

Abstract
Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) is due to heterozygous FOXL2 intragenic mutations in about 70% of the patients, whereas total or partial gene deletions account for a minority of cases. Alteration of FOXL2 regulatory elements has been rarely described in patients with BPES. In this study, a prepubertal girl with BPES due to a 197-kb de novo deletion of the regulatory elements upstream of FOXL2 is reported. This girl presented with additional clinical features such as a soft cleft palate and microcephaly; thus, this copy number variant might have other somatic effects. The present deletion encompasses 2 coding genes (MRPS22 and COPB2), whose homozygous mutations have been associated with microcephaly. In our case, the sequences of the non-deleted allele were normal, ruling out a compound genetic defect. Normal levels of new biomarkers of ovarian reserve (anti-müllerian hormone, inhibin B) likely indicate an early diagnosis of type 2 BPES, but an evolutive gonadal damage will be excluded only by long-term follow-up. Additional reports of microdeletions upstream of FOXL2 are needed to better define the underlying genetic mechanism and the related phenotypic spectrum; the ability of the new hormonal markers to predict ovarian function in adolescence and adulthood should be confirmed.
Keywords: Anti-müllerian hormone, array CGH, BPES, COPB2, FOXL2, Inhibin B, MRPS22, Ovarian function, Regulatory elements, 3q23 deletion
Approximately 5% of the human genome is structurally variable in the normal population; it may be part of deletions and duplications, collectively referred to as copy number variants (CNVs). CNVs have received considerable attention as a cause for genetic diseases; their pathogenicity has been mainly explained as the result of altered gene dosage [Swaminathan et al., 2012], but CNVs can also harbor regulatory elements or alter the 3D structure of the genome, influencing the expression of distantly located genes [Lupiáñez et al., 2015].
The role of regulatory elements in disease etiology is known for a restricted number of genes, associated with about 30 pathologies, including blepharophimosis, ptosis, epicanthus inversus syndrome (BPES; OMIM 110100) [Bhatia and Kleinjan, 2014]. BPES is a rare disease (prevalence <1.0/50,000) characterized by complex eyelid malformations. In females, 2 forms of BPES have been described: in type 1, eyelid abnormalities are associated with ovarian failure; however, this is not the case in BPES type 2 [Zlotogoraet al., 1983]. The discrimination between the 2 forms is usually performed at adolescence due to the quiescence of the hypophyseal-pituitary-gonadal axis before puberty [Styne and Grumbach, 2016]. On the contrary, all BPES-affected males have normal testicular function. The gene encoding forkhead transcription factor 2 (FOXL2; OMIM 605597) is known to be causative of BPES with an autosomal dominant pattern of inheritance [Crisponi et al., 2001]. In the majority of patients with BPES, FOXL2 intragenic point mutations are causative, but deletions covering the entire gene have also been reported [Beysen et al., 2009]. Patients with BPES due to deletions upstream and downstream of the transcription unit of FOXL2, encompassing regulatory elements, are extremely rare [Beysen et al., 2005; D'haene et al., 2009; Verdin et al., 2013].
Here, a girl with BPES and a de novo 197-kb microdeletion upstream of the transcription unit of FOXL2 is reported. She also presented with some additional somatic features and mild growth impairment. Non-steroid gonadal hormone assessment suggests preserved ovarian function at least in prepuberty.
Patient and Methods
Clinical Report
The girl was born at 39 weeks of gestation after a second uncomplicated pregnancy and an uneventful delivery from nonconsanguineous Italian healthy parents; family history was unremarkable.
At birth, the following auxological parameters were recorded: weight, 2,580 g (−1.83 SD); length, 47 cm (−1.50 SD), and head circumference, 32 cm (−1.79 SD). Apgar scores were 9 and 10 at 1 and 5 min, respectively. After birth, a soft cleft palate was detected, requiring surgery at 6 months of age. During infancy, hearing screening, cranial ultrasound, echocardiogram, and abdomen ultrasound examinations yielded normal results. When referred to our department for the first time at 3 years, 10/12 months, her gross motor development, receptive and expressive speech, and social skills were normal for age assessed by using adaptive behavioral and descriptive clinical criteria. She had a high forehead, arched eyebrows, blepharophimosis, left ptosis, bilateral epicanthus inversus, telecanthus, columella below alae nasi, a long and smooth philtrum, thin upper lip, and a pointed chin (Fig. 1). No functional visual deficit was found.
Fig. 1.
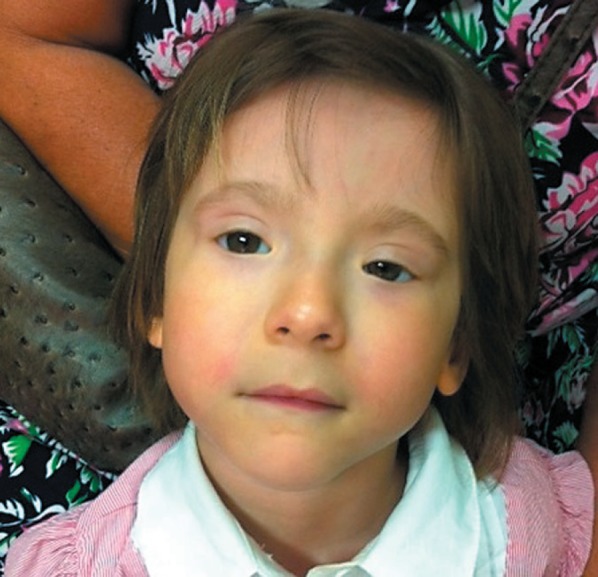
Patient with BPES at the age of 3 years and 10 months.
Methods
SD scores of birth weight, length, and head circumference were calculated according to Italian reference standards [Bertino et al., 2010]; postnatal height and weight were expressed as SD according to WHO standards (http://www.who.int/childgrowth/software/en/). Midparental height was calculated using measured parental heights adjusted for female sex: father's height + mother's height/2 minus 6.5 cm. Baseline blood samples were obtained in the fasting state between 8:00 and 9:00 a.m. to assess reserve of ovarian function by anti-müllerian hormone (AMH), inhibin B, LH, and FSH measurements. Normative levels were derived from values reported in a recent extensive review [Fanelli et al., 2018]. Ovarian volume was calculated by the formula A × B × C × 0.52 and expressed as SD according to Badouraki et al. [2008].
Array CGH was performed according to standard methods. Sex-matched normal control DNAs were obtained from Agilent, Santa Clara, CA, USA. 500 ng of genomic DNA from both the patient and her parents (test sample) and from the controls (reference sample) were differentially labeled with Cy5-dCTP or with Cy3-dCTP using random primer labeling according to the manufacturer's protocol (Agilent). The labeling reactions were applied to the array and incubated in an oven for 24 h at 67°C. The array-CGH platform was a 180K SurePrint G3 Human CGH Microarray (Agilent), which has a 13-kb overall median probe spacing (11 kb in Refseq genes). In order to better define the deletion breakpoints, we also used a 180K Sure Print G3 Cancer Microarray (Agilent) that has a similar resolution, but a different probe distribution. The slides were washed and scanned using the Agilent scanner. Identification of individual spots on scanned arrays was performed with the Agilent Cytogenomics dedicated software, as well as filtration, normalization and exclusion of spots with aberrant morphology or high background. Any polymorphisms were analyzed using the Database of Genomic Variants (DGV; http://dgv.tcag.ca/dgv/app/home). Sequencing of FOXL2 (NM_023067.3), mitochondrial ribosomal protein S22 (MRPS22; NM_605810) and coatomer protein complex subunit beta 2 (COPB2; NM_606990) were performed according to standard methods.
Results
The girl showed slightly reduced height and weight growth patterns; in particular, her height SD resulted −1.2 to −1.4 SD below midparental height. Microcephaly persisted during follow-up (Table 1). Mean ovarian volume was in the low-normal range for age (0.9 mL; n.v. 0.6-2.8 mL). Both AMH (19.3 pmol/L; n.v. 1.9-39.2 pmol/L) and inhibin B levels (9 pg/mL; n.v. <7-33.2 pg/mL) were within normal range; no other biochemical or endocrine abnormalities were found (data not shown).
Table 1.
Auxological data of the present case
| Chronological age |
|||
|---|---|---|---|
| 2.2 years | 3.8 years | 5.9 years | |
| Height | −0.78 | −1.60 | −1.41 |
| Weight for age | −1.10 | −1.20 | −1.31 |
| Weight for height | −1.01 | −0.75 | −0.65 |
| Body mass index | −0.95 | −0.66 | |
| Head circumference | −2.16 | −2.31 | −2.03 |
| Midparental heighta | −0.20 | −0.20 | −0.20 |
Values represent SD.
14 familial cases.
FOXL2 sequencing demonstrated no intragenic mutations in 5′ UTR or the coding sequence. Array-CGH data detected an interstitial deletion in 3q23. The centromeric breakpoint is comprised between probe in position 138,925,589 (not deleted) and probe in position 138,944,012 (deleted), whereas the telomeric breakpoint is comprised between probe in position 139,141,037 (deleted) and the probe in position 139,157,532 (not deleted) (Fig. 2). The minimum deletion size is 197 kb (GRCh37/hg19 reference map) located 278 kb upstream of FOXL2. The deletion was not present in the parents.
Fig. 2.

A Detailed structure of the microdeletion. The genes harbored are indicated by blue boxes; the intervals between the deleted and non-deleted probes of the distal and proximal breakpoints are indicated by striped boxes. Probe and gene positions are referred to GRCh37/hg19. B Schematic representation of microdeletions upstream of FOXL2 in patients with BPES. The numbers 1-9 correspond with those reported in Table 2. The shortest region of deletion overlap (SRO) and FOXL2 are indicated.
No genetic variants were found in the coding genes of the deleted region (MRPS22 and COPB2).
Discussion
A prepubertal girl with a de novo 197-kb microdeletion in 3q23 is reported. She presented with features of BPES associated with microcephaly, soft cleft palate, and some mild facial dysmorphisms (Fig. 1); mild growth impairment was also found. This deletion was unreported in the DGV database collecting data on healthy people. The deleted region harbors 2 protein-coding genes (MRPS22, COPB2) and the non-protein-coding gene, polled intersex syndrome regulated transcript 1 (PIRST1) (Fig. 2). MRPS22 (OMIM 605810) encodes the 28S subunit protein of mitochondrial ribosomes; COPB2 codes for a subunit of the Golgi coatomer complex, essential for Golgi budding and vesicular trafficking [De Baere et al., 1998].
The 3q23 deletion maps 278 kb upstream of FOXL2, the only gene whose heterozygous loss of function is known to cause BPES [Beysen et al., 2009]. Expression of FOXL2 is tightly regulated by distant cis-regulatory elements located both upstream and downstream of the transcription unit [Beysen et al., 2005]. The existence of 5′ regulatory elements was postulated even before the cloning of this gene since the breakpoints of 3 translocations in patients with BPES were found 130, 160, and 171 kb upstream of FOXL2, respectively [De Baere et al., 2000; Praphanphoj et al., 2000; Crisponi et al., 2004]. To date, a total of 9 microdeletions upstream of FOXL2 was described in both familial and in de novo cases of BPES (Table 2) [Beysen et al., 2005; D'haene et al., 2009; Verdin et al., 2013]. These deletions range from 7.4 kb to ∼3 Mb; the shortest region of deletion overlap (SRO) involves a 7.4-kb segment located 283 kb 5′ to FOXL2 (Fig. 2). This 7.4-kb deletion is sufficient per se to cause BPES because it was deleted in a 7-year-old sporadic male with the classic phenotype [D'haene et al., 2009]. In our case, the deletion encompasses SRO (Fig. 2), while no genetic variants were detected in the FOXL2 transcription unit. Thus, the girl is a very rare patient with BPES due to a deletion of upstream regulatory elements (Table 2).
Table 2.
Genetic and clinical data in patients with BPES due to deletion of upstream regulatory elements
| Patient No. | Reference | Deletion position (GRCh37/hg19) | Size, kb | Sex | Age, yrs | Microcephaly | Other clinical signs |
|---|---|---|---|---|---|---|---|
| Present case | - | 138,944,012–139,141,037 | 197 | F | 3 | Yes | Soft cleft palate, mild facial dysmorphisms, mild growth impairment |
| 1 | D'haene et al., 2009 (Pt. D) Verdin et al., 2013 (Pt. A) | 138,949,150–138,956,510 | 7.4 | M | 7 | No | - |
| 2 | Verdin et al., 2013 (Pt. B) Beysen et al., 2005 (Pt. BPES 15) | 138,912,808–139,012,600 | 99.8 | 7 M 7 F | ? | No | BPES type 2 II:6, III:2, III:5 e IV:3 had children |
| 3 | Verdin et al., 2013 (Pt. C) Beysen et al., 2005 (Pt. BPES 14)a |
138,867,472–139,048,942 | 181.5 | 2 F | ? | No | BPES type 2 I:2 three pregnancies, oligomenorrhea at age 40 yrs, and amenorrhea at age 47 yrs, astigmatism; II:1 normal menarche at age 12 yrs and oligomenorrhea at age 38 yrs, strabismus |
| 4 | Verdin et al., 2013 (Pt. E) D'haene et al., 2009 (Pt. C) | 138,805,920–139,012,140 | 206 | ? | ? | No | - |
| 5 | Verdin et al., 2013 (Pt. F) D'haene et al., 2009 (Pt. A) | 138,938,973–139,294,473 | 356 | ? | ? | ? | - |
| 6 | Beysen et al., 2005 (Pt. BPES 13) | 138,767,142–139,334,105 | Max 567 Min 356 | F | 2 | Yes | Stenosis of lacrimal duct, renal reflux |
| 7 | Verdin et al., 2013 (Pt. G) | 138,745,991–140,393,036 | 1,647 | ? | ? | ? | - |
| 8 | Beysen et al., 2005 (Pt. BPES 12) | 138,767,142–140,726,314 | Max 1,959 Min 860 | M | 15 | Yes | Intrauterine and postnatal growth retardation, ventricular septum defect, contracture of interphalangeal joints, right iris heterochromia |
| 9 | Verdin et al., 2013 (Pt. H) | 138,741,281–141,762,242 | 3,021 | ? | ? | ? | - |
BPES, blepharophimosis, ptosis, and epicanthus inversus syndrome; Pt, patient; yrs, years.
2 familial cases.
Some of her clinical features - such as soft cleft palate and microcephaly - may suggest that the 197-kb microdeletion may have other somatic effects besides BPES. Cleft palate was never reported in the other few patients sharing 5′ FOXL2 deletions (Table 2). Indeed, cleft palate and BPES have been described in 2 cases, both carriers of FOXL2 intragenic mutations (family FOXL2_00227 and patient FOXL2_00222) [Beysen et al., 2009], but additional reports are needed to establish whether this finding is an independent feature or a rare consequence of the altered FOXL2 expression. Microcephaly was found in our case and 2 other patients with deletion of upstream regulatory elements (patient 6 and 8 in Table 2). It has also been described in about 20 patients with BPES; however, it has never been associated with FOXL2 intragenic mutations, only with 3q23 deletions encompassing the gene [de Ru et al., 2005; D'haene et al., 2010; Dean et al., 2014]. Patients without microcephaly but with overlapping deletions have also been described [de Ru et al., 2005; D'haene et al., 2010].
The 2 genes in the deleted region, MRPS22 and COPB2, may have a role in causing microcephaly. MRPS22 homozygous gene alterations have been associated with a single case of Cornelia de Lange-like phenotype, where microcephaly was reported along with other clinical signs [Smits et al., 2011]. In addition, antenatal mitochondrial disease caused by homozygous MRPS22 mutation has been reported in 3 children born to the same set of consanguineous parents, who presented with antenatal skin oedema, hypotonia, cardiomyopathy, and tubulopathy [Saada et al, 2007]. A homozygous recessive variant in COPB2 was identified in a pedigree with 2 affected siblings showing primary microcephaly, severe developmental delay, failure to thrive, cortical blindness, and spasticity [DiStasio et al., 2017]. Even if the phenotypic pictures of these patients are completely different from those of our case, sequencing of MRPS22 and COPB2 was performed to exclude point mutations in the non-deleted allele. No genetic variants were found; thus, these genes are unlikely involved in our patient's microcephaly. Nothing is known about the function of the non-protein-coding RNA, PIRST1.
It is worth noting that focusing on those patients whose imbalances were analyzed by molecular techniques, the present deletion is the smallest one, and it partially overlaps all the others (Fig. 3). Thus, it represents a bridge between the more proximal and the more distal deletions. It could be hypothesized that this small region may be involved in the development of microcephaly through a control of 3D genome structure.
Fig. 3.

Schematic representation of microdeletions characterized by molecular techniques in patients with BPES and microcephaly. The relative references are reported. Deletion sizes were all converted to GRCh37/hg19.
In females, one of the challenges of BPES is the establishment of genotype-phenotype correlations, mainly with respect to the ovarian phenotype. A clear differentiation between type 1 and type 2 BPES is usually performed at adolescence because in prepubertal girls, gonadotropin and steroid hormone levels are extremely low or undetectable [Styne and Grumbach, 2016], so invasive endocrine evaluation was used to reach early diagnosis of type 1 BPES [Ogata et al., 1998]. Recently, Lunding et al. [2015] demonstrated that low levels of AMH (<-2 SD) are able to predict failure to enter puberty or imminent premature ovarian failure in girls with Turner syndrome, while ongoing ovarian function was shown in early adulthood by patients with AMH within the reference range [Lunding et al., 2015]. To the best of our knowledge, we evaluated this new ovarian marker in a prepubertal girl with BPES for the first time. The results suggested an early diagnosis of type 2 BPES. Normal ovarian volume and normal inhibin B levels reinforced this hypothesis. In fact, low levels of inhibin B demonstrated 100% sensitivity and specificity for the detection of impaired pubertal development due to hypogonadotropic hypogonadism [Binder et al., 2015]. Albeit present hormonal data in a single girl and their predictive value must be confirmed in additional patients, they agree with previous reports, showing that women with BPES due to deletion of upstream regulatory elements showed normal ovarian function (patients 2 and 3, Table 2). Finally, an evolutive gonadal damage cannot be excluded; thus, a long-term follow-up with serial measurements of the new ovarian markers is needed before reaching definitive conclusions.
Additional reports of microdeletions upstream of FOXL2 will permit to better define the underlying genetic mechanism and related phenotypic spectrum as well as the true predictive values of the new hormonal markers of ovarian function during childhood.
Statement of Ethics
Informed consent and the permission for the publication of the accompanying images were obtained from the parents before any diagnostic or therapeutic intervention. The study was conducted according to the Declaration of Helsinki.
Disclosure Statement
The authors declare that they have no conflicts of interest.
Funding Sources
No external funding from public, commercial, or not-for-profit sectors was obtained for this paper.
Acknowledgments
We would like to thank Wendy Doherty (Lecturer in English Language, University of Pisa) for her critical revision of the manuscript and English editing. We would also like to acknowledge Dr. Elfride De Baere and Dr. Hannah Verdin for providing their support and advice.
References
- 1.Badouraki M, Christoforidis A, Economou I, Dimitriadis AS, Katzos G. Sonographic assessment of uterine and ovarian development in normal girls aged 1 to 12 years. J Clin Ultrasound. 2008;36:539–544. doi: 10.1002/jcu.20522. [DOI] [PubMed] [Google Scholar]
- 2.Bertino E, Spada E, Occhi L, Coscia A, Giuliani F, et al. Neonatal anthropometric charts: the Italian neonatal study compared with other European studies. J Pediatr Gastroenterol Nutr. 2010;51:353–361. doi: 10.1097/MPG.0b013e3181da213e. [DOI] [PubMed] [Google Scholar]
- 3.Beysen D, Raes J, Leroy BP, Lucassen A, Yates JR, et al. Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am J Hum Genet. 2005;77:205–218. doi: 10.1086/432083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beysen D, De Paepe A, De Baere. E FOXL2 mutations and genomic rearrangements in BPES. Hum Mutat. 2009;30:158–169. doi: 10.1002/humu.20807. [DOI] [PubMed] [Google Scholar]
- 5.Bhatia S, Kleinjan DA. Disruption of long-range gene regulation in human genetic disease: a kaleidoscope of general principles, diverse mechanisms and unique phenotypic consequences. Hum Genet. 2014;133:81–845. doi: 10.1007/s00439-014-1424-6. [DOI] [PubMed] [Google Scholar]
- 6.Binder G, Schweizer R, Haber P, Blumenstock G, Braun R. Accuracy of endocrine tests for detecting hypogonadotropic hypogonadism in girls. J Pediatr. 2015;167:674–678. doi: 10.1016/j.jpeds.2015.05.039. [DOI] [PubMed] [Google Scholar]
- 7.Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27:159–166. doi: 10.1038/84781. [DOI] [PubMed] [Google Scholar]
- 8.Crisponi L, Uda M, Deiana M, Loi A, Nagaraja R, et al. FOXL2 inactivation by a translocation 171 kb away: analysis of 500 kb of chromosome 3 for candidate long-range regulatory sequences. Genomics. 2004;83:757–764. doi: 10.1016/j.ygeno.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Dean SJ, Holden KR, Dwivedi A, Dupont BR, Lyons MJ. Acquired microcephaly in blepharophimosis-ptosis-epicanthus inversus syndrome because of an interstitial 3q22.3q23 deletion. Pediatr Neurol. 2014;50:636–639. doi: 10.1016/j.pediatrneurol.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 10.De Baere E, Speleman F, Van Roy N, De Paepe A, Messiaen L. Assignment of the cellular retinol-binding protein 1 gene (RBP1) and of the coatomer beta subunit gene (COPB2) to human chromosome band 3q23 by in situ hybridization. Cytogenet Cell Genet. 1998;82:226–227. doi: 10.1159/000015107. [DOI] [PubMed] [Google Scholar]
- 11.De Baere E, Fukushima Y, Small K, Udar N, Van Camp G, et al. Identification of BPESC1, a novel gene disrupted by a balanced chromosomal translocation, t(3;4)(q23;p15.2), in a patient with BPES. Genomics. 2000;68:296–304. doi: 10.1006/geno.2000.6304. [DOI] [PubMed] [Google Scholar]
- 12.de Ru MH, Gille JJ, Nieuwint AW, Bijlsma JB, Van der Blij JF, et al. Interstitial deletion in 3q in a patient with blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) and microcephaly, mild mental retardation, and growth delay: clinical report and review of the literature. Am J Med Genet A. 2005;137:81–87. doi: 10.1002/ajmg.a.30786. [DOI] [PubMed] [Google Scholar]
- 13.D'haene B, Attanasio C, Dostie J, Lemire E, et al. Disease causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: implications for mutation screening. PLoS Genet. 2009;5:e1000522. doi: 10.1371/journal.pgen.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'haene B, Nevado J, Pugeat M, Pierquin G, Lowry RB, et al. FOXL2 copy number changes in the molecular pathogenesis of BPES: unique cohort of 17 deletions. Hum Mutat. 2010;3:E1332–E1347. doi: 10.1002/humu.21233. [DOI] [PubMed] [Google Scholar]
- 15.DiStasio A, Driver A, Sund K, Donlin M, Muraleedharan RM, et al. Copb2 is essential for embryogenesis and hypomorphicmutations cause human microcephaly. Hum Mol Genet. 2017;26:4836–4848. doi: 10.1093/hmg/ddx362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanelli F, Baronio F, Ortolano R, Mezzullo M, Cassio A, et al. Normative basal values of hormones and proteins of gonadal and adrenal functions from birth to adulthood. Sex Dev. 2018;12:50–94. doi: 10.1159/000486840. [DOI] [PubMed] [Google Scholar]
- 17.Lunding SA, Aksglaede L, Anderson RA, Main KM, Juul A, et al. AMH as predictor of premature ovarian insufficiency: a longitudinal study of 120 Turner syndrome patients. J Clin Endocrinol Metab. 2015;100:E1030–E1038. doi: 10.1210/jc.2015-1621. [DOI] [PubMed] [Google Scholar]
- 18.Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogata T, Hasegawa T, Tamai S, Sato S, Hasegawa Y, Matsuo N. Hypergonadotropichypogonadism in a 3-year-old girl with blepharophimosis, ptosis, and epicanthus inversus syndrome. Horm Res. 1998;50:190–192. doi: 10.1159/000023272. [DOI] [PubMed] [Google Scholar]
- 20.Praphanphoj V, Goodman BK, Thomas GH, Niel KM, Toomes C, et al. Molecular cytogenetic evaluation in a patient with a translocation (3;21) associated with blepharophimosis, ptosis, epicanthus inversus syndrome (BPES) Genomics. 2000;65:67–69. doi: 10.1006/geno.2000.6157. [DOI] [PubMed] [Google Scholar]
- 21.Saada A, Shaag A, Arnon S, Dolfin T, Miller C, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44:784–786. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smits P, Saada A, Wortmann SB, Heister AJ, Brink M, et al. Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur J Hum Genet. 2011;19:394–399. doi: 10.1038/ejhg.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Styne DM, Grumbach MM. Physiology and disorders of puberty: Williams Textbook of Endocrinology. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, editors. Philadelphia: WB Saunders Company; 2016. pp. pp 1074–1218. [Google Scholar]
- 24.Swaminathan GJ, Bragin E, Chatzimichali EA, Corpas M, Bevan AP, et al. DECIPHER: web-based, community resource for clinical interpretation of rare variants in developmental disorders. Hum Mol Genet. 2012;21:R37–R44. doi: 10.1093/hmg/dds362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verdin H, D'haene B, Beysen D, Novikova Y, Menten B, et al. Microhomology-mediated mechanisms underlie non-recurrent disease-causing microdeletions of the FOXL2 gene or its regulatory domain. PLoS Genet. 2013;9:e1003358. doi: 10.1371/journal.pgen.1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zlotogora J, Sagi M, Cohen T. The blepharophimosis, ptosis, and epicanthus inversus syndrome: delineation of two types. Am J Hum Genet. 1983;35:1020–1027. [PMC free article] [PubMed] [Google Scholar]