

Abstract
Inducing post-translational protein knockdown is an important approach to probe biology and validate drug targets. An efficient strategy to achieve this involves expression of a protein of interest fused to an exogenous tag, allowing tag-directed chemical degraders to mediate protein ubiquitylation and proteasomal degradation. Here, we combine improved HaloPROTAC degrader probes with CRISPR/Cas9 genome editing technology to trigger rapid degradation of endogenous target proteins. Our optimized probe, HaloPROTAC-E, a chloroalkane conjugate of high-affinity VHL binder VH298, induced reversible degradation of two endosomally localized proteins, SGK3 and VPS34, with a DC50 of 3–10 nM. HaloPROTAC-E induced rapid (∼50% degradation after 30 min) and complete (Dmax of ∼95% at 48 h) depletion of Halo-tagged SGK3, blocking downstream phosphorylation of the SGK3 substrate NDRG1. HaloPROTAC-E more potently induced greater steady state degradation of Halo tagged endogenous VPS34 than the previously reported HaloPROTAC3 compound. Quantitative global proteomics revealed that HaloPROTAC-E is remarkably selective inducing only degradation of the Halo tagged endogenous VPS34 complex (VPS34, VPS15, Beclin1, and ATG14) and no other proteins were significantly degraded. This study exemplifies the combination of HaloPROTACs with CRISPR/Cas9 endogenous protein tagging as a useful method to induce rapid and reversible degradation of endogenous proteins to interrogate their function.
Technologies that enable the post-translational degradation of proteins allow interrogation of protein function and facilitate validation of targets for therapeutics development.1 One approach to induce post-translational protein knockdown is by means of proteolysis targeting chimeras (PROTACs),2 molecules capable of binding and recruiting an E3 ligase machinery to target proteins, to induce their ubiquitylation and proteasome-mediated degradation.3−5 A key advantage of PROTAC degraders is their ability to induce time and dose-dependent, reversible, and often complete depletion of target proteins inside cells. PROTACs, however, require a ligand capable of interacting with the desired target, and high-affinity ligands are lacking for the vast majority of human proteins. To circumvent this, alternative strategies have been developed that involve attaching various tags to target proteins, to enable these to be targeted by a compound capable of inducing interaction with an E3 ligase machinery.1,6,7 One of the first approaches made use of the plant E3 ligase TIR1, exogenously expressed in non-plant cells, to trigger ubiquitylation and degradation of target proteins fused to an Auxin-inducible degron (AID) tag on addition of the plant hormone Auxin.8−10 A more recent approach exploits the ability of a phthalimide-based chimeric compound called dTAG to bind proteins fused with a mutant FKBP12 tag and induce degradation via the cereblon (CRBN) E3 ligase.11−14 CRBN is endogenously expressed in most mammalian cells and, therefore, does not need to be overexpressed. Other methods include deGradFP and AdPROM, which rely on the overexpression of the von Hippel–Lindau (VHL) E3 ligase fused to a GFP binding nanobody capable of inducing degradation of GFP-tagged proteins,15−17 or the Trim–AWAY method that enables the TRIM21 E3 ligase to be recruited to targets via a specific antibody.18 These technologies each have their own caveats; for example, AID is prone to leakage without addition of its chemical inducer,19 and phthalimide-based conjugates that target CRBN are prone to chemical instability and off-target effects.20,21 The AID, ADPPROM, and Trim–AWAY methods all involve overexpression of E3 ligases that could ubiquitylate unknown off target proteins. Antibodies employed for the TRIM–AWAY method need to be introduced into cells, which is not straightforward for many applications, and furthermore these antibodies could also interact with unknown targets in cells. An important aspect to all of these approaches is the ability to introduce the various tags into the endogenous locus via gene editing, for example using CRISPR/Cas9,22 in order to then study the effect of the targeted degradation on the endogenous protein. However, this advance has only been achieved very recently and in only a few cases.9,12 The use of these methods to induce degradation of proteins of specific subcellular localization also remains understudied.
To address these issues, we decided to further develop the “HaloPROTAC” method. HaloPROTAC exploits a VHL ligand23 or a cIAP ligand24 linked to a chloroalkane moiety capable of forming a covalent bond with a tag termed HaloTag7, a bacterial dehalogenase made more stable by introducing multiple point mutations. As such, HaloPROTACs are designed to induce ubiquitylation and degradation of HaloTag7 fusion proteins. All work undertaken with the HaloPROTAC method to date has been performed on over-expressed proteins, and has not been demonstrated against endogenous targets. We have recently described optimized VHL ligands, with improved potency and specificity than previously described ligands, both as inhibitors25,26 and conjugated into PROTACs.27−29 We hypothesized that these optimized VHL ligands could be incorporated into novel HaloPROTAC probes to permit efficient degradation of endogenous proteins fused to HaloTag7 employing CRISPR gene editing technology.
We were interested in investigating ligand-induced knockdown of two target proteins localized at endosomes, namely SGK3 (serum and glucocorticoid kinase-3) and VPS34 (Class III PI 3-kinase). SGK3 is a member of the AGC protein kinase family, bearing similarity and overlapping substrate specificity to Akt.30 Like Akt, SGK3 is activated by phosphorylation on its T-loop by PDK1 and hydrophobic motif by mTORC2. While Akt activation is solely mediated by PI3K Class I, SGK3 is in addition switched on downstream of the Class III PI3-kinase termed VPS34.30,31 SGK3 interacts with PtdIns3P generated by VPS34 at the endosomal membrane and this promotes phosphorylation and activation by PDK1 and mTORC2.31 Recent studies suggest that the VPS34-SGK3 signaling axis contributes to resistance of breast cancer cells to class I PI3K and Akt inhibitor therapy.32 VPS34 also plays important roles in endosomal membrane trafficking and autophagy.33
Results and Discussion
Generation on Halo-VPS34 and SGK3-Halo Knock-in Cell Lines
To generate endogenous HaloTag7 fusion proteins, we utilized CRISPR/Cas9 gene-editing technology to attach the HaloTag7 variant to the N-terminus of VPS34 and C-terminus of SGK3 (Figure 1 and Methods). The tag was fused to the C-terminus of SGK3, as previous work revealed that an N-terminal tag impacted on PtdIns3P binding to the N-terminal PX domain.30 Previous studies have located epitope tags at the N-terminus of VPS34 and reported that this did not affect ability to activate SGK3.31 This also allowed comparison of whether the HaloPROTAC method could be used to degrade endogenous proteins coupled with N-terminal as well as C-terminal HaloTag7 fusions. The CRISPR gene-editing was undertaken in HEK293 cells that have been extensively deployed for the analysis of the VPS34-SGK3 signaling pathway.30,31 These cells are pseudotriploid,34 and carry three copies of SGK3 and VPS34 (HEK293genome.org). A total of four cell lines of each knock-in were selected from Immunoblot analysis and subjected to DNA sequencing analysis (Figure 1 and Supporting Information (SI) Figure S1). All the selected Halo-VPS34 cell lines were homozygous at the protein level (Figure 1), and DNA sequencing revealed clones 42 and 56 to be homozygous, with clones 55 and 80 containing knockout mutations in at least one allele. In the case of SGK3-Halo, we failed to identify any homozygous clones. Three clones contained one or two copies of SGK3 fused to HaloTag7 with remaining allele(s) wild type, and the fourth clone (Clone 2) possessed two copies of the SGK3-Halo knockin allele, with remaining copy of the SGK3 gene possessing a C-terminal truncation two residues away from the functionally important hydrophobic motif, predicted to destabilize the protein. The work described in this paper was undertaken with Halo-VPS34 Clone 56 and SGK3-Halo Clone 2.
Figure 1.

Generation of HaloTag7 endogenous fusion proteins by CRISPR/Cas9. The D10A Nickase form of Cas9 was chosen for increased specificity, and two guides were designed for each cell line.22 The SGK3-Halo donor also included an IRES2-eGFP cassette, allowing successfully integrated cells to be selected through FACs sorting for GFP positive cells. The Halo-VPS34 donor contained HaloTag7 only. Cells were selected through single cell sorting. Clones were selected and lysates screened by Immunoblot analysis, using antibodies against the endogenous proteins and a mouse monoclonal antibody against HaloTag7. The clones were characterized by DNA sequencing. Clone wt (wild type) refers to parental HEK293 cells, and ko (knockout) refers to previously described SGK3-knockout cell line31
HaloTag7 Fusions Do Not Interfere with VPS34 and SGK3 Function
To investigate to what extent the HaloTag7 fusion might impact on protein function, we initially studied the localization of the Halo-VPS34 and SGK3-Halo employing the HaloTag TMR Ligand from Promega, a fluorescently labeled probe that covalently reacts with HaloTag7 fusion proteins. In the wild type HEK293 cells, as expected no signal above background was detected (Figure 2A). In contrast in the Halo-VPS34 (Figure 2B) and SGK3-Halo (Figure 2C) knock-in cells, strong punctate staining co-localizing with the Rab5 early endosomal marker was observed, consistent with previous studies showing that VPS34 and SGK3 are located at the endosome.30,31 We also treated cells with the highly selective VPS34 inhibitor, VPS34-IN1,30 to reduce PtdIns-3P levels. Consistent with previous work,30 VPS34-IN1 induced relocalization of SGK3-Halo to the cytosol (Figure 2C). VPS34-IN1 appeared to strengthen endosomal localization of Halo-VPS34 (Figure 2B). We also analyzed PtdIns3P levels employing a previously reported method35 in which fixed cells are incubated with the PtdIns-3P binding 2XFYVE-probe labeled with Alexa Fluor-594 fluorescent dye. This confirmed endosomal localization of PtdIns-3P was suppressed following treatment with VPS34-IN1 (Figure 2D and SI Figure S2). The levels of PtdIns3P were similar in the wild type and Halo-VPS34 knock-in cells, suggesting that the Halo tag was not interfering with VPS34 activity. To further verify that HaloTag does not impact VPS34 complex stability, we immunoprecipitated VPS34 from wild type and Halo-VPS34 knock-in cells with a previously characterized VPS34 antibody and undertook Immunoblot analysis for the other subunits. These experiments confirmed that the levels of co-immunoprecipitating VPS15, Beclin1, Atg14, and UVRAG were unaffected by the Halo tag (Figure 2E). In order to determine potential effects of the HaloTag on SGK3 activity, we analyzed phosphorylation of the well characterized SGK3 substrate NDRG1 at Thr34631 in HEK293 wildtype, SGK3-Halo KI and SGK3 KO cells. As NDRG1 is also phosphorylated on this site by the Akt kinases, we assessed phosphorylation upon treatment with selective Akt inhibitor (3 μM AZD536336) in the presence or absence of the selective SGK3 inhibitor (1 μM 14H37). This assay revealed that SGK3 mediated phosphorylation of NDRG1 in wild type and SGK3-Halo cells was similar and higher than observed in SGK3 knock-out cells (Figure 2F). Taken together, this evidence suggests that the added Halo tag does not alter the function and activity of SGK3 within the cell.
Figure 2.

Localization and function of VPS34 and SGK3 are unaffected by fusion to HaloTag7. A–C. Parental 293 (A), Halo-VPS34 (B), and SGK3-Halo (C) cells were treated for 15 min with HaloTag-TMR Ligand, and the ligand was washed out for 15 min. Cells were stained with Rab5 early endosomal marker to detect localization of HaloTag fusion proteins on endosome. Assays were performed in the presence or absence of 1 μM VPS34-IN1. D. Quantification of PtdIns3P, measured using a 2XFYVE domain probe, colocalizing to Rab5 expressing endosomes in Parental 293 and Halo-VPS34 cells. E. VPS34 was immunoprecipitated from Parental 293 and Halo-VPS34 cells, and immunoprecipitates were blotted for VPS34 complex components. F. Parental 293, SGK3-KO, and SGK3-Halo cells were treated for 1 h with either Akt inhibitor AZD5363 (3 μM), SGK3 inhibitor 14H (1 μM), or both. Cells were lysed and immunoblotted for the targets described.
Elaboration of HaloPROTAC Compounds
We began our ligand design by considering HaloPROTAC probes previously described by Buckley et al. that include isoindolinone-based HaloPROTAC3 (Figure 3a).23 Based on information from our extensive structure–activity relationships and structure-based design of VHL ligands,25,38 we hypothesized that substitution of the isoindolinone moiety of HaloPROTAC3 with other groups, optimized for VHL binding affinity could improve degradation activity (Figure 3b). In a first set of compounds, we replaced the isoindolinone group with N-acylamides of l-tert-leucine on the left-hand side of the molecule (as in VHL ligand VH032),38 maintaining the chloroalkane linker attachment point at the phenyl ring on the right-hand side (Figure 3c). We therefore synthesized compounds A–B bearing linkers of 15 and 21 atoms, respectively. In a second compound set, the acetyl group at the left-hand side was replaced with a cyclopropylcyanoacetic moiety (as in VHL inhibitor VH29825) in compounds E, F, while maintaining chloroalkane linkers of the same length as HaloPROTACs A, B (Figure 3c). Additionally, we prepared two HaloPROTACs (C, D) having linkers of 16 and 22 atoms length respectively attached at the N-terminal acyl amine moiety of VH032, a conjugation pattern explored previously albeit with different linker lengths (13 and 19 atoms) (Figure 3d).23
Figure 3.

Chemical structures of HaloPROTAC3 and Compounds A–F. A. Chemical structure of previously described HaloPROTAC3. B. Chemical structure of VHL inhibitors VH032 and VH298. C–D General chemical structures of HaloPROTAC compounds A–F.
Evaluation of HaloPROTAC Compounds’ Ability to induce Degradation of Halo-VPS34 and SGK3-Halo
We treated the SGK3-Halo and Halo-VPS34 cell lines with 0.001-1 μM HaloPROTACs A–F for 48 h. Protein levels were analyzed and quantified by Immunoblot analysis employing a mouse monoclonal antibody that detects HaloTag7 specifically (Figure 4a–c). Three compounds, A, C, and E, induced degradation of HaloTag fusion proteins under these conditions in a dose-dependent manner (Figure 4a–c). These compounds contained a three PEG unit in the linker between the chloroalkane and VHL-binding moiety, whereas the non-active compounds possessed a five PEG linker, in agreement with previous observations that a three PEG linker is often optimal.23 Of the three active compounds, HaloPROTACs A and E, derived from the phenyl position were much more active than HaloPROTAC-C, derived from the acetyl position. HaloPROTAC-E contains a modified VHL-binding moiety derived from our latest optimized VHL ligand VH298.25 HaloPROTAC-E achieved a maximum degradation of 95% of both SGK3 and VPS34 target proteins at 300 nM, with a DC50 between 3 and 10 nM (Figure 4a–c). In assays undertaken up to 10 μM HaloPROTAC-E we did not observe a “hook effect” (data not shown), previously observed with some other degraders, where degradation is decreased at high concentrations as formation of binary complexes outcompetes the active ternary complex.39 Additionally, HEK293 wildtype, Halo-VPS3, and Halo-SGK3 cells were treated for 48 h with 0.001-1 μM HaloPROTAC-E and no effect on cell viability was observed by MTS assay (SI Figure S3A).
Figure 4.

HaloPROTAC-mediated degradation of HaloTag7-fusion proteins. A. SGK3-Halo and Halo-VPS34 cell lines were treated for 48h with increasing concentrations of each HaloPROTAC in parallel. Degradation of target protein measured by Immunoblot analysis for HaloTag7. B and C. Quantification of Western Blot intensity from A for SGK3-Halo (B) or Halo-VPS34 (C). Protein intensity was quantified and presented relative to an untreated control. D. SGK3-Halo and Halo-VPS34 cells were treated for up to 24 h with 300 nM HaloPROTAC-E. One hour before lysis, all conditions were treated with 1 μM AZD5363 to inhibit Akt activity. Cells were lysed and lysates analyzed by Immunoblot with the antibodies specified.
In order to visualize how HaloPROTAC-E mediated degradation of Halo-VPS34 and SGK3-Halo affected the SGK3 signaling pathway, we monitored NDGR1 phosphorylation at Thr346 on treatment with HaloPROTAC-E. All conditions were performed in the presence of Akt inhibitors to remove the effect of Akt activity on NDRG1. This revealed that treatment of SGK3-Halo and Halo-VPS34 HEK293 cells with HaloPROTAC-E induced dephosphorylation of NDRG1 at Thr346 (Figure 4D), confirming that degradation of SGK3 has a functional effect.
Characterization of HaloPROTAC-E
We next sought to further characterize HaloPROTAC-E by assessing kinetics of degradation and validating the Cullin2-VHL mediated mechanism of degradation. At 300 nM treatment, 50% degradation of SGK3-Halo was achieved within 20–30 min and 50% Halo-VPS34 within 1–2 h (Figure 5a). To study whether degradation is reversible, we treated cells with HaloPROTAC-E for 24 h, and after washout of the compound, quantified expression of Halo-tagged SGK3 and VPS34. Increased expressions of SGK3 and VPS34 were observed 4 h after removal of HaloPROTAC-E. Virtually normal levels of SGK3-Halo expression were observed after 24 h (Figure 5b), but in the case of VPS34, levels were still ∼2-fold reduced after 48 h (SI Figure S3b). To confirm HaloPROTAC-E-mediated degradation of Halo-tagged SGK3 and VPS34 occurs through the Cullin2-VHL E3 Ligase,40,41 we pre-treated cells with the Cullin E3 ligase Neddylation inhibitor, MLN4924 (3 μM for 3 h prior to HaloPROTAC-E treatment) and found that this blocked the ability of HaloPROTAC-E (300 nM, 4 h) to induce degradation of Halo-tagged SGK3 (Figure 5c) and VPS34 (SI Figure S3c), with PROTAC-induced protein depletion reduced from 70% to 30%. Treatment of cells with the proteasome inhibitor MG132 (50 μM, 0.5 h prior to HaloPROTAC-E administration) had a similar effect (Figure 5C and SI Figure S1B). Hif1α, the physiological substrate of VHL, is stabilized on treatment with MLN4924 and MG132 due to blockade of Hif1α ubiquitination and degradation. However, Hif1α stabilization was not induced by treatment with 300 nM HaloPROTAC-E (Figure 5C). This data demonstrates that HaloPROTAC-E does not inhibit VHL when used at concentrations (nanomolar range) where it is active for ligand-induced protein degradation, in line with previous observations with other VHL-based PROTAC compounds.4,27 To further confirm degradation is VHL-dependent, we similarly pretreated SGK3-Halo and Halo-VPS34 cells for 15 min with 50 μM VH298 before HaloPROTAC-E treatment, and again observed substantial blockade of SGK3-Halo and HaloVPS34 degradation (Figure 5D).
Figure 5.

Mechanistic characterization of HaloPROTAC-E. A. SGK3-Halo and Halo-VPS34 cells were treated for up to 4 h with 300 nM HaloPROTAC-E. Cells were lysed and probed by Western Blot for HaloTag7, and quantified for percentage of remaining HaloTag7 protein. B. SGK3-Halo cells were treated for 24 h with 300 nM HaloPROTAC-E. After 24 h, cells were washed three times with DMEM and replaced with fresh media for the times indicated. Recovery of SGK3-Halo was analyzed by Immunoblot C. Halo-VPS34 cells were treated for 6 h with 300 nM HaloPROTAC-E, after pre-inhibition of Cullin Neddylation by MLN4924 (3 μM for 3h) or Proteasome by MG132 (50 μM for 30 min). Cells were lysed and remaining HaloTag7 fusion protein was analyzed by Immunoblot. C. SGK3-Halo and HaloVPS34 cells were treated for 6 h with 300 nM HaloPROTAC-E, after pre-inhibition of VHL by 50 μM VH298 for 15 min.
We next undertook a side-by-side comparison on the ability of HaloPROTAC-E and the previously reported HaloPROTAC323 to induce dose (Figure 6A) and time (Figure 6B) dependent degradation of Halo-VPS34. This revealed that HaloPROTAC-E exhibited a greater potency at inducing degradation of Halo-VPS34 than HaloPROTAC3 at any concentrations tested below 300 nM. For example, 10 nM HaloPROTAC-E for 24 h induced 65% degradation of Halo-VPS34 compared to 50% of the same concentration of HaloPROTAC3. Dose response degradation profiles revealed an approximately 2-fold reduction in estimated DC50. At concentrations 300 nM and above HaloPROTAC3 and HaloPROTAC-E induced similar degradation at the 24 h time point (Figure 6A). Time course analysis revealed that the rate of degradation of Halo-VPS34 up to 30 min was very similar to HaloPROTAC-E and HaloPROTAC3 (Figure 6B). However, after 30 min, significantly greater degradation of Halo-VPS34 was observed with HaloPROTAC-E. At 1–4 h, ∼75% degradation of Halo-VPS34 is observed with HaloPROTAC-E compared to ∼50% with HaloPROTAC3 (Figure 6B).
Figure 6.

Comparison of HaloPROTAC-E to previously reported HaloPROTAC3. A. Halo-VPS34 cells were treated in parallel for 24 h with 1-1000 nM HaloPROTAC-E or HaloPROTAC3. Cells were lysed and remaining HaloTag7 fusion protein was analyzed by Immunoblot. B. Halo-VPS34 cells were treated for up to 4 h with 300 nM HaloPROTAC-E or HaloPROTAC3. Cells were lysed and probed by Western Blot for HaloTag7, and quantified for percentage of remaining HaloTag7 protein.
As shown in Figure 2C, SGK3-Halo is endosomally localized, and on treatment with VPS34-IN1 translocates to the cytosol. We compared the rate at which endosomal (DMSO) versus cytosolic (1 μM VPS34-IN1) SGK3-Halo was degraded by HaloPROTAC-E and observed that both rate of degradation and Dmax at 24 h were similar in both conditions (Figure 7A). We were similarly able to visualize degradation of endosomal and cytosolic SGK3-Halo by immunofluorescence (Figure 7B–F).
Figure 7.

HaloPROTAC-E induces degradation of endosomally localized proteins. A. SGK3-Halo cells were treated for up to 24 h with 300 nM HaloPROTAC-E alone, or in combination with VPS34IN1 at 1 μM. Cells were lysed, and remaining SGK3-Halo was analyzed by Immunoblot. B–F. HEK293 cells were transfected for 24 h for SGK3-Halo expression, and treated for 8h with 1 μM HaloPROTAC-E in the presence or absence of 1 μM VPS34-IN1 before fixing and staining for HaloTag7 and endosomal marker Rab5. SGK3-Halo was overexpressed to facilitate detection and quantitation. Anti-HaloTag7 antibody was detected with Alexa Fluor-594 coupled secondary, and Rab5 by Alexa Fluor-488 coupled secondary antibodies.
Striking Specificity of HaloPROTAC-E
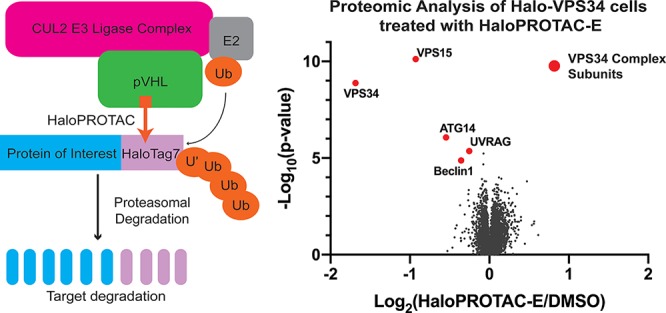
To establish the specificity of HaloPROTAC-E-induced protein degradation, we performed quantitative tandem-mass-tag (TMT)-labeled global proteomic analysis of Halo-VPS34 cells treated in the presence or absence of 300 nM HaloPROTAC-E for 4 h. Experiments undertaken in quadruplicate and analyzed in Proteome Discoverer v2.2 using Mascot search engine allowed relative quantification of 9786 proteins. This unbiased analysis revealed that HaloPROTAC-E was remarkably selective. Only protein levels of Halo-VPS34 (70% reduction) and its known regulatory subunits VPS15 (50% reduction), Beclin1 (20% reduction), ATG14 (30% reduction), and UVRAG (15% reduction) were impacted to statistically significant level (p value <10–4) (Figure 8A and SI Table S1). It is indeed striking that no other cellular protein was reduced by HaloPROTAC-E. Immunoblot analysis confirmed reduction of Halo-VPS34, VPS15, and Beclin1 expression induced by HaloPROTAC-E (Figure 8B). Time course analysis of VPS34 degradation revealed that at very early time points of 10–20 min, VPS15 was reduced at a similar rate to VPS34, whereas the reduction in Beclin1 expression was slower (Figure 8C–D). It should be noted that a previous study also observed degradation of VPS15 and Beclin1 when VPS34 expression was reduced using the ADPROM method.31 The VPS34 regulatory subunits could be degraded due to loss of stability when they are not in a complex, a hypothesis that is supported by previous siRNA knock-down data.42 In addition, we cannot rule out that these subunits could be degraded through a “by-stander” effect, by receiving collateral ubiquitination by the VHL ligase as a result of being part of the same complex.29
Figure 8.

Degradation of HaloTag7 fusions is highly specific and has a biological impact. A. Volcano plot quantifying proteins significantly downregulated on 4 h HaloPROTAC-E treatment in Halo-VPS34 cells. B. Immunoblot analysis of lysates taken for mass spectrometry analysis. C and D. HaloVPS34 cells were treated with 0.3 μM HaloPROTAC-E for up to 4 h. C. Immunoblot analysis of these lysates for VPS34 complex members. D. Quantification of relative protein levels of VPS34 complex members from 8C.
Conclusion
In summary, we describe the design and synthesis of a novel potent HaloPROTAC compound, termed HaloPROTAC-E, and show that this induces the rapid and efficient degradation of two endogenously HaloTag fusion proteins, SGK3 and VPS34. HaloPROTAC-E induced potent degradation, with a DC50 between 3 and 10 nM. Our quantitative mass spectrometry studies reveal that this is markedly selective, not degrading any other proteins other than the Halo-VPS34 and its known regulatory subunits. The rate of degradation of SGK3 (20–30 min for 50% degradation) was slightly more rapid than observed with VPS34 (1–2 h for 50% degradation) which is part of a complex. The dynamics of degradation were similar for both N-terminally tagged VPS34 and C-terminally tagged SGK3 suggesting that the tag can be attached to either end of a protein and that the approach is generalizable to other proteins. The HaloPROTAC compound described in this study was also shown to be more effective than the previously reported compound HaloPROTAC3. As VPS34 and SGK3 are localized at the endosome, our data demonstrate that the HaloPROTAC approach is suitable for degrading endosomal proteins. We would expect this technology to be applicable to cells beyond HEK293 cells. The limitation of the HaloPROTAC approach lies in the ability to generate homozygous HaloTag7 knock-in fusions. Using the current gene editing technology there are still challenges with obtaining the desired knock-in in all alleles. Screening of a large number of positive clones may be required, but with rapidly improving efficiency of gene editing technology, it should become more possible to generate these mutations rapidly. Some compromise may be needed, as in the case of SGK3-Halo (2 knock-in and 1 destabilized allele).
Because the chloroalkane handle reacts covalently with HaloTag7, a potential limitation of HaloPROTACs is that stoichiometric occupancy of the tagged protein is required to achieve complete degradation of the target protein. This is not the case of PROTACs bearing non-covalent target ligands, which instead can function catalytically at sub-stoichiometric concentrations relative to the target protein. Despite the clear strengths of non-covalent protein degradation modalities, potent ligands are often required, and structure-guided PROTAC design requires structural information on ternary complexes.28 Even with this knowledge, many variables affecting efficacy of PROTACs are still not thoroughly understood, and are highly target dependent. The HaloPROTAC approach removes these issues, allowing for exquisite selectivity and high affinity. Our improved HaloPROTAC approach, combining HaloPROTAC-E with CRISPR/Cas9 mediated endogenous protein tagging, provides a useful tool to interrogate an endogenous system and validate the therapeutic potential of degrading its protein target.
Methods
Biology
Materials
Triton X-100, EDTA, EGTA, sodium orthovanadate, sodium glycerophosphate, sodium fluoride, sodium pyrophosphate, 2-mercaptoethanol, sucrose, benzamidine, Tween 20, Tris-HCl, and sodium chloride were from Sigma. Tissue culture reagents, Novex 4-12% Bis-Tris gels and NuPAGE LDS sample buffer were from Invitrogen. Polyethylenimine was from Polysciences. Ampicillin was from Merck. CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) was from Promega. Plasmids used in the present study were generated by the MRC-PPU reagents and Services team (https://mrcppureagents.dundee.ac.uk/). All DNA constructs were verified by DNA sequencing, performed by the MRC-PPU DNA Sequencing and Service (http://www.dnaseq.co.uk). All constructs are available to request from the MRC-PPU reagents webpage (http://mrcppureagents.dundee.ac.uk), and the unique identifier (DU) numbers indicated provide direct links to the cloning and sequence details.
Cell Culture, Treatments, and Cell Lysis
HEK293 cells were purchased from the American Tissue Culture Collection and cultured in DMEM supplemented with 10% (v/v) fetal bovine serum, 2 mM l-glutamine, 100 U/mL penicillin, and 0.1 mg mL–1 streptomycin. Cell treatments were carried out as described in figure legends. The cells were lysed in buffer containing 50 mM Tris-HCl (pH 7.5), 1 mM EGTA, 1 mM EDTA, 1% (v/v) Triton X-100, 1 mM sodium orthovanadate, 50 mM NaF, 5 mM sodium pyrophosphate, 0.27 M sucrose, 10 mM sodium 2-glycerophosphate, 0.2 mM phenylmethylsulfonyl fluoride, and 1 mM benzamidine. Lysates were clarified by centrifugation at 16 000g for 10 min at 4 °C. Protein concentration was calculated using the Bradford assay (Thermo Scientific). Immunoblotting was performed using standard procedures. The signal was detected using a Licor Biosciences Odessey System and signal quantified in Image Studio Lite.
Antibodies
The following antibodies were raised in sheep, by the MRC-PPU reagents and Services team (https://mrcppureagents.dundee.ac.uk/) and affinity-purified against the indicated antigens: anti-Vps34 (S672B; third bleed; raised against full-length human Vps34) (DU3303), anti-Beclin1 (S900B; first bleed; raised against full-length human Beclin1) (DU7159), anti-UV-RAG (S323D; third bleed; raised against full-length human UV-RAG) (DU 36785), anti-SGK3 (S848D, sixth bleed; raised against human SGK3 PX domain comprising residues 1–130 of SGK3) (DU2034).
Anti-HaloTag7 was from Promega (G9211, G9281), anti-GAPDH was from Santa Cruz (sc–32233), Anti-VPS15 (14580S) and Rab5 (E6N8S) was purchased from Cell Signaling Technology. Anti-ATG14 was from MBL Life Science (PD026) Secondary antibodies coupled to IRDye680LT or IRDye800CW were obtained from Licor Biosciences. Secondary antibodies coupled to Horseradish Peroxidase (HRP) were obtained from Thermo Scientific. Secondary antibodies coupled to Alexa Fluor 488 and Alexa Fluor 594 were obtained from Thermo Scientific.
Generation of HaloTag7 Knock-in Cell Lines Using CRISPR-Cas9 Genome Editing
A modified Cas9 nickase system22 was used for the generation of N-terminal HaloTag7-VPS34, and C-terminal SGK3-HaloTag7 knock-in mutation. Optimal sgRNA pairs were identified (as close as possible to point of HaloTag7 insertion, with a low combined off-targeting score; (VPS34-sgRNA1: GCTACATCTATAGTTGTGACC (DU52071); sgRNA2: GCCCCATCGCACCGTCTGCAA (DU52082); SGK3-sgRNA1: GAGCAAAATAAGTCTATAGA (DU52684)); sgRNA2: GAAAAATAAGTCTTCTGAAGG (DU52662)) using the Sanger Institute CRISPR web tool (http://www.sanger.ac.uk/htgt/wge/find_crisprs). Complementary oligos with BbsI compatible overhangs were designed for each, annealed, and the dsDNA guide inserts ligated into BbsI-digested target vectors; the antisense guides (sgRNA2) were cloned onto the spCas9 D10A-expressing pX335 vector (Addgene plasmid no. 42335) and the sense guides (sgRNA1) into the puromycin-selectable pBABED P U6 plasmid (Dundee-modified version of the original Cell Biolabs pBABE plasmid). Donor constructs (VPS34-DU57077 and SGK3-DU52689) consisting of HaloTag7 or HaloTag7-IRES2-GFP flanked by ∼500 bp homology arms were synthesized by GeneArt (Life Technologies); each donor was engineered to contain sufficient silent mutations to prevent recognition and cleavage by Cas9 nuclease.
HEK293 knock-in cell lines were generated using 1ug each of appropriate guide plasmids and an additional 3 μg of donor plasmid. Sixteen hours post-transfection, cell selection was carried out using 2 μg/mL puromycin for 2 days. Transfections were repeated without puro selection prior to single-cell sorting by FACS, SGK3-Halo-IRES2-GFP cells were additionally sorted for GFP expression. Single cells were plated in individual wells of 96-well plates and viable clones were expanded. Integration of HaloTag7 at the target locus for knock-in clones was verified by Western blotting and genomic DNA sequencing of the targeted locus.
Immunofluorescence and PtdIns3P 2XFYVE Domain Staining
For visualization of endogenous Halo-VPS34 and SGK3-Halo, in-cell labeling of HaloTag7 fusion proteins was performed by adding HaloTag TMR Ligand to a final concentration of 5 μM for 15 min, followed by a 15-minute washout of unbound ligand with fresh DMEM. Following treatments described in figure legends, cells were fixed with 4% (v/v) paraformaldehyde and permeabilized with 1% (v/v) NP-40. Cells were blocked using 1% Bovine Serum Albumin (BSA) in PBS, then incubated for 1 h with primary antibody, washed three times in 0.2% BSA in PBS, and incubated for 1 h again with secondary antibody. For localization to endosomal compartments, Rab5 was stained with anti-Rab5 antibody and secondary anti-mouse secondary conjugated to Alexa Fluor 488. For detection of overexpressed SGK3-Halo protein, HaloTag7 was stained with anti-HaloTag7 pAb and anti-rabbit secondary conjugated to Alexa Fluor 594. Coverslips were washed once more in water and mounted using ProLong Gold Antifade (ThermoFisher #P36931).
For selective PtdIns3P staining, the GST-tagged HRS 2XFYVE domain probe, coupled to Alexa Fluor 594 was kindly donated by the Ganley laboratory. In short, the GST-tagged HRS 2× FYVE domain (residues 147-223) were expressed in Escherichia coli (BL21) and purified over a glutathione column using standard procedures. The recombinant protein was chemically conjugated to Alexa Fluor 594 using the Alexa Fluor Microscale Protein Labeling Kit (no. A30008) as per the manufacturer’s protocol. For staining, a similar protocol described earlier was followed.43 Following treatment described in the figure legends, cells were washed once on ice with phosphate-buffered saline and glutamate buffer (25 mM HEPES (pH 7.4), 25 mM KCl, 2.5 mM Mg acetate, 5 mM EGTA, 150 mM potassium glutamate). Coverslips were then immediately snap frozen in liquid nitrogen and thawed at RT for 0.5 min prior to two further washes with ice cold glutamate buffer prior to fixing by incubating cells in 3.7% (w/v) formaldehyde, 200 mM HEPES (pH 7.4). After 30 min at RT, fixed cells were quenched by incubating twice for 10 min in 10 mM HEPES, pH 7.4, and DMEM at RT. Coverslips were then blocked and stained as described above.
The images were collected on an LSM710 laser scanning confocal microscope (Carl Zeiss) using the ×63 Plan-Apochromat objective (NA 1.4), using a pinhole chosen to provide a uniform 0.8 um optical section thickness in all the fluorescence channels. For quantification, images from the microscope were imported into Volocity image processing software (PerkinElmer) and batch-processed using the same custom written programmes for all the images in an experimental group. For example, in each image, endosomes were identified from the Rab5 antibody staining, and the intensity of HaloTag7 protein or PtdIns3P in these objects was collected as the sum of the pixel intensities, normalized for the number of cells in each image. The graphs show the average sum of the intensity per cell in arbitrary units. Each treatment was repeated three times, and graphs shown are from representative experiments.
CoImmunoprecipitation
Cells were lysed in standard lysis buffer as described above. Endogenous VPS34 was Immunoprecipitated with a specific VPS34 antibody (S672B; third bleed, MRCPPU Reagents and Services), and beads were washed twice in the same lysis buffer +0.15 M NaCl (final). Protein was eluted from beads using 2X LDS Sample Buffer with 5% v/v Beta-Mercaptoethanol, and coimmunoprecipitating proteins detected by Western Blot.
Supplementary Methods
Full Chemistry and Mass Spectrometry Methods are provided in the Supporting Information
Acknowledgments
We thank the excellent technical support of the MRC-Protein Phosphorylation and Ubiquitylation Unit (PPU) DNA Sequencing Service, the MRC PPU tissue culture team (coordinated by E. Allen), MRC PPU Reagents and Services antibody purification teams (coordinated by H. McLauchlan and J. Hastie).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b01016.
Author Present Address
∥ (C.M.) University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford OX1 3TA, U.K.
This work was supported by the Medical Research Council (grant number MC_UU_12016/2 (to D.R.A.)); the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP7/2007–2013) as a Starting Grant to A.C. (grant agreement No. ERC–2012–StG–311460 DrugE3CRLs); the pharmaceutical companies supporting the Division of Signal Transduction Therapy Unit (Boehringer Ingelheim, GlaxoSmithKline, and Merck KGaA, to D.R.A.). H.T. is supported by an AstraZeneca BBSRC Studentship.
The authors declare the following competing financial interest(s): The A.C. laboratory receives sponsored research support from Boehringer Ingelheim and Nurix, Inc. A.C. is a scientific founder, director, and shareholder of Amphista Therapeutics, a company that is developing targeted protein degradation therapeutic platforms.
Supplementary Material
References
- Natsume T.; Kanemaki M. T. (2017) Conditional Degrons for Controlling Protein Expression at the Protein Level. Annu. Rev. Genet. 51, 83–102. 10.1146/annurev-genet-120116-024656. [DOI] [PubMed] [Google Scholar]
- Gu S.; Cui D.; Chen X.; Xiong X.; Zhao Y. (2018) PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. BioEssays 40, e1700247. 10.1002/bies.201700247. [DOI] [PubMed] [Google Scholar]
- Sakamoto K. M.; et al. (2001) Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 98, 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; et al. (2015) Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runcie A. C.; Chan K.-H.; Zengerle M.; Ciulli A. (2016) Chemical genetics approaches for selective intervention in epigenetics. Curr. Opin. Chem. Biol. 33, 186–194. 10.1016/j.cbpa.2016.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinjiyan F. A.; Carbonneau S.; Ross N. T. (2017) Lead discovery and chemical biology approaches targeting the ubiquitin proteasome system. Bioorg. Med. Chem. Lett. 27, 4589–4596. 10.1016/j.bmcl.2017.08.058. [DOI] [PubMed] [Google Scholar]
- Yesbolatova A.; Tominari Y.; Kanemaki M. T. (2018) Ligand-induced genetic degradation as a tool for target validation. Drug Discovery Today: Technol. 10.1016/j.ddtec.2018.11.001. [DOI] [PubMed] [Google Scholar]
- Nishimura K.; Fukagawa T.; Takisawa H.; Kakimoto T.; Kanemaki M. (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922. 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Natsume T.; Kiyomitsu T.; Saga Y.; Kanemaki M. T. (2016) Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep. 15, 210–218. 10.1016/j.celrep.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Nora E. P.; et al. (2017) Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944. 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; e22
- Erb M. A.; et al. (2017) Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B.; et al. (2018) The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441. 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub A. S.; et al. (2017) YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588. 10.1016/j.cell.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; e28
- Brunetti L.; et al. (2018) Mutant NPM1Maintains the Leukemic State through HOX Expression. Cancer Cell 34, 499–512. 10.1016/j.ccell.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; e9
- Fulcher L. J. (2016) An affinity-directed protein missile system for targeted proteolysis. Open Biol. 6, 160255. 10.1098/rsob.160255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussinus E.; Kanca O.; Affolter M. (2012) Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 19, 117–121. 10.1038/nsmb.2180. [DOI] [PubMed] [Google Scholar]
- Caussinus E.; Affolter M. (2016) deGradFP: A System to Knockdown GFP-Tagged Proteins. Methods Mol. Biol. 1478, 177–187. 10.1007/978-1-4939-6371-3_9. [DOI] [PubMed] [Google Scholar]
- Clift D.; et al. (2017) A Method for the Acute and Rapid Degradation of Endogenous Proteins. Cell 171, 1692–1706. 10.1016/j.cell.2017.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; e18
- Lambrus B. G.; Moyer T. C.; Holland A. J. (2018) Applying the auxin-inducible degradation system for rapid protein depletion in mammalian cells. Methods Cell Biol. 144, 107–135. 10.1016/bs.mcb.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Winter G. E.; et al. (2015) DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishoey M.; et al. (2018) The translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol. 13, 553. 10.1021/acschembio.7b00969. [DOI] [PubMed] [Google Scholar]
- Ran F. A.; et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; et al. (2015) HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 10, 1831–1837. 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoshige S.; Hashimoto Y.; Ishikawa M. (2016) Efficient protein knockdown of HaloTag-fused proteins using hybrid molecules consisting of IAP antagonist and HaloTag ligand. Bioorg. Med. Chem. 24, 3144–3148. 10.1016/j.bmc.2016.05.035. [DOI] [PubMed] [Google Scholar]
- Frost J.; et al. (2016) Potent and selective chemical probe of hypoxic signalling downstream of HIF-α hydroxylation via VHL inhibition. Nat. Commun. 7, 13312. 10.1038/ncomms13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P.; et al. (2018) Group-Based Optimization of Potent and Cell-Active Inhibitors of the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase: Structure–Activity Relationships Leading to the Chemical Probe (2S,4R)-1-((S)-2-(1-Cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazl-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 61, 599–618. 10.1021/acs.jmedchem.7b00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K.-H.; Ciulli A. (2015) Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; et al. (2017) Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C.; et al. (2017) Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 8, 830. 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bago R.; et al. (2014) Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J. 463, 413–427. 10.1042/BJ20140889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N.; et al. (2018) Mechanism of activation of SGK3 by growth factors via the Class 1 and Class 3 PI3Ks. Biochem. J. 475, 117–135. 10.1042/BCJ20170650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bago R.; et al. (2016) The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J. 35, 1902–1922. 10.15252/embj.201693929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backer J. M. (2008) The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem. J. 410, 1–17. 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- Bylund L.; Kytola S.; Lui W.-O.; Larsson C.; Weber G. (2004) Analysis of the cytogenetic stability of the human embryonal kidney cell line 293 by cytogenetic and STR profiling approaches. Cytogenet. Genome Res. 106, 28–32. 10.1159/000078556. [DOI] [PubMed] [Google Scholar]
- Nascimbeni A. C.; Codogno P.; Morel E. (2017) Local detection of PtdIns3P at autophagosome biogenesis membrane platforms. Autophagy 13, 1602–1612. 10.1080/15548627.2017.1341465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B. R.; et al. (2012) Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 11, 873–887. 10.1158/1535-7163.MCT-11-0824-T. [DOI] [PubMed] [Google Scholar]
- Halland N.; et al. (2015) Discovery of N-[4-(1H-Pyrazolo[3,4-b]pyrazin-6-yl)-phenyl]-sulfonamides as Highly Active and Selective SGK1 Inhibitors. ACS Med. Chem. Lett. 6, 73–78. 10.1021/ml5003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; et al. (2014) Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 57, 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass E. F. J.; Miller C. J.; Sparer G.; Shapiro H.; Spiegel D. A. (2013) A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc. 135, 6092–6099. 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K.; et al. (1999) Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. U. S. A. 96, 12436–12441. 10.1073/pnas.96.22.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W.; Yang H. (2016) The structure and regulation of Cullin 2 based E3 ubiquitin ligases and their biological functions. Cell Div. 11, 7. 10.1186/s13008-016-0020-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E.; Kishi C.; Inoue K.; Mizushima N. (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 19, 5360–5372. 10.1091/mbc.e08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson M. J.; et al. (2015) mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 34, 2272–2290. 10.15252/embj.201590992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.