

Abstract
Cisplatin (CP)-induced nephrotoxicity is widely accepted as a model for acute kidney injury (AKI). Although cisplatin-induced chronic kidney disease (CKD) in rodent has been reported, the role of phosphate in the cisplatin-induced CKD progression is not described. In this study, we gave a single peritoneal injection of CP followed by high (2%) phosphate diet for 20 weeks. High dose CP (20 mg/Kg) led to high mortality; whereas a lower dose (10 mg/Kg) resulted in a full spectrum of AKI with tubular necrosis, azotemia, and 0% mortality 7 days after CP injection. After consuming a high phosphate diet, mice developed CKD characterized by low creatinine clearance, interstitial fibrosis, hyperphosphatemia, high plasma PTH and FGF23, low plasma 1,25(OH)2 Vitamin D3 and αKlotho, and classic uremic cardiovasculopathy. The CP model was robust in demonstrating the effect of aging, sexual dimorphism, and dietary phosphate on AKI and also AKI-to-CKD progression. Finally, we used the CP-high phosphate model to examine previously validated methods of genetically manipulated high αKlotho and therapy using exogenous soluble αKlotho protein supplementation. In this CP CKD model, αKlotho mitigated CKD progression, improved mineral homeostasis, and ameliorated cardiovascular disease. Taken together, CP and high phosphate nephrotoxicity is a reproducible and technically very simple model for the study of AKI, AKI-to-CKD progression, extrarenal complications of CKD, and for evaluation of therapeutic efficacy.
Introduction
Acute kidney injury (AKI) is a serious public health problem. Using the Kidney Disease Improving Global Outcomes (KDIGO) criteria and staging system, global incidence of AKI in hospitalized patients is 22% in adults and 34% in children [1–3]. In hospitalized patients, AKI-associated mortality rates are 24% in adults and 14% in children [2]. In intensive care unit, the incidence of AKI is 50–70% with greater than 50% mortality [4]. A population-based study showed full renal recovery after AKI based on serum creatinine to be ~68% [5]. Even when patients survive AKI, the long-term outcome is far from benign with substantial risk of progression to chronic kidney disease (CKD) and end-stage renal disease [6, 7]. Thus far, the precise risk of AKI-to-CKD progression is not yet well known [1], but the hazard ratio of developing CKD post-AKI is estimated to be about two-fold [7]. Although there is increasing evidence from both animal experiments and clinical observations indicating that AKI is an independent risk for CKD, effective retardation of AKI-to-CKD progression is not available, mainly due to lack of understanding of the pathogenic mechanism underlying AKI-to-CKD progression. An animal model of AKI-to-CKD that permits pre-clinical study is indispensable.
Some rodent models of AKI-to-CKD progression have been generated by either kidney mass ablation surgery, induction of ischemia-reperfusion, or exposure to nephrotoxins. Ischemia-reperfusion by temporally clamping the renal artery or pedicle is one model widely used for studying the molecular and cellular mechanisms of AKI-to-CKD progression. We recently reported that mice undergoing either bilateral IRI or unilateral nephrectomy plus contralateral IRI followed by high (2%) phosphate diet develop CKD with azotemia, hyperphosphatemia, low plasma, αKlotho and 1,25-(OH)2-vitamin D3, high plasma PTH, phosphate and fibroblast growth factor (FGF)23, renal fibrosis, vascular calcification, and uremic cardiomyopathy [8–11]. However, preparation of IRI requires rather complex animal surgery and this model cannot mimic nephrotoxic injuries, which is the second most common cause of AKI in both children and adults [1, 12].
Cisplatin (CP) nephrotoxicity is a severe renal complication in patients receiving CP-based chemotherapy, but the incidence of AKI following CP chemotherapy is not well defined in humans. Recent studies showed that AKI occurs in 21–31.5% of patients receiving CP-containing chemotherapy [13, 14]. Acute CP nephrotoxicity has been used as an AKI model to study pathogenesis of AKI and to test new therapeutic options [15]. In contrast, the description of acute CP nephrotoxicity progression to CKD in rodent models is relatively limited without consensus protocols [16–21], and effect of high Pi on the CKD progression from acute CP nephrotoxicity is not reported yet.
In this manuscript, we describe a technically simple model for AKI followed by AKI-to-CKD transition in rodents. Acute nephrotoxicity is induced by single bolus injection of CP without the need for surgery, such as IRI, partial nephrectomy, or unilateral ureteral obstruction. After recovery from AKI, chronic high dietary phosphate promotes CKD progression with extrarenal complications with clinical relevance, such as mineral and bone disorder (CKD-MBD) and uremic cardiovasculopathy.
Materials and methods
Rodent
All animal work was conducted strictly following the guide for the care and use of laboratory animals by the National Institutes of Health and was approved by the institutional animal care and use committee at the University of Texas Southwestern Medical Center. Wild type (WT) 129 S1/SVlmJ (129 SV) mice were purchased from Jackson laboratory (Bar Harbor ME, USA) and Spraque-Dawley (SD) rats from Charles River Laboratories (San Diego, CA). Mice and rats were housed in a temperature-controlled room with a 12:12 h light-dark cycle and were given ad libitum access to tap water and standard rodent chow prior to the experiment. Equal male and female animals were used. Usually 3-months-old mice and 2-months-old rats were used unless specifically indicated. The heterozygous Klotho hypomorphic (kl/+) mice and transgenic Klotho overexpressing (Tg-Kl) mice were cross-mated with WT 129 SV mice for more than 10 generations and genotyped with standard PCR protocol described previously [8, 10, 11, 22].
CP nephrotoxicity and high phosphate diet
CP (Sigma-Aldrich St. Louis, MO) was dissolved in sterile 0.9% NaCl, and intraperitoneally injected once. For evaluation of AKI and acute mortality, 30 mice (15 female:15 male mice) were randomized into three groups: vehicle (0.9% NaCl), low dose (10mg/Kg), and high dose (20 mg/Kg) CP. After injection, mice were allowed free access to standard rodent chow (Teklad 2016, Harlan, Madison, WI) and tap water unless stated otherwise.
For the study of AKI-to-CKD progression, CP was given only with low dose (10 mg/Kg), as high-dose (20 mg/Kg) CP-injected mice could not survive more than one month. When started with 10 mice, we typically had ~8 mice at 20 weeks after CP 10 mg/Kg injection.
To promote AKI-to-CKD progression, two weeks after vehicle or CP injection, mice were switched from normal rodent chow to 2.0% (w:w) phosphate diet (Teklad 08020, Harlan, Madison, WI) for another 18 weeks.
Recombinant αKlotho protein supplementation
Soluble αKlotho protein containing the ectodomain of mouse αKlotho (amino acid number 31–982) with C-terminal V5 and 6×His tags was generated and purified in our laboratory in mammalian cells as described previously [11, 22, 23]. For the experiment of αKlotho administration in CP-treated mice, CP nephrotoxicity was induced with low dose (10 mg/kg) or vehicle [24] and high phosphate (2%) diet was given 2 weeks after CP injection [8–10]. At 4 weeks after injection, recombinant αKlotho protein (0.3 mg/kg body weight/month) or vehicle (normal saline) was given for 16 weeks via ALZET® 1004 osmotic minipumps (DURECT Corporation, Cupertino, CA). The effective lifespan for this type of minipump is 4 weeks and a new one was implanted to replace every 4 weeks resulting in 4 minipumps per animal [11]. There were 10 mice (equal male and female) at the beginning and ~8 mice at 20 weeks after CP injection.
Blood, urine, and kidney samples collection
At predetermined times post-AKI, 24-h urine was collected in individual metabolic cage. Mice were then anesthetized with Isoflurane and blood were collected in heparinized tubes, centrifuged at 3000×g for 5 min at 4 °C, and plasma was separated and stored at −80 °C until analysis. At termination, mice were sacrificed under anesthesia, and the kidneys were isolated and sliced. One slice was fixed with 4% paraformaldehyde and embedded in paraffin block for histological and immunohistological studies; the remaining renal tissue were snap-frozen in liquid N2 and stored at −80 °C until RNA or protein extraction.
Plasma and urine chemistry were analyzed by a Vitros Chemistry Analyzer (Ortho-Clinical Diagnosis, Rochester, NY). Plasma and urine creatinine concentrations were measured using a P/ACE MDQ Capillary Electrophoresis System and photodiode detector (Beckman-Coulter, Fullerton, CA) at 214 nm [10].
Measurement of plasma αKlotho, full-length FGF23, PTH, and 1,25-dihydroxyvitamin D3
αKlotho was determined by immunoprecipitation–immunoblot as described [9, 10, 25]. Intact PTH was quantified by ELIZA (Alpco, Salem, NH); 1,25-(OH)2 vitamin D3 by EIA using (Immunodiagnostic Systems, Scottsdale, AZ); and Intact FGF23 by ELIZA (Kainos, Japan) following manufactures’ instructions.
Antibodies
The following antibodies were used for immunoblotting and/or immunohistochemistry (IHC): rat monoclonal anti-αKlotho antibody (KM2076) (TransGenics, Kobe, Japan), mouse polyclonal antibody against connective tissue growth factor (CTGF) (Abcam Inc., Cambridge, MA), mouse monoclonal antibody against β-actin (Sigma-Aldrich, St. Louis, MO), mouse monoclonal antibody against α-smooth muscle actin (α-SMA) (Sigma-Aldrich, St. Louis, MO), and goat polyclonal antibody against neutrophil gelatinase-associated lipocalin-2 (NGAL) (R&D Systems Inc., Minneapolis, MN). Secondary antibodies coupled to horseradish peroxidase for immunoblotting, or to FITC, Alexa Fluor, and Syto 61 infrared fluorescent nuclear acid, as well as wheat germ agglutinin (WGA) conjugated to Alexa Fluor 555 for IHC were purchased from Molecular Probes/Invitrogen (Molecular Probes Inc., Eugene, OR).
Kidney histology and histopathology
Kidney tissues were fixed in 4% paraformaldehyde for 16 h at 4 °C, and 4 μm sections of paraffin embedded kidney tissues were stained with Hematoxylin and Eosin (HE), Trichrome (TC), and Picrosirius red (PSR). Kidney histology was examined and photographed by a histopathologist blinded to the experimental protocols. We used a semi-quantitative pathological score systems for the evaluation of chronic kidney histologic damage based on the literature with minor modification [1, 2, 22, 24, 26]. The images were reviewed for four types of changes: glomeruli (sclerosis, dilated, or collapsing Bowman’ s space), tubulointerstitial fibrosis, chronic inflammatory cells infiltration, and renal tubules (necrosis, casts, or atrophy) at ×40 magnification on HE-stained kidney sections. Each item was scored from 0 to 4 based on percentage involvement (<25, 25–50, 50 > 75, or >75% respectively). Therefore, there might be 16 at maximum in each field. We randomly examined 10 fields at ×40, and the final score will be from 0 to 160.
To evaluate renal fibrosis, 4 μm paraffin-embedded kidney sections were stained with TC and PSR, examined, and photographed by a histopathologist blinded to the experimental protocol. Renal fibrosis score is expressed in arbitrary units, and reflects both fibrotic area and severity of fibrosis in PSR-stained sections, which were quantified with Image J software with published methods [8, 10, 11, 27]. If the photo micrograph contains glomeruli, we removed it manually with Image J software. Tubulointerstitial fibrosis is, hereinafter, simply referred as for renal fibrosis.
Immunohistochemistry and immunoblotting in the kidney
Four-micrometers sections of paraffin-embedded kidney were subjected to IHC with established protocols [8, 10, 11, 27]. Total kidney lysate covering all kidney zones was prepared as described [8, 10, 11, 27]. Thirty-micrograms protein of kidney lysate was solubilized in Laemmli’ s sample buffer and subjected to SDS-PAGE. After transferring to PVDF membranes, proteins were immunoblotted with different primary antibodies and β-actin for loading control. Signal was visualized using the ECL kit (Perkin-Elmer LAS, Inc., Boston, MA).
Assessment of cardiac fibrosis and hypertrophy
Paraffin-embedded heart sections were stained with TC to assess fibrosis. Images were scanned with Zeiss laser confocal scanning microscope (Carl Zeiss Micro-Imaging, Inc., Thornwood, NY), and analyzed using the Image J program [10, 11, 28]. Cardiac fibrosis score is also expressed in arbitrary unit, which reflects both fibrotic area and severity of fibrosis in TC-stained heart sections. To measure the surface area of cardiomyocytes, cardiomycte membranes in heart sections were labeled with Alexa Fluor 555-conjugated to WGA (Invitrogen, Carlsbad, CA) as described [10, 11]. Immunofluorescent images were taken on a Zeiss laser confocal scanning microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY). Image J software was used to quantify cross-sectional cell surface area along the mid-chamber free wall based on WGA-positive staining [10, 11].
Von Kossa staining and calcium concentration measurement
The aorta was stained for calcification with Von Kossa [9]. Tissue sections were incubated with 1% silver nitrate solution under ultraviolet light for 30 min followed by incubation with 5% sodium thiosulfate for 10 min to remove the unreacted silver. Aortic sections were counterstained with nuclear fast red, photographed blindly by two investigators using Axioplan 2 Imaging (Carl Zeiss MicroImaging, Inc., Thornwood, NY). The calcium content in thoracic aortas was measured using the o-cresolphthalein complexone method (Sigma-Aldrich, St. Louis, MO) [9], which was quantified by normalized concentration to protein concentration (μg/mg protein) determined by Bradford protein assay.
Real-time reverse transcription–polymerase chain reaction (RT–PCR)
Total RNA was extracted by RNAeasy kit (Qiagen, Germantown, MD) from mouse kidneys. Complimentary DNA (cDNA) was generated with oligo-dT primers using SuperScript III First Strand Synthesis System (Invitrogen, Carlsbad, CA) according to manufacturer’s protocol. Primers used for qPCR are shown in Supplementary Table 1 with conditions described [8, 11, 22, 24]. The reaction was performed in triplicate for each sample. Data are expressed at amplification number of 2−ΔΔCt by normalization of cyclophilin and comparison to controls.
Statistical analyses
Data are expressed as means ± SD. Analysis was performed with Sigma Stat software (Systat Software, Inc., San Jose, CA). As appropriate, statistical analysis was performed using unpaired Student-t-test, or one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls post hoc test when applicable. A value of P ≤ 0.05 was considered statistically significant.
Results
Acute dose-dependent CP nephrotoxicity
As the severity of kidney injury is an important independent contributor to AKI-to-CKD progression [29], we tested the effect of CP dosage on kidney damage and mortality. Mice given higher dose (20 mg/Kg) CP had higher mortality compared to lower dose (10 mg/Kg) (Fig. 1a). The higher dose CP also caused more significant elevation of plasma creatinine (PCr) (Fig. 1b) and plasma phosphate (Pi) (Fig. 1c) at early phase (2 weeks after CP injection) compared to the lower dose. All of the mice injected with high dose CP died ~3 weeks post-injection, which may be useful for studying AKI and mortality but renders the study of CKD progression impossible.
Fig. 1.
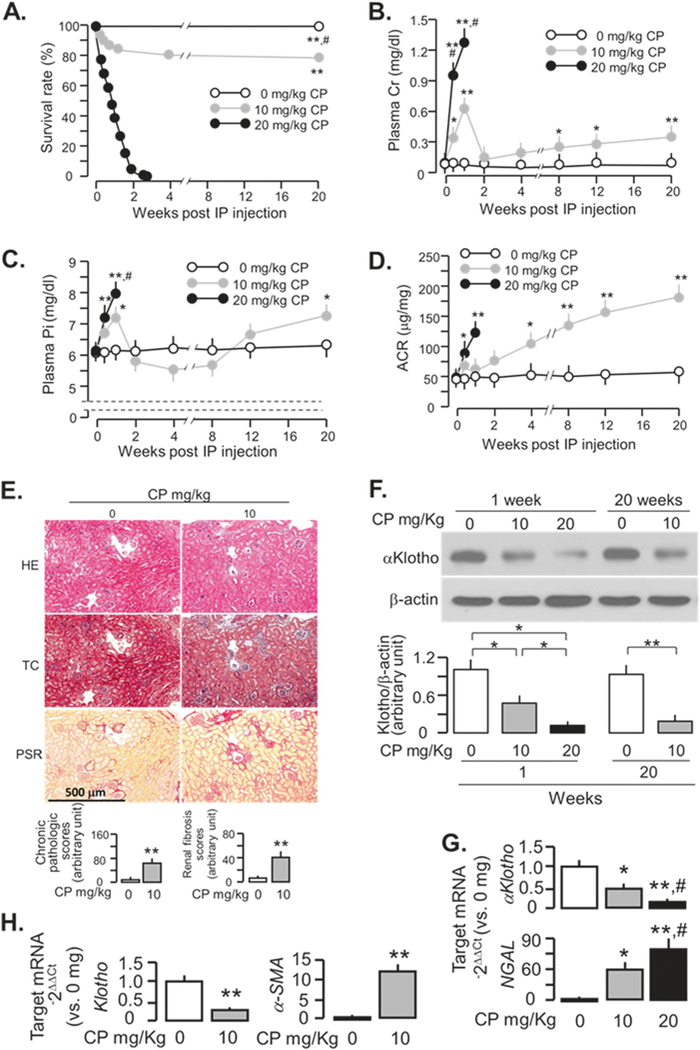
Dose dependent cisplatin (CP)-induced kidney injury and mortality in WT mice. 129 sv WT mice were intraperitoneally injected once with 0, 10, 20 mg/Kg CP and followed for up to 20 weeks after the injection. a Survival rate. b Plasma Cr. c Plasma Pi. d ACR. e Kidney histology assessed by HE, TC, and PSR staining (scale bar = 500 μm); and semi-quantitative assessment (bottom panel including chronic pathologic score based on HE stain and renal fibrosis score based on PSR stain). f Representative immunoblots for αKlotho protein in total kidney lysates (upper panel). Summary of all immunoblots (bottom panel). g αKlotho and NGAL mRNA expression in kidney lysates at one week after CP injection. Transcripts in the kidneys were analyzed with qPCR and results are expressed as 2−ΔΔCt (Ct = cycle number) by normalization to cyclophilin and compared to the 0 mg/Kg group. Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student–Newman–Keuls post hoc test, and significance was accepted when *P < 0.05; **P < 0.01 vs. 0mg/Kg, #P < 0.05 vs. 10mg/Kg for a–d, g; or when *P < 0.05; **P < 0.01 between two groups for f. h αKlotho and α-SMA mRNA expression in kidney lysates at 20 weeks after the injection. Data are expressed as means ± SD from each group and statistical significance was evaluated by unpaired Student-t-test and significance was accepted when *P < 0.05; **P < 0.01 between two groups for e and h. BUN blood urea nitrogen; Cr creatinine; CP cisplatin; HE Hematoxylin and Eosin stain; IP intraperitoneal injection; Pi plasma phosphate; TC Trichrome stain; PSR Picrosirius red stain
Gender effect on AKI
To explore the effect of gender on severity of AKI, we examined for differences of blood urea nitrogen (BUN) and PCr between male and female mice at 7th day after CP (10 mg/Kg) injection and found more severe kidney injury in male mice compared to female mice (Table 1), which is consistent with a previous study of CP nephrotoxicity in rats [30].
Table 1.
Comparison of differences of the manifestations of acute kidney injury between male and female mice injected with cisplatin at 3 months of age
| Male | Female | P-value | |
|---|---|---|---|
| Animal number | 4 | 5 | |
| BUN (mg/dl) | 91.5 ± 5.4 | 80.4 ± 4.6 | 0.0125 |
| Cr (mg/dl) | 0.71 ± 0.05 | 0.64 ± 0.04 | 0.05 |
| CrCl (ml/min/100g BW) | 0.22 ± 0.01 | 0.26 ± 0.02 | <0.05 |
Data were presented as means ± SD at 7 days after cisplatin injection (10 mg/Kg). The statistical significance was analyzed by unpaired Student t-test
Chronic kidney disease from CP
By 20 weeks after low dose of CP injection (10 mg/Kg), mice developed moderate renal insufficiency as evident by elevation of plasma Cr (Fig. 1b), Pi (Fig. 1c), urinary albumin/creatinine ratio (ACR) (Fig. 1d), and alteration of renal histology including dilated or collapsed Bowman’s space, glomerular sclerosis, renal tubular degeneration, vacuolation or necrosis, and interstitial infiltration and fibrosis (Fig. 1e, and Supplementary Fig. 1). In addition, CP-injected mice had lower renal αKlotho (Fig. 1f, g) at 1 week and sustained lower αKlotho and higher α-SMA in the kidney (Fig. 1f, h) at 20 weeks, which are typical for CKD [8–11, 31].
Effect of age on AKI and AKI-to-CKD transition
Older rats had more severe acute kidney damage after CP injection [32], which differs from human study [33]. To further define the age effect on the severity of CP nephrotoxicity in mice, we injected CP into three groups of mice at ages: 3, 6, and 12 months old. Clearly, after CP injection, 12-months-old mice had more severe decline in ClCr (Fig. 2a) and significantly higher BUN (Fig. 2b), plasma Pi (Fig. 2c), and ACR (Fig. 2d) than 3-months-old mice. There was more kidney morphologic changes and renal fibrosis in 12-months-old mice injected with CP compared to 3 months old (Fig. 2e). There was more reduction of αKlotho and increase in fibrotic markers in the kidneys in 12-months-old mice compared to those in 3-months-old mice (Fig. 2f). The CP-induced changes in 6-months-old mice were intermediate between the 3 and 12-months-old-mice (Fig. 2), indicating older age is associated with more severe CKD with less αKlotho expression, and more renal fibrosis. To explore whether the age dependence is generalized in rodents, we also injected 10mg/Kg CP into 2 and 12-months-old SD rats respectively. Similarly, more severe kidney injury was found in older compared to younger rats (Supplementary Fig. 2) supporting that this is an universal finding.
Fig. 2.
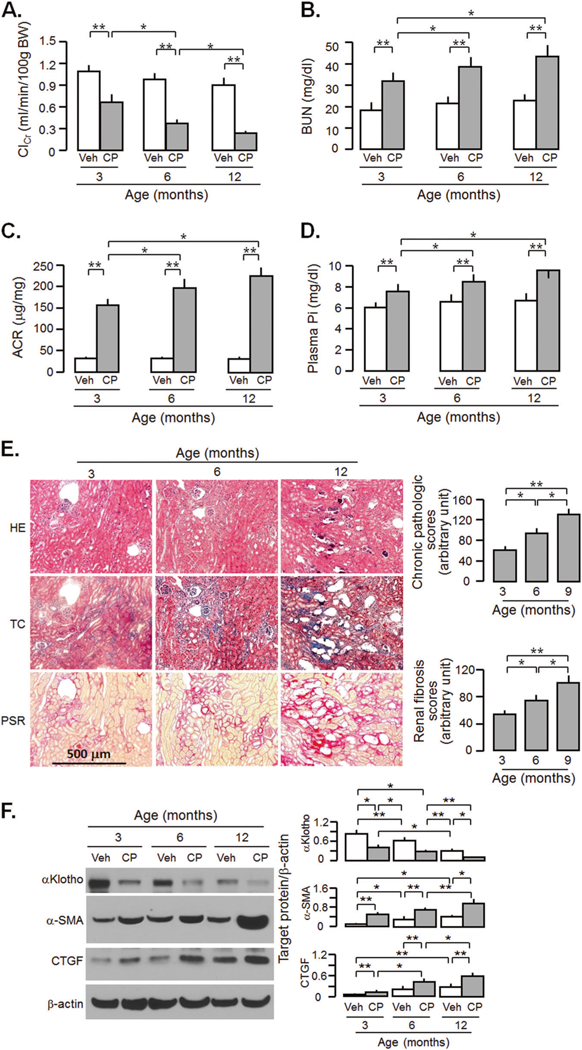
Aged mice are more vulnerable to acute cisplatin (CP) nephrotoxicity and at higher risk for CKD. WT mice at 3, 6, and 12 months old were injected with CP (10mg/Kg) or normal saline as vehicle (veh) and sacrificed at 20 weeks. a Creatinine clearance (ClCr). b Blood urea nitrogen (BUN). c Plasma phosphate (Pi). d Albumin-to-creatinine ratio (ACR). e Kidney histology assessed by HE, TC, and PSR stain (scale bar = 500 μm); and semi-quantitative assessment (right panel including chronic pathologic score based on HE stain and renal fibrosis score based on PSR stain). f αKlotho and fibrotic markers in the kidney. Left panel: representative immunoblot for αKlotho, α-SMA, and CTGF in total kidney lysates. Right panel: summary of all immunoblots. Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student–Newman–Keuls post hoc test, and significance was accepted when *P < 0.05; **P < 0.01 between two groups. HE Hematoxylin and Eosin stain; TC Trichrome stain; PSR Picrosirius red stain
High phosphate promotes AKI-to-CKD progression after CP-induced AKI
Human observational studies showed a relationship between excessive phosphate and abnormal phosphate metabolism and cardiovascular disease (CVD) in the general population and CKD population [34–40]. However, evidence for direct pathogenic effects of phosphate on cardiovascular events and kidney disease progression were strictly derived from rodent experimental data [10, 11, 41–43]. High dietary phosphate modestly, but significantly reduced ClCr and increased plasma Pi in vehicle-injected mice, but remarkably exacerbated these changes in CP-injected mice compared to CP-injected mice fed a normal phosphate diet (Fig. 3a, b). Furthermore, the levels of BUN, and plasma Cr and Pi was much higher in CP-injected and high Pi-fed mice compared to other three groups starting at 10 weeks after CP injection (Supplementary Fig. 3), indicating that high dietary phosphate accelerated AKI-to-CKD progression. Kidney histology study showed more changes in kidney structure and more visible renal fibrosis in high phosphate fed mice in both vehicle and CP-injected mice (Fig. 3c, and Supplementary Fig. 4). The highest level of fibrotic markers were in the kidneys of CP-injected mice fed a high phosphate diet, and moderate elevations of those markers in either high phosphate fed mice alone or CP-injected mice alone, compared to vehicle-injected mice fed a normal phosphate diet (Fig. 3d). High phosphate diet did not only decrease renal αKlotho protein expression in vehicle-injected mice, but also exacerbated the decline in renal αKlotho protein levels in CP-injected mice (Fig. 3d).
Fig. 3.
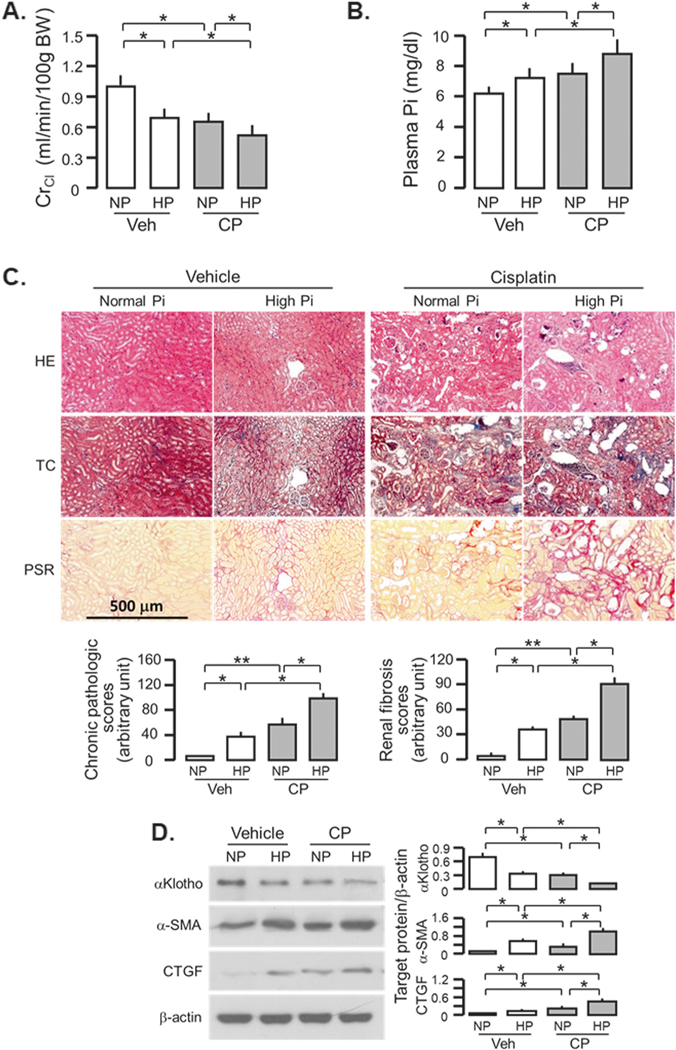
High phosphate (Pi) diet exacerbates renal fibrosis and renal αKlotho reduction in CKD mice. WT mice at 3 months old were injected with CP (10 mg/Kg) or normal saline as vehicle (veh) followed by normal (NP) or high Pi (HP) diet (2.0% Pi), respectively, for 18 weeks starting 2 weeks after CP injection. a Creatinine clearance (ClCr). b Plasma phosphate (Pi). c Kidney histology assessed by HE, TC, and PSR stain (scale bar = 500 μm); and semi-quantitative assessment (bottom panel including chronic pathologic score based on HE stain and renal fibrosis score based on PSR stain). d αKlotho and fibrotic markers in the kidney. Left panel: representative immunoblot for αKlotho, α-SMA, and CTGF protein in total kidney lysates. Right panel: summary of all immunoblots. Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student–Newman–Keuls post hoc test and significance was accepted when *P < 0.05; **P < 0.01 between two groups. HE Hematoxylin and Eosin stain; TC Trichrome stain; PSR Picrosirius red stain
Effect of αKlotho levels on the severity of AKI and CKD after CP injection
We have shown that αKlotho levels prior to renal insults influence renal outcome and the speed of progression to CKD after IRI-induced AKI [8]. As expected, heterozygous αKlotho-deficient (KI/+) mice had lower ClCr (Fig. 4a), higher plasma Pi levels (Fig. 4b), and ACR (Fig. 4c) compared to WT mice. Transgenic mice with Klotho overexpression (Tg-Kl) had the opposite changes, supporting our previous findings in IRI model [8]. Kidney histology showed more alterations of kidney structure and severe fibrosis in Kl/+ and milder changes in Tg-Kl mice compared to WT mice at 20 weeks after CP injection (Fig. 4d, and Supplementary Fig. 4). The higher levels of α-SMA and CTGF protein expression in Kl/+ mice and lower levels in Tg-Kl mice compared to WT mice (Fig. 4e), are in line with the changes in TC and PSR stain. As anticipated, αKlotho levels were higher in Tg-Kl mice and lower in Kl/+ mice compared to WT mice in both vehicle and CP-treated conditions (Fig. 4e). Because Tg-Kl mice have milder kidney damage at acute stage [24], the better renal outcome at chronic stage (Fig. 4) could not be solely interpreted as retardation of CKD progression. Moreover, after CP injection, Tg-Kl mice had lower renal Klotho protein than prior to injection, but still maintained higher Klotho compared to WT mice received CP, thus less kidney damage and mild renal fibrosis in Tg-Kl mice receiving CP is likely attributable to higher renal and circulating Klotho.
Fig. 4.
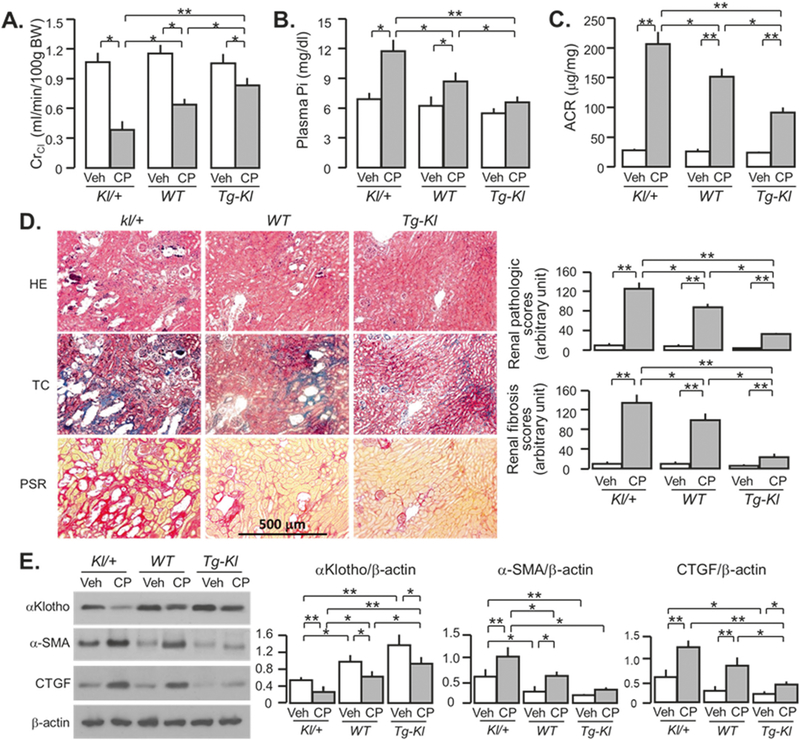
αKlotho-deficient mice progress more rapidly to CKD after cisplatin (CP) injection. Mice with three different levels of αKlotho (low kl/+; normal WT; and high Tg-Kl) at 3 months old were intraperitoneally injected with normal saline as vehicle or CP (10mg/Kg), and fed high phosphate (Pi) rodent chow 2 weeks after the injection for 18 weeks. a Creatinine clearance (ClCr). b Plasma phosphate (Pi). c Albumin-to-creatinine ratio (ACR). d Kidney histology assessed by HE, TC, and PSR stain (scale bar = 500 μm); and semi-quantitative assessment (right panel including chronic pathologic score based on HE stain and renal fibrosis score based on PSR stain). e αKlotho and fibrotic markers in the kidney. Left panel: representative immune-blotting for αKlotho, α-SMA, and CTGF protein in total kidney lysates. Right panel: summary of immunoblots in arbitrary units from all immunoblots. Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student–Newman–Keuls post hoc test, and significance was accepted when *P < 0.05; **P < 0.01 between two groups. HE Hematoxylin and Eosin stain; TC Trichrome stain; PSR Picrosirius red stain
Cardiovascular disease in CP-induced CKD
As CVD is the major cause of mortality in CKD; a relevant experimental AKI-to-CKD transition model should exhibit CVD. CP-injected mice had heavier hearts (Fig. 5a) and larger cardiomyocytes (Fig. 5b) than vehicle-injected mice after high dietary phosphate intake. We also found that 18-week high Pi diet alone was sufficient to initiate cardiac hypertrophy in WT mice with normal kidney function (Fig. 5a, b). Furthermore, high phosphate diet-induced cardiac fibrosis were more appreciable in CP-injected mice than vehicle-injected mice (Fig. 5c).
Fig. 5.
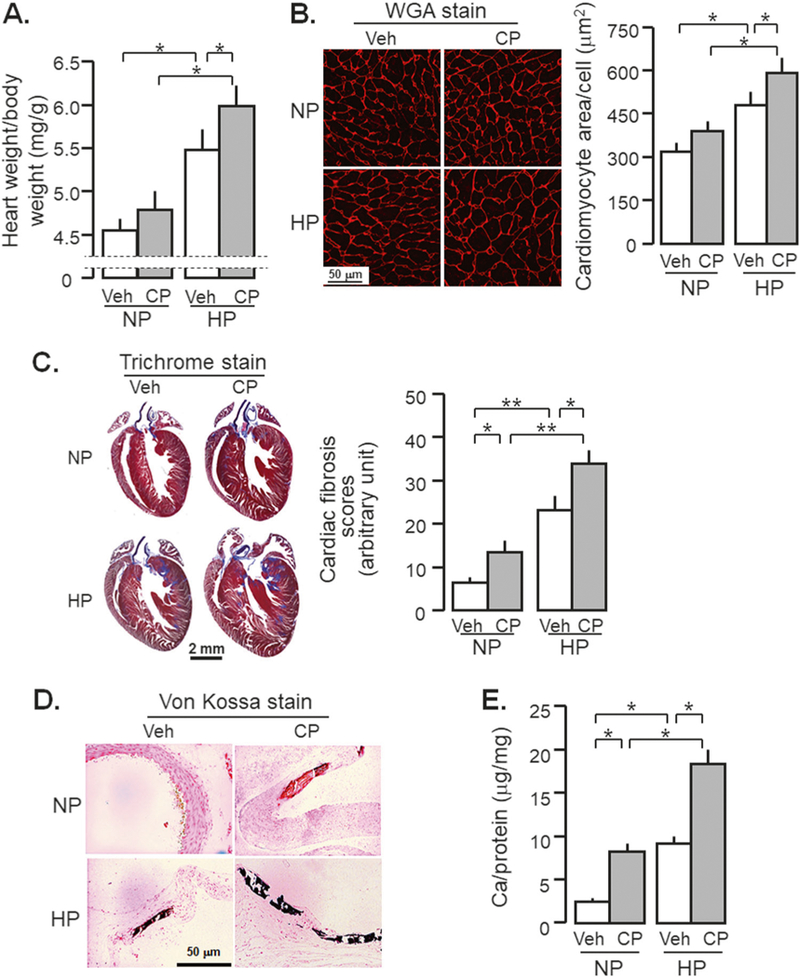
High phosphate (Pi) diet promotes cardiovascular disease in CKD mice. WT mice at 3 months old were injected with CP (10 mg/Kg) or normal saline as vehicle (veh) followed by normal (NP) or high phosphate diet (HP, 2.0% Pi) for 18 weeks starting 2 weeks after CP injection. At 20 weeks after the injection, mice were sacrificed and the hearts and aorta were collected for morphology and molecular studies. a Heart weight/body weight. b Wheat germ agglutinin (WGA) stain: Left panel: representative microscopic images with WGA in heart section, scale bar = 50 μm. Right panel: sizes of cardiomyocytes were obtained from WGA-stained section with Image J program. c Cardiac fibrosis. Left panel: representative Trichrome staining in a heart sections, scale bar = 2 mm. Right panel: cardiac fibrosis score. d Von Kossa stain: there were 0/10 positive stain in Veh-NP, 2/6 positive stain in CP-NP, 2/10 positive stain in Veh-HP, and 5/8 positive stain in CP-HP group. Representative micrographs of positive stain in the aortic roots. Scale bar = 50 μm. e Calcium content in the thoracic aorta with o-cresolphthalein complexone (OCPC) assay. Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student–Newman–Keuls post hoc test, and significance was accepted when *P < 0.05; **P < 0.01 between two groups
In addition to cardiac remodeling, severe vascular calcification was found in CP-injected mice after high dietary phosphate intake. Much less vascular calcification was seen in CP-injected mice without high phosphate diet (Fig. 5d). Quantitative determination of calcium content in aorta showed the highest levels of calcium in CP-treated mice with high phosphate diet, moderate levels in either CP-injected mice with normal phosphate diet or vehicle-injected mice with high phosphate diet (Fig. 5e).
Therefore, CP-injected mice developed AKI in acute stage and progressed to CKD with the full range of disturbed mineral homeostasis (Table 2), renal fibrosis, cardiac remodeling, and vascular calcification after chronic high dietary phosphate loading (Fig. 5).
Table 2.
Association of cisplatin doses with the changes in mineral metabolism and renal function in WT mice at 20 weeks after cisplatin or vehicle injection at 3 months of age
| Cisplatin-0 mg/kg |
Cisplatin-10 mg/kg |
|||
|---|---|---|---|---|
| NP | HP | NP | HP | |
| Animal number | 10 | 10 | 9 | 8 |
| BUN (mg/dl) | 19.3 ± 0.8 | 28.6 ± 1.3* | 31.7 ± 1.5** | 43.9 ± 2.2**,#,$ |
| Cr (mg/dl) | 0.08 ± 0.01 | 0.19 ± 0.07* | 0.31 ± 0.18**,# | 0.52 ± 0.23**,##,$ |
| CrCl (ml/min/100 g BW) | 1.11 ± 0.06 | 0.90 ± 0.07 | 0.73 ± 0.05 | 0.58 ± 0.03**,##,$ |
| Plasma Pi (mg/dl) | 6.38 ± 0.21 | 7.03 ± 0.85 | 7.91 ± 0.28 | 8.47 ± 0.69**,##,$ |
| Plasma Ca (mg/dl) | 9.25 ± 0.05 | 9.09 ± 0.08 | 9.15 ± 0.07 | 9.01 ± 0.05* |
| Plasma PTH (pg/ml) | 103 ± 31 | 219 ± 19* | 305 ± 47**,# | 439 ± 54**,##,$ $ |
| Plasma 1,25-(OH)2D3 (pg/ml) | 109.2 ± 4.8 | 78.7 ± 4.5* | 67.9 ± 5.8* | 49.2 ± 3.6**,# |
| Plasma FGF23 (pg/ml) | 163 ± 39 | 647 ± 358** | 1832 ± 237**,## | 2390 ± 494**,##,$ |
NP normal Pi diet, HP high Pi diet
Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student–Newman–Keuls post hoc test, and significance was accepted when
P < 0.05;
P < 0.01 vs. NP at 0 mg/Kg;
P < 0.05;
P < 0.01 vs. HP at 0mg/Kg;
P < 0.05;
P < 0.01 vs. NP at 10mg/Kg
Effect of αKlotho administration on AKI-to-CKD progression
To examine the αKlotho effect on AKI-to-CKD transition after acute CP nephrotoxicity, we administered αKlotho intraperitoneally to mice after exposure to CP and high phosphate diet (Fig. 6a). We found better renal function (Fig. 6b), better phosphate homeostasis (Fig. 6c), and less renal fibrosis (Fig. 6d, e, and Supplementary Fig. 6) compared to CP-injected mice without αKlotho administration. In addition, we further confirmed the efficacy of recombinant αKlotho protein in retardation of AKI-to-CKD progression [8]. αKlotho-treated mice had lesser kidney injury and fibrosis, and higher native αKlotho protein in the kidney compared to vehicle-treated mice (Fig. 6e). Higher levels of renal αKlotho might result from better preservation of renal function and lower plasma Pi (Fig. 6f). Furthermore, αKlotho treatment significantly alleviated uremic cardiomyopathy (Fig. 6g). Obviously, this study design cannot discern whether better cardiac histology results from αKlotho’s direct cardioprotective effect, secondary effects due to renoprotection and better renal function, or both.
Fig. 6.
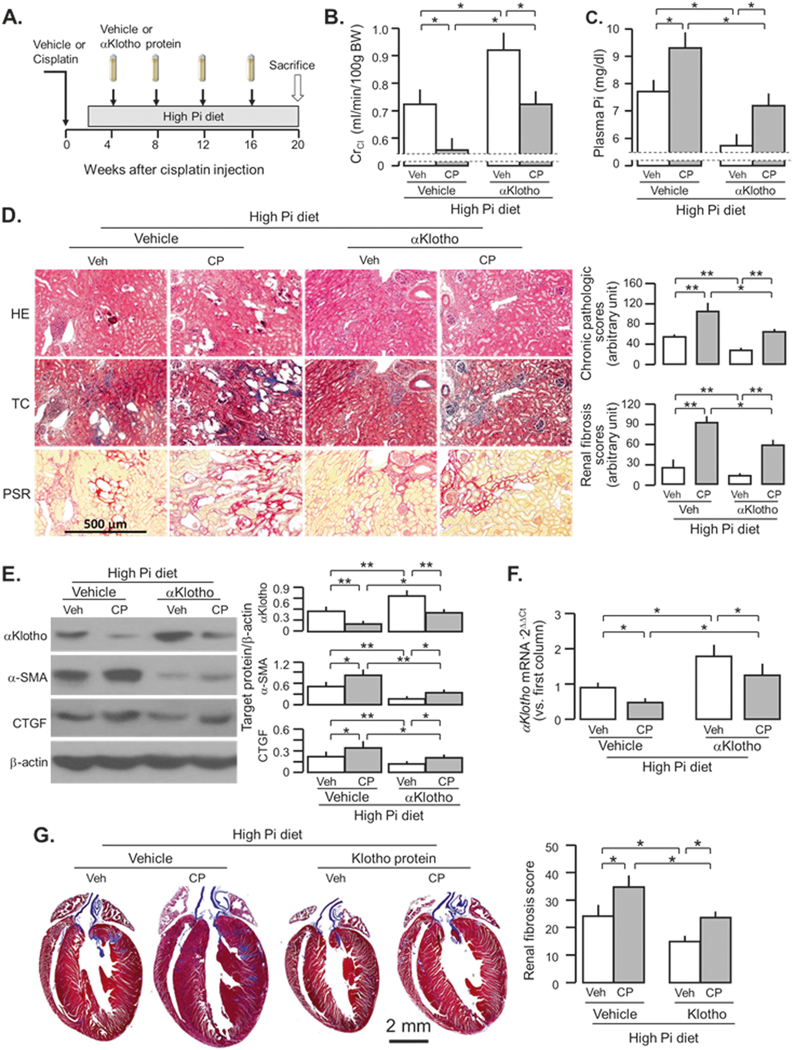
αKlotho administration post-cisplatin (CP) injection retards AKI-to-CKD progression. a WT mice at 3 months old were intraperitoneally injected with CP (10mg/Kg) or normal saline as vehicle (veh) followed by feeding with high Pi diet in 2 weeks after the injection for 18 weeks. At 4 weeks after the injection, αKlotho protein or control buffer (normal saline) was given through osmotic mini-pumps for 16 weeks. At 20 weeks after the injection, mice were sacrificed and the kidneys, and urine and plasma samples were collected for further studies. b Creatinine clearance (ClCr). c Plasma phosphate (Pi). d Kidney histology assessed by HE, TC, and PSR stain (scale bar = 500 μm); and semi-quantitative assessment (right panel including chronic pathologic score based on HE stain and renal fibrosis score based on PSR stain). e αKlotho protein and fibrotic markers in the kidney. Left panel: representative immunoblot for αKlotho, α-SMA, and CTGF protein in total kidney lysates. Right panel: summary of all immunoblots. f αKlotho mRNA expression in the kidney lysate. g Cardiac fibrosis at 20 weeks after the injection. Left panel: representative Trichrome staining in heart sections, scale bar = 2 mm. Right panel: cardiac fibrosis score. Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student–Newman–Keuls post hoc test, and significance was accepted when *P < 0.05; **P < 0.01 between two groups. HE Hematoxylin and Eosin stain; TC Trichrome stain; PSR Picrosirius red stain
Gender effect on CKD progression
We compared the difference in severity of CKD and cardiac hypertrophy between male and female mice given CP followed by normal or high Pi diet. Male mice had faster decline in renal function and rise in plasma Pi (Fig. 7a, b, and Table 3) in WT mice after CP-induced AKI, and were more sensitive to high Pi challenge with more severe loss of renal function and higher plasma Pi (Table 4 and Fig. 7c, d), more disturbed mineral metabolism and more cardiac hypertrophy than female mice (Table 4). These results are similar to clinical observations and some animal studies [44–48]. Similar to WT mice, male Kl/+ mice had worse renal function and higher plasma Pi than female mice, indicating that gender influences AKI progression to CKD (Fig. 7e, f). Furthermore, on the comparison of ClCr and plasma Pi between WT CKD mice (Fig. 7c, d, and Table 3) and αKlotho-treated WT CKD mice (Fig. 7g, h, and Table 5), exogenous αKlotho administration was more effective in improvement of kidney function and Pi metabolism in male CKD mice, because male mice had 33.0% elevation of ClCr from vehicle-treated CKD mice and 13.0% reduction of plasma Pi from vehicle-treated CKD mice (Tables 3, 5), while female mice only had 23% elevation of ClCr and 8.9% reduction of plasma Pi. Similarly, cardiac hypertrophy was attenuated by 26.4% in male CKD mice vs. 11.1% in female CKD mice when αKlotho was given. Therefore, male CKD mice benefit more from αKlotho protein administration compared to females, although male CKD mice still had worse renal function, Pi metabolism, and cardiomyopathy than female CKD mice.
Fig. 7.
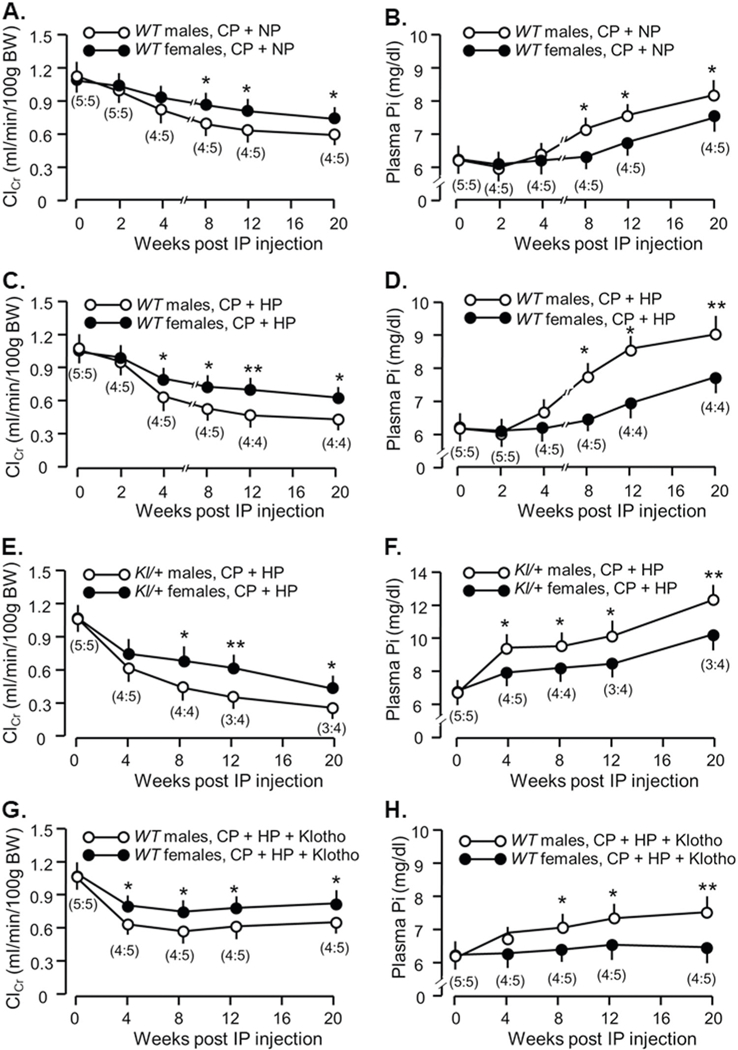
Sexual dimorphism in CP-induced CKD progression and in the response to αKlotho treatment. WT mice at 3 months old were injected with CP (10 mg/Kg) or normal saline as vehicle (veh) followed by normal (NP) or high phosphate diet (HP, 2.0% Pi) for 18 weeks starting 2 weeks after CP injection. At 20 weeks after the injection, mice were sacrificed. a Creatinine clearance (ClCr) and b plasma phosphate (Pi) in male and female mice fed with normal Pi diet. c Creatinine clearance (ClCr) and d plasma phosphate (Pi) in male and female mice fed with high Pi diet. Mice with low levels of αKlotho (kl/+) at 3 months old were intraperitoneally injected with CP (10 mg/Kg), and fed high phosphate (Pi) rodent chow 2 weeks after the injection for 18 weeks. e Creatinine clearance (ClCr) and f plasma phosphate (Pi) in male and female Kl/+mice. WT mice at 3 months old were intraperitoneally injected with CP (10 mg/Kg) followed by feeding with high Pi diet in 2 weeks after the injection for 18 weeks. At 4 weeks after the injection, αKlotho protein was given through osmotic minipumps for 16 weeks. g Creatinine clearance (ClCr) and h plasma phosphate (Pi) in male and female WT mice. Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student–Newman–Keuls post hoc test, and significance was accepted when *P < 0.05; **P < 0.01 between two groups at same time point. The number in brackets is male:female number in each group at different time point
Table 3.
Comparison of differences in manifestations of CKD and cardiac hypertrophy between male and female mice fed with a normal Pi diet
| Male | Female | P-value | |
|---|---|---|---|
| Animal number | 4 | 5 | |
| BUN (mg/dl) | 36.3 ± 1.3 | 31.7 ± 1.9 | <0.01 |
| Cr (mg/dl) | 0.34 ± 0.09 | 0.31 ± 0.07 | <0.05 |
| CrCl (ml/min/100g BW) | 0.69 ± 0.04 | 0.77 ± 0.06 | <0.05 |
| Plasma Pi (mg/dl) | 8.21 ± 0.32 | 7.62 ± 0.24 | <0.05 |
| Plasma Ca (mg/dl) | 9.21 ± 0.10 | 9.11 ± 0.07 | >0.05 |
| Plasma PTH (pg/ml) | 321 ± 52 | 291 ± 42 | >0.05 |
| Plasma 1,25-(OH)2D3 (pg/ml) | 70.4 ± 6.2 | 65.5 ± 5.3 | >0.05 |
| Plasma FGF23 (pg/ml) | 2016 ± 330 | 1757 ± 139 | >0.05 |
| Heart weight/body weight (mg/g) | 5.69 ± 0.32 | 5.28 ± 0.31 | 0.08 |
Data presented in Table 3 are derived from mice injected cisplatin and treated with normal Pi diet presented in Table 2 and were shown as means ± SD at 20 weeks. The statistical significance between male and female mice was analyzed by unpaired Student t-test
Table 4.
Comparison of differences in manifestations of CKD and cardiac hypertrophy between male and female mice fed with a high Pi diet
| Male | Female | P-value | |
|---|---|---|---|
| Animal number | 4 | 4 | |
| BUN (mg/dl) | 48.4 ± 3.4 | 39.5 ± 2.4 | <0.05 |
| Cr (mg/dl) | 0.61 ± 0.04 | 0.43 ± 0.03 | <0.01 |
| CrCl (ml/min/100 g BW) | 0.51 ± 0.04 | 0.64 ± 0.05 | <0.05 |
| Plasma Pi (mg/dl) | 9.04 ± 0.71 | 7.78 ± 0.65 | 0.058 |
| Plasma Ca (mg/dl) | 8.97 ± 0.75 | 9.06 ± 0.79 | >0.05 |
| Plasma PTH (pg/ml) | 516 ± 49 | 390 ± 23 | <0.01 |
| Plasma 1,25-(OH)2D3 (pg/ml) | 43.1 ± 3.8 | 56.4 ± 4.9 | <0.05 |
| Plasma FGF23 (pg/ml) | 2700 ± 325 | 2081 ± 196 | <0.05 |
| Heart weight/body weight (mg/g) | 6.61 ± 0.42 | 5.68 ± 0.29 | <0.05 |
Data presented in Table 4 are derived from mice injected cisplatin and treated with high Pi diet presented in Table 2 and were shown as means ± SD at 20 weeks. The statistical significance was analyzed by unpaired Student t-test
Table 5.
Comparison of differences in responses to exogenous Klotho protein between male and female CKD mice fed with a high Pi diet
| Vehicle |
Cisplatin |
|||||
|---|---|---|---|---|---|---|
| Male | Female | P-value | Male | Female | P-value | |
| Animal number | 5 | 5 | 4 | 5 | ||
| BUN (mg/dl) | 22.3 ± 1.2 | 20.9 ± 1.1 | >0.05 | 31.2 ± 2.2 | 22.9 ± 1.5 | <0.01 |
| Cr (mg/dl) | 0.14 ± 0.03 | 0.11 ± 0.02 | >0.05 | 0.27 ± 0.03 | 0.21 ± 0.01 | <0.01 |
| CrCl (ml/min/100g BW) | 1.01 ± 0.06 | 0.94 ± 0.05 | 0.08 | 0.75 ± 0.03 | 0.85 ± 0.04 | <0.05 |
| Plasma Pi (mg/dl) | 6.92 ± 0.48 | 6.67 ± 0.31 | >0.05 | 7.41 ± 0.27 | 6.38 ± 0.21 | <0.01 |
| Plasma Ca (mg/dl) | 9.31 ± 0.45 | 9.23 ± 0.32 | >0.05 | 9.12 ± 0.45 | 9.19 ± 0.32 | >0.05 |
| Plasma PTH (pg/ml) | 165 ± 16 | 147 ± 10 | >0.05 | 303 ± 36 | 290 ± 19 | >0.05 |
| Plasma 1,25-(OH)2D3 (pg/ml) | 78.2 ± 6.4 | 89.8 ± 11.7 | 0.087 | 27.1 ± 2.7 | 34.6 ± 3.6 | <0.05 |
| Plasma FGF23 (pg/ml) | 155 ± 78 | 140 ± 45 | >0.05 | 1308 ± 219 | 883 ± 102 | <0.01 |
| Heart weight/body weight (mg/g) | 5.13 ± 0.29 | 4.89 ± 0.14 | >0.05 | 5.23 ± 0.39 | 5.11 ± 0.21 | >0.05 |
Data were presented as means ± SD at 20 weeks. The statistical significance was analyzed by unpaired Student t-test
Discussion
Most of the present knowledge on renal physiology and pathophysiology comes from the experimental study in laboratory animals. Discovery of novel therapeutic or diagnostic approaches primarily begins in animal models. Therefore, having a technically easy, reproducible, and stable experimental models of CKD is an essential step. We present a rodent model of a single bolus intraperitoneal injection of CP followed by high dietary phosphate intake that produces CKD in both mice and rats. The rodents develop AKI, as previously noted [24, 49–56], but importantly, one does not lose animals from AKI, and acute CP nephrotoxicity progresses to CKD with CVD and disturbed mineral metabolism relevant to human CKD. Therefore, this rodent model is suitable for the study of AKI, or AKI-to-CKD progression or chronic complication, such as cardiovascular disease in CKD.
Phenotypes of CP-model of AKI, AKI-to-CKD transition, and CKD-associated CVD
Up to thirty-two percent of the patients after CP treatment develop AKI constituting the major limiting factor to human use of CP [13, 14, 57]. Animal studies have shown that CP-induced AKI can progress to CKD in certain scenarios [16–20, 58, 59]. Basically, there are two strategies to induce CKD with CP (referred to [21] for detailed information). One is multiple injections. Decades ago, Marcussen reported that repeated low dose CP injection (2 mg/Kg/week for 10 weeks followed by 3 times/week for two weeks) induced chronic renal failure with atubular glomeruli, massive tubular dilation, and interstitial fibrosis in rats [60]. Sharp and colleagues showed that repeatedly weekly injection of moderate dose of CP (7–9 mg/Kg body weight) consecutively for 4 weeks induced CKD [20]. Interestingly, repeated low doses of CP (1 mg/Kg, twice weekly) for 10 weeks still effectively induced an increase in serum Cr, interstitial fibrosis, tubular dilation, and atrophy in rats [18]. Torres and colleagues reported two injections of high dose CP (15 mg/Kg body weight) 2 weeks apart induced CKD around 9 weeks, and CKD progression by 25 weeks [19]. Okada showed that 5 mg/Kg CP injection followed by repeating injection twice in 21-day interval dramatically decrease ClCr by 50% in rats [61]. When CP was repeatedly given weekly at 10 mg/Kg, all mice died within 4 weeks [17]. So, if one elects repeated injection, the dose should be low enough or interval of injection should be longer to prevent animal death. The advantage of repeating low dose is that there is low cell injury or death, but high level of renal fibrosis [20, 21]. Second method is high dose of CP injection (≥20 mg/Kg), which induces serious kidney injury and inevitably ends with high mortality that prevents chronic studies (Fig. 1a) [20]. The CP-induced CKD is characterized by tubular atrophy and dilation, massive interstitial fibrosis and collagen deposition in the kidney, as well as peritubular capillary rarefaction [19]. But most, if not all, of these studies primarily focused on the changes in the kidney and none of them examined extra-renal complications in mineral metabolism and cardiovascular pathology [19, 20]. In addition, none of those experiments tested the role of high Pi in acceleration of CKD progression from acute CP nephrotoxicity. The current study clearly showed that mice do not only develop CKD with renal fibrosis, uremia, and CKD-MBD, but also uremic cardiomyopathy and vascular calcification after a single injection of moderate dose of CP (10 mg/Kg) followed by high Pi diet. No surgical skills are required to generate this model.
Gender effect
Epidemiologic observations showed more dramatic kidney damage in male AKI patients and higher risk of progression to CKD in male than female patients [44, 47, 62–64]. There are limited animal experiments to address the gender difference in AKI and CKD. We showed higher plasma Cr and BUN, and lower ClCr in male mice than female mice after one time CP injection even after normalization of animal body weights. We also observed more renal function loss, higher cardiac hypertrophy, and worse mineral metabolism in male mice than female mice after CP injection and high Pi diet challenge. But we cannot conclude that gender accounts for faster progression to CKD in male than female since male mice had more severe acute kidney damage at the onset. Further experiment is required to reduce acute kidney damage in male by lower CP dose and compare decline rate of renal function between male and female mice when both have same trough ClCr at acute phase, which may allow us to examine if gender makes the difference in CKD progression.
αKlotho treatment prevents CKD progression, improves Pi metabolism, and cardiac hypertrophy in CKD more efficiently in male mice than female mice, suggesting that CKD male benefits more from Klotho treatment than CKD females. These findings are consistent with previous reports of transgenic male mice with overexpressing Klotho having prolonged lifespan compared to WT male mice, but transgenic female mice do not [65]. These results provide exciting evidence to support that Klotho protein might be highly promising strategy for CKD subjects.
Age effect
We showed that old mice had more severe kidney injury and cardiomyopathy after exposure to IRI followed by 2.0% Pi diet than young mice [10]. The current study further confirmed that the aging renders mice more susceptible to CP nephrotoxicity, which is in agreement with clinical observation [14]. Furthermore, older mice develop worse CVD in the CP nephrotoxicity model compared to young mice. The molecular mechanisms of how aging affects kidney and CVD remain to be established and are likely complicated. Factors such as low renal and circulating αKlotho [10], oxidative stress [66], endothelial dysfunction [67, 68], and low autophagy [69] may particulate in contributing to poor renal and cardiovascular outcome in aged mice.
αKlotho effect on the severity of CP-induced nephrotoxicity
αKlotho deficiency is a feature of CKD and is a critical pathogenic intermediate for CKD progression and CVD development in CKD [8–11, 35, 41, 70, 71]. Low αKlotho levels in the kidney and the circulation were not only present at acute period of CP nephrotoxicity, but persisted way beyond the apparent “recovery” of plasma Cr. CKD mice had high plasma Pi, PTH, and FGF23, as well as low 1,25-(OH)2 vitamin D3, all of them might be contributors to Klotho deficiency. As we previously showed the effect of αKlotho on the IRI model [8, 11], αKlotho supplementation post-CP injection was effective in the retardation of CKD progression, as well as attenuation of uremic cardiomyopathy and vascular calcification, confirming the efficacy of αKlotho treatment in the CP-induced CKD model fed with high Pi diet.
In conclusion, CP leads to not only acute nephrotoxicity-AKI, and it causes AKI-to-CKD transition when coupled with high dietary phosphate. CKD rodents have disturbed mineral metabolism and uremic cardiovasculopathy. It is a technically easy and reproducible model that can be utilized for the study of AKI-to-CKD transition and CKD-associated complications, and test therapeutic maneuvers.
Supplementary Material
Acknowledgements
These studies were supported by the National Institutes of Health (NIDDK-R01-DK091392, NIDDK-R01-DK092461, NIDDK-R01-DK092461–6A1), the George O’Brien Kidney Research Center at University of Texas Southwestern Medical Center (NIDDK-P30-DK079328), the Charles and Jane Pak Center Innovative Research Support, the Pak-Seldin Center for Metabolic and Clinical Research, and C. Y Pak Foundation. The authors particularly thank Ms. Ao Bian, Ms. Han Cho, and Ms. Jean Paek for expert technical assistance.
Footnotes
Compliance with ethical standards
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41374–018-0063–2) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Cerda J, Lameire N, Eggers P, et al. Epidemiology of acute kidney injury. Clin J Am Soc Nephrol. 2008;3:881–6. [DOI] [PubMed] [Google Scholar]
- 2.Susantitaphong P, Cruz DN, Cerda J, et al. World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol. 2013;8:1482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouchard J, Mehta RL. Acute kidney injury in western countries. Kidney Dis. 2016;2:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Case J, Khan S, Khalid R, Khan A. Epidemiology of acute kidney injury in the intensive care unit. Crit Care Res Pract. 2013;2013: 479730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali T, Khan I, Simpson W, et al. Incidence and outcomes in acute kidney injury: a comprehensive population-based study. J Am Soc Nephrol. 2007;18:1292–8. [DOI] [PubMed] [Google Scholar]
- 6.Hsu CY. Yes, AKI truly leads to CKD. J Am Soc Nephrol. 2012;23:967–9. [DOI] [PubMed] [Google Scholar]
- 7.Bucaloiu ID, Kirchner HL, Norfolk ER, Hartle JE 2nd, Perkins RM. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012;81:477–85. [DOI] [PubMed] [Google Scholar]
- 8.Shi M, Flores B, Gillings N, et al. AlphaKlotho mitigates progression of AKI to CKD through activation of autophagy. J Am Soc Nephrol. 2016;27:2331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu MC, Shi M, Cho HJ, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol. 2015;26:1290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu MC, Shi M, Gillings N, et al. Recombinant alpha-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 2017;91:1104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faught LN, Greff MJ, Rieder MJ, Koren G. Drug-induced acute kidney injury in children. Br J Clin Pharmacol. 2015;80:901–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato K, Watanabe S, Ohtsubo A, et al. Nephrotoxicity of cisplatin combination chemotherapy in thoracic malignancy patients with CKD risk factors. BMC Cancer. 2016;16:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latcha S, Jaimes EA, Patil S, et al. Long-term renal outcomes after cisplatin treatment. Clin J Am Soc Nephrol. 2016;11:1173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker EM Jr., Gale GR. Methods of reduction of cisplatin nephrotoxicity. Ann Clin Lab Sci. 1981;11:397–410. [PubMed] [Google Scholar]
- 16.Ravichandran K, Wang Q, Ozkok A, et al. CD4 T cell knockout does not protect against kidney injury and worsens cancer. J Mol Med. 2016;94:443–55. [DOI] [PubMed] [Google Scholar]
- 17.Katagiri D, Hamasaki Y, Doi K, et al. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 2016;89:374–85. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez R, Romay C, Borrego A, et al. Lipid peroxides and antioxidant enzymes in cisplatin-induced chronic nephrotoxicity in rats. Mediat Inflamm. 2005;2005:139–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torres R, Velazquez H, Chang JJ, et al. Three-dimensional morphology by multiphoton microscopy with clearing in a model of cisplatin-induced CKD. J Am Soc Nephrol. 2016;27:1102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharp CN, Doll MA, Dupre TV, et al. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Ren Physiol. 2016;310:F560–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp CN, Siskind LJ. Developing better mouse models to study cisplatin-induced kidney injury. Am J Physiol Ren Physiol. 2017;313:F835–F41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu MC, Shi M, Zhang J, et al. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010;78:1240–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu MC, Shi M, Zhang J, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panesso MC, Shi M, Cho HJ, et al. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int. 2014;85:855–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barker SL, Pastor J, Carranza D, et al. The demonstration of alphaKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol Dial Transplant. 2015;30:223–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moghal NE, Ferreira MA, Howie AJ, et al. The late histologic findings in diarrhea-associated hemolytic uremic syndrome. J Pediatr. 1998;133:220–3. [DOI] [PubMed] [Google Scholar]
- 27.Hu MC, Moe OW. Klotho as a potential biomarker and therapy for acute kidney injury. Nat Rev Nephrol. 2012;8:423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kramann R, Tanaka M, Humphreys BD. Fluorescence micro-angiography for quantitative assessment of peritubular capillary changes after AKI in mice. J Am Soc Nephrol. 2014;25:1924–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nematbakhsh M, Ebrahimian S, Tooyserkani M, et al. Gender difference in cisplatin-induced nephrotoxicity in a rat model: greater intensity of damage in male than female. Nephrourol Mon. 2013;5:818–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J Am Soc Nephrol. 2013;24:771–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Appenroth D, Braunlich H. Age differences in cisplatinum nephrotoxicity. Toxicology. 1984;32:343–53. [DOI] [PubMed] [Google Scholar]
- 33.Hrushesky WJ, Shimp W, Kennedy BJ. Lack of age-dependent cisplatin nephrotoxicity. Am J Med. 1984;76:579–84. [DOI] [PubMed] [Google Scholar]
- 34.Kanbay M, Goldsmith D, Akcay A, Covic A. Phosphate-the silent stealthy cardiorenal culprit in all stages of chronic kidney disease: a systematic review. Blood Purif. 2009;27:220–30. [DOI] [PubMed] [Google Scholar]
- 35.Foley RN. Phosphate levels and cardiovascular disease in the general population. Clin J Am Soc Nephrol. 2009;4:1136–9. [DOI] [PubMed] [Google Scholar]
- 36.Striker GE, Grosjean F, Vlassara H. Phosphate and CVD: it’s all in what’s on the table. Arterioscler Thromb Vasc Biol. 2011;31:1951–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toussaint ND, Pedagogos E, Tan SJ, et al. Phosphate in early chronic kidney disease: associations with clinical outcomes and a target to reduce cardiovascular risk. Nephrology. 2012;17:433–44. [DOI] [PubMed] [Google Scholar]
- 38.Li JW, Xu C, Fan Y, Wang Y, Xiao YB. Can serum levels of alkaline phosphatase and phosphate predict cardiovascular diseases and total mortality in individuals with preserved renal function? A systemic review and meta-analysis. PLoS ONE. 2014;9:e102276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Isakova T, Ix JH, Sprague SM, et al. Rationale and approaches to phosphate and fibroblast growth factor 23 reduction in CKD. J Am Soc Nephrol. 2015;26:2328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jozefacki A, White CA, Shobeiri NS, et al. Phosphate excretion is decreased in older cardiac patients with normal kidney function: an emerging dietary risk factor? Appl Physiol Nutr Metab. 2016;41:452–5. [DOI] [PubMed] [Google Scholar]
- 41.Shen ZJ, Hu J, Shiizaki K, Kuro-o M, Malter JS. Phosphate-induced renal fibrosis requires the prolyl isomerase pin1. PLoS ONE. 2016;11:e0150093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vervloet MG, Sezer S, Massy ZA, et al. The role of phosphate in kidney disease. Nat Rev Nephrol. 2017;13:27–38. [DOI] [PubMed] [Google Scholar]
- 43.Bian A, Xing C, Hu MC. Alpha Klotho and phosphate homeostasis. J Endocrinol Invest. 2014;37:1121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kollerits B, Fliser D, Heid IM, et al. Gender-specific association of adiponectin as a predictor of progression of chronic kidney disease: the mild to moderate kidney disease study. Kidney Int. 2007;71:1279–86. [DOI] [PubMed] [Google Scholar]
- 45.Diwan V, Small D, Kauter K, Gobe GC, Brown L. Gender differences in adenine-induced chronic kidney disease and cardiovascular complications in rats. Am J Physiol Ren Physiol. 2014;307:F1169–78. [DOI] [PubMed] [Google Scholar]
- 46.Cobo G, Hecking M, Port FK, et al. Sex and gender differences in chronic kidney disease: progression to end-stage renal disease and haemodialysis. Clin Sci. 2016;130:1147–63. [DOI] [PubMed] [Google Scholar]
- 47.Chang PY, Chien LN, Lin YF, et al. Risk factors of gender for renal progression in patients with early chronic kidney disease. Medicine. 2016;95:e4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carrero JJ, Hecking M, Ulasi I, Sola L, Thomas B. Chronic kidney disease, gender, and access to care: a global perspective. Semin Nephrol. 2017;37:296–308. [DOI] [PubMed] [Google Scholar]
- 49.Jones TW, Chopra S, Kaufman JS, Flamenbaum W, Trump BF. Cis-diamminedichloroplatinum (II)-induced acute renal failure in the rat. Correlation of structural and functional alterations. Lab Invest. 1985;52:363–74. [PubMed] [Google Scholar]
- 50.Chopra S, Kaufman JS, Jones TW, et al. Cis-diamminedichlorplatinum-induced acute renal failure in the rat. Kidney Int. 1982;21:54–64. [DOI] [PubMed] [Google Scholar]
- 51.Bhatt K, Zhou L, Mi QS, et al. MicroRNA-34a is induced via p53 during cisplatin nephrotoxicity and contributes to cell survival. Mol Med. 2010;16:409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang M, Dong Z. Self-eating for death or survival during cisplatin nephrotoxicity? Clin Exp Nephrol. 2010;14:516–7. [DOI] [PubMed] [Google Scholar]
- 53.Yang Y, Liu H, Liu F, Dong Z. Mitochondrial dysregulation and protection in cisplatin nephrotoxicity. Arch Toxicol. 2014;88:1249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Safirstein R, Miller P, Dikman S, Lyman N, Shapiro C. Cisplatin nephrotoxicity in rats: defect in papillary hypertonicity. Am J Physiol. 1981;241:F175–85. [DOI] [PubMed] [Google Scholar]
- 55.Nakada J, Machida T, Endou H. Nephrotoxicity of cisplatin in rats. Dev Toxicol Environ Sci. 1986;14:179–82. [PubMed] [Google Scholar]
- 56.Trujillo J, Molina-Jijon E, Medina-Campos ON, et al. Renal tight junction proteins are decreased in cisplatin-induced nephrotoxicity in rats. Toxicol Mech Methods. 2014;24:520–8. [DOI] [PubMed] [Google Scholar]
- 57.Campbell AB, Kalman SM, Jacobs C. Plasma platinum levels: relationship to cisplatin dose and nephrotoxicity. Cancer Treat Rep. 1983;67:169–72. [PubMed] [Google Scholar]
- 58.Mulder RL, Knijnenburg SL, Geskus RB, et al. Glomerular function time trends in long-term survivors of childhood cancer: a longitudinal study. Cancer Epidemiol Biomark Prev. 2013;22:1736–46. [DOI] [PubMed] [Google Scholar]
- 59.Dekkers IA, Blijdorp K, Cransberg K, et al. Long-term nephrotoxicity in adult survivors of childhood cancer. Clin J Am Soc Nephrol. 2013;8:922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marcussen N Atubular glomeruli in cisplatin-induced chronic interstitial nephropathy. Exp Stereol Investig APMIS. 1990;98:1087–97. [DOI] [PubMed] [Google Scholar]
- 61.Okada A, Fukushima K, Fujita M, et al. Alterations in cisplatin pharmacokinetics and its acute/sub-chronic kidney injury over multiple cycles of cisplatin treatment in rats. Biol Pharm Bull. 2017;40:1948–55. [DOI] [PubMed] [Google Scholar]
- 62.Reyes D, Lew SQ, Kimmel PL. Gender differences in hypertension and kidney disease. Med Clin North Am. 2005;89:613–30. [DOI] [PubMed] [Google Scholar]
- 63.Kummer S, von Gersdorff G, Kemper MJ, Oh J. The influence of gender and sexual hormones on incidence and outcome of chronic kidney disease. Pediatr Nephrol. 2012;27:1213–9. [DOI] [PubMed] [Google Scholar]
- 64.Eriksen BO, Ingebretsen OC. The progression of chronic kidney disease: a 10-year population-based study of the effects of gender and age. Kidney Int. 2006;69:375–82. [DOI] [PubMed] [Google Scholar]
- 65.Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fan LM, Cahill-Smith S, Geng L, et al. Aging-associated metabolic disorder induces Nox2 activation and oxidative damage of endothelial function. Free Radic Biol Med. 2017;108:940–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wulfert FM, van Meurs M, Kurniati NF, et al. Age-dependent role of microvascular endothelial and polymorphonuclear cells in lipopolysaccharide-induced acute kidney injury. Anesthesiology. 2012;117:126–36. [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Bonventre JV, Parrish AR. The aging kidney: increased susceptibility to nephrotoxicity. Int J Mol Sci. 2014;15:15358–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou J, Chong SY, Lim A, et al. Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging (Albany NY). 2017;9:583–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu X, Hu MC. Klotho/FGF23 axis in chronic kidney disease and cardiovascular disease. Kidney Dis. 2017;3:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neyra JA, Hu MC. Potential application of klotho in human chronic kidney disease. Bone. 2017;100:41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.