

 This work is licensed under a
This work is licensed under a Abstract
Broncho-pulmonary neuroendocrine neoplasms (BP-NENs) are neoplasms orphan of an efficient therapy. Available medical treatments derived from clinical trials are not specific for the management of this malignancy. Sunitinib is a multi-receptor tyrosine-kinases (RTKs) inhibitor that has already shown its efficacy in NENs, but there are no available data about its action in BP-NENs. Therefore, our aim was to understand the effects of RTKs inhibition promoted by sunitinib in order to evaluate new putative targets useful in malignancy treatment. Since our results underlined a role for EGFR and IGF1R in modulating sunitinib antiproliferative action, we investigated the effects of erlotinib, an EGFR inhibitor, and linsitinib, an IGF1R inhibitor, in order to understand their function in regulating cells behaviour. Cell viability and caspase activation were evaluated on two immortalised human BP-NEN cell lines and primary cultures. Our results showed that after treatment with sunitinib and/or IGF1, EGF and VEGF, the antiproliferative effect of sunitinib was counteracted by EGF and IGF1 but not by VEGF. Therefore, we evaluated with AlphaScreen technology the phosphorylated EGFR and IGF1R levels in primary cultures treated with sunitinib and/or EGF and IGF1. Results showed a decrease of p-IGF1R after treatment with sunitinib and an increase after co-treatment with IGF1. Then, we assessed cell viability and caspase activation on BP-NEN cell lines after treatment with linsitinib and/or erlotinib. Results demonstrate that these two agents have a stronger antiproliferative effect compared to sunitinib. In conclusion, our results suggest that IGF1R and EGF1R could represent putative molecular targets in BP-NENs treatment.
Keywords: BP-NENs, sunitinib, IGF1R, EGFR, erlotinib, linsitinib
Introduction
Neuroendocrine neoplasms (NENs) are neoplasms with an estimated annual incidence of 6.9/100,000 people in Europe per year, arising from cells of the diffuse endocrine system (DNES) (1, 2). NENs represent a wide group of malignancies originating from different parts of the human body. More than one half (about 70%) occurs in the gastrointestinal tract, while the other 25% occurs mostly in the broncho-pulmonary system (3, 4). NENs that affect the bronchial tree are called broncho-pulmonary NENs (BP-NENs) and represents approximately 25/30% of all NENs with an incidence of 1.2/100,000 people/year in European countries (5). The World Health Organization defines BP-NENs as an heterogeneous group that ranges from well-differentiated neoplasms characterised by an indolent behaviour to poorly differentiated forms with a high aggressive potential (6, 7, 8). The clinical management of BP-NENs needs a multidisciplinary approach but to date the only available curative treatment remains surgery that is not feasible in most of the cases due to metastatic disease (6, 9, 10). Therefore, medical therapies, including chemotherapy, radiation, drugs acting on hormone receptors and molecular target therapy, gain a key role in the clinical management of BP-NENs (11). Chemotherapy and radiation have already been demonstrated to be ineffective in the treatment of BP-NENs, while somatostatin analogues have shown a great effect in controlling the eventual secretory pattern of carcinoids but not the ability of reducing the tumour mass (12, 13, 14). Recent studies have led to advances in the knowledge of the molecular mechanisms of NENs allowing the development of novel targeted therapies that are now included in daily clinical practice (1, 15). Everolimus and sunitinib are two targeted agents approved by the Food and Drugs Administration (FDA) for the treatment of different types of NENs (16). Everolimus is an mTOR inhibitor capable to slow down the tumour growth and arrest tumour cells proliferation; this agent has already been approved for renal cell carcinoma, breast cancer and neuroendocrine carcinomas such as gastroenteropancreatic and lung neuroendocrine neoplasms (17, 18, 19). Clinical trials have demonstrated that everolimus prolongs progression-free survival (PFS) in patients with advanced, nonfunctional, progressive NENs of lung or gastrointestinal origin. However, despite its initial efficacy, most of the patients exhibit progressive disease after a period of treatment (4, 20). Everolimus resistance can vary according to the patient and, in some cases, can be attributed to rebound AKT activation due to the activity of one of the two mTOR subunits: mTORC2 complex (21, 22). Nonetheless, everolimus still represents the key therapeutic agent in the treatment of BP-NENs but, since patients can be insensitive or develop resistance after a period of treatment, there is an increasing need of new molecular targeted therapies (20). Sunitinib is a multi-tyrosine kinase growth factor receptor (RTKs) inhibitor indicated for the treatment of several NENs, such as gastrointestinal stromal tumours, advanced renal cell carcinoma and pancreatic neuroendocrine neoplasms (23). Its efficacy has been well described especially in pancreatic neuroendocrine tumours, due to the frequent overexpression of RTKs and the associated growth factors in this tumour (1, 24, 25, 26). Sunitinib was shown to have an anti-tumoural and antiangiogenic effect in patients with well-differentiated pancreatic NEN and to improve PFS of patients in a phase III clinical trial (3, 27). Despite its role in the treatment of pancreatic neuroendocrine neoplasms, there is still no clear evidence of its efficacy in NENs with a nonpancreatic origin as only few cases of BP-NENs were included in sunitinib’s previous trials (27, 28, 29). The lack of knowledge between the tumour type and drug efficacy highlights the importance of new in vitro studies useful to understand the possible role of sunitinib in BP-NENs treatment and find new potential molecular targets. Therefore, the aim of our study was to elucidate sunitinib’s mechanism of action in two BP-NEN cell lines and primary cultures, in order to identify new potential therapeutic targets in the treatment of BP-NENs. Moreover, we have also assessed whether other RTK inhibitors, erlotinib and linsitinib, could be effective in malignancy management.
Materials and methods
Drugs and chemicals
Sunitinib, erlotinib and linsitinib were purchased from Selleckchem. Compounds were dissolved in DMSO and stored at −80°C as 10 mM stock solutions.
EGF and IGF1 were purchased from ProSpec protein specialists (East Brunswick, NJ, USA); VEGF was purchased from Peprotech Inc (Rocky Hill, NJ, USA). EGF, IGF1 and VEGF were resuspended in sterile PBS with 0.1% BSA. All other reagents, if not specified, were purchased from Sigma.
In vitro immortalised human cell lines
NCI-H727 and NCI-H720 cell lines, derived from human BP-NEN, were purchased from the American Type Culture Collection and were cultured in RPMI-1640 medium (Euroclone, Milan, Italy) supplemented with 10% fetal bovine serum, at 37°C in a humidified atmosphere with 5% CO2. Experiments were performed within the seventh passage.
Cell viability
Variations in viable cell number were assessed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). 2 × 104 cells were seeded for each well in 96-well black plates, as previously described, and treated with sunitinib 5 μM, linsitinib 5 μM and elotinib with or without growth factors (IGF1 100 nM, EGF 30 nM and VEGF 50 ng/ml) for 72 h (30). Control cells were treated with vehicle alone (DMSO). After incubation, the revealing solution was added, and the luminescent output (relative luminescence units (RLUs)) was recorded using the Envision Multilable Reader (Perkin Elmer). Results are expressed as mean value ± standard error percentage RLU vs the vehicle-treated control cells from three independent experiments in six replicates.
Caspase activation
Caspase activity was measured using Caspase-Glo 3/7 assay (Promega), as previously described (31). Briefly, 2 × 104 cells/well were seeded in 96-well black plates and treated with the indicated compounds for 72 h. Then, the Caspase-Glo 3/7 reagent was added at room temperature directly to the cell culture plates, which were then shaken at 12.7 g for 30 s, incubated for 1 h and measured for luminescent output (relative luminescence unit (RLU)) using the Envision Multilabel Reader (Perkin Elmer). Results are expressed as mean value ± s.e.m. percentage RLU vs vehicle-treated control cells from three independent experiments in six replicates.
3D model
3D spheroids were obtained as previously described (32). Briefly, 30 μL of cells suspension with 2.4 × 103 cells per well in an ultra-low attachment 96-well plate (Corning 96-well Clear Round Bottom Ultra-Low Attachment Microplate, NY, USA) in RPMI complete medium. After seeding, the plates were centrifuged at 300 g for 3 min. Plates were treated with the indicated compounds 72 h after seeding.
Human BP-NEN tissues collection and primary culture
Samples were derived from nine patients diagnosed with BP-NEN, whose characteristics are shown in Table 1 (four females and five males; age = 21/81, mean = 50.44; ES ± 7.14), as previously described with minor modifications (30). All patients underwent surgical resection, and all had histological and immunohistochemical diagnosis of BP-NENs, according to the WHO classification. Primary cultures were prepared as previously described and treated with the indicated compounds (31, 32, 33). In order to prevent fibroblast overgrowth and subsequent cell viability decrease, which is observed after 4 days of culture, experiments were performed within 3 days. All the primary culture experiments were performed in accordance with the regulation of the University of Ferrara. The study was approved by the University of Ferrara Ethics Committee. Informed consents were obtained for disclosing clinical investigation and performing the in vitro study, in accordance with the local ethical committee.
Table 1.
BP-NEN patients.
| No. | Sex | Age | Dimension (mm) | Histology |
|---|---|---|---|---|
| 1 | F | 61 | 190 | Typical carcinoid |
| 2 | F | 21 | 140 | Typical carcinoid |
| 3 | M | 24 | 300 | Typical carcinoid |
| 4 | M | 69 | 18 | Typical carcinoid |
| 5 | M | 43 | 350 | Atypical carcinoid |
| 6 | M | 33 | 120 | Typical carcinoid |
| 7 | F | 53 | 9 × 10 × 5 | Typical carcinoid |
| 8 | F | 69 | 22 × 7 × 20 | Typical carcinoid |
| 9 | M | 81 | 10 × 5 × 4 | Atypical carcinoid |
| 10 | F | 57 | 330 | Typical carcinoid |
Kinase activity assay
Phosphorylated EGFR (p-Tyr1068) and IGFR (p-Y1135/1136), p-VEGFR 2 (Tyr1175) levels were measured using the AlphaScreen SureFire assays (Perkin Elmer). Briefly, 2 × 104 cells/well were seeded in complete medium, in 96-well plates and, after overnight attachment, were incubated for 24 h with or without sunitinib and EGF, alone and in combination, or sunitinib and IGF1, alone or in combination. AlphaScreen SureFire assays were performed as previously described (30), and evaluated as per the manufacturer’s protocol. The plates were measured on an Envison plate reader (Perkin Elmer), using standard AlphaScreen settings, and the output was recorded as counts per second (cps).
Live/dead cells assay
Viability or cell death was determined using the Ready Probes Cell Viability Imaging Kit (Blue/Green) (Life Technologies). BP-NEN spheroids were treated with the indicated compounds 3 days after seeding. Seventy-two hours after treatment, 100 μL of cell stain mix (two drops of each stain per mL of culture media) was added to each well and incubated for 15 min. The different luminescence was observed using the EVOS FL Cell imaging System (Thermo Fisher Scientific). Fluorescence intensity was measured using ImageJ software.
Statistical analysis
Results are expressed as the mean ± s.e.m. Statistical analyses were carried out using ANOVA after proof of homogeneity of variances and normality tests, Tukey’s test was used for multiple comparisons. Data were analysed using GraphPad (Prism v-7.0); P values <0.05 were considered significant (*). For all the other experiments, if not otherwise indicated, Student’s paired or unpaired t-test was used to evaluate the individual differences between the means, and P values <0.05 were considered significant.
Results
Influence of EGF, IGF1, VEGF and sunitinib on BP-NEN cell lines viability and caspase activation in 2D and 3D culture systems
In order to understand how treatments with sunitinib and growth factors can influence cell viability and caspase activation of NCI-H720 and NCI-H727 cells, we performed viability and caspase activation assays using 2D system culture model. NCI-H720 and NCI-H727 cells were treated with EGF 30 nM, VEGF 50 ng/mL, IGF1 100 nM and/or sunitinib 5 μM. The concentration of sunitinib was chosen on the basis of preliminary experiment (data non shown), while growth factors concentration was chosen based on patients reported plasma concentration (25). As shown in Fig. 1A, EGF and VEGF did not affect cell viability of NCI-H720, while IGF1 significantly increased this parameter by 11% (P < 0.05 vs vehicle-treated cells). Sunitinib decreased cell viability by 40% (P < 0.05 vs vehicle cells). EGF and VEGF did not influence the effect of sunitinib, while IGF1 was able to counteract the antiproliferative effects of sunitinib (P < 0.05 vs sunitinib-treated cells). In NCI-H727 cells, VEGF did not affect cell viability, while both EGF and IGF1 significantly increased this parameter by 10 and 15% respectively (P < 0.05 vs vehicle cells). Sunitinib decreased cell viability by 20% (P < 0.05 vs vehicle cells). VEGF did not influence the effect of sunitinib that was instead decreased by co-treatment with IGF1 and EGF (P < 0.05 vs sunitinib-treated cells).
Figure 1.

Effects of sunitinib and growth factors on cell viability, caspase activation and spheroids structure in human BP-NEN cell lines. NCI-H720 and NCI-H727 cells were incubated in 96-well plates for 72 h in culture medium supplemented with 5 μM sunitinib or/plus IGF1 100 nM, EGF 30 nM and VEGF 50 ng/ml; control cells were treated with a vehicle solution. (A) Cell viability was measured as luminescent output in three independent experiments with six replicates each, and it is expressed as the mean ± s.e.m. *P < 0.05 vs vehicle cells; #P < 0.05 vs cells treated with sunitinib. (B) Caspase activity was measured as luminescent output in three independent experiments with six replicates each, and it is expressed as the mean ± s.e.m. *P < 0.05 vs vehicle cells; #P < 0.05 vs cells treated with sunitinib. (C) Representative spheroids pictures were taken with EVOS FL Cell imaging System 72 h after treatment. NCI-H720 and NCI-H727 cells spheroids were treated as described earlier; pictures were taken without and with the fluorescent staining. The second and fourth columns in each plot represent the merge between the two fluorescence detected. The blue dye stains the nuclei of all cells (excitation/emission maxima: 360/460 nm), while the green dye stains only the nuclei of dead cells with compromised plasma membranes (excitation/emission maxima: 504/523 nm). (D) Green fluorescence from spheroids was analysed using ImageJ software.
In order to verify if the modulation of cell viability was also due to apoptosis, caspase 3/7 activation was evaluated. As shown in Fig. 1B, in NCI-H720 cells, none of the growth factors was able to significantly affect caspase 3/7 activation while 5 μM sunitinib was able to increase the apoptotic activity by 100% (P < 0.05 vs vehicle cells). On the other hand, this effect was partially affected by IGF1 (−34% vs sunitinib-treated cells). In NCI-H727 cells (Fig. 1B), EGF was able to significantly reduce caspase 3/7 activity by 13%. Sunitinib increased the apoptotic activation by 22% (P < 0.05 vs vehicle cells); however, this effect was significantly abrogated by co-treatment with both EGF and IGF1.
Moreover, we have cultured BP-NEN cell lines in 3D in order to observe the possible changes in cell viability/death in a more realistic solid tumour model using two different fluorescent dyes. As shown in Fig. 1C, NCI-H720 and NCI-H727 spheroids treated with sunitinib were characterised by a stronger green fluorescence, mostly focused in the middle of the spheroid, in comparison with cells treated with vehicle solution and growth factors. Green fluorescence (which indicates cell death) was evaluated, and the results overlap with those obtained with cell viability assay (Fig. 1D). The predominance of green fluorescence indicates a more extensive cell death in spheroids treated with sunitinib counteract at least in part by co-treatment with growth factors.
Influence of EGF, IGF1 and sunitinib on BP-NEN primary cell lines viability and caspase activation
In order to verify data obtained in immortalised cell lines in primary tumours, we tested the effects of EGF and IGF1 on cell viability in BP-NEN primary cultures. As shown in Fig. 2A, EGF 30 nM was able to increase cell viability, and partially counteract the effects of sunitinib in reducing of cell viability. Similarly, IGF1 (Fig. 2B) was able to significantly increase cell viability (P < 0.05) and to counteract, at least partially, the effects of sunitinib.
Figure 2.

Effects of sunitinib and growth factors on cell viability and caspase activation in human BP-NEN primary cultures. Cells were derived from patients samples and incubated in 96-well plates for 48 h in culture medium supplemented with 5 μM sunitinib or/plus EGF 30 nM (A and C) and IGF1 100 nM (B and D); control cells were treated with a vehicle solution. (A and B) Cell viability was measured as luminescent output and evaluated in one experiment with six replicates; each point represents the mean of single sample. Data are expressed as the mean ± s.e.m. *P < 0.05, ****P < 0.0001 vs CT. (C and D) Caspase activity was measured as a luminescent output and evaluated in one experiment with six replicates each point represents the mean of single sample. Data are expressed as the mean ± s.e.m. ****P < 0.0001; ***P < 0.001 vs CT; ####P < 0.00001 vs cells treated with sunitinib.
As concerns apoptotic activation (Fig. 2C and D), both EGF and IGF1 were able to significantly counteract caspase 3/7 activity promoted by 5 μM sunitinib (P < 0.00001).
Effects of sunitinib on EGFR and IGF1R phosphorylation
To further investigate the mechanisms of action by which sunitinib exerts its effects on BP-NEN cells, phosphorylation levels of EGFR and IGF1R were evaluated by a kinase activity assay. Results concerning EGFR phosphorylation after treatments were not univocal (Fig. 3A). On the other hand, as shown in Fig. 3B, sunitinib was able to significantly decrease IGF1R phosphorylation levels and its co-treatment with the corresponding growth factor IGF1 partially restored receptor phosphorylation. pVEGFR2 levels were evaluated after treatment with sunitinib on primary culture, however, a modest and not significant reduction was observed in all sample analysed.
Figure 3.

Evaluation of phosphorylated EGFR and IGF1R in primary cultures after treatments. (A and B) Cells were treated with sunitinib 5 μM or/plus EGF 30 nM (A) and IGF1 100 nM (B). Cells were incubated for 24 h and control cells were treated with a vehicle solution. (A) Primary culture cells were lysed and processed for phosphorylated EGFR SureFire assays in one experiment with three replicates; in each graph is shown the global mean. Data are expressed as percentage of p-EGFR vs the control, considering as the control sample the untreated primary culture cells. (B) Primary culture cells were lysed and processed for phosphorylated IGF1R SureFire assays in one experiment with six replicates; in each graph is shown the global mean. Data are expressed as percentage of p-IGF1R vs vehicle-treated cells. (C) Primary culture cells were lysed and processed for phosphorylated VEGFR2 SureFire assays in one experiment with six replicates; in each graph is shown the global mean. Data are expressed as percentage of p-VEGFR vs vehicle-treated cells.
Influence of erlotinib and linsitinib on BP-NEN cell viability and caspase activation
Since our data indicate that EGF and IGF1 influence sunitinib activity, we assessed cell viability and caspase activation using erlotinib, an EGFR inhibitor and linsitinib, an IGF1R inhibitor, on NCI-H720 and NCI-H727 cells. Cells were treated with erlotinib 5 μM alone and in combination with linsitinib 5 μM. As shown in Fig. 4A, erlotinib and linsitinib showed an antiproliferative action in both cell lines. Erlotinib decreased cell viability of both cell lines by ~about a 20% (P < 0.01 vs vehicle cells) while treatment with linsitinib was able to reduce cell viability by 30% (P < 0.001 vs vehicle cells). The combination of the two agents showed a greater effect in comparison with single treatments in both NCI-H720 and NCI-H727 cells reducing cell viability by ~ 40% vs vehicle-treated cells and by 20% vs treatment with erlotinib (P < 0.05 for NCI-H727 and P < 0.01 for NCI-H720 cells).
Figure 4.

Effects of erlotinib and linsitinib on cell viability, caspase activation and spheroids structure in human BP-NEN cell lines. NCI-H720 and NCI-H727 cells were incubated in 96-well plates for 72 h in culture medium supplemented with 5 μM erlotinib and/or 5 μM linsitinib; control cells were treated with a vehicle solution. (A) Cell viability was measured as luminescent output in three independent experiments with six replicates each, and it is expressed as the mean ± s.e.m. *P < 0.05 vs vehicle cells or erlotinib; **P < 0.01 vs vehicle cells or erlotinib; ***P < 0.001 vs vehicle cells or erlotinib; ****P < 0.0001 vs vehicle cells or erlotinib. (B) Caspases activity was measured as luminescent output in three independent experiments with six replicates each and it is expressed as the mean ± s.e.m. *P < 0.05 vs vehicle cells or erlotinib; **P < 0.01 vs vehicle cells or erlotinib; ***P < 0.001 vs vehicle cells or erlotinib; ****P < 00001 vs vehicle cells or erlotinib. (C) Representative spheroids pictures were taken with EVOS FL Cell imaging System 72 h after treatment. NCI-H720 and NCI-H727 were treated as described above; pictures were taken without and with the fluorescent staining. The second and fourth columns in each plot represent the merge between the two fluorescence dyes detected. The blue dye stains the nuclei of all cells (excitation/emission maxima: 360/460 nm) while the green dye stains only the nuclei of dead cells with compromised plasma membranes (excitation/emission maxima: 504/523 nm). (D) Green fluorescence from spheroids was analysed using ImageJ software.
With regard to caspase activation, treatment with erlotinib did not significantly affect apoptosis in NCI-H720 cells, while linsitinib caused a significant increase by 40% (P < 0.01% vs vehicle cells). The combination of the two agents strongly activated caspase causing an increase of the 250% vs vehicle cells (P < 0.01).
In NCI-H727 cells, caspase activation was significant vs vehicle cells for both erlotinib and linsitinib alone and the combination of the two agents resulted in an increase in caspase activation of 300% (P < 0.01% vs vehicle cells). In both cell lines, the combination of erlotinib and linsitinib, significantly improve the activation of caspase 3/7 as compare to both single agent (P < 0.001).
Furthermore, we cultured BP-NEN cell lines in 3D in order to observe the possible changes in cell viability/death in a more realistic solid tumour model using two different fluorescent dyes. As shown in Fig. 1C, NCI-H720 and NCI-H727 spheroids treated with erlotinib and linsitinib were characterised by a stronger green fluorescence in comparison with cells treated with vehicle solution. Green fluorescence (which indicate cell death) was evaluated, and the results overlap with those obtained with cell viability assay (Fig. 4D). In order to confirm this data, we performed erlotinib and linsitinib treatment in BP-NEN primary culture, as shown in Fig. 5 both agents were able to reduce cell viability (P < 0.01) and to induce caspase 3/7 activation (significantly just for erlotinib P < 0.01).
Figure 5.
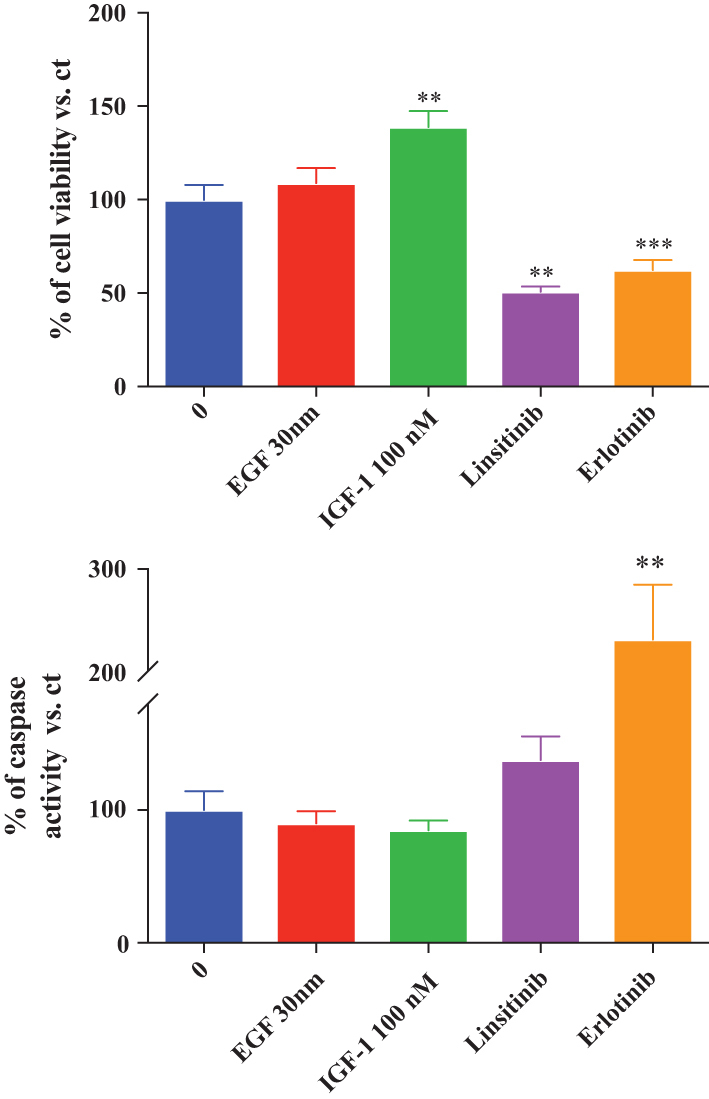
Effects of erlotinib and linsitinib on cell viability, caspase activation in human BP-NEN primary culture. Cells were incubated in 96-well plates for 72 h in culture medium supplemented with 5 µM erlotinib, 5 µM linsitinib, IGF1 100 nM or EGF 30 nM; control cells were treated with vehicle solution. (A) Cell viability was measured as luminescent output in three independent experiments with six replicates each, and it is expressed as the mean ± s.e.m. **P < 0.01, ***P < 0.001 vs vehicle-treated cells. (B) Caspases activity was measured as luminescent output in three independent experiments with six replicates each and it is expressed as the mean ± s.e.m. **P < 0.01 vs vehicle-treated cells.
Discussion
BP-NENs are a group of malignancies increasing in incidence without a specific/effective therapeutic strategy (27). Everolimus is the only available molecular targeted therapy approved for this malignancy but recurrence of resistance after a period of treatment has already been demonstrated (21). Sunitinib, a small multi-RTKs inhibitor, has been approved for the treatment of some NENs such as pancreatic neuroendocrine neoplasms (23). Even if during sunitinib phase I and II clinical trials several NENs have been included, such as BP-NENs, during phase III, only patients with well-differentiated pancreatic endocrine tumours were included (34). Previously published evidence demonstrated the efficacy of sunitinib on GEP-NEN cell lines and primary culture (35, 36). However, the lack of data about sunitinib’s action and effectiveness in NENs of extra-pancreatic origin leads to the crucial necessity to study in vitro the possible role of sunitinib on BP-NENs and identify possible markers that may be useful in the treatment of a malignancy lacking of original designed trials. In this study, we have shown for the first time the effects of Sunitinib on BP-NEN using two cell lines and primary cultures. Our findings are consistent with other studies that have already demonstrated the efficacy of the drug also in malignancies for which sunitinib is not approved (25, 37). Our data suggest that EGF and IGF1 may be involved in sunitinib mechanism of action since co-treatment with these factors counteracts, at least in part, the drug antiproliferative and pro apoptotic action. Therefore, VEGFR, described in literature as the main sunitinib target, may not represent a key regulator of its mechanism of action in BP-NEN. Our results also show how IGF1 significantly counteracts the effects of sunitinib on cell viability and caspase activation in both NCI-H720 and NCI-H727 cells. This trend could also be observed in NCI-H727 cells with EGF and sunitinib combined treatment and, with less impact, on primary cultures. These findings are in contrast with literature where VEGFR is described as the main target of sunitinib. Only few studies have explored the possible involvement of others RTKs in the drug mechanism of action, whereas our data suggest IGF1R and EGFR as key target in BP-NENs (1, 38, 39). In addition, we employed a 3D culture system in order to better characterise the role of EGFR and IGF1R as key regulators of sunitinib mechanism of action. Impact of sunitinib and growth factors on three-dimensional cell structure and cell viability were evaluated using a staining assay and our findings indicate that even in a 3D system sunitinib has a strong antiproliferative effect that it’s focused in the centre of the spheroids. Furthermore, sunitinib effects have been deeply evaluated in primary cultures. The drug has been tested alone and in combination with EGF and IGF1 and our findings indicate that treatment with the drug decreases IGF1R phosphorylation levels that are partially restored with co-treatment with IGF1. On the other hand, the correlated experiments with sunitinib and EGF are not univocal probably due to the small number of available samples. Since our results indicated that IGF1R and EGFR are involved in modulating sunitinib effects, we decided to better characterise the role of these two receptors in regulating BP-NEN cell lines proliferation employing linsitinib and erlotinib, respectively, IGF1R and EGFR inhibitors. Results indicate that both erlotinib and linsitinib have an antiproliferative action on the two cell lines and that treatments generate a strong caspase activation.
Our findings indicated also that the combination of erlotinib and linsitinib decreased cell viability to a greater extent as compared to single treatments and to treatment with sunitinib. These results are mirrored by caspase activation that, particularly for NCI-H727 cells, is induced by almost three fold as compared to treatments with linsitinib, erlotinib and even sunitinib alone. Analysis of 3D spheroids structure and cell viability revealed that these drugs seem to act mostly in the centre of the spheroids and that linsitinib and erlotinib decrease cell viability with a greater effect in combination. The importance of EGFR and IGF1R in NENs treatment is supported by several hypotheses. Gilbert et al. have demonstrated that NENs samples derived from patients exhibit high EGFR levels and that high IGF1R levels are present in BP-NEN cell lines (40, 41). Erlotinib decrease NCI-H720 cell viability of a 20% and its effect is visible also in caspase activation (+20%) even if the result is not significant. This can be due to the small, but still present, effect of erlotinib in this cell line. Moreover, erlotinib effect is evident once in combination with linsitinib; their combined effect is stronger than the single treatment of both drugs underling a synergistic effect of the two agents. The synergistic effect of linsitinib and erlotinib is particularly important for NCI-H727, as these cells have been shown to be resistant to Everolimus and NVP-BEZ235 (30) and they show limited response to sunitinib as compare to NCI-H720 (Fig. 1A).
Therefore, in this case, EGFR and IGF1R are suggested as new potential targets useful for the development of new therapies in NENs. Moreover, the double inhibition induced by co-treatments with erlotinib and linsitinib could lead to the block of the well-established reciprocal cross-talk between EGFR and IGF1R. This relation has been previously demonstrated by Haluska et al. to induced in many cancer types the adaptive activation of IGF1R family members upon the inactivation of EGFR and vice versa (42). These authors also showed that in ovarian and breast cancer cell lines, the co-treatment with IGF1R and EGFR inhibitors results in a synergic antiproliferative effect followed by the decrease of several important proteins involved in cancer progression, morphological changes and caspase activation in comparison to single treatments. These results are in agreement with our findings supporting the hypothesis that more specific RTK inhibitors could be important in the management of NENs characterised by IGF1R and EGFR overexpression.
Our results show for the first time the effectiveness of sunitinib in BP-NENs cells and that the VEGF/VEGFR system may not be crucial in modulating BP-NENs responsiveness to the drug. Evidence that IGF1 and EGF counteract sunitinib action indicates that IGF1R and EGFR may be key regulators of sunitinib resistance that might be overcome using a combination of more specific IGF1R and EGFR inhibitors. In conclusion, our data suggest that RTKs inhibition can open new therapeutic horizons in BP-NENs treatment and that specific targets need to be inhibited to overcome resistance.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author contribution statement
Conceptualization, T G and G B; methodology, G B; software, V V; validation, A D and C C; formal analysis, G B; investigation, G B; resources, F R, N G C and M S; data curation, G G; writing – original draft preparation, G B; writing – review and editing, T G; visualisation, G B and T G; supervision, T G, G G and M C Z. All authors reviewed the manuscript.
Acknowledgements
The authors would like to thank the Company of Biologists for providing a travel grant to Giulia Bresciani, the European Neuroendocrine Tumor Society for supporting T G and G B continuous education in the field and Professor Susan Ayers for proofreading the manuscript.
References
- 1.Nigri G, Petrucciani N, Debs T, Mangogna LM, Crovetto A, Moschetta G, Persechino R, Aurello P, Ramacciato G. Treatment options for PNET liver metastases: a systematic review. World Journal of Surgical Oncology 2018. 16 142 ( 10.1186/s12957-018-1446-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uri I, Grozinsky-Glasberg S. Current treatment strategies for patients with advanced gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Clinical Diabetes and Endocrinology 2018. 4 16 ( 10.1186/s40842-018-0066-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martins D, Spada F, Lambrescu I, Rubino M, Cella C, Gibelli B, Grana C, Ribero D, Bertani E, Ravizza D, et al Predictive markers of response to everolimus and sunitinib in neuroendocrine tumors. Targeted Oncology 2017. 12 611–622. ( 10.1007/s11523-017-0506-5) [DOI] [PubMed] [Google Scholar]
- 4.Barbieri F, Albertelli M, Grillo F, Mohamed A, Saveanu A, Barlier A, Ferone D, Florio T. Neuroendocrine tumors: insights into innovative therapeutic options and rational development of targeted therapies. Drug Discovery Today 2014. 19 458–468. ( 10.1016/j.drudis.2013.10.015) [DOI] [PubMed] [Google Scholar]
- 5.Peri M, Botteri E, Pisa E, De Marinis F, Ungaro A, Spada F, Grana CM, Gasparri R, Spaggiari L, Romentz N, et al A single-institution retrospective analysis of metachronous and synchronous metastatic bronchial neuroendocrine tumors. Journal of Thoracic Disease 2018. 10 3928–3939. ( 10.21037/jtd.2018.06.78) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferolla P. Medical treatment of advanced thoracic neuroendocrine tumors. Thoracic Surgery Clinics 2014. 24 351–355. ( 10.1016/j.thorsurg.2014.05.006) [DOI] [PubMed] [Google Scholar]
- 7.Öberg K, Hellman P, Ferolla P, Papotti M. & ESMO Guidelines Working Group. Neuroendocrine bronchial and thymic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology 2012. 23 (Supplement 7) vii120–vii123. ( 10.1093/annonc/mds267) [DOI] [PubMed] [Google Scholar]
- 8.Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E, Flieder DB, et al The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. Journal of Thoracic Oncology 2015. 10 1243–1260. ( 10.1097/JTO.0000000000000630) [DOI] [PubMed] [Google Scholar]
- 9.Carretta A, Ceresoli GL, Arrigoni G, Canneto B, Reni M, Cigala C, Zannini P. Diagnostic and therapeutic management of neuroendocrine lung tumors: a clinical study of 44 cases. Lung Cancer 2000. 29 217–225. ( 10.1016/S0169-5002(00)00119-7) [DOI] [PubMed] [Google Scholar]
- 10.Fazio N, Ungaro A, Spada F, Cella CA, Pisa E, Barberis M, Grana C, Zerini D, Bertani E, Ribero D, et al The role of multimodal treatment in patients with advanced lung neuroendocrine tumors. Journal of Thoracic Disease 2017. 9 S1501–S1510. ( 10.21037/jtd.2017.06.14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer 2008. 113 5–21. ( 10.1002/cncr.23542) [DOI] [PubMed] [Google Scholar]
- 12.Grozinsky-Glasberg S, Grossman AB, Korbonits M. The role of somatostatin analogues in the treatment of neuroendocrine tumours. Molecular and Cellular Endocrinology 2008. 286 238–250. ( 10.1016/j.mce.2007.10.006) [DOI] [PubMed] [Google Scholar]
- 13.Srirajaskanthan R, Toumpanakis C, Karpathakis A, Marelli L, Quigley AM, Dusmet M, Meyer T, Caplin ME. Surgical management and palliative treatment in bronchial neuroendocrine tumours: a clinical study of 45 patients. Lung Cancer 2009. 65 68–73. ( 10.1016/j.lungcan.2008.10.025) [DOI] [PubMed] [Google Scholar]
- 14.Wolin EM. The expanding role of somatostatin analogs in the management of neuroendocrine tumors. Gastrointestinal Cancer Research 2012. 5 161–168. [PMC free article] [PubMed] [Google Scholar]
- 15.Marotta V, Franzese MD, Del Prete M, Chiofalo MG, Ramundo V, Esposito R, Marciello F, Pezzullo L, Carratù A, Vitale M, et al Targeted therapy with kinase inhibitors in aggressive endocrine tumors. Expert Opinion on Pharmacotherapy 2013. 14 1187–1203. ( 10.1517/14656566.2013.796931) [DOI] [PubMed] [Google Scholar]
- 16.Yoo C, Cho H, Song MJ, Hong SM, Kim KP, Chang HM, Chae H, Kim TW, Hong YS, Ryu MH, et al Efficacy and safety of everolimus and sunitinib in patients with gastroenteropancreatic neuroendocrine tumor. Cancer Chemotherapy and Pharmacology 2017. 79 139–146. ( 10.1007/s00280-016-3215-3) [DOI] [PubMed] [Google Scholar]
- 17.Hasskarl J. Everolimus. Recent Results in Cancer Research 2018. 211 101–123. ( 10.1007/978-3-319-91442-8_8) [DOI] [PubMed] [Google Scholar]
- 18.Yao JC, Pavel M, Lombard-Bohas C, Van Cutsem E, Voi M, Brandt U, He W, Chen D, Capdevila J, de Vries EGE, et al Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. Journal of Clinical Oncology 2016. 34 3906–3913. ( 10.1200/JCO.2016.68.0702) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pusceddu S, Verzoni E, Prinzi N, Mennitto A, Femia D, Grassi P, Concas L, Vernieri C, Lo Russo G, Procopio G. Everolimus treatment for neuroendocrine tumors: latest results and clinical potential. Therapeutic Advances in Medical Oncology 2017. 9 183–188. ( 10.1177/1758834016683905) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee L, Ito T, Jensen RT. Everolimus in the treatment of neuroendocrine tumors: efficacy, side-effects, resistance, and factors affecting its place in the treatment sequence. Expert Opinion on Pharmacotherapy 2018. 19 909–928. ( 10.1080/14656566.2018.1476492) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Research 2006. 66 1500–1508. ( 10.1158/0008-5472.CAN-05-2925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Svejda B, Kidd M, Kazberouk A, Lawrence B, Pfragner R, Modlin IM. Limitations in small intestinal neuroendocrine tumor therapy by mTor kinase inhibition reflect growth factor-mediated PI3K feedback loop activation via ERK1/2 and AKT. Cancer 2011. 117 4141–4154. ( 10.1002/cncr.26011) [DOI] [PubMed] [Google Scholar]
- 23.Blumenthal GM, Cortazar P, Zhang JJ, Tang S, Sridhara R, Murgo A, Justice R, Pazdur R. FDA approval summary: sunitinib for the treatment of progressive well-differentiated locally advanced or metastatic pancreatic neuroendocrine tumors. Oncologist 2012. 17 1108–1113. ( 10.1634/theoncologist.2012-0044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen H, Fang Y, Dong W, Mu X, Liu Q, Du J. IGF-1 receptor is down-regulated by sunitinib induces MDM2-dependent ubiquitination. FEBS Open Bio 2012. 2 1–5. ( 10.1016/j.fob.2011.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gagliano T, Gentilin E, Tagliati F, Benfini K, Di Pasquale C, Feo C, Falletta S, Riva E, degli Uberti E, Zatelli MC. Inhibition of epithelial growth factor receptor can play an important role in reducing cell growth and survival in adrenocortical tumors. Biochemical Pharmacology 2015. 98 639–648. ( 10.1016/j.bcp.2015.10.012) [DOI] [PubMed] [Google Scholar]
- 26.Hao Z, Sadek I. Sunitinib: the antiangiogenic effects and beyond. OncoTargets and Therapy 2016. 9 5495–5505. ( 10.2147/OTT.S112242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uri I, Avniel-Polak S, Gross DJ, Grozinsky-Glasberg S. Update in the therapy of advanced neuroendocrine tumors. Current Treatment Options in Oncology 2017. 18 72 ( 10.1007/s11864-017-0514-9) [DOI] [PubMed] [Google Scholar]
- 28.Kulke MH, Lenz HJ, Meropol NJ, Posey J, Ryan DP, Picus J, Bergsland E, Stuart K, Tye L, Huang X, et al Activity of sunitinib in patients with advanced neuroendocrine tumors. Journal of Clinical Oncology 2008. 26 3403–3410. ( 10.1200/JCO.2007.15.9020) [DOI] [PubMed] [Google Scholar]
- 29.Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, et al Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. Journal of Clinical Oncology 2006. 24 25–35. ( 10.1200/JCO.2005.02.2194) [DOI] [PubMed] [Google Scholar]
- 30.Gagliano T, Bellio M, Gentilin E, Molè D, Tagliati F, Schiavon M, Cavallesco NG, Andriolo LG, Ambrosio MR, Rea F, et al mTOR, p70s6K, AKT, and ERK1/2 levels predict sensitivity to mTOR and PI3K/mTOR inhibitors in human bronchial carcinoids. Endocrine-Related Cancer 2013. 20 463–475. ( 10.1530/ERC-13-0042) [DOI] [PubMed] [Google Scholar]
- 31.Zatelli MC, Gentilin E, Daffara F, Tagliati F, Reimondo G, Carandina G, Ambrosio MR, Terzolo M, Degli Uberti EC. Therapeutic concentrations of mitotane (o,p′-DDD) inhibit thyrotroph cell viability and TSH expression and secretion in a mouse cell line model. Endocrinology 2010. 151 2453–2461. ( 10.1210/en.2009-1404) [DOI] [PubMed] [Google Scholar]
- 32.Stebbing J, Shah K, Lit LC, Gagliano T, Ditsiou A, Wang T, Wendler F, Simon T, Szabó KS, O'Hanlon T, et al LMTK3 confers chemo-resistance in breast cancer. Oncogene 2018. 37 531–541. ( 10.1038/s41388-018-0197-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zatelli MC, Maffei P, Piccin D, Martini C, Rea F, Rubello D, Margutti A, Culler MD, Sicolo N, degli Uberti EC. Somatostatin analogs in vitro effects in a growth hormone-releasing hormone-secreting bronchial carcinoid. Journal of Clinical Endocrinology and Metabolism 2005. 90 3113–3130. ( 10.1210/jc.2004-2156) [DOI] [PubMed] [Google Scholar]
- 34.Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, et al Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. New England Journal of Medicine 2011. 364 501–513. ( 10.1056/NEJMoa1003825) [DOI] [PubMed] [Google Scholar]
- 35.Elgendy M, Abdel-Aziz AK, Renne SL, Bornaghi V, Procopio G, Colecchia M, Kanesvaran R, Toh CK, Bossi D, Pallavicini I, et al Dual modulation of MCL-1 and mTOR determines the response to sunitinib. Journal of Clinical Investigation 2017. 127 153–168. ( 10.1172/JCI84386) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiedmer T, Blank A, Pantasis S, Normand L, Bill R, Krebs P, Tschan MP, Marinoni I, Perren A. Autophagy inhibition improves sunitinib efficacy in pancreatic neuroendocrine tumors via a lysosome-dependent mechanism. Molecular Cancer Therapeutics 2017. 16 2502–2515. ( 10.1158/1535-7163.MCT-17-0136) [DOI] [PubMed] [Google Scholar]
- 37.Ketola K, Kallioniemi O, Iljin K. Chemical biology drug sensitivity screen identifies sunitinib as synergistic agent with disulfiram in prostate cancer cells. PLoS ONE 2012. 7 e51470 ( 10.1371/journal.pone.0051470) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roskoski R. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochemical and Biophysical Research Communications 2007. 356 323–328. ( 10.1016/j.bbrc.2007.02.156) [DOI] [PubMed] [Google Scholar]
- 39.Bergerot P, Burns K, Prajapati D, Fox R, Salgia M, Pal SK. Advances in the treatment of metastatic renal cell carcinoma. Cancer Treatment and Research 2018. 175 127–137. ( 10.1007/978-3-319-93339-9_6) [DOI] [PubMed] [Google Scholar]
- 40.Gilbert JA, Adhikari LJ, Lloyd RV, Halfdanarson TR, Muders MH, Ames MM. Molecular markers for novel therapeutic strategies in pancreatic endocrine tumors. Pancreas 2013. 42 411–421. ( 10.1097/MPA.0b013e31826cb243) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert JA, Adhikari LJ, Lloyd RV, Rubin J, Haluska P, Carboni JM, Gottardis MM, Ames MM. Molecular markers for novel therapies in neuroendocrine (carcinoid) tumors. Endocrine-Related Cancer 2010. 17 623–636. ( 10.1677/ERC-09-0318) [DOI] [PubMed] [Google Scholar]
- 42.Haluska P, Carboni JM, TenEyck C, Attar RM, Hou X, Yu C, Sagar M, Wong TW, Gottardis MM, Erlichman C. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Molecular Cancer Therapeutics 2008. 7 2589–2598. ( 10.1158/1535-7163.MCT-08-0493) [DOI] [PMC free article] [PubMed] [Google Scholar]