gli2 rescues delays in brain development induced by Kif3a dysfunction. Cereb. Cortex 29 751–764. 10.1093/cercor/bhx356 [DOI] [PubMed] [Google Scholar]
- Chizhikov V. V., Davenport J., Zhang Q., Shih E. K., Cabello O. A., Fuchs J. L., et al. (2007). Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J. Neurosci. 27 9780–9789. 10.1523/jneurosci.5586-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., Reiter J. F. (2005). Vertebrate smoothened functions at the primary cilium. Nature 437 1018–1021. 10.1038/nature04117 [DOI] [PubMed] [Google Scholar]
- Corbit K. C., Shyer A. E., Dowdle W. E., Gaulden J., Singla V., Chen M. H., et al. (2008). Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 10 70–76. 10.1038/ncb1670 [DOI] [PubMed] [Google Scholar]
- Davenport J. R., Yoder B. K. (2005). An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am. J. Physiol. Renal Physiol. 289 F1159–F1169. [DOI] [PubMed] [Google Scholar]
- Dessaud E., Mcmahon A. P., Briscoe J. (2008). Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development 135 2489–2503. 10.1242/dev.009324 [DOI] [PubMed] [Google Scholar]
- Echelard Y., Epstein D. J., St-Jacques B., Shen L., Mohler J., Mcmahon J. A., et al. (1993). Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75 1417–1430. 10.1016/0092-8674(93)90627-3 [DOI] [PubMed] [Google Scholar]
- Eguether T., Hahne M. (2018). Mixed signals from the cell’s antennae: primary cilia in cancer. EMBO Rep. 19:e46589. 10.15252/embr.201846589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekker S. C., Ungar A. R., Greenstein P., Von Kessler D. P., Porter J. A., Moon R. T., et al. (1995). Patterning activities of vertebrate hedgehog proteins in the developing eye and brain. Curr. Biol. 5 944–955. 10.1016/s0960-9822(95)00185-0 [DOI] [PubMed] [Google Scholar]
- Ferent J., Traiffort E. (2015). Hedgehog: multiple paths for multiple roles in shaping the brain and spinal cord. Neuroscientist 21 356–371. 10.1177/1073858414531457 [DOI] [PubMed] [Google Scholar]
- Foerster P., Daclin M., Asm S., Faucourt M., Boletta A., Genovesio A., et al. (2017). mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development 144 201–210. 10.1242/dev.138271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazea M., Tasouri E., Tolve M., Bosch V., Kabanova A., Gojak C., et al. (2016). Primary cilia are critical for Sonic hedgehog-mediated dopaminergic neurogenesis in the embryonic midbrain. Dev. Biol. 409 55–71. 10.1016/j.ydbio.2015.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes J. M., Katsanis N. (2008). Ciliary function and Wnt signal modulation. Curr. Top. Dev. Biol. 85 175–195. 10.1016/s0070-2153(08)00807-7 [DOI] [PubMed] [Google Scholar]
- Gerdes J. M., Liu Y., Zaghloul N. A., Leitch C. C., Lawson S. S., Kato M., et al. (2007). Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39 1350–1360. 10.1038/ng.2007.12 [DOI] [PubMed] [Google Scholar]
- Gherman A., Davis E. E., Katsanis N. (2006). The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 38 961–962. 10.1038/ng0906-961 [DOI] [PubMed] [Google Scholar]
- Goetz S. C., Anderson K. V. (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11 331–344. 10.1038/nrg2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz S. C., Ocbina P. J., Anderson K. V. (2009). The primary cilium as a Hedgehog signal transduction machine. Methods Cell Biol. 94 199–222. 10.1016/s0091-679x(08)94010-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorivodsky M., Mukhopadhyay M., Wilsch-Braeuninger M., Phillips M., Teufel A., Kim C., et al. (2009). Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev. Biol. 325 24–32. 10.1016/j.ydbio.2008.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Anton E. S. (2014). Decision making during interneuron migration in the developing cerebral cortex. Trends Cell Biol. 24 342–351. 10.1016/j.tcb.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Higginbotham H., Li J., Nichols J., Hirt J., Ghukasyan V., et al. (2015). Developmental disruptions underlying brain abnormalities in ciliopathies. Nat. Commun. 6:7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldipur P., Bharti U., Govindan S., Sarkar C., Iyengar S., Gressens P., et al. (2012). Expression of Sonic hedgehog during cell proliferation in the human cerebellum. Stem Cells Dev. 21 1059–1068. 10.1089/scd.2011.0206 [DOI] [PubMed] [Google Scholar]
- Han Y. G., Spassky N., Romaguera-Ros M., Garcia-Verdugo J. M., Aguilar A., Schneider-Maunoury S., et al. (2008). Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci. 11 277–284. 10.1038/nn2059 [DOI] [PubMed] [Google Scholar]
- Hartman T. R., Liu D., Zilfou J. T., Robb V., Morrison T., Watnick T., et al. (2009). The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 18 151–163. 10.1093/hmg/ddn325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten M. E. (1999). Central nervous system neuronal migration. Annu. Rev. Neurosci. 22 511–539. 10.1146/annurev.neuro.22.1.511 [DOI] [PubMed] [Google Scholar]
- Higginbotham H., Eom T. Y., Mariani L. E., Bachleda A., Hirt J., Gukassyan V., et al. (2012). Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex. Dev. Cell 23 925–938. 10.1016/j.devcel.2012.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H., Guo J., Yokota Y., Umberger N. L., Su C. Y., Li J., et al. (2013). Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat. Neurosci. 16 1000–1007. 10.1038/nn.3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F., Benzing T., Katsanis N. (2011). Ciliopathies. N. Engl. J. Med. 364 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang-Minh L. B., Deleyrolle L. P., Nakamura N. S., Parker A. K., Martuscello R. T., Reynolds B. A., et al. (2016a). PCM1 depletion inhibits glioblastoma cell ciliogenesis and increases cell death and sensitivity to temozolomide. Transl. Oncol. 9 392–402. 10.1016/j.tranon.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang-Minh L. B., Deleyrolle L. P., Siebzehnrubl D., Ugartemendia G., Futch H., Griffith B., et al. (2016b). Disruption of KIF3A in patient-derived glioblastoma cells: effects on ciliogenesis, hedgehog sensitivity, and tumorigenesis. Oncotarget 7 7029–7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P., Schier A. F. (2009). Dampened Hedgehog signaling but normal Wnt signaling in zebrafish without cilia. Development 136 3089–3098. 10.1242/dev.041343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D., Anderson K. V. (2005). Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. U.S.A. 102 11325–11330. 10.1073/pnas.0505328102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliszewski M., Knott A. B., Bossy-Wetzel E. (2015). Primary cilia and autophagic dysfunction in Huntington’s disease. Cell Death Differ. 22 1413–1424. 10.1038/cdd.2015.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara K., Miyoshi K., Murakami S., Miyazaki I., Asanuma M. (2014). Visualization of astrocytic primary cilia in the mouse brain by immunofluorescent analysis using the cilia marker Arl13b. Acta Med. Okayama 68 317–322. [DOI] [PubMed] [Google Scholar]
- Keryer G., Pineda J. R., Liot G., Kim J., Dietrich P., Benstaali C., et al. (2011). Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J. Clin. Invest. 121 4372–4382. 10.1172/jci57552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada M., Saitsu H., Kinboshi M., Miura T., Shiota K., Ishibashi M. (2008). Hedgehog signaling is involved in development of the neocortex. Development 135 2717–2727. 10.1242/dev.015891 [DOI] [PubMed] [Google Scholar]
- Kumamoto N., Gu Y., Wang J., Janoschka S., Takemaru K., Levine J., et al. (2012). A role for primary cilia in glutamatergic synaptic integration of adult-born neurons. Nat. Neurosci. 15:S391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Yi S., Kang Y. E., Chang J. Y., Kim J. T., Sul H. J., et al. (2016). Defective ciliogenesis in thyroid hurthle cell tumors is associated with increased autophagy. Oncotarget 7 79117–79130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. M., Hwang S. K., Lee J. A. (2013). Neuronal autophagy and neurodevelopmental disorders. Exp. Neurobiol. 22 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Kroemer G. (2008). Autophagy in the pathogenesis of disease. Cell 132 27–42. 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis P. M., Gritli-Linde A., Smeyne R., Kottmann A., Mcmahon A. P. (2004). Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev. Biol. 270 393–410. 10.1016/j.ydbio.2004.03.007 [DOI] [PubMed] [Google Scholar]
- Lim K. C., Crino P. B. (2013). Focal malformations of cortical development: new vistas for molecular pathogenesis. Neuroscience 252 262–276. 10.1016/j.neuroscience.2013.07.037 [DOI] [PubMed] [Google Scholar]
- Lin F., Hiesberger T., Cordes K., Sinclair A. M., Goldstein L. S., Somlo S., et al. (2003). Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. U.S.A. 100 5286–5291. 10.1073/pnas.0836980100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton J. O., Sahin M. (2014). The neurology of mTOR. Neuron 84 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loskutov Y. V., Griffin C. L., Marinak K. M., Bobko A., Margaryan N. V., Geldenhuys W. J., et al. (2018). LPA signaling is regulated through the primary cilium: a novel target in glioblastoma. Oncogene 37 1457–1471. 10.1038/s41388-017-0049-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold R., Hayashi S., Rutlin M., Muzumdar M. D., Nery S., Corbin J. G., et al. (2003). Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron 39 937–950. 10.1016/s0896-6273(03)00561-0 [DOI] [PubMed] [Google Scholar]
- Marsh D., Dragich J. M. (2018). Autophagy in mammalian neurodevelopment and implications for childhood neurological disorders. Neurosci. Lett. 697 29–33. 10.1016/j.neulet.2018.04.017 [DOI] [PubMed] [Google Scholar]
- Marshall W. F. (2008). Basal bodies platforms for building cilia. Curr. Top. Dev. Biol. 85 1–22. [DOI] [PubMed] [Google Scholar]
- May S. R., Ashique A. M., Karlen M., Wang B., Shen Y., Zarbalis K., et al. (2005). Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 287 378–389. 10.1016/j.ydbio.2005.08.050 [DOI] [PubMed] [Google Scholar]
- May-Simera H. L., Kelley M. W. (2012). Cilia, Wnt signaling, and the cytoskeleton. Cilia 1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. (2007). Autophagy: process and function. Genes Dev. 21 2861–2873. 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- Moser J. J., Fritzler M. J., Rattner J. B. (2014). Ultrastructural characterization of primary cilia in pathologically characterized human glioblastoma multiforme (GBM) tumors. BMC Clin. Pathol. 14:40. 10.1186/1472-6890-14-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa N., Li J., Yabuno-Nakagawa K., Eom T. Y., Cowles M., Mapp T., et al. (2017). APC sets the Wnt tone necessary for cerebral cortical progenitor development. Genes Dev. 31 1679–1692. 10.1101/gad.302679.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina P. J., Tuson M., Anderson K. V. (2009). Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PLoS One 4:e6839. 10.1371/journal.pone.0006839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E. C., Katsanis N. (2013). Context-dependent regulation of Wnt signaling through the primary cilium. J. Am. Soc. Nephrol. 24 10–18. 10.1681/asn.2012050526 [DOI] [PubMed] [Google Scholar]
- Orhon I., Dupont N., Zaidan M., Boitez V., Burtin M., Schmitt A., et al. (2016). Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat. Cell Biol. 18 657–667. 10.1038/ncb3360 [DOI] [PubMed] [Google Scholar]
- Pal K., Mukhopadhyay S. (2015). Primary cilium and sonic hedgehog signaling during neural tube patterning: role of GPCRs and second messengers. Dev. Neurobiol. 75 337–348. 10.1002/dneu.22193 [DOI] [PubMed] [Google Scholar]
- Pampliega O., Orhon I., Patel B., Sridhar S., Diaz-Carretero A., Beau I., et al. (2013). Functional interaction between autophagy and ciliogenesis. Nature 502 194–200. 10.1038/nature12639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. M., Lim J. S., Ramakrishina S., Kim S. H., Kim W. K., Lee J., et al. (2018). Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron 99:e87. [DOI] [PubMed] [Google Scholar]
- Petralia R. S., Schwartz C. M., Wang Y. X., Kawamoto E. M., Mattson M. P., Yao P. J. (2013). Sonic hedgehog promotes autophagy in hippocampal neurons. Biol. Open 2 499–504. 10.1242/bio.20134275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J. F., Blacque O. E., Leroux M. R. (2012). The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 13 608–618. 10.1038/embor.2012.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J. F., Leroux M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 18 533–547. 10.1038/nrm.2017.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R., Milenkovic L., Scott M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317 372–376. 10.1126/science.1139740 [DOI] [PubMed] [Google Scholar]
- Rosengren T., Larsen L. J., Pedersen L. B., Christensen S. T., Moller L. B. (2018). TSC1 and TSC2 regulate cilia length and canonical Hedgehog signaling via different mechanisms. Cell. Mol. Life Sci. 75 2663–2680. 10.1007/s00018-018-2761-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross A. J., May-Simera H., Eichers E. R., Kai M., Hill J., Jagger D. J., et al. (2005). Disruption of bardet-biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 37 1135–1140. 10.1038/ng1644 [DOI] [PubMed] [Google Scholar]
- Rui Y. N., Xu Z., Patel B., Chen Z., Chen D., Tito A., et al. (2015). Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17 262–275. 10.1038/ncb3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai N., Briscoe J. (2012). Primary cilia and graded Sonic Hedgehog signaling. Wiley Interdiscip. Rev. Dev. Biol. 1 753–772. 10.1002/wdev.43 [DOI] [PubMed] [Google Scholar]
- Saxton R. A., Sabatini D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales S. J., de Sauvage F. J. (2009). Mechanisms of hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 30 303–312. 10.1016/j.tips.2009.03.007 [DOI] [PubMed] [Google Scholar]
- Shillingford J. M., Murcia N. S., Larson C. H., Low S. H., Hedgepeth R., Brown N., et al. (2006). The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. U.S.A. 103 5466–5471. 10.1073/pnas.0509694103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M., Gloy J., Ganner A., Bullerkotte A., Bashkurov M., Kronig C., et al. (2005). Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 37 537–543. 10.1038/ng1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snedeker J., Schock E. N., Struve J. N., Chang C. F., Cionni M., Tran P. V., et al. (2017). Unique spatiotemporal requirements for intraflagellar transport genes during forebrain development. PLoS One 12:e0173258. 10.1371/journal.pone.0173258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotthibundhu A., Li Q. X., Thangnipon W., Coulson E. J. (2009). Abeta(1-42) stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol. Aging 30 1975–1985. 10.1016/j.neurobiolaging.2008.02.004 [DOI] [PubMed] [Google Scholar]
- Spassky N., Han Y. G., Aguilar A., Strehl L., Besse L., Laclef C., et al. (2008). Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev. Biol. 317 246–259. 10.1016/j.ydbio.2008.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottmann R. W., Tran P. V., Turbe-Doan A., Beier D. R. (2009). Ttc21b is required to restrict sonic hedgehog activity in the developing mouse forebrain. Dev. Biol. 335 166–178. 10.1016/j.ydbio.2009.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z., Lin M. G., Stowe T. R., Chen S., Zhu M., Stearns T., et al. (2013). Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502 254–257. 10.1038/nature12606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong C. K., Han Y. G., Shah J. K., Obernier K., Guinto C. D., Alvarez-Buylla A. (2014). Primary cilia are required in a unique subpopulation of neural progenitors. Proc. Natl. Acad. Sci. U.S.A. 111 12438–12443. 10.1073/pnas.1321425111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T., Breunig J. J., Sarkisian M. R., Spilianakis C., Ayoub A. E., Liu X., et al. (2008). The stumpy gene is required for mammalian ciliogenesis. Proc. Natl. Acad. Sci. U.S.A. 105 2853–2858. 10.1073/pnas.0712385105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran P. V., Haycraft C. J., Besschetnova T. Y., Turbe-Doan A., Stottmann R. W., Herron B. J., et al. (2008). THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 40 403–410. 10.1038/ng.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukachinsky H., Lopez L. V., Salic A. (2010). A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 191 415–428. 10.1083/jcb.201004108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente E. M., Rosti R. O., Gibbs E., Gleeson J. G. (2014). Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 10 27–36. 10.1038/nrneurol.2013.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R., Nusse R. (2009). Towards an integrated view of Wnt signaling in development. Development 136 3205–3214. 10.1242/dev.033910 [DOI] [PubMed] [Google Scholar]
- Vogel T. W., Carter C. S., Abode-Iyamah K., Zhang Q., Robinson S. (2012). The role of primary cilia in the pathophysiology of neural tube defects. Neurosurg. Focus 33:E2. [DOI] [PubMed] [Google Scholar]
- Wahl P. R., Serra A. L., Le Hir M., Molle K. D., Hall M. N., Wuthrich R. P. (2006). Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial Transplant. 21 598–604. 10.1093/ndt/gfi181 [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R. J., Scott M. P. (1999). Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22 103–114. 10.1016/s0896-6273(00)80682-0 [DOI] [PubMed] [Google Scholar]
- Wegiel J., Kuchna I., Nowicki K., Imaki H., Wegiel J., Marchi E., et al. (2010). The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 119 755–770. 10.1007/s00401-010-0655-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway G., Abdelhamed Z., Natarajan S., Toomes C., Inglehearn C., Johnson C. A. (2013). Aberrant Wnt signalling and cellular over-proliferation in a novel mouse model of Meckel-Gruber syndrome. Dev. Biol. 377 55–66. 10.1016/j.ydbio.2013.02.015 [DOI] [PubMed] [Google Scholar]
- Willaredt M. A., Hasenpusch-Theil K., Gardner H. A., Kitanovic I., Hirschfeld-Warneken V. C., Gojak C. P., et al. (2008). A crucial role for primary cilia in cortical morphogenesis. J. Neurosci. 28 12887–12900. 10.1523/jneurosci.2084-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P., Sieben C. J., Xu X., Harris P. C., Lin X. (2017). Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 26 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann K. W. (1898). Beiträge zur Kenntniss einiger Drüsen und Epithelien. Archiv. Mikrosk. Anat. 52 552–706. 10.1007/bf02975837 [DOI] [Google Scholar]