

Abstract
Alport syndrome (ATS) is an inherited glomerular disease caused by mutations in one of the type IV collagen novel chains (α3, α4, and α5). ATS is characterized by persistent microscopic hematuria that starts during infancy, eventually leading to either progressive nephritis or end-stage renal disease. There are 3 known genetic forms of ATS, namely X-linked ATS, autosomal recessive ATS, and autosomal dominant ATS. About 80% of patients with ATS have X-linked ATS, which is caused by mutations in the type IV collagen α5 chain gene, COL4A5. Although an 80% mutation detection rate is observed in men with X-linked ATS, some difficulties do exist in the genetic diagnosis of ATS. Most mutations are point mutations without hotspots in the COL4A3, COL4A4, and COL4A5 genes. Further, there are insufficient data on the detection of COL4A3 and COL4A4 mutations for their comparison between patients with autosomal recessive or dominant ATS. Therefore, diagnosis of ATS in female patients with no apparent family history can be challenging. Therefore, in this study, we used whole-exome sequencing (WES) to identify mutations in type IV collagen in 2 girls with glomerular basement membrane structural changes suspected to be associated with ATS; these patients had no relevant family history. Our results revealed de novo c.4688G>A (p.Arg1563Gln) and c.2714G>A (p.Gly905Asp) mutations in COL4A5. Therefore, we suggest that WES is an effective approach to obtain genetic information in ATS, particularly in female patients without a relevant family history, to detect unexpected DNA variations.
Keywords: Alport syndrome, Whole exome sequencing, Child, Collagen type IV
Introduction
Alport syndrome (ATS; OMIM #301050, #104200, #203780) is an inherited glomerular disease caused by mutations in one of the novel chains (α3, α4, and α5) of type IV collagen, which is the major protein component of the glomerular basement membrane (GBM) [1]. ATS is characterized by persistent microscopic hematuria starting from infancy, eventually leading to either progressive nephritis or end-stage renal disease (ESRD). Extrarenal symptoms, including cochlear dysfunction and ocular anomalies, are also present in proportion to the severity of renal manifestations [1].
There are 3 known genetic forms of ATS: X-linked ATS, autosomal recessive ATS, and autosomal dominant ATS. About 80% of patients with ATS have X-linked ATS, caused by mutations in the type IV collagen gene α5 chain gene, COL4A5 [1,2]. In about 15% of cases, autosomal recessive ATS results from either compound heterozygous or homozygous mutations in COL4A3 or COL4A4. Approximately 5% of ATS cases exhibit autosomal dominant inheritance caused by heterozygous mutations in either COL4A3 or COL4A4 [1,2].
The clinical manifestations of X-linked ATS differ between males and females [1]. The classic presentation, including hematuria and sensorineural hearing loss, is fully seen in male patients with X-linked ATS. The majority of these cases show GBM structural changes in renal biopsy and progression to ESRD [3]. However, female patients with X-linked ATS have a mild clinical presentation. Furthermore, the diagnostic ultrastructural changes in the GBM, such as lamellation, splitting, and duplication of the lamina densa, are often not seen, and diffuse thinning of the GBM is the predominant finding in young or female patients [1,3]. Therefore, pathological diagnosis is sometimes difficult in females with isolated hematuria in the absence of a family history of either hematuria or ESRD.
Lack of expression of the novel chains of type IV collagen revealed by immunostaining can provide accurate diagnostic information even in patients with ATS who are too young to display characteristic abnormalities in the GBM ultrastructure [1]. Immunostaining may also provide useful clues for the differentiation of X-linked ATS from autosomal recessive ATS. Expression of the novel chains is completely absent in ~80% of males with X-linked ATS, and 60%–70% of females with X-linked ATS show a mosaic pattern of expression of these chains [1]. On the other hand, immunostaining for the α5 chain in Bowman’s capsules and tubular basement membranes is positive in most patients with autosomal recessive ATS [1]. However, immunostaining is not feasible in hospitals with few cases of renal biopsy in Korea, because of problems such as difficulty in obtaining antibodies or in reading the immunofluorescence pathology results to be uncovered by health insurance.
A COL4A5 mutation test has been commercially available in Europe and the United States. Mutation detection rates are about 80% in males with X-linked ATS [1]. However, there are some difficulties in the genetic diagnosis of ATS. Most mutations are point mutations without hot spots in the COL4A3, COL4A4, and COL4A5 genes [4]. Further, there is insufficient data on COL4A3 and COL4A4 mutation detection for the mutations to be compared between patients with either autosomal recessive or dominant ATS [1].
The next generation sequencing (NGS) technique has enabled the sequencing of an entire human genome within a day and superseded conventional Sanger sequencing. Moreover, the increased sensitivity of NGS allows detection of unexpected DNA variations [5]. There have been studies on the identification of ATS-associated mutations using NGS [6,7]. For example, Artuso et al. [6] identified mutations in ATS patients by analyzing all the COL4A3, COL4A4, and COL4A5 genes in a single experiment using NGS. On similar lines, whole-exome sequencing (WES) allows the identification of variations in the protein-coding region of any gene (known as the sequencing of an). Chiereghin et al. [8] used a WES approach to evaluate ATS cold cases.
In this study, we used WES to identify mutations in type IV collagen in two Korean girls with suspicious GBM structural changes associated with ATS but no relevant family history.
Case reports
1. Case 1
A previously healthy 11-year-old girl visited our hospital owing to incidentally detected proteinuria in the national school-based urinary screening program. She had episodic gross hematuria, large corneal astigmatism, and mild lumbar scoliosis. Further, she had no family history of kidney disease. The urinalysis showed 1+proteinuria and 3+ occult blood with many red blood cells per high-power field. The laboratory tests revealed the following: blood urea nitrogen, 14.1 mg/dL; creatinine, 0.50 mg/dL; total protein, 7.1 g/dL; albumin, 4.1 g/dL; and serum C3 and C4, 102 mg/dL and 28 mg/dL, respectively. She tested negative for antinuclear antibody, and her renal ultrasonography result was unremarkable. Pure tone audiometry and speech audiometry results were also unremarkable. A renal biopsy was performed owing to persistent proteinuria and microscopic hematuria during the 6 months to follow-up (Fig. 1).
Fig. 1.
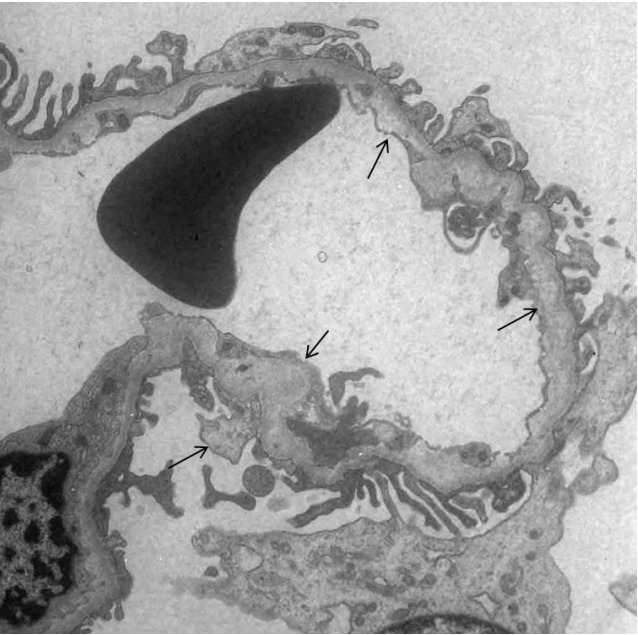
Electron microscopic examination of renal biopsy tissues in case 1. The ultrastructure of the glomerular basement membrane shows focal marked abnormalities with thickening, lysis, reticulation, and subepithelial protrusion of the lamina densa (arrow) (×8,000).
2. Case 2
A 12-year-old girl visited our hospital for a regular health check-up. She had had recurrent gross hematuria from 24 months of age. Around the age of 7 years, she was diagnosed with ATS by renal biopsy at another hospital. Further, her physician did not find a COL4A5 mutation by direct sequencing. She had neither extra-renal symptoms, including hearing loss or ocular lesions, nor any family history of kidney disease. At first visit her renal function was normal, except persistent microscopic hematuria and proteinuria.
3. WES
Genomic DNA was extracted from the patients’ and their parents’ peripheral blood leukocytes using the Wizard Genomic DNA Purification Kit following the manufacturer’s instructions (Promega, Madison, WI, USA). SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) was used for library preparation. Sequencing was performed using the Illumina NextSeq500 platform (Illumina Inc., San Diego, CA, USA), generating 2×150-bp paired-end reads. The variants that passed the quality filters were screened against public databases, such as the Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/) and the Korean Reference Genome Database (KRGDB) (http://152.99.75.168/KRGDB/) for a minor allele frequency of <5.0%. Protein-altering variants were then selected. The variants derived from the variant-filtering strategy were then prioritized on the basis of their likelihood to affect protein function and to totally or partially match the patient's phenotype using public algorithms, such as SIFT. Finally, a gene-specific analysis was performed with an in-silico gene panel comprised of 31 genes, including COL4A3, COL4A4, and COL4A5. These genes were obtained by searching previous publications and disease databases (Human Gene Mutation Database, HGMD: http://hgmd.cf.ac.uk and Online Mendelian Inheritance in Man, OMIM: https://www.omim.org/).
The variants identified in the proband were classified according to the Standards and Guidelines for the Interpretation of Sequence Variants by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [9]. Pathogenic variants (PVs) or likely PVs were confirmed by Sanger sequencing. A familial study was performed using a targeted PV analysis with Sanger sequencing.
Case 1 had a likely pathogenic c.4688G>A (p.Arg1563Gln) variant in COL4A5 (Fig. 2C). The c.4688G>A variant was not observed in either parent. The c.4688G>A variant has previously been reported in a family with ATS [10]. Our patient also had one variant of uncertain significance (c.4817G>A; p.Gly1606Glu) in COL4A4. This variant was heterozygous in the mother (not seen). Case 2 had a likely pathogenic c.2714G>A (p.Gly905Asp) variant, as shown in Fig. 2D. The c.2714G>A variant was not found in either parent. The c.2714G>A variant has been detected previously in a patient with ATS [11].
Fig. 2.
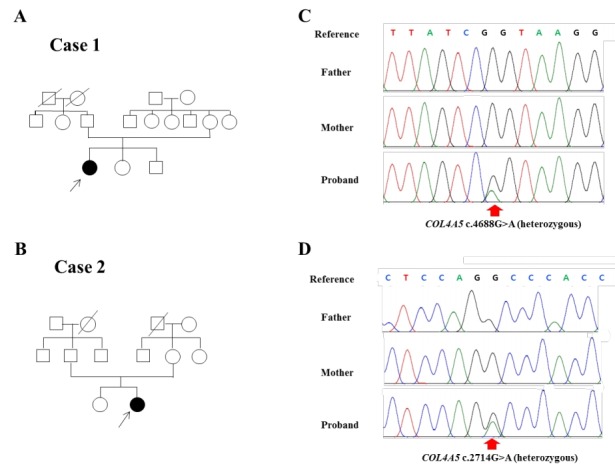
Identification of candidate COL4A5 variants in 2 Korean families with Alport syndrome. (A, B) Pedigree of cases 1 and 2, where filled symbols indicate Alport syndrome and open symbols indicate normal subjects. The proband is marked with an arrow. (C, D) Electropherograms are shown. A heterozygous c.4688G>A mutation causing p.Arg1563Gln in exon 48 and a heterozygous c.2714G>A mutation causing p.Gly905Asp in exon 32 were identified in cases 1 and 2, respectively (NM_000495.4).
This study was approved by the Institutional Review Boards of the Jeju National University Hospital (IRB No.2013-11-019-007). Written informed consent for the genetic study was obtained from all patients and their parents.
Discussion
Type IV collagen is known to form the networks for cell-cell and cell-matrix interactions required for proper glomerular structure and function [1]. There are six α chains in the type IV collagen protein family, and 3 of them form a triple helical structure, resulting in 3 triplet species: α1α1α2, α3α4α5, and α5α5α6 [12]. Each chain contains a collagenous domain containing triplet sequence Gly-X-Y repeats (X and Y representing other amino acids), interrupted by short noncollagenous sequences [12]. A missense mutation replaces the glycine residue with another amino acid in the Gly-X-Y repeats, which accounts for approximately 30% of the COL4A5 mutations in ATS [1]. The glycine substitution in the collagenous domain of the α5 (IV) chain leads to a less robust network in the triple helical conformation of type IV collagen [13]. Wang et al. [13] demonstrated that glycine substitutions on the α5 (IV) chain cause different structural changes of the GBM and could determine the clinical severity of ATS. Using WES, we identified a COL4A5 missense mutation within exon 32 (NM_000495.4: c.2714G>A) in case 2, causing the substitution of glycine with an aspartic acid (p.Gly905Asp). The patient is now 12 years old; mild proteinuria, microscopic hematuria, and intermittent episodes of gross hematuria are apparent, but extrarenal symptoms are not clear. This is similar to a previously reported clinical manifestation, in which missense mutations in the Gly-X-Y repeats resulted in less severe ocular changes and hearing loss [14].
The mutation found in case 1 was a nonglycine substitution that has previously been reported [10]. No specific hot spot has been identified in ATS, but the distance of the mutation from the C- terminus could affect the phenotype in terms of the zipper-like folding mechanism. Missense mutations located within exons 1– 20 are associated with less severe phenotypes compared with those associated with mutations located in exons 21–47 [14]. Typical visual abnormalities in patients with ATS are mainly retinopathy and anterior lenticonus, which do not usually occur during childhood [15]. However, this patient had corneal astigmatism as a visual abnormality. These ocular changes are still mild, but it is necessary to follow up on whether the severity becomes more evident as a typical ophthalmological change including anterior lenticonus in ATS, because the subject carries a mutation located in exon 48.
Large deletions and duplications in either COL4A3 or COL4A4, disease-causing intronic variants, and digenic mutations have been uncovered by Sanger sequencing [2], and thus, researchers studying ATS have begun to introduce newer genetic techniques, such as NGS and exome sequencing [2,6,8]. These studies were all based on a positive family history of hematuria, chronic kidney disease, sensory hearing loss, or specific ocular changes. Genetic diagnosis is more difficult for female patients with ATS in the absence of a relevant family history than for male patients with a positive family history, despite a renal biopsy result that is compatible with ATS. WES is an alternative technique to identify an unknown genetic cause [8]. In these cases, however, additional functional studies are needed to verify the causal relationship between the mutation and phenotype. We must acknowledge here that the lack of functional studies is a limitation of our study.
In conclusion, we identified mutations in type IV collagen in two Korean girls with ATS without a relevant family history, using a WES approach. Our results suggest that WES will be beneficial for genetic analyses of ATS.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Kashtan CE, Gubler MC. Inherited glomerular diseases: type IV collagen disorders. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, editors. Pediatric nephrology. 6th ed. Springer; 2009. pp. 621–8. [Google Scholar]
- 2.Weber S, Strasser K, Rath S, Kittke A, Beicht S, Alberer M, et al. Identification of 47 novel mutations in patients with Alport syndrome and thin basement membrane nephropathy. Pediatr Nephrol. 2016;31:941–55. doi: 10.1007/s00467-015-3302-4. [DOI] [PubMed] [Google Scholar]
- 3.Meleg-Smith S, Magliato S, Cheles M, Garola RE, Kashtan CE. X- linked Alport syndrome in females. Hum Pathol. 1998;29:404–8. doi: 10.1016/s0046-8177(98)90123-x. [DOI] [PubMed] [Google Scholar]
- 4.Cheong HI, Park HW, Ha IS, Choi Y. Mutational analysis of COL4A5 gene in Korean Alport syndrome. Pediatr Nephrol. 2000;14:117–21. doi: 10.1007/s004670050025. [DOI] [PubMed] [Google Scholar]
- 5.Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Child Educ Pract Ed. 2013;98:236–8. doi: 10.1136/archdischild-2013-304340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Artuso R, Fallerini C, Dosa L, Scionti F, Clementi M, Garosi G, et al. Advances in Alport syndrome diagnosis using next-generation sequencing. Eur J Hum Genet. 2012;20:50–7. doi: 10.1038/ejhg.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beicht S, Strobl-Wildemann G, Rath S, Wachter O, Alberer M, Kaminsky E, et al. Next generation sequencing as a useful tool in the diagnostics of mosaicism in Alport syndrome. Gene. 2013;526:474–7. doi: 10.1016/j.gene.2013.05.045. [DOI] [PubMed] [Google Scholar]
- 8.Chiereghin C, Robusto M, Mastrangelo A, Castorina P, Montini G, Giani M, et al. Alport syndrome cold cases: Missing mutations identified by exome sequencing and functional analysis. PLoS One. 2017;12:e0178630. doi: 10.1371/journal.pone.0178630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou J, Gregory MC, Hertz JM, Barker DF, Atkin C, Spencer ES, et al. Mutations in the codon for a conserved arginine-1563 in the COL4A5 collagen gene in Alport syndrome. Kidney Int. 1993;43:722–9. doi: 10.1038/ki.1993.103. [DOI] [PubMed] [Google Scholar]
- 11.Wang F, Zhao D, Ding J, Zhang H, Zhang Y, Yu L, et al. Skin biopsy is a practical approach for the clinical diagnosis and molecular genetic analysis of X-linked Alport's syndrome. J Mol Diagn. 2012;14:586–93. doi: 10.1016/j.jmoldx.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Hudson BG. The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J Am Soc Nephrol. 2004;15:2514–27. doi: 10.1097/01.ASN.0000141462.00630.76. [DOI] [PubMed] [Google Scholar]
- 13.Wang YF, Ding J, Wang F, Bu DF. Effect of glycine substitutions on alpha5(IV) chain structure and structure-phenotype correlations in Alport syndrome. Biochem Biophys Res Commun. 2004;316:1143–9. doi: 10.1016/j.bbrc.2004.02.168. [DOI] [PubMed] [Google Scholar]
- 14.Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transplant. 2002;17:1218–27. doi: 10.1093/ndt/17.7.1218. [DOI] [PubMed] [Google Scholar]
- 15.Savige J, Sheth S, Leys A, Nicholson A, Mack HG, Colville D. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol. 2015;10:703–9. doi: 10.2215/CJN.10581014. [DOI] [PMC free article] [PubMed] [Google Scholar]