

Abstract
Euphrasia (Orobanchaceae) is a genus which is widely distributed in temperate regions of the southern and northern hemisphere. The taxonomy of Euphrasia is still controversial due to the similarity of morphological characters and a lack of genomic resources. Here, we present the first complete chloroplast (cp) genome of this taxonomically challenging genus. The cp genome of Euphrasia regelii consists of 153,026 bp, including a large single-copy region (83,893 bp), a small single-copy region (15,801 bp) and two inverted repeats (26,666 bp). There are 105 unique genes, including 71 protein-coding genes, 30 tRNA and 4 rRNA genes. Although the structure and gene order is comparable to the one in other angiosperm cp genomes, genes encoding the NAD(P)H dehydrogenase complex are widely pseudogenized due to mutations resulting in frameshifts, and stop codon positions. We detected 36 dispersed repeats, 7 tandem repeats and 65 simple sequence repeat loci in the E. regelii plastome. Comparative analyses indicated that the cp genome of E. regelii is more conserved compared to other hemiparasitic taxa in the Pedicularideae and Buchnereae. No structural rearrangements or loss of genes were detected. Our analyses suggested that three genes (clpP, ycf2 and rps14) were under positive selection and other genes under purifying selection. Phylogenetic analysis of monophyletic Orobanchaceae based on 45 plastomes indicated a close relationship between E. regelii and Neobartsia inaequalis. In addition, autotrophic lineages occupied the earliest diverging branches in our phylogeny, suggesting that autotrophy is the ancestral trait in this parasitic family.
Keywords: Euphrasia regelii, hemiparasite, chloroplast genome, pseudogenization, phylogenetic analyses
Introduction
The chloroplast (cp) is the most important organelle for green plants as it is the place where photosynthesis and carbon fixation occurs. The cp genome is uniparentally inherited and generally has a quadripartite structure consisting of one large single-copy (LSC) region, one small single-copy (SSC) region, and two inverted repeat regions (IRs) of the same length (Bendich, 2004). The cp genome is more conserved than the nuclear and mitochondrial genomes in terms of gene structure and composition (Asaf et al., 2017a). Due to the highly conserved and non-recombinant nature of the cp genome, it has been shown to be a very useful genetic resource for inferring evolutionary relationships at different taxonomic levels (Caron et al., 2000; Cho et al., 2015). Recently, with the advent of next generation sequencing, it has become comparatively easy to sequence the complete cp genome of non-model taxa and infer phylogenetic relationships based on whole plastomes (Ruhsam et al., 2015; Guo et al., 2017; Saarela et al., 2018).
The genus Euphrasia (Orobanchaceae) is widely distributed throughout temperate regions of the southern and northern hemispheres, and contains about 458 species and subspecies, most of which occur in the northern hemisphere (Gussarova et al., 2008; Secretariat, 2017; Moura et al., 2018). Euphrasia plants are either perennial or annual herbs which mainly parasitise the roots of Gramineae species (Wu et al., 2005; Gussarova et al., 2008). Some species in this genus are used as folk medicine to treat diseases such as blepharitis, conjunctivitis and coughs (Li and Wang, 2003). Euphrasia was once included in the tribe Rhinantheae of the Scrophulariaceae but based on molecular data, was moved with all other parasitic plants in this family to Orobanchaceae (Olmstead et al., 2001). Due to frequent autogamy as well as interspecific hybridization and morphological diversity, Euphrasia comprises a taxonomically complex group of taxa where species delimitation remains challenging (Vitek, 1998; French et al., 2008; Gussarova et al., 2008).
In China, 11 species of Euphrasia are currently recognized which are divided into two sections based on morphological characteristics, namely Sect. Semicalcaratae and Sect. Paradoxae (Hong et al., 1998). The annual herb Euphrasia regelii Wettst., belongs to Sect. Semicalcaratae, and is used for the treatment of hyperglycemia, inflammation, hay fever, conjunctivitis, colds, influenza and coughs (Shuya et al., 2004). Due to the medicinal value of E. regelii, research has mainly focused on identifying the effective chemical constituents of this species (Li and Wang, 2003; Shuya et al., 2004). Few studies have been conducted to infer the phylogenetic position of E. regelii or its genetic diversity due to a lack of informative genetic markers. Additionally, research has been hampered because E. regelii is difficult to distinguish from other Euphrasia species due to morphological similarities. Therefore, more discriminating genetic markers are needed to infer the phylogenetic relationship of E. regelii with other Euphrasia taxa and to facilitate reliable genetic authentication of this important medicinal herb. Although the cp genome of some Orobanchaceae species has been sequenced and utilized in phylogenetic studies (Wicke et al., 2013; Samigullin et al., 2016; Zeng et al., 2017), no cp genome which could have been used for the development of new and variable markers has been published for the genus Euphrasia until now.
In this study, we characterize the complete cp genome of E. regelii and compare it with the available cp genomes of Orobanchaceae taxa. Our results will be useful for marker development, species discrimination, and the inference of phylogenetic relationships in the genus Euphrasia.
Materials and Methods
Plant Material and DNA Extraction
Euphrasia regelii was collected from Taibai mountain (107°16′47.172″ E, 33°59′27.1068″ N) in the Chinese province of Shaanxi. Young leaves were put into silica gel for DNA extraction and a voucher specimen was deposited at the herbarium of Xi’an Jiaotong University (XJTU) (Xi’an, China). Total genomic DNA was extracted using a modified CTAB protocol (Doyle, 1987), and the quantity and quality of the extracted DNA was determined by gel electrophoresis and a NanoDrop 2000 Spectrophotometer.
Chloroplast Genome Sequencing and Assembly
The DNA Library with an insert size of 270 bp was constructed using TruSeq DNA sample preparation kits and sequenced on an Illumina HiSeq X Ten platform with an average paired end read length of 150 bp. The raw reads were filtered to obtain high-quality reads by removing adapters, low-quality sequences such as reads with unknown bases (“N”), and reads with more than 50% low-quality bases (quality value ≤ 10) using the NGS QC Toolkit v2.3.3 (Patel and Jain, 2012). To filter reads from the chloroplast genome, paired-end high quality reads were mapped to the previously published cp genomes (NC_034308, NC_027838, NC_022859, KF922718, NC_022859; Supplementary Table S1) in the Orobanchaceae using Bowtie v2.2.6 with default parameter (Langmead and Salzberg, 2012). Matched paired-end reads were de novo assembled using SPAdes v3.6.0 (Bankevich et al., 2012), and the longest contig was selected as Seed sequence for further assembly using NOVOPlasty v2.6.2 (Dierckxsens et al., 2017). Finally, all the clean reads were mapped to the unannotated cp genome using Geneious v10.1 with bowtie 2 algorithm (Biomatters, Ltd., Auckland, New Zealand) in order to avoid assembly errors. Seven regions with low coverage were Sanger sequenced (Supplementary Table S2). The cp genome was aligned to its reverse complement to determine inverted repeat regions. The boundaries of the inverted repeats and single copy regions were also verified by Sanger sequencing (Supplementary Table S2).
Genome Annotation, Codon Usage, and Repeat Structure
The complete cp genome was annotated using the automatic annotator DOGMA (Wyman et al., 2004) with manual verification via BLAST searches against the cp genomes of other Orobanchaceae species. During the annotation process, open reading frames (ORFs) that can be matched with known cp genes were annotated, and the remaining ORFs lacking protein evidence were disregarded. Genes that contained one or more frameshift mutations or premature stop codons were considered potential pseudogenes. The circular annotated plastid genome map was drawn using the online program OrganellarGenome DRAW (Lohse et al., 2013) and deposited in GenBank (MK070895). The codon usage frequency was calculated based on protein-coding genes using MEGA v6 (Tamura et al., 2013). Tandem repeat sequences were searched for using the Tandem Repeats Finder program (Benson, 1999) with the following parameters: 2 for the alignment parameter match and 7 for mismatch and indels. Dispersed and palindromic repeats were identified using REPuter with a minimum repeat size of 30 bp and sequence identity of no less than 90% (hamming distance equal to 3) (Kurtz et al., 2001). Simple sequence repeats (SSRs) were identified using the software MISA (Thiel et al., 2003) with the following minimum number of repeats: 10 for mono, 5 for di-, 4 for tri-, and 3 for tetra-, penta, and hexa-nucleotide SSRs.
Genome Comparison and Sequence Divergence
Eleven plastome sequences, including two from non-parasitic taxa (Rehmannia glutinosa, NC_034308; Lindenbergia philippensis, NC_022859), three from facultative hemiparasites (Triphysaria versicolor, KU212369 Aureolaria virginica, MF780870; Buchnera americana, MF780871), four from obligate hemiparasites (Neobartsia inaequalis, KF922718; Schwalbea americana NC_023115; Castilleja paramensis, NC_031805; Pedicularis cheilanthifolia, NC_036010; Striga aspera, MF780872) and one from a holoparasite (Lathraea squamaria, NC_027838), were retrieved from GenBank and used in the subsequent analyses. Comparative Genomics of 12 Orobanchaceae plastomes was performed and visualized using the mVISTA software (Frazer et al., 2004) with the annotation of R. glutinosa as a reference. Any large structural changes such as gene order rearrangements were recorded using Mauve v1.1.1 with default settings (Darling et al., 2004). IR expansion/contraction of these plastomes were also analyzed. The nucleotide diversity (Pi) and sequence polymorphism of Rhinantheae species were analyzed using DNAsp v6.0 (Rozas et al., 2017). In order to detect whether plastid genes were under selection, the non-synonymous (dN), synonymous (dS), and dN/dS values of 64 protein coding gene from Rhinantheae species were calculated using the PAML package v 4.0 with YN algorithm (Yang, 2007). Nucleotide substitution rates were not calculated for pseudogenes due to the existence of premature stop codons.
Phylogenetic Analysis
To infer phylogenetic relationships within Orobanchaceae a total of 42 cp genomes were used with Salvia miltiorrhiza (Lamiaceae), Tectona grandis (Lamiaceae), and Solanum lycopersicum (Solanaceae) as outgroup (Supplementary Table S1). All cp genome sequences were aligned using MAFFT v7.402 (Katoh and Standley, 2013) and the most variable positions were excluded from the alignment using Gblocks v0.91b (Talavera and Castresana, 2007). A maximum likelihood (ML) and a Bayesian inference (BI) approach were used to infer phylogenetic relationships. The Maximum likelihood analyses were conducted using IQ-TREE v1.6.1 (Nguyen et al., 2015) with the best best-fit model selected by ModelFinder and 1,000 bootstrap replicates. Bayesian inference was conducted using MrBayes v3.2.6 (Ronquist et al., 2012) with a nucleotide substitution model inferred by Modeltest v3.7 (Posada and Crandall, 1998) (Supplementary Table S3). The Markov chain Monte Carlo (MCMC) algorithm was run for 1 million generations and sampled every 100 generations. The first 25% of resultant trees were discarded and the remaining trees were used to build a majority-rule consensus tree with posterior probability (PP) values for each node. As gene loss from the cp genome is a common phenomenon in the parasitic family of Orobanchaceae, the most conserved regions (TMCRs) of the cp genomes were retrieved using HomBlocks (Bi et al., 2018). TMCRs were then used to construct the phylogenetic trees using the two methods specified above. Additionally, the phylogeny of the genus Euphrasia was inferred using the following chloroplast regions: trnL, trnL-trnF, and atpB-rbcL. The sequences of 39 Euphrasia species were downloaded from TreeBase with the Accession No. 224921.
Results
The Chloroplast Genome of Euphrasia regelii
A total of 7,867,077 paired-end reads were retrieved with a sequence length of 150 bp. A total of 7,861,321 of high quality reads were used for the cp genome assembly. The raw reads were deposited in NCBI SRA database under the Accession No. SRR8237421. Based on a combination of de novo and reference guided assemblies, the cp genome of E. regelii was obtained with the average coverage of 956×. The complete cp genome of E. regelii is 153,026 bp in length and possesses the typical quadripartite structure including a LSC region of 83,893 bp separated from the 15,801 bp long SSC region by two inverted repeats (IRs), each 26,666 bp (Figure 1 and Table 1).
FIGURE 1.
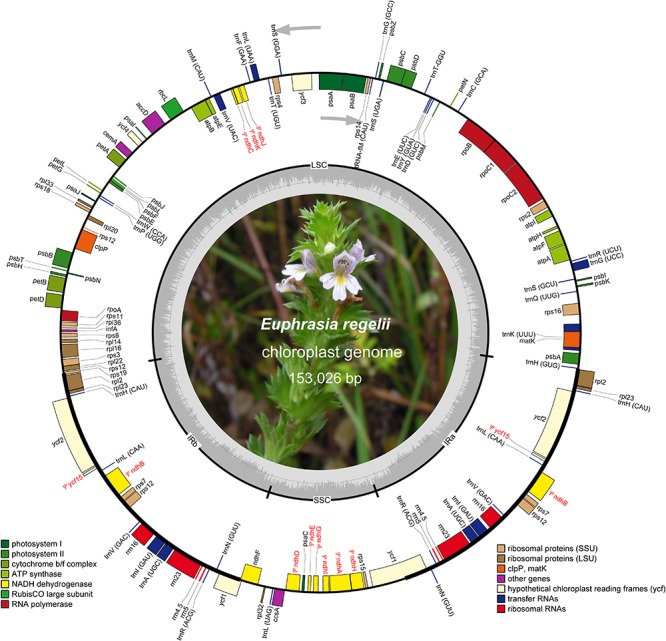
Map of the chloroplast genome of Euphrasia regelii. Genes belonging to different functional groups are highlighted in different colors. Gray arrows indicate the direction of gene transcription. Small single copy (SSC), large single copy (LSC), and inverted repeats (IRa, IRb) are indicated. Pseudogenes are marked by Ψ.
TABLE 1.
Statistics of the chloroplast genomes of Euphrasia regelii and seven other Orobanchaceae species.
| E. regelii | L. squamaria | N. inaequalis | S. americana | T. versicolor | L. philippensis | R. glutinosa | A. fasciculatum | |
|---|---|---|---|---|---|---|---|---|
| Genome length (bp) | 153,026 | 150,504 | 151,349 | 160,910 | 152,448 | 155,103 | 153,622 | 106,796 |
| LSC length (bp) | 83,893 | 81,981 | 83,806 | 84,756 | 83,650 | 85,606 | 84,605 | 43970 |
| SSC length (bp) | 15,801 | 16,061 | 16,327 | 6,517 | 17,520 | 17,885 | 17,579 | 530 |
| IR length (bp) | 26,666 | 26,231 | 25,566 | 34,818 | 25,639 | 25,800 | 25,719 | 31148 |
| No. of different genes | 107 | 78 | 102 | 108 | 106 | 117 | 121 | 66 |
| No. of different protein-coding genes | 69 | 46 | 69 | 74 | 73 | 80 | 82 | 28 |
| No. of different tRNA genes (duplicated in IR) | 30 (7) | 30 (7) | 27 (5) | 30 (7) | 28 (7) | 30 (7) | 30 (7) | 29 (7) |
| No. of different rRNA genes (duplicated in IR) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) |
| No. of genes duplicated in IR | 15 | 14 | 10 | 19 | 15 | 16 | 12 | 17 |
| No. of different genes with introns | 16 | 12 | 18 | 17 | 15 | 18 | 18 | 10 |
| No. of pseudogenes | 11 | 32 | 0 | 2 | 1 | 4 | 0 | 8 |
| GC content (%) | 38.4 | 38.1 | 37.5 | 38.1 | 38.2 | 37.8 | 38.0 | 34.7 |
The plastome of E. regelii was predicted to contain 105 unique genes, including a set of 71 protein-coding genes, 30 tRNA genes and 4 rRNA genes (Table 1 and Supplementary Table S4). Unexpectedly, 10 plastid genes encoding the subunits of the NAD(P)H dehydrogenase complex (ndh genes) were pseudogenized, and only the intact ORF of ndhF existed. Ycf15 was also found to be a pseudogene due to an internal stop codon in its ORF frame. Of 105 genes, four protein-coding genes (rpl2, ycf2, rpl23, rps7), seven tRNA genes (trnH-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-GAC, trnR-ACG, trnN-GUU), and four rRNA genes (rrn16, rrn23, rrn4.5, rrn5) were duplicated in the IR regions. Sixteen intron-containing genes were detected in the E. regelii cp genome, including seven protein-coding genes and six tRNA genes with one intron, whereas the remaining three protein-coding genes (clpP, rps12, ycf3) had two introns (Table 2). We found that trnK-UUU had the largest intron (2,472 bp) and included the gene matK. The tRNA gene trnL-UAA had the smallest intron (462 bp) (Table 2). The overall GC content of 38.4% of the E. regeli cp genome was generally low (LSC, SSC, and IR regions had 36.2, 33.9, and 42.9% GC content, respectively).
TABLE 2.
Genes with introns in the chloroplast genome of Euphrasia regeli.
| Exon I | Intron I | Exon II | Intron II | Exon III | ||
|---|---|---|---|---|---|---|
| Gene | Location | (bp) | (bp) | (bp) | (bp) | (bp) |
| trnK-UUU | LSC | 37 | 2,472 | 35 | ||
| rps16 | LSC | 40 | 839 | 194 | ||
| trnG-UCC | LSC | 23 | 663 | 48 | ||
| atpF | LSC | 234 | 687 | 411 | ||
| rpoC1 | LSC | 456 | 767 | 1,668 | ||
| ycf3 | LSC | 134 | 698 | 229 | 705 | 153 |
| trnL-UAA | LSC | 35 | 462 | 50 | ||
| trnV-UAC | LSC | 38 | 582 | 35 | ||
| clpP | LSC | 71 | 728 | 292 | 627 | 228 |
| petB | LSC | 6 | 728 | 642 | ||
| petD | LSC | 8 | 765 | 475 | ||
| rpl16 | LSC | 9 | 865 | 399 | ||
| rpl2 | IR | 394 | 669 | 434 | ||
| rps12* | IR/LSC | 114 | 232 | 538 | 26 | |
| trnI-GAU | IR | 37 | 946 | 35 | ||
| trnA-UGC | IR | 38 | 812 | 35 |
*rps12 gene is trans-spliced gene with the two duplicated 3′ end exons in IR regions and 5′ end exon in the LSC region.
Codon Usage Bias of E. regelii cp Genome
The frequency of codons in the E. regelii cp genome was calculated based on protein-coding genes (Table 3). In total, all genes were encoded by 23,629 codons. We found that leucine was the most frequent amino acid (2,427 codons, 10.27%) and cysteine (265 codons, 1.1%) the least frequent in the cp genome (Table 3). Similar to other angiosperms cp genomes, codon usage in the E. regelii plastome was biased toward a high representation of U and A at the third codon position [relative synonymous codon usage values (RSCU) > 1].
TABLE 3.
Codon–anticodon recognition pattern and codon usage in the Euphrasia regeli chloroplast genome.
| Codon | Amino acid | Count | RSCU | tRNA | Codon | Amino acid | Count | RSCU | tRNA |
| UUU | F | 882 | 1.33 | trnF-GAA | UAU | Y | 667 | 1.62 | trnY-GUA |
| UUC | F | 441 | 0.67 | UAC | Y | 156 | 0.38 | ||
| UUA | L | 747 | 1.85 | trnL-UAA | UAA | 42 | 1.68 | ||
| UUG | L | 511 | 1.26 | trnL-CAA | UAG | 17 | 0.68 | ||
| CUU | L | 510 | 1.26 | trnL-UAG | CAU | H | 435 | 1.48 | trnH-GUG |
| CUC | L | 157 | 0.39 | CAC | H | 152 | 0.52 | ||
| CUA | L | 330 | 0.82 | CAA | Q | 649 | 1.5 | trnQ-UUG | |
| CUG | L | 172 | 0.43 | CAG | Q | 216 | 0.5 | ||
| AUU | I | 952 | 1.48 | trnI-GAU | AAU | N | 927 | 1.55 | trnN-GUU |
| AUC | I | 405 | 0.63 | AAC | N | 272 | 0.45 | ||
| AUA | I | 569 | 0.89 | trnI-CAU | AAA | K | 1034 | 1.5 | trnK-UUU |
| AUG | M | 506 | 1 | trnM-CAU | AAG | K | 342 | 0.5 | |
| GUU | V | 452 | 1.41 | trnV-GAC | GAU | D | 753 | 1.6 | trnD-GUC |
| GUC | V | 163 | 0.51 | GAC | D | 187 | 0.4 | ||
| GUA | V | 479 | 1.49 | GAA | E | 933 | 1.5 | trnE-UUC | |
| GUG | V | 192 | 0.6 | trnV-UAC | GAG | E | 314 | 0.5 | |
| UCU | S | 499 | 1.63 | trnS-GGA | UGU | C | 198 | 1.49 | trnC-GCA |
| UCC | S | 302 | 0.99 | UGC | C | 67 | 0.51 | ||
| UCA | S | 326 | 1.07 | UGA | 16 | 0.64 | |||
| UCG | S | 214 | 0.7 | trnS-UGA | UGG | W | 400 | 1 | trnW-CCA |
| CCU | P | 334 | 1.37 | trnP-UGG | CGU | R | 312 | 1.25 | trnR-ACG |
| CCC | P | 205 | 0.84 | CGC | R | 119 | 0.48 | trnR-UCU | |
| CCA | P | 276 | 1.13 | CGA | R | 327 | 1.31 | ||
| CCG | P | 160 | 0.66 | CGG | R | 130 | 0.52 | ||
| ACU | T | 507 | 1.63 | AGA | R | 450 | 1.8 | ||
| ACC | T | 223 | 0.72 | AGG | R | 163 | 0.65 | ||
| ACA | T | 365 | 1.17 | trnT-UGU | AGU | S | 385 | 1.26 | trnS-GCU |
| ACG | T | 149 | 0.48 | trnT-GGU | AGC | S | 106 | 0.35 | |
| GCU | A | 533 | 1.72 | trnA-UGC | GGU | G | 503 | 1.27 | trnG-GCC |
| GCC | A | 200 | 0.64 | GGC | G | 165 | 0.42 | ||
| GCA | A | 363 | 1.17 | GGG | G | 339 | 0.85 | ||
| GCG | A | 147 | 0.47 | GGA | G | 582 | 1.47 | trnG-UCC |
*indicates the stop codon.
Repeat Analysis
Of the E. regelii cp genome, 19 forward repeats, 17 palindromic repeats, and 7 tandem repeats were detected (Figure 2). More than half of the repeats (58.3%) were found in intergenic regions and introns, and 74.4% of these repeats have a repeat length between 30 and 50 bp (Figure 2). Within the CDS region, only two genes (ycf1 and ycf2) contained six forward repeats, six palindromic repeats and two tandem repeats, respectively (Supplementary Tables S5, S6). A total of 44 SSRs were detected in the E. regelii cp genome, the majority of which were mononucleotide repeats (22), followed by dinucleotide (12), tetranucleotide (6), and trinucleotide (4) repeats. Most SSRs (29) were distributed in non-coding regions with the remaining 15 SSRs located in genic regions including rpoC2, psbC, atpB, rpoA, ycf1, ccsA (Figure 2). Just over half (54.5%) of the SSRs were located in the LSC region, whereas 36.4 and 9.1% were found in the SSC and the IR regions (Figure 2).
FIGURE 2.

Statistics of repeat elements in the chloroplast genome of Euphrasia regeli. (A) Number of palindromic repeats, forward repeats and tandem repeats. (B) Frequency of identified SSR motifs. (C) Frequency of SSRs in the LSC, SSC, and IR. (D) Total numbers of identified SSRs in coding regions.
Genome Comparison and Selective Pressure Analyses
To investigate cp genome divergence between E. regelii and other Orobanchaceae species, sequence alignment of 12 cp genomes were conducted using the annotated cp genome of R. glutinosa as a reference. The results indicated that the IR regions are more conserved than the SC regions and that the divergence in intergenic regions is higher than in genic regions (Figure 3). Many differences were found in the SSC regions of these plastomes, and the LSC regions of B. americana and S. aspera differed markedly from other autotrophic and parasitic species (Figure 4). The cp genome of E. regelii is very similar to the plastomes of R. glutinosa, L. philippensis, T. versicolor, L. squamaria, and N. inaequalis. All other plastomes contained multiple rearrangements, especially in B. americana and S. aspera. No rearrangements were detected in the three included Rhinantheae species (E. regelii, L. squamaria, N. inaequalis) except that some genes within the SSC region of N. inaequalis were lost. However, the orientation of the SSC region of S. americana was inverted and showed a reverse gene order compared to the other three Rhinantheae species. A sliding window analysis indicated that most of the variation in the cp genomes of the three Rhinantheae species occurred in the LSC and SSC regions (Figure 5). The most divergent non-coding regions among the four Rhinantheae cp genomes were trnH (GUG) – psbA, rps16 – trnQ (UUG), trnS (GCU) – trnG (UCC), atpH-atpI, petN – psbM, trnT (GGU) – psbD, ndhC – trnV (UAC), rbcL – accD, petA – psbJ, clpP – psbB, ndhF – rpl32, rpl32 – trnL (UAG). Although coding regions were conserved in these cp genomes, minor sequence variation was observed among the four cp genomes in the rpoC2, rpoC1, ndhF, ycf1, and ycf2 gene.
FIGURE 3.
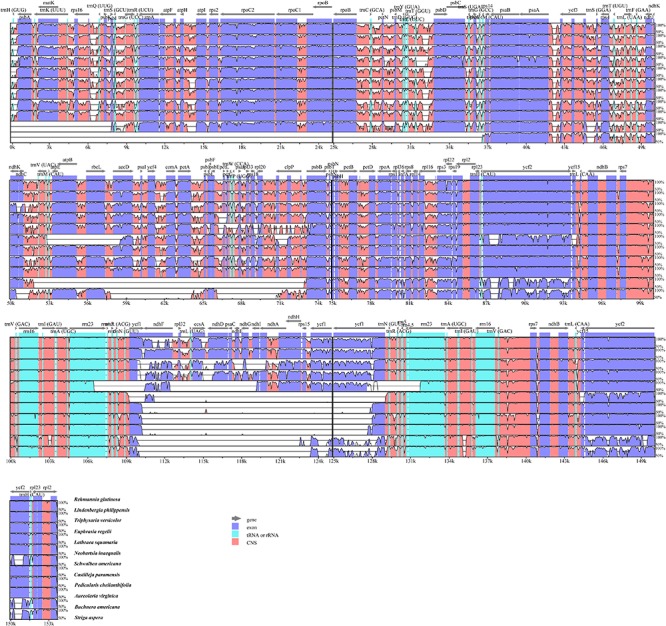
Percentages of identity comparing 12 chloroplast genomes from Orobanchaceae to the reference Rehmannia glutinosa (mVISTA). The y-axis represents the percent identity within 50–100%. Genome regions are color-coded as protein coding (purple), rRNA or tRNA coding genes (blue), and non-coding sequences (pink).
FIGURE 4.
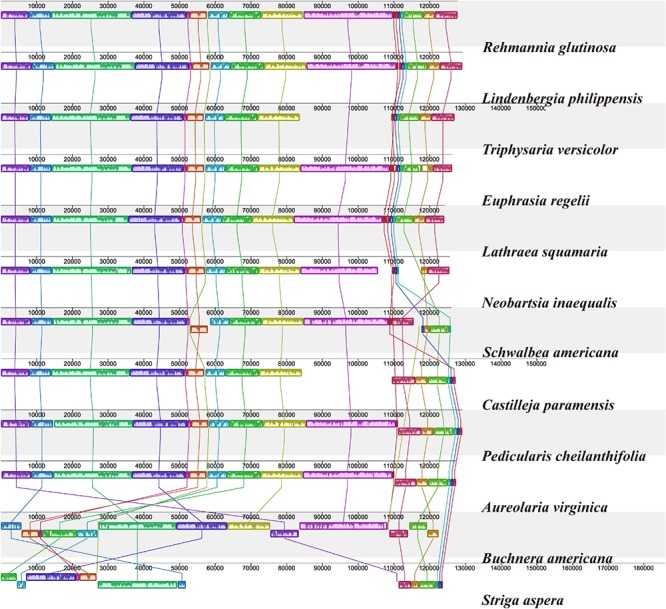
Plastome alignment of 12 species in the Orobanchaceae family. Alignment was carried out with only one copy of the IR taken out in Mauve v1.1.1 using R. glutinosa as a reference. Blocks on the top row are in the same orientation, while blocks on the bottom row are in inverse orientation.
FIGURE 5.

Nucleotide diversity (Pi) in the complete cp genomes of four Orobanchaceae species (E. regelii, Lathraea squamaria, Neobartsia inaequalis, and Schwalbea americana). Sliding window analysis with a window length of 600 bp and a step size of 200 bp.
Genomic structure and size varied in the 12 Orobanchaceae cp genomes and the IR/SC border regions of these species were also different (Figure 6). Fifteen genes including rps19, ycf1, rpl2, ndhF, ndhE, rpl23, rpl32, psbK, ndhA, ndhG, atpF, atpA, psbI, petL and trnH, were found in the LSC/IR and SSC/IR borders of the 12 plastomes. Of these, S. aspera, B. americana, and S. americana all exhibited larger plastome sizes due to the increased IR length, and the corresponding genes distributed in the SSC/IR border were quite different from other plastomes. Apart from the above three cp genomes, the IRs of E. regelii were much longer than in other cp genomes, especially in the area of the LSC/IRb and the IRb/SSC regions (Figure 6). The ndhF 3′-end sequence in the cp genomes of E. regelii and L. squamaria shared the region in the IRb with the rest of the ycf1 3′-end sequence, while the IRb-SSC border of L. philippensis, R. glutinosa, and N. inaequalis
FIGURE 6.

Chloroplast genome borders in 12 Orobanchaceae species. LSC (large single copy region), SSC (small single copy region), and IR (inverted repeat region). Ψ indicates a pseudogene. * indicates a region similar to a plastid gene.
were separated from the stop codon of ndhF by 32, 73, and 82 bp, respectively. Notably, genes located at the IR/SSC border of Castilleja paramensis, P. cheilanthifolia, and A. virginica showed a reverse gene order compared to E. regelii.
In order to detect whether the protein-coding genes of four Rhinantheae cp genomes (E. regelii, L. squamaria, N. inaequalis, and S. americana) were under selective pressure, rates of synonymous (dS) and non-synonymous (dN) substitutions, and the dN/dS ratios were calculated. As many pseudogenes were found in the cp genomes of E. regelii and L. squamaria, only 64 cp genes could be used for this analysis. The average dS values between paired Rhinantheae species (E. regelii-S. americana/E. regelii-L. squamaria/S. americana-L. squamaria/E. regelii-N. inaequalis/S. americana-N. inaequalis/L. squamaria-N. inaequalis) were 0.2175/0.1006/0.2131/0.0781/0.1861/0.0711 and the dN values ranged from 0 to 1.1435, with an average of 0.0718/0.0224/0.0857/0.0113/0.0639/0.0146, respectively (Supplementary Table S7). 305 paired dN/dS values were obtained most of which were less than 1, indicating that cp genes were under purifying selection. Only three genes (clpP, ycf2, rps14) had dN/dS values > 1, indicating that these genes had undergone positive selection.
Phylogenetic Analyses Based on Chloroplast Genome Sequence
Forty-five complete chloroplast genomes were used to infer the phylogenetic position of E. regelii (Supplementary Table S1). Phylogenetic analyses were performed using Maximum likelihood (ML) and Bayesian inference (BI) with Salvia miltiorrhiza, Tectona grandis, and Solanum lycopersicum as outgroup. Two datasets were used to infer phylogenetic relationships, one dataset included the complete cp genome and the other dataset only TMCRs of the 45 cp genomes. Both datasets yielded a consistent phylogenetic signal (Figure 7 and Supplementary Figure S1) Except for P. cheilanthifolia, which clustered with the outgroup, all other species of the Orobanchaceae formed a monophyletic group with high bootstrap and BI support. Similarly, E. regelii and two other Rhinantheae species (L. squamaria and N. inaequalis) formed a highly supported clade, with E. regelii being sister to N. inaequalis. Unexpectedly, S. americana, another species in the Rhinantheae tribe, clustered with Buchnera and Striga. Apart from L. squamaria, all holoparasitic species clustered in the same clade which also was the most derived in Orobanchaceae. Autotrophic genera including Lindenbergia and Rehmannia belonged to the earliest diverging groups, suggesting that autotrophic lineages may be the ancestors of parasitic lineages in Orobanchaceae.
FIGURE 7.

Maximum Likelihood tree based on the complete chloroplast genome. Numbers or asterisks above branches are statistical support values for maximum likelihood/Bayesian inference analyses, respectively, with * denoting maximum support in both analyses.
The phylogenetic relationship of 39 Euphrasia species was infered based on three cpDNA makers. All Euphrasia species formed a highly supported clade, however, species relationships remained unresolved (Supplementary Figure S2).
Discussion
Here we present the complete chloroplast genome of E. regelii which is the first complete plastome for this hemiparasitic genus. The chloroplast genome of E. regelii displays the typical quadripartite structure with a LSC and a SSC region which are separated by two inverted repeat regions. The structure is comparable to the one of other hemiparasitic species in Orobanchaceae (Wicke et al., 2013, 2016; Cho et al., 2018). In the plastome of E. regelii, only 71 protein-coding genes are retained due to pseudogenization of some plastid genes, especially ndhA – E, ndhG – K, and ycf15. Previous studies indicated that relaxed selective constraints in relation to photosynthesis resulted in extensive pseudogenization of ndh genes in some parasitic genera such as Lathraea, Pedicularis, and Schwalbea (Barrett et al., 2013; Wicke et al., 2013; Cho et al., 2018). In contrast, the ycf15 gene is usually truncated as a pseudogene in many angiosperm chloroplast genomes (Dong et al., 2013; Fajardo et al., 2013; Hu et al., 2016; Lu et al., 2016; Ge et al., 2018). Gene loss or plastome reduction is a common phenomenon in most parasitic plant species (Wicke et al., 2013; Samigullin et al., 2016), however, this was not observed in the cp genome of E. regelii as ndh genes were only pseudogenized but not lost. It has been shown that ndh genes were pseudogenized or lost entirely several times during land plant evolution, which is largely related to a heterotrophic lifestyle (Wicke et al., 2011; Barrett and Davis, 2012; Barrett et al., 2014; Graham et al., 2017; Wicke and Naumann, 2018). Euphrasia species are facultative hemiparasites which can complete their lifecycle without a host, however, they grow much better attached to a suitable host (Twyford et al., 2019). This facultative lifestyle probably accounts for the retention and subsequent pseudogenization of ndh genes. Similar to most angiosperm cp genomes, the overall GC content and the codon usage of E. regelii cp genome is heavily biased.
Repeat elements in plastomes were shown to play an important role in genomic rearrangements and recombination (Asano et al., 2004; Weng et al., 2013). Low number of repeat elements were found in the cp genome of E. regelii compared to the previously published Rehmannia plastome (Zeng et al., 2017). Most repeats were located in intergenic regions or ycf genes (ycf1 and ycf2) which is similar to the situation in other angiosperm lineages (Curci et al., 2015; Yang et al., 2016; Zhou et al., 2016). Chloroplast simple sequence repeats (cpSSRs) have been proven to be an important molecular marker for distinguishing species at lower taxonomic levels, and are therefore potentially useful marker for population genetics (Provan et al., 2001; Yang et al., 2011; Xue et al., 2012; Hu et al., 2016; Ruhsam et al., 2016). In the present study, 44 SSRs were detected in the E. regelii cp genome with mononucleotide repeats (A/T) being the most abundant type. Poly (A)/(T) SSRs are usually more common than other SSR repeat types in many plant cp gnomes (Yang et al., 2016; Asaf et al., 2017b; Dong et al., 2017; Li et al., 2018; Wang et al., 2018b; Ye et al., 2018; Zhou et al., 2018). Likewise, most cpSSRs were observed in non-coding regions, and only a small proportion was found in coding regions. CpSSRs located in non-coding regions are generally short mononucleotide tandem repeats and commonly show intraspecific variation in repeat numbers (Eguiluz et al., 2017). Therefore, cpSSR loci detected in this study will be useful tools for investigating levels of genetic diversity in Euphrasia and might even be able to discriminate between species.
Morphological similarity renders the reliable identification of many Euphrasia species challenging. In addition, standard DNA plant barcodes (Techen et al., 2014; Li et al., 2015) have failed to discriminate between Euphrasia species (Wang et al., 2018a). Therefore, it is necessary to develop Euphrasia specific DNA barcodes. Here, several highly variable cpDNA markers were obtained based on the comparative chloroplast genome analyses of Orobanchaceae species which could be tested as Euphrasia specific DNA barcodes. These regions might also provide sufficient genetic variation for resolving the phylogenetic relationships between Euphrasia species. Compared to two photoautotrophic species our results indicated that there are no structural rearrangements in the cp genome of E. regelii which is probably related to the facultative hemiparasitic life form of this species (Frailey et al., 2018). No major gene rearrangements were detected among four Rhinantheae species, except for B. americana which had a reversed SSC region. The SSC region is usually flipped in plastomes and the reversed SSC often show in a 50:50 ratio in plant cells (Palmer, 1985; Frailey et al., 2018). A similar situation was also detected in A. virginica and two other Pedicularideae species.
Size variability in cp genomes is usually due to the contraction and expansion of the IRs (He et al., 2017). This was apparent in the plastomes of S. aspera, B. americana, and S. americana where the IRs were much longer. Interestingly, the cp IR borders of B. americana are quite different from other Rhinantheae species as most of the repeat region extended into the SSC region. A previous study showed that B. americana belongs to an early diverging lineage in the Rhinantheae clade (McNeal et al., 2013), which suggests that the repeat expansion occurred independently in the B. americana lineage. The IR length of E. regelii was the longest out of the other three above cp genomes sequenced and was expanded much more than cp genomes of L. squamaria and N. inaequalis. Generally, ycf1 in the IRb is often pseudogenized in several angiosperm cp genomes (Daniell et al., 2006; Yao et al., 2016). However, no internal stop codons were detected in the coding sequence of ycf1 in E. regelii, thus the additional length of ycf1 affected the IR length and the gene distribution at the SC/IR borders. We hypothesize that the expansion of the IR caused a duplication of ycf1, like it has been reported for Eucommia ulmoides and Fagopyrum dibotrys (Wang et al., 2016, 2018b).
The results from the sequence divergence analysis of protein coding genes in four Rhinantheae plastomes indicated low sequence divergence and purifying selection (dN/dS < 1) for most genes which is consistent with the results from other studies (Rousseau-Gueutin et al., 2015; Xu et al., 2015; Zhou et al., 2016; Yin et al., 2018). Only three protein-coding genes (clpP, ycf2, rps14) were under positive selection. ClpP, which encodes a proteolytic subunit of the ATP-dependent protease, is very important for chloroplast biogenesis (Shikanai et al., 2001). Clp proteases are highly conserved in many organisms (Schirmer et al., 1996; Shikanai et al., 2001) but previous studies indicated that clpP genes showed significantly accelerated substitution rates and were under positive selection in Pelargonium plastid genomes (Weng et al., 2017). It is likely that clpP may have higher substitution rates in parasitic plant species. Ycf2 is one of the largest genes encoding for a putative membrane protein (Drescher et al., 2000; Kikuchi et al., 2013) and has rapidly evolved in several species of Fagopyrum, Ipomoea, Ophrys, and Mimosoideae (Cho et al., 2015; Mensous et al., 2017; Park et al., 2018; Roma et al., 2018). Likewise, ycf2 may have evolved at a faster rate in the Rhinantheae plastomes.
Chloroplast genomes which contain sufficient informative sites have been proven to be effective in resolving phylogenetic relationships among angiosperms even at lower taxonomic levels (Ma et al., 2014; Carbonell-Caballero et al., 2015; Yang et al., 2016; Dong et al., 2017; Zhang et al., 2017; Zhao et al., 2018). We retrieved the available cp genomes of non-parasitic (autotrophic) and parasitic species in the Orobanchaceae and inferred the phylogeny of Orobanchaceae based on ML and Bayesian methods. Our results were consistent with the results of previous studies based on nuclear and plastid markers (McNeal et al., 2013) as well as 17 cp genomes (Samigullin et al., 2016). Except for the placement of P. cheilanthifolia, all the parasitic species formed a highly supported clade. Unexpectedly, the overall genomic structure of P. cheilanthifolia is more similar to the cp genome of autotrophic species than to that of the closely related Pedicularis species. Thus, high sequence divergence of the P. cheilanthifolia plastome resulted in a discordant phylogenetic position. Previous phylogenetic analyses based on a few cpDNA markers did not support the monophyly of Rhinantheae (Olmstead et al., 2001) which is consistent with our results of four Rhinantheae species where S. americana was not included in Rhinantheae but was sister to Buchnereae. Also, Euphrasia was more closely related to Neobartsia than to Lathraea which is consistent with previous phylogenetic studies in the Rhinantheae tribe (McNeal et al., 2013; Pinto-Carrasco et al., 2017). Our results suggested that all non-parasitic species belonged to the earliest diverging lineages in Orobanchaceae indicating that autotrophy was the ancestral state in this mainly parasitic family. This has also been highlighted by previous studies (Bennett and Mathews, 2006; McNeal et al., 2013). However, to obtain a reliable inference of ancestral states a comprehensive sampling of all taxa in Orobanchaceae is necessary as limited taxon sampling can result in different tree topologies (Leebens-Mack et al., 2005; Eguiluz et al., 2017).
Due to the recent divergence of many Euphrasia species (Gussarova et al., 2008), the commonly used standard DNA barcodes are not variable enough to resolve phylogenetic relationships in Euphrasia which is obvious from our results based on three cpDNA fragments as well as previous phylogenetic studies (Gussarova et al., 2008; Wang et al., 2018a). However, even the complete chloroplast genome might not substantially raise species discriminatory power in evolutionarily young lineages, and very large numbers of characters from the nuclear genome are likely to be required for this task (Ruhsam et al., 2015).
Conclusion
The complete chloroplast genome of E. regelii, which is the first published cp genome in Euphrasia, provides a valuable genomic resource for this important medicinal plant and other Euphrasia species. The structure and gene content of the cp genome are comparable to other hemiparasitic and two photoautotrophic species in Orobanchaceae. No structural rearrangements were detected, however, 10 genes encoding the NAD(P)H dehydrogenase complex were widely pseudogenized but not lost. Coding gene sequence divergence analyses indicated that only three plastid genes were under positive selection. We also identified cpSSRs that could be used for population genetic studies in Euphrasia and whole cp genome comparison of E. regelii with other Orobanchaceae species indicated several variable hotspots, which could be used to develop DNA markers suitable for the discrimination between Euphrasia species, and for the inference of phylogenetic relationships.
Author Contributions
XW and TZ conceived and designed the experiments. JW, WL, YX, HZ, and FX performed the experiments and analyzed the data. TZ, XZ, XW, and MR wrote the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This research was supported by the Scientific Research Supporting Project for New Teacher of Xi’an Jiaotong University (Grant Nos. YX1K105 and 1191319802). The Royal Botanic Garden Edinburgh was supported by the Scottish Government’s Rural and Environment Science and Analytical Services Division.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00444/full#supplementary-material
Phylogenetic relationship inferred from Maximum Likelihood/Bayesian Inference analysis based on the most conserved regions (TMCRs) of the chloroplast genome. The numbers associated with each node are bootstrap support and posterior probability values, respectively. Asterisks indicate support values of 100/1.0.
Phylogenetic relationships of Euphrasia using cpDNA trnL intron, trnL-trnF, and atpB-rbcL. (A) Phylogenetic tree inferred from ML analysis. (B) Phylogenetic tree inferred from BI analysis. The numbers associated with each node are bootstrap support and posterior probability values.
List of plastome sequences included in the phylogenetic analyses.
Primers used for Sanger re-sequencing.
Models in ML and BI analysis based on different datasets.
Genes encoded in the Euphrasia regelii chloroplast genome.
Forward and Palindromic repeats in the Euphrasia regelii chloroplast genome.
Tandem repeats in the Euphrasia regelii chloroplast genome.
dN/dS ratio between pairwise of four Rhinantheae species protein coding sequences.
References
- Asaf S., Khan A. L., Aaqil Khan M., Muhammad Imran Q., Kang S.-M., Al-Hosni K., et al. (2017a). Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLoS One 12:e0182281. 10.1371/journal.pone.0182281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaf S., Waqas M., Khan A. L., Khan M. A., Kang S.-M., Imran Q. M., et al. (2017b). The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 8:304. 10.3389/fpls.2017.00304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T., Tsudzuki T., Takahashi S., Shimada H., Kadowaki K. (2004). Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res. 11 93–99. 10.1093/dnares/11.2.93 [DOI] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett C. F., Davis J. I. (2012). The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am. J. Bot. 99 1513–1523. 10.3732/ajb.1200256 [DOI] [PubMed] [Google Scholar]
- Barrett C. F., Davis J. I., Leebens-Mack J., Conran J. G., Stevenson D. W. (2013). Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 29 65–87. 10.1111/j.1096-0031.2012.00418.x [DOI] [PubMed] [Google Scholar]
- Barrett C. F., Freudenstein J. V., Li J., Mayfield-Jones D. R., Perez L., Pires J. C., et al. (2014). Investigating the path of plastid genome degradation in an early-transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Mol. Biol. Evol. 31 3095–3112. 10.1093/molbev/msu252 [DOI] [PubMed] [Google Scholar]
- Bendich A. J. (2004). Circular chloroplast chromosomes: the grand illusion. Plant Cell 16 1661–1666. 10.1105/tpc.160771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J. R., Mathews S. (2006). Phylogeny of the parasitic plant family Orobanchaceae inferred from phytochrome A. Am. J. Bot. 93 1039–1051. 10.3732/ajb.93.7.1039 [DOI] [PubMed] [Google Scholar]
- Benson G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi G., Mao Y., Xing Q., Cao M. (2018). HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics 110 18–22. 10.1016/j.ygeno.2017.08.001 [DOI] [PubMed] [Google Scholar]
- Carbonell-Caballero J., Alonso R., Ibañez V., Terol J., Talon M., Dopazo J. (2015). A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 32 2015–2035. 10.1093/molbev/msv082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron H., Dumas S., Marque G., Messier C., Bandou E., Petit R. J., et al. (2000). Spatial and temporal distribution of chloroplast DNA polymorphism in a tropical tree species. Mol. Ecol. 9 1089–1098. 10.1046/j.1365-294x.2000.00970.x [DOI] [PubMed] [Google Scholar]
- Cho K.-S., Yun B.-K., Yoon Y.-H., Hong S.-Y., Mekapogu M., Kim K.-H., et al. (2015). Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS One 10:e0125332. 10.1371/journal.pone.0125332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W.-B., Lee D.-H., Choi I.-S., Lee J.-H. (2018). The complete chloroplast genome of hemi-parasitic Pedicularis hallaisanensis (Orobanchaceae). Mitochondrial DNA Part B 3 235–236. 10.1080/23802359.2018.1437820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curci P. L., De Paola D., Danzi D., Vendramin G. G., Sonnante G. (2015). Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other asteraceae. PLoS One 10:e0120589. 10.1371/journal.pone.0120589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniell H., Lee S.-B., Grevich J., Saski C., Quesada-Vargas T., Guda C., et al. (2006). Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes. Theor. Appl. Genet. 112: 1503. 10.1007/s00122-006-0254-x [DOI] [PubMed] [Google Scholar]
- Darling A. C. E., Mau B., Blattner F. R., Perna N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14 1394–1403. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierckxsens N., Mardulyn P., Smits G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45: e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W., Xu C., Cheng T., Zhou S. (2013). Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS One 8: e77965. 10.1371/journal.pone.0077965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W., Xu C., Li W., Xie X., Lu Y., Liu Y., et al. (2017). Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences. Front. Plant Sci. 8: 1148. 10.3389/fpls.2017.01148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle J. J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19 11–15. [Google Scholar]
- Drescher A., Ruf S., Calsa T., Carrer H., Bock R. (2000). The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22 97–104. 10.1046/j.1365-313x.2000.00722.x [DOI] [PubMed] [Google Scholar]
- Eguiluz M., Rodrigues N. F., Guzman F., Yuyama P., Margis R. (2017). The chloroplast genome sequence from Eugenia uniflora, a myrtaceae from neotropics. Plant Syst. Evol. 303 1199–1212. 10.1007/s00606-017-1431-x [DOI] [Google Scholar]
- Fajardo D., Senalik D., Ames M., Zhu H., Steffan S. A., Harbut R., et al. (2013). Complete plastid genome sequence of Vaccinium macrocarpon: structure, gene content, and rearrangements revealed by next generation sequencing. Tree Genet. Genomes 9 489–498. 10.1007/s11295-012-0573-9 [DOI] [Google Scholar]
- Frailey D. C., Chaluvadi S. R., Vaughn J. N., Coatney C. G., Bennetzen J. L. (2018). Gene loss and genome rearrangement in the plastids of five hemiparasites in the family orobanchaceae. BMC Plant Biol. 18: 30. 10.1186/s12870-018-1249-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer K. A., Pachter L., Poliakov A., Rubin E. M., Dubchak I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32 W273–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French G. C., Hollingsworth P. M., Silverside A. J., Ennos R. A. (2008). Genetics, taxonomy and the conservation of British Euphrasia. Conserv. Genet. 9 1547–1562. 10.1007/s10592-007-9494-9 [DOI] [Google Scholar]
- Ge J., Cai L., Bi G.-Q., Chen G., Sun W. (2018). Characterization of the complete chloroplast genomes of Buddleja colvilei and B. sessilifolia: implications for the taxonomy of Buddleja L. Molecules 23 1248. 10.3390/molecules23061248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham S. W., Lam V. K. Y., Merckx V. S. F. T. (2017). Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol. 214 48–55. 10.1111/nph.14398 [DOI] [PubMed] [Google Scholar]
- Guo X., Liu J., Hao G., Zhang L., Mao K., Wang X., et al. (2017). Plastome phylogeny and early diversification of Brassicaceae. BMC Genomics 18:176. 10.1186/s12864-017-3555-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussarova G., Popp M., Vitek E., Brochmann C. (2008). Molecular phylogeny and biogeography of the bipolar Euphrasia (Orobanchaceae): recent radiations in an old genus. Mol. Phylogenet. Evol. 48 444–460. 10.1016/j.ympev.2008.05.002 [DOI] [PubMed] [Google Scholar]
- He L., Qian J., Li X., Sun Z., Xu X., Chen S. (2017). Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22:249. 10.3390/molecules22020249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D., Yang H., Jin C., Noel H. (1998). Scrophulariaceae Flora of China. Beijing: Science Press. [Google Scholar]
- Hu Y., Woeste K. E., Zhao P. (2016). Completion of the chloroplast genomes of five chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7: 1955. 10.3389/fpls.2016.01955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S., Bédard J., Hirano M., Hirabayashi Y., Oishi M., Imai M., et al. (2013). Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 339 571–574. 10.1126/science.1229262 [DOI] [PubMed] [Google Scholar]
- Kurtz S., Choudhuri J. V., Ohlebusch E., Schleiermacher C., Stoye J., Giegerich R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 4633–4642. 10.1093/nar/29.22.4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leebens-Mack J., Raubeson L. A., Cui L., Kuehl J. V., Fourcade M. H., Chumley T. W., et al. (2005). Identifying the basal angiosperm node in chloroplast genome phylogenies: sampling one’s way out of the felsenstein zone. Mol. Biol. Evol. 22 1948–1963. 10.1093/molbev/msi191 [DOI] [PubMed] [Google Scholar]
- Li L., Wang H. (2003). Studies on the chemical constituents from the water-soluble part of Euphrasia regelii. China J. Chin. Mater. Med. 28 733–734. [PubMed] [Google Scholar]
- Li X., Li Y., Zang M., Li M., Fang Y. (2018). Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int. J. Mol. Sci. 19: 2443. 10.3390/ijms19082443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Yang Y., Henry R. J., Rossetto M., Wang Y., Chen S. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. 90 157–166. 10.1111/brv.12104 [DOI] [PubMed] [Google Scholar]
- Lohse M., Drechsel O., Kahlau S., Bock R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41 W575–W581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S., Hou M., Du F. K., Li J., Yin K. (2016). Complete chloroplast genome of the oriental white oak: Quercus aliena blume. Mitochondrial DNA Part A 27 2802–2804. [DOI] [PubMed] [Google Scholar]
- Ma P.-F., Zhang Y.-X., Zeng C.-X., Guo Z.-H., Li D.-Z. (2014). Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 63 933–950. 10.1093/sysbio/syu054 [DOI] [PubMed] [Google Scholar]
- McNeal J. R., Bennett J. R., Wolfe A. D., Mathews S. (2013). Phylogeny and origins of holoparasitism in Orobanchaceae. Am. J. Bot. 100 971–983. 10.3732/ajb.1200448 [DOI] [PubMed] [Google Scholar]
- Mensous M., Van De Paer C., Manzi S., Bouchez O., Baâli-Cherif D., Besnard G. (2017). Diversity and evolution of plastomes in Saharan mimosoids: potential use for phylogenetic and population genetic studies. Tree Genet. Genomes 13: 48. [Google Scholar]
- Moura M., Dias E. F., Belo Maciel M. G. (2018). Conservation genetics of the highly endangered Azorean endemics Euphrasia azorica and Euphrasia grandiflora using new SSR data. Conserv. Genet. 19 1211–1222. 10.1007/s10592-018-1089-0 [DOI] [Google Scholar]
- Nguyen L.-T., Schmidt H. A., Von Haeseler A., Minh B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmstead R. G., Pamphilis C. W., Wolfe A. D., Young N. D., Elisons W. J., Reeves P. A. (2001). Disintegration of the Scrophulariaceae. Am. J. Bot. 88 348–361. 10.2307/2657024 [DOI] [PubMed] [Google Scholar]
- Palmer J. D. (1985). Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19 325–354. 10.1146/annurev.genet.19.1.325 [DOI] [PubMed] [Google Scholar]
- Park I., Yang S., Kim W. J., Noh P., Lee H. O., Moon B. C. (2018). The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic Pharbitidis Semen (Seeds of I. nil or I. purpurea). Front. Plant Sci. 9:965. 10.3389/fpls.2018.00965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R. K., Jain M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7: e30619. 10.1371/journal.pone.0030619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Carrasco D., Scheunert A., Heubl G., Rico E., Martínez-Ortega M. M. (2017). Unravelling the phylogeny of the root-hemiparasitic genus Odontites (tribe Rhinantheae, Orobanchaceae): evidence for five main lineages. Taxon 66 886–908. 10.12705/664.6 [DOI] [Google Scholar]
- Posada D., Crandall K. A. (1998). Modeltest: testing the model of DNA substitution. Bioinformatics 14 817–818. 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- Provan J., Powell W., Hollingsworth P. M. (2001). Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol. Evol. 16 142–147. 10.1016/s0169-5347(00)02097-8 [DOI] [PubMed] [Google Scholar]
- Roma L., Cozzolino S., Schlüter P. M., Scopece G., Cafasso D. (2018). The complete plastid genomes of Ophrys iricolor and O. sphegodes (Orchidaceae) and comparative analyses with other orchids. PLoS One 13: e0204174. 10.1371/journal.pone.0204174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F., Teslenko M., Van Der Mark P., Ayres D. L., Darling A., Höhna S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Boil. 61 539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau-Gueutin M., Bellot S., Martin G. E., Boutte J., Chelaifa H., Lima O., et al. (2015). The chloroplast genome of the hexaploid Spartina maritima (Poaceae, Chloridoideae): comparative analyses and molecular dating. Mol. Phylogenet. Evol. 93 5–16. 10.1016/j.ympev.2015.06.013 [DOI] [PubMed] [Google Scholar]
- Rozas J., Ferrer-Mata A., Sánchez-Delbarrio J. C., Guirao-Rico S., Librado P., Ramos-Onsins S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34 3299–3302. 10.1093/molbev/msx248 [DOI] [PubMed] [Google Scholar]
- Ruhsam M., Clark A., Finger A., Wulff A. S., Mill R. R., Thomas P. I., et al. (2016). Hidden in plain view: cryptic diversity in the emblematic Araucaria of New Caledonia. Am. J. Bot. 103 888–898. 10.3732/ajb.1500487 [DOI] [PubMed] [Google Scholar]
- Ruhsam M., Rai H. S., Mathews S., Ross T. G., Graham S. W., Raubeson L. A., et al. (2015). Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Mol. Ecol. Resour. 15 1067–1078. 10.1111/1755-0998.12375 [DOI] [PubMed] [Google Scholar]
- Saarela J. M., Burke S. V., Wysocki W. P., Barrett M. D., Clark L. G., Craine J. M., et al. (2018). A 250 plastome phylogeny of the grass family (Poaceae): topological support under different data partitions. PeerJ 6: e4299. 10.7717/peerj.4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samigullin T. H., Logacheva M. D., Penin A. A., Vallejo-Roman C. M. (2016). Complete plastid genome of the recent holoparasite Lathraea squamaria reveals earliest stages of plastome reduction in Orobanchaceae. PLoS One 11: 0150718. 10.1371/journal.pone.0150718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer E. C., Glover J. R., Singer M. A., Lindquist S. (1996). HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem. Sci. 21 289–296. 10.1016/s0968-0004(96)10038-4 [DOI] [PubMed] [Google Scholar]
- Secretariat G. (2017). GBIF Backbone Taxonomy Checklist dataset. Available at: https://www.gbif.org/dataset/d7dddbf4-2cf0-4f39-9b2a-bb099caae36c (accessed October 16, 2018). [Google Scholar]
- Shikanai T., Shimizu K., Ueda K., Nishimura Y., Kuroiwa T., Hashimoto T. (2001). The chloroplast clpP gene, encoding a proteolytic subunit of ATP-Dependent protease, is indispensable for chloroplast development in tobacco. Plant Cell Physiol. 42 264–273. 10.1093/pcp/pce031 [DOI] [PubMed] [Google Scholar]
- Shuya C., Shengda Q., Xingguo C., Zhide H. (2004). Identification and determination of effective components in Euphrasia regelii by capillary zone electrophoresis. Biomed. Chromatogr. 18 857–861. 10.1002/bmc.401 [DOI] [PubMed] [Google Scholar]
- Talavera G., Castresana J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56 564–577. 10.1080/10635150701472164 [DOI] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techen N., Parveen I., Pan Z., Khan I. A. (2014). DNA barcoding of medicinal plant material for identification. Curr. Opin. Biotechnol. 25 103–110. 10.1016/j.copbio.2013.09.010 [DOI] [PubMed] [Google Scholar]
- Thiel T., Michalek W., Varshney R., Graner A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106 411–422. 10.1007/s00122-002-1031-0 [DOI] [PubMed] [Google Scholar]
- Twyford A. D., Frachon N., Wong E. L. Y., Metherell C., Brown M. R. (2019). Life history evolution and phenotypic plasticity in parasitic eyebrights (Euphrasia, Orobanchaceae). bioRxiv [Preprint]. 10.1101/362400 [DOI] [PubMed] [Google Scholar]
- Vitek E. (1998). Are the taxonomic concepts of agamospermous genera useful for autogamous groups – A critical discussion using the example of Euphrasia (Scrophulariaceae). Folia Geobot. 33 349–352. 10.1007/bf03216211 [DOI] [Google Scholar]
- Wang L., Wuyun T.-N., Du H., Wang D., Cao D. (2016). Complete chloroplast genome sequences of Eucommia ulmoides: genome structure and evolution. Tree Genet. Genomes 12 1–15. [Google Scholar]
- Wang X., Gussarova G., Ruhsam M., Hollingsworth P. M., De Vere N., Metherell C., et al. (2018a). DNA barcoding a taxonomically complex hemiparasitic genus reveals deep divergence between ploidy levels but lack of species-level resolution. AoB Plants 10: ly026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhou T., Bai G., Zhao Y. (2018b). Complete chloroplast genome sequence of Fagopyrum dibotrys: genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 8: 12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M.-L., Blazier J. C., Govindu M., Jansen R. K. (2013). Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Mol. Biol. Evol. 31 645–659. 10.1093/molbev/mst257 [DOI] [PubMed] [Google Scholar]
- Weng M.-L., Ruhlman T. A., Jansen R. K. (2017). Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 214 842–851. 10.1111/nph.14375 [DOI] [PubMed] [Google Scholar]
- Wicke S., Muller K. F., De Pamphilis C. W., Quandt D., Wickett N. J., Zhang Y., et al. (2013). Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell 25 3711–3725. 10.1105/tpc.113.113373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S., Müller K. F., Depamphilis C. W., Quandt D., Bellot S., Schneeweiss G. M. (2016). Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proc. Natl. Acad. Sci. U.S.A. 113 9045–9050. 10.1073/pnas.1607576113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S., Naumann J. (2018). “Chapter eleven – Molecular evolution of plastid genomes in parasitic flowering plants,” in Advances in Botanical Research, eds Chaw S.-M., Jansen R. K. (Cambridge, MA: Academic Press; ), 315–347. 10.1016/bs.abr.2017.11.014 [DOI] [Google Scholar]
- Wicke S., Schneeweiss G. M., Depamphilis C. W., Müller K. F., Quandt D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76 273–297. 10.1007/s11103-011-9762-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M. J., Huang S. F., Huang T. C., Lee P. F., Lin T. P. (2005). Evolution of the Euphrasia transmorrisonensis complex (Orobanchaceae) in alpine areas of Taiwan. J. Biogeogr. 32 1921–1929. 10.1111/j.1365-2699.2005.01327.x [DOI] [Google Scholar]
- Wyman S. K., Jansen R. K., Boore J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20 3252–3255. 10.1093/bioinformatics/bth352 [DOI] [PubMed] [Google Scholar]
- Xu J.-H., Liu Q., Hu W., Wang T., Xue Q., Messing J. (2015). Dynamics of chloroplast genomes in green plants. Genomics 106 221–231. 10.1016/j.ygeno.2015.07.004 [DOI] [PubMed] [Google Scholar]
- Xue J., Wang S., Zhou S.-L. (2012). Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. 99 e240–e244. 10.3732/ajb.1100547 [DOI] [PubMed] [Google Scholar]
- Yang A.-H., Zhang J.-J., Yao X.-H., Huang H.-W. (2011). Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 98 e123–e126. 10.3732/ajb.1000532 [DOI] [PubMed] [Google Scholar]
- Yang Y., Zhou T., Duan D., Yang J., Feng L., Zhao G. (2016). Comparative analysis of the complete chloroplast Genomes of five Quercus species. Front. Plant Sci. 7: 959. 10.3389/fpls.2016.00959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 1586–1591. 10.1093/molbev/msm088 [DOI] [PubMed] [Google Scholar]
- Yao X., Tan Y.-H., Liu Y.-Y., Song Y., Yang J.-B., Corlett R. T. (2016). Chloroplast genome structure in Ilex (Aquifoliaceae). Sci. Rep. 6: 28559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W.-Q., Yap Z.-Y., Li P., Comes H. P., Qiu Y.-X. (2018). Plastome organization, genome-based phylogeny and evolution of plastid genes in Podophylloideae (Berberidaceae). Mol. Phylogenet. Evol. 127 978–987. 10.1016/j.ympev.2018.07.001 [DOI] [PubMed] [Google Scholar]
- Yin K., Zhang Y., Li Y., Du F. (2018). Different natural selection pressures on the atpF gene in evergreen sclerophyllous and deciduous oak species: evidence from comparative analysis of the complete chloroplast genome of Quercus aquifolioides with other oak species. Int. J. Mol. Sci. 19: 1042. 10.3390/ijms19041042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng S., Zhou T., Han K., Yang Y., Zhao J., Liu Z.-L. (2017). The complete chloroplast genome sequences of six Rehmannia species. Genes 8:103. 10.3390/genes8030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhou T., Kanwal N., Zhao Y., Bai G., Zhao G. (2017). Completion of eight Gynostemma BL. (Cucurbitaceae) chloroplast genomes: characterization, comparative analysis, and phylogenetic relationships. Front. Plant Sci. 8:1583. 10.3389/fpls.2017.01583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M.-L., Song Y., Ni J., Yao X., Tan Y.-H., Xu Z.-F. (2018). Comparative chloroplast genomics and phylogenetics of nine Lindera species (Lauraceae). Sci. Rep. 8: 8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Chen C., Wei Y., Chang Y., Bai G., Li Z., et al. (2016). Comparative transcriptome and chloroplast genome analyses of two related Dipteronia Species. Front. Plant Sci. 7: 1512. 10.3389/fpls.2016.01512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Wang J., Jia Y., Li W., Xu F., Wang X. (2018). Comparative chloroplast genome analyses of species in Gentiana section Cruciata (Gentianaceae) and the development of authentication markers. Int. J. Mol. Sci. 19: 1962. 10.3390/ijms19071962 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic relationship inferred from Maximum Likelihood/Bayesian Inference analysis based on the most conserved regions (TMCRs) of the chloroplast genome. The numbers associated with each node are bootstrap support and posterior probability values, respectively. Asterisks indicate support values of 100/1.0.
Phylogenetic relationships of Euphrasia using cpDNA trnL intron, trnL-trnF, and atpB-rbcL. (A) Phylogenetic tree inferred from ML analysis. (B) Phylogenetic tree inferred from BI analysis. The numbers associated with each node are bootstrap support and posterior probability values.
List of plastome sequences included in the phylogenetic analyses.
Primers used for Sanger re-sequencing.
Models in ML and BI analysis based on different datasets.
Genes encoded in the Euphrasia regelii chloroplast genome.
Forward and Palindromic repeats in the Euphrasia regelii chloroplast genome.
Tandem repeats in the Euphrasia regelii chloroplast genome.
dN/dS ratio between pairwise of four Rhinantheae species protein coding sequences.