

Abstract
TRPV4, a nonselective calcium permeable ion channel, is expressed broadly in many organs including bone and neurons. Pathogenic variants in TRPV4 are known to cause both a spectrum of skeletal dysplasias and neuropathies. Recent publications have documented a few patients who have a combined phenotype of skeletal dysplasia and neuropathy secondary to TRPV4 pathogenic variants. We present an additional patient who has an overlapping neuromuscular and skeletal phenotype secondary to a TRPV4 pathogenic variant. The patient has spondylometaphyseal dysplasia-Kozlowski type and Charcot-Marie-Tooth disease type 2C. This and prior reports illustrate that TRPV4-related skeletal dysplasias and TRPV4-related neuropathies are not fully distinct disorders secondary to unique sets of pathogenic variants as originally postulated, but rather are 2 phenotypes on the same spectrum that may or may not overlap. We suggest that evaluation for patients presenting with any TRPV4-related disorder include assessment for both skeletal and neurological findings.
Keywords: Charcot-Marie-Tooth, CMT2C, SMD-K, Spondlyometaphyseal dysplasia Kozlowski, TRPV4
Spondylometaphyseal dysplasia (SMD) is a group of skeletal dysplasias characterized by spine and metaphyseal changes [Kozlowski et al., 1982]. Distinctions between types are made based on severity and specific radiographic findings. In 1967, SMD-Kozlowski type (SMD-K) was characterized as a distinct type of SMD with patients often coming to medical attention due to short stature. SMD-K includes scoliosis, metaphyseal changes observed in the pelvis, and platyspondyly of the vertebrae [Kozlowski, 1967]. A decade ago, Krakow et al. [2009] demonstrated that SMD-K was caused by pathogenic variants in transient receptor potential cation channel, subfamily V, member 4 (TRPV4).
Charcot-Marie-Tooth disease (CMT) is a group of disorders affecting peripheral nerves. In 1994, Charcot-Marie-Tooth disease type 2C (CMT2C) was identified as a distinct peripheral axonal neuropathy which includes diaphragm and vocal cord paresis [Dyck et al., 1994], and in 2010, pathogenic variants in TRPV4 were identified as causative for CMT2C [Auer Grumbach et al., 2010; Deng et al., 2010; Landouré et al.,2010].
Most TRPV4 variants have apparently led to a clinical presentation as either a skeletal dysplasia or a neuropathy. However, there are a few descriptions of patients who have combined phenotypes [Chen et al., 2010; Zimoń et al., 2010; Unger et al., 2011; Cho et al., 2012; Evangelista et al., 2015]. TRPV4 mutations are inherited in a dominant/heterozygous manner, and the clinical phenotype tends to be similar in families.
TRPV4 is a nonselective calcium permeable ion channel. TRPV4 is expressed broadly in many organs, including bone and neurons. Its expression contributes to the generation of a Ca2+ signal [Everaerts et al., 2010].
TRPV4 seems to play a role in multiple regulatory pathways involved in skeletal development [Guilak et al., 2010]. It is expressed in bone in the osteoblasts and osteoclasts modulating bone remodeling [Masuyama et al., 2008]. Muramatsu et al. [2007] demonstrated that TRPV4 expression in chondrocytes may regulate the SOX9 pathway and play a role in endochondral osteogenesis. The TRPV4 spectrum of skeletal dysplasias from mildest to most severe includes familial digital arthropathy-brachydactyly, autosomal dominant brachyolmia, SMD-K, spondyloepiphyseal dysplasia-Maroteaux type, parastremmatic dysplasia, and metatropic dysplasia [Rock et al., 2008]. The severity of the disease is thought to be directly related to the channel open probability; the more likely the ion channel is open, the more severe the phenotype [Loukin et al., 2011].
Pathogenic variants in TRPV4 also have been shown to cause a variety of neuropathies [Deng et al., 2010] including CMT2C, scapuloperitoneal spinal muscular atrophy, and congenital distal spinal muscular atrophy. Several studies have suggested that a gain-of-function mechanism is responsible for the TRPV4-associated neuropathies due to cytotoxicity from increased intracellular calcium concentration [Landouré et al., 2010; Fecto et al., 2011]. Jang et al. [2012] found that TRPV4 promotes neuritogenesis suggesting that if there is a breakdown in communication between target tissues and nerves, survival of the sensory nerves is compromised and a neuropathy results. It has been proposed that the spectrum of neuromuscular disorders is caused specifically by arginine substitutions in the ankyrin repeat domains (ARD) of TRPV4 [Deng et al., 2010; Landouré et al., 2010; Sullivan et al., 2015]. However, arginine substitutions in ARD are also commonly observed in TRPV4 pathogenic variants associated with skeletal dysplasia without neurological findings [Dai et al., 2010].
We present a further case (11th) in which there is a combined neuromuscular and skeletal phenotype: SMD-K and CMT2C.
Clinical Report
The female patient is the second child born to a nonconsanguineous couple. Pregnancy was uncomplicated followed by onset of spontaneous labor at 37 weeks. She required observation for transient tachypnea and respiratory distress and was discharged in excellent health. Family history is negative for documented neuromuscular disease, peripheral neuropathy, or skeletal dysplasia. Her father is reported to be 183 cm tall and her mother and sister are reported to both be 164 cm tall.
Scoliosis and asymmetries of the patient's spine were first noted at age 18 months. Platyspondyly found on initial scoliosis radiographs led to further radiologic evaluation, which showed abnormal metaphyses of the long bones. She received a nonspecific diagnosis of SMD. Bracing for scoliosis was provided at an outside facility.
When the patient was 5 years old, the family relocated and established care with the Midwest Regional Bone Dysplasia Clinic (MRBDC) and pediatric orthopedics at the University of Wisconsin School of Medicine and Public Health. Her diagnosis was further defined as SMD-K based on significant platyspondyly, anterior vertebral body abnormalities, abnormal odontoid development, and metaphyseal changes of the long bones with particular abnormality at the hips with short femoral necks and proximal femoral metaphyseal irregularity (Fig. 1, 2). She also had asymmetric femoral neck-shaft angles of 130 degrees on the left (normal) and 155 degrees on the right (coxa valga) (Fig. 2). Hand radiograph obtained was also consistent with SMD-K (Fig. 3).
Fig. 1.
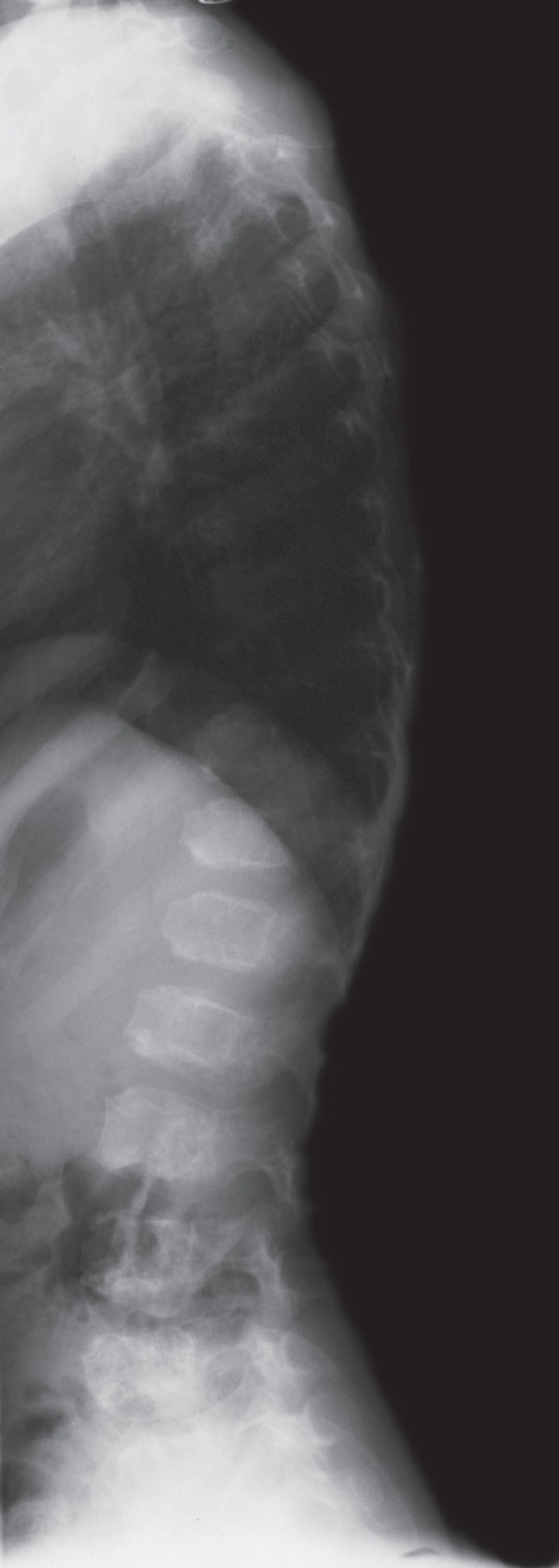
Lateral spine radiograph at 5 years of age demonstrating generalized platyspondyly and anterior vertebral abnormalities consistent with spondylometaphyseal dysplasia-Kozlowski type.
Fig. 2.

Radiograph of the pelvis and lower spine at 5 years of age. There is moderate to marked metaphyseal irregularity at the proximal femurs and of the greater trochanters, very short femoral necks, and asymmetric neck shaft femoral angles. The ilia are mildly broad.
Fig. 3.
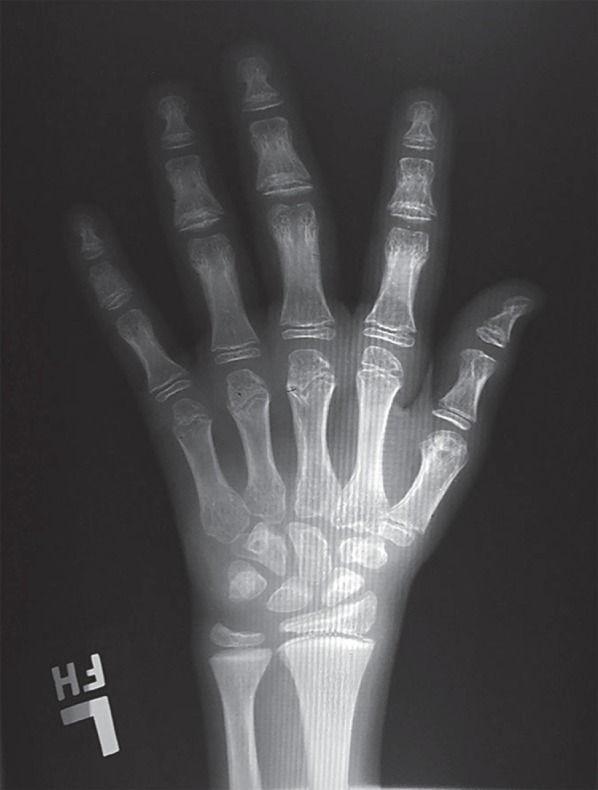
Hand radiograph at 10 years of age. There is moderate to marked brachydactyly with shortening particularly of the distal phalanges. Metaphyseal cupping as is seen in spondylometaphyseal dysplasia-Kozlowski type is also evident.
Concerns regarding weakness and gait differences led to pediatric neurology assessment at 5 years of age. Neurological abnormalities included asymmetry of bulk and strength of vastus medialis and gastrocnemius as well as asymmetric but bilateral pes cavus (Fig. 4). Patellar and Achilles reflexes were absent. Responses to light touch, temperature, pinprick, or vibration were normal. Electromyography of the legs showed active denervation as well as chronic changes of denervation/reinnervation both proximally and distally. Nerve conduction studies showed normal sensory velocities. Based on these findings and normal spinal cord anatomy on MRI, her clinical picture was thought suggestive of either distal spinal muscular atrophy or a predominately motor neuropathy. She ultimately received a diagnosis of CMT2C.
Fig. 4.

A, B Patient's hands at age 27 showing characteristics consistent with Charcot-Marie-Tooth disease type 2. Note decreased muscle bulk and clawing. C Patient's feet at age 27. Note residual pes cavus and metatarsus adductus. D Frontal view of patient at age 27. Note relatively short trunk and distal extremity muscle wasting.
Between 8 and 12 years of age, the patient underwent 3 foot surgeries to treat bilateral pes cavus and foot position abnormalities (Fig. 4), including medial foot releases, multiple osteotomies, and tendon transfers. Due to progressive kyphoscoliosis, at 10 years of age, she underwent spinal fusion with anterior discectomy of T3-T8 and posterior spinal instrumentation. Evaluation by neurology at 10 years of age showed the patient had persistent weakness and remained uncoordinated but did not have any deterioration of motor function. As she progressed through adolescence, she developed some sensory deficits including loss of vibration sense. Repeat electromyography/nerve conduction studies showed evidence of a generalized, chronic, motor greater than sensory, axonal polyneuropathy.
Regular pulmonary function monitoring was performed due to her scoliosis and early fusion surgery. These have shown no deterioration.
CMT2C findings affected her function more than her skeletal findings. Writing skills were slightly delayed, and she utilized a combination of writing and keyboarding. Fine motor skills required therapeutic intervention. She had apparent normal functioning in speech, language, memory, and other higher cognitive functioning. The patient ambulates independently without the use of assistive devices. In the past, the patient had bilateral ankle-foot orthoses but stopped wearing them prior to high school. At 26 years of age, the patient sustained a left pelvic iliac wing fracture when she lost her balance and fell backwards. Bone density appeared normal on radiographs, and the fracture healed normally.
At last contact, the patient was 27 years old; her height was 148.8 cm (Z = −2.2) weight was 45.65 kg (4th percentile), and OFC was 56.0 cm (Z = 1.52). The patient has no facial dysmorphisms. She has a residual left thoracic kyphoscoliosis and a shortened trunk with relatively long extremities (Fig. 4D). She continues to have decreased muscle bulk in distal forearms and hands, mild brachydactyly, and significant hypermobility throughout her hands (Fig. 4A). Additionally, she has decreased muscle bulk throughout the distal lower extremities and feet, knee valgus, relatively wide feet, residual pes cavus, metatarsus adductus, and hypermobile toes (Fig. 4C). Strength remains decreased. Upper extremities show decreased grip strength in digits 1, 2, and 3 as well as decreased interossei strength (Fig. 4B). Lower limbs have decreased hip flexion and abduction with active movement. She walks with a waddling gait on the lateral aspect of her feet and has a pelvic tilt secondary to her scoliosis (Fig. 4D). Upper extremity sensation remains intact to sharp/dull and vibration. In lower extremities, her sensation is intact to sharp/dull and vibration, but it is decreased compared to the upper extremities. The patient has no subjective hearing issues. She remains cognitively normal and has completed a Master's degree.
The patient's bone dysplasia and peripheral neuropathy had largely been considered 2 separate processes and treated symptomatically by neurology and orthopedic professionals.
Results
Molecular Findings
Prior molecular genetic evaluation of the patient's peripheral neuropathy included normal CMT1A testing of PMP22, CMT1B testing of MPZ (CMT2 testing was not available at that time), and spinal muscular atrophy testing to rule out an exon 7 or 8 deletion of the SMN gene. In 2014, because of recognition that she might have an “overlapping” phenotype, genetic testing of TRPV4 was completed. TRPV4 was amplified by PCR and sequenced using ABI 3730 sequencers and analyzed for copy number variation using high-density targeted array. Genome build GRCh37/hg19 was used. DNA sequencing revealed 2 changes in exon 6, one of which had been previously described as pathogenic and one variant of unknown significance. The pathogenic variant c.943C>T converts a codon for arginine (CGG) to a codon for tryptophan (TGG) (R315W). The second change is a c. 904A>T variant (N302Y). Parental testing was performed and showed the patient's father has the same TRPV4 c.904A>T, N302Y variant of unknown significance. The patient's mother was negative for any TRPV4 variants; thus the patient's c.943C>T, R315W pathogenic variant is de novo.
Discussion
This is the 11th published individual recognized to have an overlapping phenotype including both a TRPV4-associated skeletal dysplasia and a neuropathy, presumably both being caused by the TRPV4 pathogenic variant.
The first presumed instance of the combined skeletal and neurological phenotypes of TRPV4 pathogenic variants was described by Chen et al. [2010]. While the neurological abnormalities were reasonably well delineated, unfortunately the skeletal features were not. Affected members of both described families were more than 2 SD below mean for height. However, no radiographs were published (or described), and so it is impossible to more explicitly identify the skeletal process present in these families. One of these families is particularly relevant since they have the same pathogenic variant as is present in the patient described here. Additional overlapping phenotypes of skeletal dysplasia and neuropathy have been reported. Zimoń et al. [2010] described one patient diagnosed with hereditary motor and sensory neuropathy 2 and brachyolmia. Radiographs describe a right convex kyphoscoliosis with flattened vertebrae in the mid-thoracic segment and shortened femoral necks that were wide and slightly irregular. No brachydactyly or delay in carpal bone ossification was seen. The authors suggested that the spine changes might either have arisen as a primary result of the TRPV4 pathogenic variant or as a secondary consequence of longstanding neurological disease. Unger at al. [2011] described 4 fetuses (in 3 families) prenatally diagnosed with both metatropic dysplasia and fetal akinesia secondary to pathogenic variants in TRPV4. Cho et al. [2012] described 3 additional patients with combined phenotypes. The first patient had SMD-K and hereditary motor and sensory neuropathy 2. This patient's mother also showed platyspondyly and flaring of the iliac bones, but did not have a neuropathy phenotype. The second patient had a diagnosis consistent with spondyloepimetaphyseal dysplasia-Maroteaux type and a motor/sensory axonal neuropathy predominately in the lower limbs. The third patient had SMD-K and scapulohumeral spinal muscular atrophy. Evangelista et al. [2015] described an additional patient with scapuloperitoneal spinal muscular atrophy and metatropic dysplasia.
The R315W pathogenic variant present in the patient described here has been previously identified in individuals with isolated neuromuscular disease including CMT and distal spinal muscular atrophy which is congenital and nonprogressive (ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/variation/4999/). As noted, it has once previously also been found in a family with presumed combined phenotypes [Chen et al., 2010]. R315W has not been reported in individuals with only skeletal dysplasia.
The patient described here is the first with reasonably well-characterized SMD-K and CMT2 arising from the R315W pathogenic variant. She adds to the growing list of patients with both neuromuscular and skeletal findings secondary to TRPV4 pathogenic variants (Table 1).
Table 1.
TRPV4 pathogenic variants and corresponding diagnosis in patients with dual diagnoses
| TRPV4 variant | Skeletal dysplasia | Neuropathy | Reference |
|---|---|---|---|
| E278K | SMD-K | HMSN2 | Cho et al., patient 1 [2012] |
| P799R | SEMD-M | Motor and sensory axonal neuropathy predominately in the lower limb | Cho et al., patient 2 [2012] |
| A217S | SMD-K | Scapulohumeral spinal muscular atrophy | Cho et al., patient 3 [2012] |
| S542Y | Short stature | CMT2C | Chen et al., family A [2010] |
| R315W | Short stature | CMT2C | Chen et al., family B [2010] |
| G78W | Metatropic dysplasia | Fetal akinesia | Unger et al., case 1 [2011] |
| T740I | Metatropic dysplasia | Fetal akinesia | Unger et al., case 2, 3 [2011] |
| K276E | Metatropic dysplasia | Fetal akinesia | Unger et al., case 4 [2011] |
| R269C | Metatropic dysplasia | SPSMA | Evangelista et al., patient 1 [2015] |
| V620I | Brachyolmia | HMSN2 | Zimoń et al., [2010] |
| R315W | SMD-K | CMT2C | Current patient |
CMT2C, Charcot-Marie-Tooth disease type 2C; HMSN2, hereditary motor and sensory neuropathy 2; SEMD-M, spondyloepimetaphyseal dysplasia-Maroteaux type; SMD, spondylometaphyseal dysplasia; SMD-K, SMD-Kozlowski type; SPSMA, scapuloperitoneal spinal muscular atrophy.
This patient's R315W pathogenic variant causes an arginine to be substituted for a tryptophan in the Ankyrin repeats of TRPV4. It was initially proposed that the spectrum of neuromuscular disorders is caused by molecular variants in the ARD of TRPV4 [Landouré et al., 2010; Deng et al., 2010; Sullivan et al., 2015]. It was additionally emphasized that all of the variants involved an arginine substitution. However, arginine substitutions in ARD are also commonly observed in TRPV4 pathogenic variants resulting in skeletal dysplasia without neurological findings [Sullivan et al., 2015]. Thus, arginine substitutions are not specific to the spectrum of TRPV4 neuromuscular disorders [Dai et al., 2010]. Based on this report in addition to previous reports, the domain location of TRPV4 pathogenic variants does not appear to exclusively predict the phenotype.
Neurological issues affected this individual's function more than did her skeletal dysplasia, although she did require surgical intervention for scoliosis. Previously reported patients with this pathogenic variant having both skeletal and neurological phenotypes also seem to have experienced greater symptoms related to their neuromuscular disorder, while their skeletal presentation apparently was limited to short stature [Chen et al., 2010]. Skeletal assessments and radiographic evaluations certainly were not performed on all individuals with TRPV4-related neuromuscular disorders. Therefore, it is possible that more overlap between the 2 phenotypes exists than is currently documented.
This report supports the previously reported idea that TRPV4 pathogenic variants causing a skeletal dysplasia and a neuropathy are not mutually exclusive, and individuals may present with overlapping findings [Chen et al., 2010]. Evaluation of individuals who present with TRPV4-related neuromuscular disorders should include assessment of stature and radiologic investigations of the skeleton. Similarly, comprehensive evaluation for neurological abnormalities should be done in all patients diagnosed with TRPV4-related skeletal dysplasias.
Statement of Ethics
The authors have no ethical conflicts to declare.
Disclosure Statement
The authors have no conflicts of interest to disclose.
Acknowledgments
We thank the patient and her family for their willingness to contribute to further medical knowledge about TRPV4. The initial diagnosis of spondylopmetaphyseal dysplasia - Kozlowski was established by Leonard O. Langer Jr. (deceased). Neurological diagnoses were principally made by Drs. Christopher Inglese and Kurt Hecox.
References
- 1.Auer-Grumbach M, Olschewski A, Papic L, Kremer H, McEntagart ME, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet. 2010;42:160–164. doi: 10.1038/ng.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen DH, Sul Y, Weiss M, Hillel A, Lipe H, et al. CMT2C with vocal cord paresis associated with short stature and mutations in the TRPV4 gene. Neurology. 2010;75:1968–1975. doi: 10.1212/WNL.0b013e3181ffe4bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho TJ, Matsumoto K, Fano V, Dai J, Kim OH, et al. TRPV4-pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients. Am J Med Genet A. 2012;158A:795–802. doi: 10.1002/ajmg.a.35268. [DOI] [PubMed] [Google Scholar]
- 4.Dai J, Cho TJ, Unger S, Lausch E, Nishimura G, et al. TRPV4-pathy, a novel channelopathy affecting diverse systems. J Hum Genet. 2010;55:400–402. doi: 10.1038/jhg.2010.37. [DOI] [PubMed] [Google Scholar]
- 5.Deng HX, Klein CJ, Yan J, Shi Y, Wu Y, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet. 2010;42:165–169. doi: 10.1038/ng.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dyck PJ, Litchy WJ, Minnerath S, Bird TD, Chance PF, et al. Hereditary motor and sensory neuropathy with diaphragm and vocal cord paresis. Ann Neurol. 1994;35:608–615. doi: 10.1002/ana.410350515. [DOI] [PubMed] [Google Scholar]
- 7.Evangelista T, Bansagi B, Pyle A, Griffin H, Douroudis K, et al. Phenotypic variability of TRPV4 related neuropathies. Neuromuscul Disord. 2015;25:516–521. doi: 10.1016/j.nmd.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol. 2010;103:2–17. doi: 10.1016/j.pbiomolbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Fecto F, Shi Y, Huda R, Martina M, Siddique T, Deng HX. Mutant TRPV4-mediated toxicity is linked to increased constitutive function in axonal neuropathies. J Biol Chem. 2011;286:17281–17291. doi: 10.1074/jbc.M111.237685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guilak F, Leddy HA, Liedtke W. Transient receptor potential vanilloid 4 the sixth sense of the musculoskeletal system? Ann N Y Acad Sci. 2010;1192:404–409. doi: 10.1111/j.1749-6632.2010.05389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang Y, Jung J, Kim H, Oh J, Jeon JH, et al. Axonal europathy-associated TRPV4 regulates neurotrophic factor-derived axonal growth. J Biol Chem. 2012;287:6014–6024. doi: 10.1074/jbc.M111.316315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozlowski K, Maroteaux P, Spranger JW. La dysostose spondylometaphisaire. Presse Med. 1967;75:2769. [Google Scholar]
- 13.Kozlowski K, Beemer FA, Bens G, Dijkstra PF, Iannaccone G, et al. Spondylo-metaphyseal dysplasia. (Report of 7 cases and essay of classification). Prog Clin Biol Res. 1982;104:89–101. [PubMed] [Google Scholar]
- 14.Krakow D, Vriens J, Camacho N, Luong P, Deixler H, et al. Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am J Hum Genet. 2009;84:307–315. doi: 10.1016/j.ajhg.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landouré G, Zdebik AA, Martinez TL, Burnett BG, Stanescu HC, et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet. 2010;42:170–174. doi: 10.1038/ng.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loukin S, Su Z, Kung C. Increased basal activity is a key determinant in the severity of human skeletal dysplasia caused by TRPV4 mutations. PLoS One. 2011;6:e19533. doi: 10.1371/journal.pone.0019533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8:257–265. doi: 10.1016/j.cmet.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Muramatsu S, Wakabayashi M, Ohno T, Amano K, Ooish R, et al. Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. J Biol Chem. 2007;282:32158–32167. doi: 10.1074/jbc.M706158200. [DOI] [PubMed] [Google Scholar]
- 19.Rock MJ, Prenen J, Funari VA, Funari TL, Merriman B, et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet. 2008;40:999–1003. doi: 10.1038/ng.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sullivan JM, Zimanyi CM, Aisenberg W, Bears B, Chen D, et al. Novel mutations highlight the ankyrin repeat domain in the TRPV4-mediated neuropathy. Neurol Genet. 2015;1:e29. doi: 10.1212/NXG.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unger S, Lausch E, Stanzial F, Gillessen-Kaesbach G, Stefanova I, et al. Fetal akinesia in metatropic dysplasia: the combined phenotype of chondrodysplasia and neuropathy? Am J Med Genet A. 2011;155A:2860–2864. doi: 10.1002/ajmg.a.34268. [DOI] [PubMed] [Google Scholar]
- 22.Zimoń M, Baets J, Auer-Grumbach M, Berciana J, Garcia A, et al. Dominant mutations in the cation channel gene transient receptor potential vanilloid 4 cause an unusual spectrum of neuropathies. Brain. 2010;133:1798–1809. doi: 10.1093/brain/awq109. [DOI] [PMC free article] [PubMed] [Google Scholar]