

Abstract
Objectives
Thalassemia is a highly prevalent monogenic inherited disease in southern China. It is important to collect epidemiological data comprehensively for proper prevention and treatment.
Methods
In this study, blood samples collected from 15 807 residents of Chenzhou were primarily screened by hematological tests. A total of 3973 samples of suspected thalassemia carriers were further characterized by combined next‐generation sequencing (NGS) and Gap‐PCR.
Results
In total, 1704 subjects were diagnosed as thalassemia carriers with a total prevalence rate of 10.78%, including 943 α‐thalassemia carriers, 708 β‐thalassemia carriers, and 53 composite α and β‐thalassemia carriers. The prevalence rates of α‐thalassemia, β‐thalassemia, and composite α and β‐thalassemia were 5.97%, 4.48%, and 0.34%, respectively. Meanwhile, we characterized 19 α‐thalassemia variations and 21 β‐thalassemia variations in thalassemia carriers. Approximately 2.88% of thalassemia carriers would be missed by traditional genetic analysis. In addition, four novel thalassemia mutations and one novel abnormal hemoglobin mutation were identified.
Conclusions
Our data suggest a high prevalence of thalassemia and a diverse spectrum of thalassemia‐associated variations in Chenzhou. Also, combined NGS and Gap‐PCR is an effective thalassemia screening method. Our findings might be helpful for prevention and treatment of thalassemia in this region.
Keywords: mutation, next‐generation sequencing, prevalence, thalassemia, variant
1. INTRODUCTION
Thalassemia is one of the most prevalent monogenic inherited disorders in the world. Approximately 5% of the population worldwide are thalassemia carriers. Due to population growth in recent decades, the number of births suffered from thalassemia is increasing, especially in developing and low‐income regions.1, 2
Thalassemia is characterized by reduced or even absent production of one of the subunits of hemoglobin. The majority of adult hemoglobin is composed of two α‐globin and two β‐globin subunits, while fetal hemoglobin is composed of two α‐globin and two γ‐globin subunits. Thalassemia mainly consists of α and β‐thalassemia. For α‐thalassemia, because of the absence or reduced production of α‐globin chains, excess β chains or γ chains form non‐functional tetramers, which are called hemoglobin H and hemoglobin Bart's, respectively. Hemoglobin H could form inclusion bodies which are harmful to erythrocytes. On the contrary, β‐thalassemia is caused by little or reduced production of β‐globin chains. Hence, erythrocytes would be damaged by insoluble aggregates formed by excess free α‐globin chains.1, 2, 4
Clinically, thalassemia has variable manifestations ranging from absence of symptoms to fatal. Thalassemia is mainly classified as thalassemia trait, thalassemia intermedia, and thalassemia major according to clinical severity. The latter two subgroups are also diagnosed as thalassemia patients. The phenotypic severity of the disease mainly correlates with degree of imbalance of α:non‐α chains.5
Although prognosis for thalassemia has been markedly improved, lifelong care is required for many cases.6, 7 Proper treatment brings substantial financial burden to patients as well as society in prevalent areas.11 Accordingly, prevention of births with thalassemia is particularly important. Comprehensive molecular epidemiological data of the disease are necessary for proper prevention and treatment. At present, combined reverse dot blot (RDB) and Gap‐PCR is the most commonly used method in identifying thalassemia mutations.12 The major limitation of these methods is that only common variations could be identified. Therefore, it is required to develop novel technology to screen mutations comprehensively. Recently, next‐generation sequencing (NGS) was used to screen thalassemia carriers in a few studies of China.13, 14 These studies indicated that the spectrum of thalassemia‐associated variations was much broader than previously reported and suggested that NGS was an effective method in screening thalassemia‐associated variations to facilitate diagnosis.
Thalassemia is popular in tropical and subtropical regions, including South China. Chenzhou is the southernmost city of Hunan Province, People's Republic of China, and sits on the border of Hunan and Guangdong provinces. In China, Guangdong and Guangxi provinces have the highest prevalence of thalassemia. Our previous results showed a high prevalence rate of thalassemia in Chenzhou by RDB and Gap‐PCR.16 However, the spectrum of thalassemia variations was not comprehensive. We speculated that many types of thalassemia variations could be missed. Here, we firstly combined NGS and Gap‐PCR in screening thalassemia variations in Chenzhou Region to assess thalassemia variation burden comprehensively and its potential application in preventing births with thalassemia.
2. MATERIALS AND METHODS
2.1. Participants
This study was approved by BGI's institutional review board on bioethics and biosafety and the Ethic Committee of Chenzhou No. 1 People's Hospital. Written informed consents of all participants were obtained. A total of 15 807 participants (6164 men and 9643 women), who visited hospital for routine medical examination between April 2015 and February 2017, were enrolled in this study. Participants with following cases were excluded from the study: (a) incomplete information, (b) consanguinity, and (c) lack of informed consent. The age distribution of the participants was as follows: 1‐15 years old, 2901; 16‐25 years old, 3390; 26‐35 years old, 6819; 36‐45 years old, 2540; and 46‐53 years old, 157. The family members of novel thalassemia mutation carriers were directly screened by hematological tests and genetic analysis and were not included in this cohort.
2.2. Primary hematological screening
Peripheral venous blood samples were obtained from all participants. All samples were primarily screened with routine blood examination and/or hemoglobin electrophoresis. Subjects were considered as suspected thalassemia carriers if either of the following parameters was tested positive: (a) mean corpuscular volume (MCV) <82 fl and/or mean corpuscular hemoglobin (MCH) <27 pg, (b) Hb A2 concentration < 2.5% and Hb F concentration < 2%, and (c) Hb A2 concentration > 3.5% and Hb F concentration at >3.5%. Suspected thalassemia subjects were subjected to further genetic analysis.
Routine blood examinations were performed with ADVIA 2120i Hematology System (Siemens Healthineers, Erlangen, Germany). Hemoglobin electrophoresis was performed with Capillarys 2 Flex Piercing (SEBIA, Lisses, France).
2.3. DNA extraction
Genomic DNA of suspected thalassemia carriers was extracted from whole blood using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The concentration of DNA samples was quantified by Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
2.4. Gap‐PCR
The three most common α‐thalassemia‐associated deletions (‐‐SEA, ‐α3.7, and ‐α4.2) and two rare deletions (‐‐FIL and ‐‐THAI) in China were characterized by the Gap‐PCR. Two common β‐globin gene deletions (SEA‐HPFH and Gγ+(Asγδβ)) were also analyzed by Gap‐PCR.
2.5. NGS screening
The full length of HBA1, HBA2, and HBB was amplified by PCR. The amplicons spanned all the exons and introns of HBA1, HBA2, and HBB genes, which ensured that most thalassemia‐associated mutations and CNVs in the HbVar database could be detected. Sequencing libraries were constructed according the Illumina HiSeq sequencing library preparation protocol. These libraries were further paired‐end‐sequenced for 100 base pairs (PE100) with an Illumina HiSeq 2000 machine. The protocol of bioinformatic analysis of identifying hemoglobin gene variations was described previously.14 All variations were validated with Sanger sequencing.
3. RESULTS
3.1. Thalassemia carriers found by NGS
In total, 15 807 subjects were primarily screened by hematological examinations and 3973 suspected subjects were further analyzed by NGS and Gap‐PCR. Among these subjects, 1704 subjects were diagnosed as thalassemia carriers, including 943 α‐thalassemia carriers, 708 β‐thalassemia carriers, and 53 composite α and β‐thalassemia carriers. The overall prevalence rate of thalassemia in Chenzhou was 10.78%, and the prevalence rates of α and β‐thalassemia were 5.97% and 4.48%, respectively. In addition, the rate of composite α and β‐thalassemia was firstly determined in Chenzhou, which was found in 0.34% of all subjects.
Among 996 carriers with α‐thalassemia variations, we identified 19 different variations with 30 distinct genotypes in this study (Table 1). ‐‐SEA/αα was the most abundant α‐thalassemia genotype with a proportion of 67.87%. ‐α3.7/αα, ‐α4.2/αα, and ‐α3.7/‐‐SEA genotypes occurred frequently and represented 14.86%, 4.42%, and 3.41% of all genotypes, respectively. Notably, rare variations such as HBA2:c.95+5_95+28delGGCTCCCTCCCCTGCTCCGACCCG, initiation codon (‐T), and IVS‐I‐117 (G>A) were firstly reported in Mainland China. In addition, two novel α‐globin mutations, including HBA2:c.6_7insTG and HBA1:c.2T>C, were discovered in three unrelated individuals.
Table 1.
Distribution of α‐thalassemia genotypes in Chenzhou Region
| Genotype | Number | Frequency (%) |
|---|---|---|
| ‐‐SEA/αα | 676 | 67.87 |
| ‐α3.7/αα | 148 | 14.86 |
| ‐α4.2//αα | 44 | 4.42 |
| ‐α3.7/‐‐SEA | 34 | 3.41 |
| αCSα/αα | 17 | 1.71 |
| αWSα/αα | 15 | 1.51 |
| αQSα/αα | 9 | 0.90 |
| Alpha2 Codon 30 del GAG/αα | 8 | 0.80 |
| αCSα/‐‐SEA | 8 | 0.80 |
| ‐α4.2/‐‐SEA | 6 | 0.60 |
| ‐α3.7/‐α3.7 | 3 | 0.30 |
| Initiation codon (‐T)/αα | 3 | 0.30 |
| ‐‐THAI/αα | 3 | 0.30 |
| Alpha2 Codon 31 (AGG>AAG)/αα | 2 | 0.20 |
| αWSα/‐‐SEA | 2 | 0.20 |
| HBA2:c.46G>A(Gly>Ser)/αα | 2 | 0.20 |
| HBA2:c.6_7insTG/αα | 2 | 0.20 |
| HBA2:c.95+5_95+28delGGCTCCCTCCCCTGCTCCGACCCG/αα | 2 | 0.20 |
| ‐α3.7/‐α4.2 | 1 | 0.10 |
| ‐α4.2/‐α4.2 | 1 | 0.10 |
| Codon 116 (G>T)/αα | 1 | 0.10 |
| αCSα/‐α3.7 | 1 | 0.10 |
| αQSα/‐‐SEA | 1 | 0.10 |
| HBA1:c.2T>C/αα | 1 | 0.10 |
| HBA1:c.429+51_429+53delCCT/αα | 1 | 0.10 |
| HBA2:c.184A>T(Lys>End)/αα | 1 | 0.10 |
| IVS‐I‐117 (G>A)/‐‐SEA | 1 | 0.10 |
| ααα3.7/‐‐SEA | 1 | 0.10 |
| HBA2:c.46G>A(Gly>Ser)/‐‐SEA | 1 | 0.10 |
| HBA2:c.95+5_95+28delGGCTCCCTCCCCTGCTCCGACCCG/‐α3.7 | 1 | 0.10 |
| Total | 996 | 100.00 |
In this cohort, we also found 21 β‐thalassemia mutations and 32 genotypes in 761 subjects (Table 2). Codons 41/42 (‐TTCT)/βN and IVS‐II‐654 (C>T)/βN are the top two most frequent genotypes. The ranking order of the two major genotypes is distinct from our previous result. The proportions of Codons 41/42 (‐TTCT)/βN and IVS‐II‐654 (C>T)/βN were 34.69% and 30.62%, respectively, in the current study. However, in our previous study, the proportions of Codons 41/42 (‐TTCT)/βN and IVS‐II‐654 (C>T)/βN were 31.00% and 37.99%, respectively.16 The remaining common genotypes were Codon 17 (A>T)/βN, −28 (A>G)/βN, and Codons 71/72 (+A)/βN with corresponding proportions of 12.61%, 6.70%, and 4.20%. Meanwhile, rare β‐thalassemia mutation IVS II‐761 A>G was identified for the first time in Mainland China. Moreover, we also characterized two novel β‐thalassemia mutations, including HBB:c.260 or 261delC and HBB:c.43delC.
Table 2.
Distribution of β‐thalassemia genotypes in Chenzhou Region
| Genotype | Number | Frequency (%) |
|---|---|---|
| Codons 41/42 (‐TTCT) | 264 | 34.69 |
| IVS‐II‐654 (C>T) | 233 | 30.62 |
| Codon 17 (A>T) | 96 | 12.61 |
| ‐28 (A>G) | 51 | 6.70 |
| Codons 71/72 (+A) | 32 | 4.20 |
| Hb E | 14 | 1.84 |
| Codons 27/28 (+C) | 11 | 1.45 |
| Codon 43 (G>T) | 11 | 1.45 |
| ‐29 (A>G) | 10 | 1.31 |
| ‐50 G>A | 7 | 0.92 |
| Codons 14/15 (+G) | 5 | 0.66 |
| Codons 41/42 (‐TTCT)/Codons 41/42 (‐TTCT) | 3 | 0.39 |
| ‐90 (C>T) | 2 | 0.26 |
| 5'UTR +43 to +40 (‐AAAC) | 2 | 0.26 |
| CD37 (TGG>TAG) | 2 | 0.26 |
| IVS‐I‐1 (G>T) | 2 | 0.26 |
| Codon 17 (A>T)/‐28 (A>G) | 1 | 0.13 |
| Codons 41/42 (‐TTCT)/‐28 (A>G) | 1 | 0.13 |
| Codons 41/42 (‐TTCT)/‐50 G>A | 1 | 0.13 |
| Codons 41/42 (‐TTCT)/Hb E | 1 | 0.13 |
| Codons 41/42 (‐TTCT)/IVS‐II‐654 (C>T) | 1 | 0.13 |
| Codons 41/42 (‐TTCT)/SEA‐HPFH | 1 | 0.13 |
| IVS‐II‐654 (C>T)/‐28 (A>G) | 1 | 0.13 |
| IVS‐II‐654 (C>T)/IVS‐II‐654 (C>T) | 1 | 0.13 |
| IVS‐II‐654 (C>T)/Hb E | 1 | 0.13 |
| ‐31 (A>C) | 1 | 0.13 |
| Hb E/Hb E | 1 | 0.13 |
| HBB:c.260 or 261delC | 1 | 0.13 |
| HBB:c.43delC | 1 | 0.13 |
| Initiation codon ATG>AGG | 1 | 0.13 |
| IVS II‐761 A>G | 1 | 0.13 |
| SEA‐HPFH | 1 | 0.13 |
| Total | 761 | 100.00 |
Fifty‐three subjects were carriers with both α‐ and β‐globin variations (Table 3). Among these carriers, 83.02% of genotypes consisted of common deletions of α‐globin gene (αα/‐‐SEA, αα/‐α3.7, αα/‐α4.2) combined with a β‐globin gene point mutation. Among these genotypes, composite αα/‐‐SEA and Codons 41/42 (‐TTCT)/βN was the most frequent genotype.
Table 3.
Genotypes of composite α and β‐thalassemia in Chenzhou Region
| α | β | Number |
|---|---|---|
| ‐‐SEA/αα | Codons 41/42 (‐TTCT)/βN | 9 |
| ‐‐SEA/αα | ‐28 (A‐>G)/βN | 5 |
| ‐‐SEA/αα | ‐50 G>A/βN | 4 |
| ‐‐SEA/αα | IVS‐II‐654 (C‐>T)/βN | 4 |
| ‐α3.7/αα | IVS‐II‐654 (C‐>T)/βN | 4 |
| ‐α3.7/αα | Codons 41/42 (‐TTCT)/βN | 3 |
| ‐α4.2/αα | IVS‐II‐654 (C‐>T)/βN | 3 |
| ‐α3.7/αα | Hb E/βN | 2 |
| ‐α4.2/αα | Codons 41/42 (‐TTCT)/βN | 2 |
| ‐‐SEA/αα | Codon 17 (A‐>T)/βN | 1 |
| ‐‐SEA/αα | Codons 14/15 (+G)/βN | 1 |
| ‐‐SEA/αα | Codons 27/28 (+C)/βN | 1 |
| ‐α3.7/αα | ‐29 (A‐>G)/βN | 1 |
| ‐α3.7/αα | 5'UTR +43 to +40 (‐AAAC)/βN | 1 |
| ‐α3.7/αα | Codon 17 (A‐>T)/βN | 1 |
| ‐α3.7/αα | Codon 43 (G‐>T)/βN | 1 |
| ‐α4.2/αα | ‐28 (A‐>G)/βN | 1 |
| ‐α3.7/‐‐SEA | IVS‐II‐654 (C‐>T)/βN | 1 |
| αWSα/αα | Codon 43 (G‐>T)/βN | 1 |
| αWSα/αα | Codons 41/42 (‐TTCT)/βN | 1 |
| HBA2:c.46G>A(Gly>Ser)/αα | Codons 41/42 (‐TTCT)/βN | 1 |
| HBA2:c.46G>A(Gly>Ser)/αα | IVS‐II‐654 (C‐>T)/βN | 1 |
| Initiation codon (‐T)/αα | ‐28 (A‐>G)/Codon 17 (A‐>T) | 1 |
| Alpha2 Codon 30 del GAG/αα | Codon 17 (A‐>T)/βN | 1 |
| Alpha2 Codon 31 (AGG>AAG)/αα | IVS‐II‐654 (C‐>T)/βN | 1 |
| HBA1:c.429+51_429+53delCCT/αα | Codons 71/72 (+A)/βN | 1 |
In addition, 13 abnormal hemoglobin variants were identified in 35 subjects with a carrier rate of 0.22% (Table 4). Among these subjects, nine subjects and five subjects were simultaneously affected by α‐thalassemia mutations and β‐thalassemia mutations, respectively. Three rare abnormal hemoglobin variants, Hb Zurich‐Langstrasse, Hb Yusa, and Hb Genova, were reported for the first time in China. Moreover, we found a new case of Hb Savaria with a novel mutation HBA2:c.150C>G, which was predicted as one of the three mutated forms of Hb Savaria before.17
Table 4.
Abnormal hemoglobin variants in this cohort
| Abnormal hemoglobin variant | Number | Frequency (%) |
|---|---|---|
| Hb New York | 8 | 0.0454 |
| Hb Hekinan II | 7 | 0.0397 |
| Hb Q‐Thailand | 4 | 0.0227 |
| Hb Zurich‐Langstrasse | 4 | 0.0227 |
| Hb G‐Taipei | 2 | 0.0113 |
| Hb J‐Bangkok | 2 | 0.0113 |
| Hb Port Phillip | 2 | 0.0113 |
| Hb Zurich‐Albisrieden | 1 | 0.0057 |
| Hb Genova | 1 | 0.0057 |
| Hb G‐Honolulu | 1 | 0.0057 |
| Hb Hamilton | 1 | 0.0057 |
| Hb Yusa | 1 | 0.0057 |
| Hb Savaria (HBA2:c.150C>G(Ser>Arg)) | 1 | 0.0057 |
| Total | 35 | 0.1985 |
3.2. Characterization of novel mutations
In this study, four novel thalassemia mutations were identified by NGS in five probands and were further confirmed by Sanger sequencing (Figure 1). Among these mutations, two novel α‐thalassemia mutations have been identified in three individuals. The mutations and their hematological parameters are listed in Table 5. The mutation HBA1:c.2T>C, which was a novel mutation of the translation initiation codon of the α1‐globin gene, was found in a woman with reduced MCV and MCH. The HBA2:c.6_7insTG, resulting in a completely different polypeptide from the original alpha‐globin peptide, was observed in two unrelated individuals. They were both associated with reduced MCV and MCH. One of them was a woman who had two children. Direct DNA sequencing showed that this mutation was inherited from her mother and her two children were both heterozygous for this mutation. They all show slightly reduced MCV and MCH.
Figure 1.
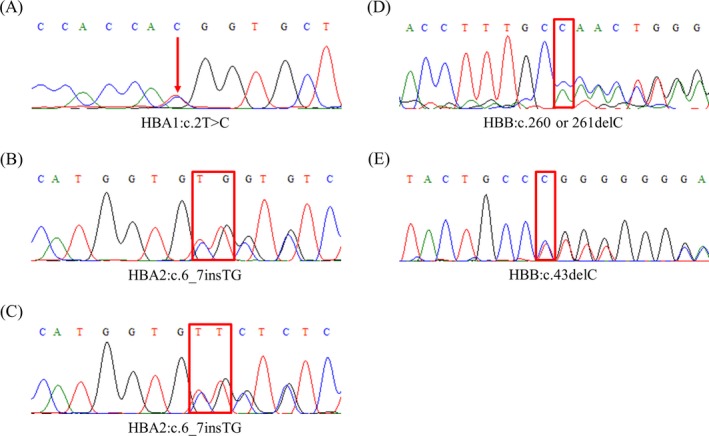
DNA sequence analysis of the probands with novel hemoglobin mutations. The point‐mutated site is labeled with red arrow, and the inserted and deleted sites are labeled with red rectangle. A, HBA1:c.2T>C in proband 1; B, HBA2:c.6_7insTG in proband 2; C, HBA2:c.6_7insTG in proband 3; D, HBB:c.260 or 261delC in proband 4; E, HBB:c.43delC in proband 5
Table 5.
Hematological data from probands with novel mutations and family members of partial probands
| Case | Age (y)/sex | Genotype | Hb (g/L) | MCV (fL) | MCH (pg) | Hb A (%) | Hb A2 (%) | Hb F (%) | |
|---|---|---|---|---|---|---|---|---|---|
| α | β | ||||||||
| Proband 1 | 37/F | HBA1:c.2T>C/αα | βN/βN | 104 | 77.2 | 25.7 | — | — | — |
| Proband 2 | 1/M | HBA2:c.6_7insTG/αα | βN/βN | 90 | 59.2 | 17.8 | 97.3 | 2.7 | 0 |
| Family A | |||||||||
| Proband 3 | 36/F | HBA2:c.6_7insTG/αα | βN/βN | 117 | 80.6 | 26 | 97.4 | 2.6 | 0 |
| Mother | 74/F | HBA2:c.6_7insTG/αα | βN/βN | 140 | 83.9 | 26.3 | 97.2 | 2.8 | 0 |
| Daughter | 1/F | HBA2:c.6_7insTG/αα | βN/βN | 103 | 70 | 21.6 | 95 | 2.7 | 2.3 |
| Daughter | 11F | HBA2:c.6_7insTG/αα | βN/βN | 111 | 77.7 | 23.4 | 97.6 | 2.4 | 0 |
| Family B | |||||||||
| Proband 4 | 22/F | αα/αα | HBB:c.260 or 261delC/βN | 74 | 72.7 | 21.3 | 86.5 | 5.4 | 0 |
| Son | 1/M | αα/αα | Codons 41/42 (‐TTCT)/HBB:c.260 or 261delC | 50 | 66.7 | 19.2 | — | — | — |
| Husband | 28/M | αα/αα | Codons 41/42 (‐TTCT)/βN | 136 | 65.3 | 19.5 | 95.65 | 4.35 | 0 |
| Family C | |||||||||
| Proband 5 | 1/F | αα/αα | HBB:c.43delC/βN | 98 | 57.2 | 18.2 | 80 | 4.5 | 15.5 |
| Father | 27/M | αα/αα | HBB:c.43delC/βN | 115 | 63.4 | 18.2 | 93.7 | 5.3 | 1 |
| Mother | 29/F | αα/αα | βN/βN | 138 | 87.4 | 29.9 | 96.7 | 2.8 | 0.5 |
—, not available; Hb A, hemoglobin A; Hb A2, hemoglobin A2; Hb F, hemoglobin F; Hb, hemoglobin; MCH, mean corpuscular hemoglobin; MCV, mean cell hemoglobin.
Two novel β‐thalassemia mutations were discovered, and the molecular and hematological parameters of each allele are listed in Table 5. These two mutations were both frameshift mutations. The HBB:c.43delC mutation was found in a 1‐year‐old girl with significantly reduced MCV (63.4 fL) and MCH (18.2 pg). This mutation was inherited from her father, who was also associated with significantly reduced MCV (63.4 fL) and MCH (18.2 pg). The second mutation HBB:c.260 or 261del was firstly found in a pregnant woman with reduced MCV (65 fL), MCH (21.4 pg), and HBA (86.5%) and increased Hb A2 (5.4%). Her husband also carried a β0 globin mutation (Codons 41/42 (‐TTCT)). Direct sequencing of the α‐ and β‐globin genes by amniocentesis showed that the fetus was doubly heterozygous for HBB:c.260 or 261del and Codons 41/42 (‐TTCT). However, this mother insisted to keep this fetus. Now this child shows serve β‐thalassemia phenotype and needs transfusion therapy every month.
4. DISCUSSION
There is a high frequency of thalassemia in southern China, particularly in Guangdong, Guangxi, and Hainan provinces.18, 19 The percentage of thalassemia carriers is relatively low in most regions in Hunan Province.21 This disorder was neglected by provincial health system in some high‐prevalence regions due to limited molecular epidemiological data.
In this study, we report molecular epidemiological data of thalassemia in Chenzhou Region comprehensively for the first time. Our data confirmed that Chenzhou had the highest overall prevalence rate of thalassemia (10.78%) in Hunan Province, which was slightly higher than that of our previous result (10.00%).16 Meanwhile, the rates of α‐thalassemia and β‐thalassemia were slightly higher and less than those of our previous results, respectively. These changes were probably caused by different genetic screening methods and/or population mobility and migration. The rate of thalassemia in Chenzhou was significantly higher than the average of Hunan Province (4.18%)22, 23 and was closer to that of Guangdong Province and southern Jiangxi Province.19, 24 Notably, the rates of α and β‐thalassemia in Chenzhou had a special distribution pattern. Generally, α‐thalassemia occurs at a much higher frequency than that of β‐thalassemia; however, the rate of α‐thalassemia carriers (5.97%) was close to that of β‐thalassemia carriers (4.48%) in Chenzhou Region. This distribution pattern was consistent with the data of Changsha Region in Hunan Province and indicated that Chenzhou had a lessened rate of α‐thalassemia and a relatively higher rate of β‐thalassemia compared with surrounding provinces of southern China.25 Moreover, the rate of composite α and β‐thalassemia (0.34%) in Chenzhou was newly determined in this study.
In contrast to six variations and 10 genotypes identified in our previous study,16 we identified 30 distinct α‐thalassemia genotypes with 19 different variations here. Among α‐thalassemia genotypes, the most common subtype was ‐‐SEA/αα with a remarkable proportion of 67.87%. The proportion was similar to that of Changsha Region in Hunan Province and higher than that of surrounding provinces.21, 25 Apart from these common variation types, a series of rare and novel variations were identified. IVS‐I‐117 (G>A) was a rare mutation which was firstly reported in Indian population.26 And HBA2:c.95+5_95+28delGGCTCCCTCCCCTGCTCCGACCCG was only reported once in Malaysia.27 Interestingly, HBA2:c.184A>T(Lys>End) was a recently reported novel mutation which was also found by NGS in Guangdong Province, China.28 Furthermore, we identified two novel α‐thalassemia‐associated mutations in this study, including HBA1:c.2T>C and HBA2:c.6_7insTG. For β‐thalassemia, we identified 21 β‐thalassemia variations with 32 genotypes in this cohort, whereas only 13 mutations and 13 genotypes were identified in our previous study.16 Of the β‐thalassemia genotypes, Codons 41/42 (‐TTCT)/βN and IVS‐II‐654 (C>T)/βN were the most two frequent β‐thalassemia subtypes, accounting for 65.31% of the genotypes. And the ranking order of the two major mutations was different from our previous result.16 We assumed that it was probably due to population mobility and/or new genetic screening methods. In addition, a rare mutation of β‐globin gene, IVS II‐761 A>G, was firstly identified in Mainland China. IVS II‐761 A>G was previously reported once in a pan‐ethnic population of America.29 Furthermore, HBB:c.260 or 261delC and HBB:c.43delC were newly characterized β‐thalassemia mutations.
In this study, the prevalence of abnormal hemoglobins (0.22%) was determined in Chenzhou Region for the first time. Thirteen abnormal hemoglobin variants were identified. Among these variants, Hb Zurich‐Langstrasse, Hb Yusa, and Hb Genova were firstly reported in China. Hb Zurich‐Langstrasse and Hb Yusa were also rarely reported worldwide.30, 31 In addition, a new form of mutation (HBA2:c.150C‐>G) referred to as Hb Savaria was identified. Theoretically, Hb Savaria has three different variants, including HBA2:c.150C‐>A, HBA2:c.148A‐>C, and HBA2:c.150C‐>G. HBA2:c.150C‐>A, which is present in HbVar, was firstly reported in a female from Kenya.32 Another variant HBA2:c.148A‐>C was recently reported in France.17 The last form of mutation HBA2:c.150C‐>G was firstly found in a woman with normal hematological parameters in our study.
With the advent of NGS techniques in recent years, NGS emerged as a popular tool in prenatal screening.33, 34 So far, many recent investigations confirmed the advantage of NGS in screening thalassemia carriers and detecting novel and complex variations.13, 14, 15, 36 An investigation of prevalence and genetic background of thalassemia in Baise Region found 24 common variations and 4 rare novel variations.13 In another study, 49.5% of Dai people were diagnosed as thalassemia carriers via direct NGS screening. By contrast, 22.0% was found by traditional methods in the same cohort14. In our study, we identified 40 genomic variations including 11 rare and novel mutations by combined NGS and Gap‐PCR. Among these variations, only three types of deletion and 20 types of mutation could be detected by combined RDB and Gap‐PCR. Twenty types of genomic variation could be missed. In other words, 2.88% of all thalassemia carriers could be missed by traditional genetic analysis in this cohort. The child with compound heterozygosity of HBB:c.260 or 261del and Codons 41/42 (‐TTCT) in our study served as a prime example of the importance of identification of novel mutations in prenatal diagnosis. This result implied that the molecular background of thalassemia was much more complex than previously reported and traditional methods are not sufficient for accurate diagnosis of thalassemia. NGS could be a better mass screening method, especially in high‐prevalence regions.
Several other limitations of this study should also be considered. Although the clinical manifestations of thalassemia mainly depend on the degree of imbalance of the ratio of α:non‐α chains, individuals with identical genotypes can exhibit variable clinical severities. Genetic modulators and cis‐regulatory elements have significant effects on clinical manifestation.5 More loci should be covered in the NGS screening, which is important for precise diagnosis and treatment of thalassemia. Recently, a designed targeted gene panel, which included all eight globin genes and validated modulators (KLF1, BCL11A, and MYB), was applied in molecular screening and clinical genotyping in thalassemia. The result showed that comprehensive NGS greatly facilitates screening and diagnosis of thalassemia.15 However, there are still some limitations of NGS techniques. Zebisch et al39 identified a novel variant of epsilon‐gamma‐delta‐beta thalassemia by using MLPA and CGH, which was missed by NGS in this study. In consideration of diverse types of thalassemia variations, combined methods are needed to detect thalassemia variations completely. On the other side, new technologies such as haplotype‐resolved genome sequencing and single‐molecule real‐time sequencing may completely solve this problem in the future.40, 41
In conclusion, we demonstrated the great diversity of thalassemia‐associated variations and a high prevalence of thalassemia in Chenzhou Region by applying combined NGS and Gap‐PCR technology. The rare and novel variations of this region were identified for the first time. Our findings would be meaningful for prevention and treatment of thalassemia in this region and other high‐prevalence areas. Our updated epidemiological data may also draw the attention of local governments in the severity of this disorder, and more public funds might be allocated for prevention. Limitations of our study, such as possible missed carriers during primary screening and complex variations, will be further addressed in the future.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the ChenZhou Municipal Science And Technology Bureau [Grant Numbers CZ2014012 and CZKJ2016037] and Shenzhen Municipal Government of China [Grant Number KJYY20170412153606214].
Zhang H, Li C, Li J, et al. Next‐generation sequencing improves molecular epidemiological characterization of thalassemia in Chenzhou Region, P.R. China. J Clin Lab Anal. 2019;33:e22845 10.1002/jcla.22845
Haoqing Zhang, Caiyun Li, and Jianbiao Li contributed equally to this work.
Contributor Information
Jian Guo, Email: guojian@genomics.cn.
Dongzhu Lei, Email: leidongzhu@sohu.com.
REFERENCES
- 1. Rund D, Rachmilewitz E. Beta‐thalassemia. N Engl J Med. 2005;353(11):1135‐1146. [DOI] [PubMed] [Google Scholar]
- 2. Piel FB, Weatherall DJ. The alpha‐thalassemias. N Engl J Med. 2014;371(20):1908‐1916. [DOI] [PubMed] [Google Scholar]
- 3. Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model‐based map and population estimates. Lancet. 2013;381(9861):142‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muncie HL Jr, Campbell J. Alpha and beta thalassemia. Am Fam Physician. 2009;80(4):339‐344. [PubMed] [Google Scholar]
- 5. Shang X, Xu X. Update in the genetics of thalassemia: what clinicians need to know. Best Pract Res Clin Obstet Gynaecol. 2017;39:3‐15. [DOI] [PubMed] [Google Scholar]
- 6. Srivastava A, Shaji RV. Cure for thalassemia major ‐ from allogeneic hematopoietic stem cell transplantation to gene therapy. Haematologica. 2017;102(2):214‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fucharoen S, Weatherall DJ. Progress toward the control and management of the thalassemias. Hematol Oncol Clin North Am. 2016;30(2):359‐371. [DOI] [PubMed] [Google Scholar]
- 8. Rund D. Thalassemia 2016: modern medicine battles an ancient disease. Am J Hematol. 2016;91(1):15‐21. [DOI] [PubMed] [Google Scholar]
- 9. Makis A, Hatzimichael E, Papassotiriou I, Voskaridou E. 2017 Clinical trials update in new treatments of beta‐thalassemia. Am J Hematol. 2016;91(11):1135‐1145. [DOI] [PubMed] [Google Scholar]
- 10. Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med. 2015;21(3):221‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331‐4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Galanello R, Cao A. Gene test review. Alpha‐thalassemia. Genet Med. 2011;13(2):83‐88. [DOI] [PubMed] [Google Scholar]
- 13. He S, Qin Q, Yi S, et al. Prevalence and genetic analysis of alpha‐ and beta‐thalassemia in Baise region, a multi‐ethnic region in southern China. Gene. 2016;619:71‐75. [DOI] [PubMed] [Google Scholar]
- 14. He J, Song W, Yang J, et al. Next‐generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: the Dai nationality, China. Genet Med. 2017;9:1022‐1031. [DOI] [PubMed] [Google Scholar]
- 15. Shang X, Peng Z, Ye Y, et al. Targeted next‐generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies . EBioMedicine. 2017;23:150‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li CY, Hou S, Zhang HQ, Chen DJ, Yan HY, Lei DZ. The prevalence and molecular spectrum of alpha‐thalassemia and beta‐thalassemia in Chenzhou, Hunan Province. Int J Lab Med. 2015;36(12):1779‐1780. [Google Scholar]
- 17. Tran Houangkeo TH, Bodereau V, Riou J, Pissard S. Hb Savaria [alpha49(CE7)Ser–>Arg; HBA2: c.150C > A]: a new case and complete description. Hemoglobin. 2016;40(4):267‐269. [DOI] [PubMed] [Google Scholar]
- 18. Xiong F, Sun M, Zhang X, et al. Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China. Clin Genet. 2010;78(2):139‐148. [DOI] [PubMed] [Google Scholar]
- 19. Yin A, Li B, Luo M, et al. The prevalence and molecular spectrum of alpha‐ and beta‐globin gene mutations in 14,332 families of Guangdong Province, China. PLoS ONE. 2014;9(2):e89855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yao H, Chen X, Lin L, et al. The spectrum of alpha‐ and beta‐thalassemia mutations of the Li people in Hainan Province of China. Blood Cells Mol Dis. 2014;53(1–2):16‐20. [DOI] [PubMed] [Google Scholar]
- 21. He J, Zeng H, Zhu L, Li H, Shi L, Hu L. Prevalence and spectrum of thalassaemia in Changsha, Hunan province, China: discussion of an innovative screening strategy. J Genet. 2017;96(2):327‐332. [DOI] [PubMed] [Google Scholar]
- 22. Wu WQ, Jin Q, Cai J, Xu XX, Geng X, Xie JS. Analysis of the frequency and molecular spectrum of alpha‐ and beta‐thalassemia in the general population of Hunan province. Chin J Birth Health Heredity. 2007;15(11):43‐44. [Google Scholar]
- 23. Li M, Nie L, Tao X, et al. Frequency and genotypes of thalassemia in Shenzhen area. J Trop Med. 2005;3:301‐303. [Google Scholar]
- 24. Lin M, Zhong TY, Chen YG, et al. Molecular epidemiological characterization and health burden of thalassemia in Jiangxi Province, P. R. China . PLoS ONE. 2014;9(7):e101505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lai K, Huang G, Su L, He Y. The prevalence of thalassemia in mainland China: evidence from epidemiological surveys. Sci Rep. 2017;7(1):920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Curuk MA, Baysal E, Gupta RB, Sharma S, Huisman TH. An IVS‐I‐117 (G–>A) acceptor splice site mutation in the alpha 1‐globin gene is a nondeletional alpha‐thalassaemia‐2 determinant in an Indian population. Br J Haematol. 1993;85(1):148‐152. [DOI] [PubMed] [Google Scholar]
- 27. Tan JA, Chin SS, Ong GB, et al. Transfusion‐dependent thalassemia in Northern Sarawak: a molecular study to identify different genotypes in the multi‐ethnic groups and the importance of genomic sequencing in unstudied populations. Public Health Genomics. 2015;18(1):60‐64. [DOI] [PubMed] [Google Scholar]
- 28. Liang Y, Peng Q, Li M, Li S, Li W, Lu X. A novel alpha‐thalassemia nonsense mutation on the alpha2‐globin gene: HBA2: c.184A>T. Hemoglobin. 2017;41:306‐307. [DOI] [PubMed] [Google Scholar]
- 29. Chan OT, Westover KD, Dietz L, Zehnder JL, Schrijver I. Comprehensive and efficient HBB mutation analysis for detection of beta‐hemoglobinopathies in a pan‐ethnic population. Am J Clin Pathol. 2010;133(5):700‐707. [DOI] [PubMed] [Google Scholar]
- 30. Kleinert P, Schmid M, Zurbriggen K, et al. Mass spectrometry: a tool for enhanced detection of hemoglobin variants. Clin Chem. 2008;54(1):69‐76. [DOI] [PubMed] [Google Scholar]
- 31. Harano T, Harano K, Ueda S, et al. Hemoglobin Yusa (beta 21 (B3) Asp leads to Tyr), a new abnormal hemoglobin found in Japan. Hemoglobin. 1981;5(2):121‐131. [DOI] [PubMed] [Google Scholar]
- 32. Ojwang PJ, Ogada T, Webber BB, Wilson JB, Huisman TH. Hb Savaria or alpha2 (49)(CE7)Ser––Arg beta2 in an indigenous female from Kenya. Hemoglobin. 1985;9(2):197‐200. [DOI] [PubMed] [Google Scholar]
- 33. Lohmann K, Klein C. Next generation sequencing and the future of genetic diagnosis. Neurotherapeutics. 2014;11(4):699‐707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stark Z, Tan TY, Chong B, et al. A prospective evaluation of whole‐exome sequencing as a first‐tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18(11):1090‐1096. [DOI] [PubMed] [Google Scholar]
- 35. Gregg AR, Van den Veyver IB, Gross SJ, Madankumar R, Rink BD, Norton ME. Noninvasive prenatal screening by next‐generation sequencing. Annu Rev Genomics Hum Genet. 2014;15:327‐347. [DOI] [PubMed] [Google Scholar]
- 36. Henderson SJ, Timbs AT, McCarthy J, et al. Ten years of routine alpha‐ and beta‐globin gene sequencing in UK hemoglobinopathy referrals reveals 60 novel mutations. Hemoglobin. 2016;40(2):75‐84. [DOI] [PubMed] [Google Scholar]
- 37. Farashi S, Rad F, Shahmohammadi B, Imanian H, Azarkeivan A, Najmabadi H. First report of a dominantly inherited beta‐thalassemia caused by a novel elongated beta‐globin chain. Hemoglobin. 2016;40(2):102‐107. [DOI] [PubMed] [Google Scholar]
- 38. Shooter C, Rooks H, Thein SL, Clark B. Next generation sequencing identifies a novel rearrangement in the HBB cluster permitting to‐the‐base characterization. Hum Mutat. 2015;36(1):142‐150. [DOI] [PubMed] [Google Scholar]
- 39. Zebisch A, Schulz E, Grosso M, et al. Identification of a novel variant of epsilon‐gamma‐delta‐beta thalassemia highlights limitations of next generation sequencing. Am J Hematol. 2015;90(3):E52‐E54. [DOI] [PubMed] [Google Scholar]
- 40. Chin CS, Peluso P, Sedlazeck FJ, et al. Phased diploid genome assembly with single‐molecule real‐time sequencing. Nat Methods. 2016;13(12):1050‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Snyder MW, Adey A, Kitzman JO, Shendure J. Haplotype‐resolved genome sequencing: experimental methods and applications. Nat Rev Genet. 2015;16(6):344‐358. [DOI] [PubMed] [Google Scholar]