

Abstract
Komagataeibacter species are well‐recognized bionanocellulose (BNC) producers. This bacterial genus, formerly assigned to Gluconacetobacter, is known for its phenotypic diversity manifested by strain‐dependent carbon source preference, BNC production rate, pellicle structure, and strain stability. Here, we performed a comparative study of nineteen Komagataeibacter genomes, three of which were newly contributed in this work. We defined the core genome of the genus, clarified phylogenetic relationships among strains, and provided genetic evidence for the distinction between the two major clades, the K. xylinus and the K. hansenii. We found genomic traits, which likely contribute to the phenotypic diversity between the Komagataeibacter strains. These features include genome flexibility, carbohydrate uptake and regulation of its metabolism, exopolysaccharides synthesis, and the c‐di‐GMP signaling network. In addition, this work provides a comprehensive functional annotation of carbohydrate metabolism pathways, such as those related to glucose, glycerol, acetan, levan, and cellulose. Findings of this multi‐genomic study expand understanding of the genetic variation within the Komagataeibacter genus and facilitate exploiting of its full potential for bionanocellulose production at the industrial scale.
Keywords: bacterial cellulose, c‐di‐GMP network, comparative genomics, exopolysaccharides, genome flexibility, Komagataeibacter
1. INTRODUCTION
The Komagataeibacter genus has recently emerged within the family of acetic acid bacteria (AAB) and consists of fourteen species (Barja, Andrés‐Barrao, Ortega Pérez, María Cabello, & Chappuis, 2016; Duardo, Ryngajllo, Jedrzejczak‐Krzepkowska, Bielecki, & Gama, 2016; Yamada, 2014, 2016; Yamada, Yukphan, Lan Vu, et al. 2012; Yamada, Yukphan, Vu, et al. 2012). This genus was named after a Japanese microbiologist, Prof. Kazuo Komagata, in appreciation of his contribution to the systematics of AAB. These species were mostly isolated from: vinegar (K. xylinus, K. europaeus, K. hansenii, K. oboediens, K. kakiaceti, K. medellinensis, K. maltaceti, Ga. entanii), fruit or fruit juice (K. xylinus, K. rhaeticus, K. swingsii, K. saccharivorans, K. sucrofermentans), tea fungus beverage—Kombucha (K. intermedius, K. rhaeticus), or from nata de coco (K. nataicola) (Yamada, 2016). This genus, apart from being an obligate aerobe and a gram‐negative alpha‐proteobacteria, is characterized by a lack of flagellation, production of acetic acid from ethanol, growth in the presence of 0.35% acetic acid (v/v), and no synthesis of 2,5‐diketo‐d‐gluconate from d‐glucose (Yamada, 2016). Komagataeibacter strains are highly resistant to acetic acid and are the dominant species of submerged vinegar processes, where acidities are highly elevated (Barja et al., 2016).
The species of Komagataeibacter have been shown to be the exceptionally efficient cellulose producers among bacterial species (Jedrzejczak‐Krzepkowska, Kubiak, Ludwicka, & Bielecki, 2016; Lin et al., 2013; Valera, Torija, Mas, & Mateo, 2015; Valera, Poehlein, et al., 2015). Ultrapure bionanocellulose (BNC) is finding applications in biotechnology, medicine, and various industry sectors. In comparison to plant‐derived cellulose, Komagataeibacter produces BNC with superior mechanical strength, purity, water‐holding capacity, and biodegradability (Cacicedo et al., 2016; Gama, Dourado, & Bielecki, 2016). Cellulose, a water‐insoluble exopolysaccharide composed of β‐1,4‐glucan chains, gives the bacterial cells protection from UV radiation or desiccation (Williams & Cannon, 1989). When growing in a liquid medium, synthesis of a cellulose membrane enables retention of cells close to the medium surface, where the amount of oxygen is high (Czaja, Young, Kawecki, & Brown, 2007; Williams & Cannon, 1989). The process of bacterial cellulose synthesis has been described only from the metabolic point of view. The cellulose synthase enzyme, which consists of four subunits, was discovered and studied in K. xylinus (Jedrzejczak‐Krzepkowska et al., 2016; Umeda et al., 1999; Wong et al., 1990). The function of subunits A and B (BcsA and BcsB), which are conserved among taxa, is well understood and proved to be responsible for β‐glucan chain formation (Morgan et al., 2016; Römling & Galperin, 2015). However, the role of the other subunits (BcsC and BcsD), which influence the efficiency of cellulose synthesis, is still under discussion (Hu et al., 2010; Iyer, Catchmark, Brown, & Tien, 2011; Saxena, Kudlicka, Okuda, & Brown, 1994). Moreover, there have been observed diversities in the structure of cellulose synthase operon (bcs) among the Komagataeibacter species (Matsutani et al., 2015; Saxena & Brown, 1995). It has also been reported that the efficiency of cellulose synthesis, the culturing conditions (e.g., the preferred carbon source), and structural and mechanical properties of cellulose (e.g., porosity and elasticity) depend on the bacterial strain (Czaja et al., 2007; Masaoka, Ohe, & Sakota, 1993; Suwanposri, Yukphan, Yamada, & Ochaikul, 2013; Toyosaki et al., 1995; Zeng, Laromaine, & Roig, 2014). Moreover, synthesis of soluble exopolysaccharides (EPS) may also influence cellulose features (Fang & Catchmark, 2014; Ishida, Sugano, Nakai, et al., 2002; Yoshinaga, Tonouchi, & Watanabe, 1997). Several of the Komagataeibacter strains were reported to synthesize acetan, derivatives of acetan, or levan (Couso, Ielpi, & Dankert, 1987; Kornmann, Duboc, Marison, & von Stockar, 2003; MacCormick, Harris, Gunning, & Morris, 1993). However, types of EPS synthesized vary not only between species but also between strains (Fang & Catchmark, 2015).
One of the well‐characterized mechanisms regulating cellulose synthesis is allosteric activation of BcsA with cyclic di‐GMP (c‐di‐GMP) molecule, a universal bacterial second messenger discovered in K. xylinus (Römling, 2012; Ross et al., 1985, 1987). Independent research revealed important role of c‐di‐GMP regulatory role for motility, virulence, biofilm formation, and the cell cycle control (Römling & Galperin, 2015). Cellular levels of c‐di‐GMP are under control of proteins with opposite enzymatic activities: diguanylate cyclases (DGCs) and c‐di‐GMP‐specific phosphodiesterases (PDEs), which catalyze c‐di‐GMP formation or degradation, respectively (Römling, Galperin, & Gomelsky, 2013; Tal et al., 1998). It has been later shown that DGC activity is associated with the presence of the GGDEF domain, whereas PDEs contain the EAL domain (Ausmees et al., 2001; Simm, Morr, Kader, Nimtz, & Römling, 2004).
Although AAB are known for their ability to oxidize ethanol, tolerance to extremely acidic conditions, and production of cellulose, these features appear to be transient, as they are often rapidly lost when cells are cultured in media without the selective pressure of acetate or ethanol (Beppu, 1993; Coucheron, 1991; Krystynowicz et al., 2002, 2005; Sokollek, Hertel, & Hammes, 1998; Takemura, Horinouchi, & Beppu, 1991). This phenotypic instability has a negative impact on the industrial performance of these species. Studies in various, mainly pathogenic bacteria, have shown that the genetic basis of phenotypic instability has been most commonly caused by spontaneous mutations and genome rearrangements, often associated with genetic elements, such as insertion sequences (IS), genomic islands (GIs), transposable elements (TEs), and transposable bacteriophages (Brzuszkiewicz, Gottschalk, Ron, Hacker, & Dobrindt, 2009; Chan et al., 2015; Kung, Ozer, & Hauser, 2010). Some IS have been found in the genomes of Komagataeibacter species, which were associated with cessation of the EPS synthesis (Coucheron, 1993; Iversen, Standal, Pedersen, & Coucheron, 1994; Standal et al., 1994). Moreover, alterations of the plasmid profile have been observed in cellulose‐negative (Cel−) cells (Coucheron, 1991). Other genetic elements of the mobile genome of the Komagataeibacter genus, however, have not been studied.
Overall, the molecular biology of Komagataeibacter has been tested to a limited extend so far and focused mainly on the metabolic pathway of cellulose production from glucose. Furthermore, different laboratories have been using various strains and typically considered only single genes in their analysis (Kubiak, Jedrzejczak‐Krzepkowska, Ludwicka, & Bielecki, 2016). In parallel, many changes in the taxonomic classification bring additional challenges in interpreting the available data. Published results appeared to be very strain‐specific thus making any conclusions about metabolism or regulatory mechanisms, responsible for the BNC production rate, unattainable at the genus level. Therefore, there is a need to broaden the genetic knowledge and to find common features of these strains that drive them to cellulose overproduction. By using the next‐generation sequencing (NGS) technology, we sequenced and assembled the genomes of three strains producing bionanocellulose. These are K. xylinus E26 from own collection, K. xylinus BCRC 12334, and K. hansenii ATCC 53582. Strain K. xylinus BCRC 12334 has been exploited mainly in Taiwan (Kuo, Chen, Liou, & Lee, 2016; Wu & Liu, 2012). The strain ATCC 53582, also known as NQ5, has been commonly used in the research worldwide, which can be emphasized by the fact that it has been sequenced by two independent teams during the time of our work (Florea, Reeve, Abbott, Freemont, & Ellis, 2016; Pfeffer, Mehta, & Brown, 2016b). Especially recently, we can observe a steep increase in the number of sequenced Komagataeibacter genomes (Pfeffer, Mehta, & Brown, 2016a; Pfeffer, Santos, Ebels, Bordbar, & Brown, 2017a,b,c; Wang et al., 2017; Zhang, Poehlein, Hollensteiner, & Daniel, 2018; Zhang et al., 2017). The growing number of genomic sequences of Komagataeibacter strains encouraged us to perform a comparative study with the aim to infer the precise phylogeny of the genus and to investigate sequence conservation in both, the core and the flexible part of the genome. By further focusing on functional diversity among the strains in carbohydrate uptake, EPS biosynthesis, and c‐di‐GMP signaling, we harness the gathered knowledge with the aim to explain the observed phenotypic variability of the in‐house strains.
2. MATERIAL AND METHODS
2.1. Bacterial strains and growth conditions
Cellulose‐producing strains (K. xylinus E26 (from the in‐house collection), K. xylinus BCRC 12334 (kind courtesy of Prof. Jyh Ming Wu, Department of Chemical and Materials Engineering, Chinese Culture University, Taipei, Taiwan), and K. hansenii ATCC 53582 American Type Culture Collection) were cultured in 10‐ml test tubes filled with 5‐ml of media at 30°C under static conditions for 3 days. One liter culture medium (Hestrin‐Schramm, HS; Hestrin & Schramm, 1954) contained 20.0 g glucose (POCh, Poland), 5.0 g yeast extract (BTL, Poland), 5.0 g bacterial peptone (BTL, Poland), 2.7 g sodium phosphate dibasic (Chempur, Poland), 1.15 g citric acid (Chempur, Poland), and 0.5 g magnesium sulfate (Chempur, Poland). The initial pH of the medium was adjusted to 5.7 with 80% acetic acid (Chempur, Poland). 1% cellulase (from Trichodrma reesei ATCC 26921, Sigma‐Aldrich) was added to the culture, and the released cells were harvested for genomic DNA purification.
2.2. Genomic DNA isolation and genome sequencing
Genomic DNA from the three strains was isolated according to the procedure published elsewhere (Ausubel et al., 1992) with modifications. Bacterial cells from 3.0 ml of liquid culture were pelleted and washed twice with TGE buffer (25 mM Tris‐Cl; 10 mM EDTA, 50 mM glucose; pH 8) prior to lysozyme (1 mg/ml in TGE) treatment (RT, 30 min). Next, SDS was added to the cells suspension (up to final conc. 0.5%), which was next treated with proteinase K (0.1 mg/ml final conc., Qiagen), at 56°C for 10 min. In the next step, incubation at 65°C for 30 min with CTAB and NaCl (final conc. 1% and 0.7 M, respectively) was applied. After cooling down, the nucleic acids were extracted twice with equal volume of phenol‐chloroform‐isoamyl alcohol (25:24:1) mixture and once with equal volume of chloroform/isoamyl alcohol (24:1) mixture. Finally, RNaseA (Qiagen) hydrolysis was done at 37°C for 20 min followed by additional extraction steps (same as above). Purified genomic DNA was precipitated with 4 M ammonium acetate and 100% ethanol at RT. The DNA pellet was washed twice with 70% ethanol, air‐dried, and suspended in TE buffer. All reagents were from Sigma‐Aldrich if not otherwise stated. NGS libraries were prepared using Nextera XT DNA Library Preparation Kit (Illumina). Genome sequencing was performed using the Illumina MiSeq platform, in 2 × 150 bp paired‐end reads mode. The genomes were sequenced at 20× coverage, on average.
2.3. Measurement of carbon source effect on the BNC yield
A pre‐culture was prepared in 5 ml of HS medium (as described above) from one isolated colony and incubated for 72 hr. The final culture was prepared, using 5% inoculum, in 6‐well plates with 10 ml of HS medium in which carbon source (glucose) was replaced with, either fructose, maltose, sucrose, or glycerol. The culture was incubated for 7 days at 30°C. BNC membranes were next treated with 2% solution of NaOH for one night and 1.5% of acetic acid for 4 hr, and then carefully washed in distilled water until neutral pH was reached. The purified membranes were pressed between two filter papers and dried at 80°C in a Gel Dryer apparatus (model 543, Bio‐Rad) until a constant weight was reached (Tiboni et al., 2012). For each strain (K. xylinus E25, K. xylinus E26, K. xylinus BCRC 12334, and K. hansenii ATCC 53582 strains) and each carbon source, six cultures (replicates) were prepared.
2.4. Scanning electron microscopy
Cellulose biofilms were harvested after 7 days of incubation in 250‐ml flasks, purified by several washes in water and 0.1% NaOH. Finally, water was exchanged into iso‐propanol. Such membranes were deep‐frozen in liquid nitrogen and then freeze‐dried. Before imaging, probes were coated with gold. Membranes’ surfaces were analyzed by Scanning Electron Microscope FEI, Quanta FEG 250, at 40,000× magnification, at Bionanopark sp. z o.o., Lodz, Poland.
2.5. Genome assembling and annotation
The sequencing reads were assembled de novo using SPAdes (with activated mismatch careful mode and otherwise default settings; v. 3.5.0; Nurk et al., 2013). These Whole Genome shotgun projects were deposited at DDBJ/EMBL/GenBank under the BioProject accession: PRJNA339514 (K. xylinus E26), PRJNA339679 (K. xylinus BCRC 12334), PRJNA339678 (K. hansenii ATCC 53582). The assemblies of the remaining seventeen genomes were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/. (Accessed January 2016); Table 1). Genome statistics for the entire set of twenty genomes were calculated using QUAST (v. 2.3; Gurevich, Saveliev, Vyahhi, & Tesler, 2013). MUM index (MUMi) between pairs of whole genome sequences was generated using Parsnp program with default cutoff settings for maximum MUMi distance (v. 1.2; Treangen, Ondov, Koren, & Phillippy, 2014).
Table 1.
Properties of the analyzed genomes
| Strain | Assembly status | Contigs | N50 | Total length [Mbp] | GC% | CDSs | tRNA | tmRNA | rRNA | Cellulose synthesis? | Reference | NCBI assembly ID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K. xylinus E26 | Contig | 366 | 20387 | 3.48 | 62.48 | 3253 | 40 | 1 | 3 | + | TW | BioProject accession: PRJNA339514 |
| K. xylinus BCRC 12334 | Contig | 306 | 29323 | 3.66 | 62.43 | 3386 | 40 | 1 | 3 | + | TW | BioProject accession: PRJNA339679 |
| K. hansenii ATCC 53582 | Contig | 252 | 49082 | 3.39 | 59.42 | 2952 | 45 | 1 | 3 | + | TW | BioProject accession: PRJNA339678 |
| Ga. diazotrophicus PAl 5 | Complete Genome | 3 | NA | 4.00 | 66.33 | 3740 | 60 | 1 | 12 | − | Bertalan et al. (2009), Valera, Torija, Mas, and Mateo (2015) and Valera, Poehlein, et al. (2015) | ASM6704v1 |
| K. europaeus 5P3 | Scaffold | 256 | 39631 | 3.99 | 61.49 | 3773 | 54 | 2 | 15 | ND | Andrés‐Barrao et al. (2011) | ASM28533v1 |
| K. europaeus CECT 8546 | Contig | 116 | 103383 | 4.11 | 61.31 | 3869 | 55 | 1 | 9 | + | Valera, Torija, Mas, and Mateo (2015) and Valera, Poehlein, et al. (2015) | ASM127364v1 |
| K. europaeus LMG 18494 | Scaffold | 216 | 64353 | 3.99 | 61.24 | 3738 | 59 | 1 | 24 | ND | Andrés‐Barrao et al. (2011) | ASM22754v1 |
| K. europaeus LMG 18890T | Scaffold | 321 | 35510 | 4.23 | 61.26 | 4050 | 51 | 2 | 12 | − | Andrés‐Barrao et al. (2011), Valera, Torija, Mas, and Mateo (2015) and Valera, Poehlein, et al. (2015) | ASM28529v1 |
| K. europaeus NBRC 3261 | Scaffold | 596 | 27771 | 3.63 | 61.6 | 3460 | 41 | 2 | 3 | ND | NP | ASM96448v1 |
| K. hansenii ATCC 23769 | Chromosome | 1 | NA | 3.64 | 59.5 | 3186 | 45 | 1 | 1 | + | Iyer, Geib, Catchmark, Kao, and Tien (2010), Valera, Torija, Mas, and Mateo (2015) and Valera, Poehlein, et al. (2015) | ASM16439v1 |
| K. hansenii JCM 7643 | Scaffold | 467 | 25111 | 3.71 | 59.29 | 3275 | 40 | 1 | 3 | + | Valera, Torija, Mas, and Mateo (2015) and Valera, Poehlein, et al. (2015) | ASM96440v1 |
| K. intermedius AF2 | Scaffold | 377 | 59913 | 4.52 | 61.34 | 4222 | 45 | 1 | 12 | + | Dos Santos et al. (2015) | ASM81725v1 |
| K. intermedius TF2 | Contig | 943 | 61064 | 3.88 | 61.67 | 3511 | 39 | 1 | 3 | ND | NP | ASM96442v1 |
| K. kakiaceti JCM 25156 | Contig | 947 | 5713 | 3.13 | 62.14 | 3859 | 35 | 1 | 3 | + | Iino et al. (2012) | ASM61330v1 |
| K. medellinensis NBRC 3288 | Complete Genome | 8 | NA | 3.51 | 60.58 | 3348 | 57 | 1 | 15 | +/− | Matsutani et al. (2015) and Ogino et al. (2011) | ASM18274v1 |
| K. oboediens 174Bp2 | Scaffold | 200 | 74982 | 4.18 | 61.26 | 3996 | 66 | 0 | 30 | + | Andrés‐Barrao et al. (2011) | ASM22756v1 |
| K. rhaeticus AF1 | Scaffold | 213 | 73183 | 3.94 | 62.49 | 3647 | 53 | 1 | 5 | + | Dos Santos et al. (2014) | GLUCORHAEAF1_v1 |
| K. sp. SXCC‐1 | Contig | 64 | 162897 | 4.23 | 62.44 | 3908 | 59 | 1 | 15 | ND | Du, Jia, Yang, and Wang (2011) | ASM20863v1 |
| K. xylinus E25 | Complete Genome | 6 | NA | 3.91 | 62.13 | 3671 | 59 | 1 | 15 | + | Kubiak et al. (2014) | ASM55076v1 |
| K. xylinus NBRC 13693 | Contig | 211 | 70122 | 3.34 | 61.95 | 3024 | 46 | 1 | 3 | + | NP | ASM96450v1 |
In bold are given genomes, which were sequenced in this work. Total assembly length for each genome was calculated by adding all genome sequences (chromosomes, plasmids, contigs, or scaffolds). Total number of predicted genes coding for rRNA (5S/16S/23S) is given in the “rRNA” column. +/−: the strain synthesizes/does not synthesize cellulose; NA: not applicable; ND: not determined; NP: no publication; TW: this work.
To check the presence of plasmid repA gene in the sequenced draft genomes, we searched them with the sequences of plasmid RepA protein (based on NCBI annotation) from the plasmids of the completely sequenced Komagataeibacter genomes (K. europaeus SRCM101446, pKE1446‐1 (WP_087609090.1); K. xylinus E25, pGX3 (WP_081749530.1); K. medellinensis NBRC 3288, pGXY020 (WP_007284615.1); K. nataicola RZS01, pKNA02 (WP_078528475.1), pKNA03 (WP_078528546.1), pKNA04 (WP_078528626.1)) by using tblastn program and applying a hit validity threshold of 50% identity and 70% coverage.
Genome sequences were next annotated using Prokka (default settings; v. 1.11; Seemann, 2014) employing: Prodigal (protein‐coding gene prediction; v. 2.6; Hyatt et al., 2010); Aragorn (transfer RNA gene prediction; v. 1.2; Laslett & Canback, 2004); Barrnap (ribosomal RNA gene prediction; v. 0.7; http://www.vicbioinformatics.com/software.barrnap.shtml); Infernal (noncoding RNA prediction; v. 1.1.2; Kolbe & Eddy, 2011). Proteins of Komagataeibacter strains were additionally annotated using software package InterProScan (v. 5.19; Jones et al., 2014) with option to scan for Pfam collection of protein families. A close inspection was given to the proteins, which had only one of the GGDEF/EAL (Pfam: PF00563/PF00990) domains. The single‐domain proteins were mostly those, which harbored a short EAL domain and lay at the start of a contig. It was verified with K. xylinus E26 and K. xylinus BCRC 12334 assemblies that for those genes, the proceeding contig matched GGDEF domain sequence, however, was not predicted by Prokka, since it lacked a stop codon, due to contig truncation, which is a commonly observed drawback of draft genomes.
2.6. Orthologous proteins analysis and core genome calling
The clusters of orthologous proteins in the analyzed genomes were generated using Proteinortho program (v. 5.11; Lechner et al., 2011). When it was necessary, gene presence was verified using the NCBI BLAST program (version 2.2.26; Altschul, Gish, Miller, Myers, & Lipman, 1990; Camacho et al., 2009). The core genome set was built by finding clusters of genes, which had an ortholog in every Komagataeibacter genome (G. diazotrophicus PAl 5 was not included). Genomes of K. kakiaceti JCM 25156 and K. intermedius TF2, were excluded from this list due to low genome quality. Hierarchical clustering and heatmap plotting was done using R (v. 3.1.0) and gplots package (release 2.17.0). CLC Sequence Viewer (v. 7.8.1) was used to generate and visualize multiple sequence alignments. SnapGene Viewer (v. 3.3.4) was used to display structure of gene clusters.
2.7. Phylogenetic analysis
The predicted 16S rRNA sequences (DNA) were aligned using MUSCLE (Edgar, 2004) and the phylogenetic analysis was conducted using MEGA (v. 6.06; Tamura, Stecher, Peterson, Filipski, & Kumar, 2013). Phylogenetic tree was constructed using the Maximum Likelihood method and by employing the Hasegawa‐Kishino‐Yano DNA substitution model with GAMMA distributed rate variation among sites model (HKY+G). The reliability of the tree was estimated using bootstrap method with 500 replicates. For the phylogenetic tree based on the orthologous protein sequences, the original orthologs list was filtered for single copy genes present in all twenty genomes, which resulted in 868 clusters. The sequences of these proteins were aligned in their ortholog sets using MAFFT (v. 7.305; Katoh, Misawa, Kuma, & Miyata, 2002). These alignments were next concatenated. The Maximum Likelihood tree was searched using RAxML (v. 8.2.69; Stamatakis, 2014) under the GAMMA model of rate heterogeneity and with automatic protein substitution model selection. Number of bootstrap replicates was selected using automatic bootstrapping criteria, as implemented in the program. The trees generated using RAxML were drawn in FigTree (v. 1.4.3; http://tree.bio.ed.ac.uk/software/figtree/).
2.8. Genomic islands, insertion sequences, prophages, and CRISPR‐Cas loci prediction
Genomic islands were predicted using IslandViewer3 online service (http://www.pathogenomics.sfu.ca/islandviewer/. (Accessed March 2016); Dhillon et al., 2015). Insertion sequences were predicted using ISsaga online service on the chromosome sequences of the complete genomes and of K. hansenii ATCC 23769 strain (http://issaga.biotoul.fr/issaga_index.php. (Accessed March 2016); Varani, Siguier, Gourbeyre, Charneau, & Chandler, 2011). Prophage prediction was done using PHASTER online service on unannotated chromosome sequences of the complete genomes and the chromosome sequence of K. hansenii ATCC 23769 strain (NZ_CM000920.1 for K. hansenii ATCC 23769; NC_010125.1 for G. diazotrophicus PAl 5; NZ_CP004360.1 for K. xylinus E25; NC_016027.1 for K. medellinensis NBRC 3288 (http://phaster.ca/ (Accessed November 2017); Arndt et al., 2016). Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) were predicted in all analyzed genomes using MinCED program (v. 0.2.0; https://github.com/ctSkennerton/minced). Additionally, predictions were made using CRISPRFinder web tool (http://crispr.i2bc.paris-saclay.fr/Server/ (Accessed March 2016); Grissa, Vergnaud, & Pourcel, 2007). Only the confirmed loci were considered. The results generated by both programs agreed in the majority of cases.
2.9. Functional enrichment of the core genome
The proteome of K. xylinus E25 was annotated using COG through WebMGA server of Wei Zhong Li Lab (http://weizhongli-lab.org/metagenomic-analysis/server/cog/ (Accessed April 2016). In total, 2,461 genes were assigned to at least one COG category, which was the case for 1,402 proteins of the core genome set. Additionally, this proteome was annotated using RAST annotation service (Aziz et al., 2008). In this case, 1,350 genes were annotated with at least one RAST category. For every COG or RAST category, assigned proteins were counted in the entire and in the core genome. Multi‐category proteins (assigned to more than one COG/RAST category) were counted in each of their categories. Functional enrichment was conducted using Fisher's exact test (one‐tailed) as implemented in R. FDR was controlled by using Benjamini & Hochberg method (1995).
3. RESULTS AND DISCUSSION
3.1. Phenotypic and genomic diversity of the Komagataeibacter strains
We have observed that four Komagataeibacter strains from the in‐house collection displayed phenotypic differences related to the produced cellulose pellicle (Figure 1a). Each of pellicles differed in microscopic arrangements of fibrils, which formed pores of various sizes. Consistent with previous reports, one of the strains, K. hansenii ATCC 53582, produced wider cellulose fibers (Czaja et al., 2007). Furthermore, the four strains from the in‐house collection varied in cellulose productivity when grown on different carbon sources with glucose and glycerol being most preferable, whereas maltose was the least preferable one (Figure 1b). Similar discrepancies have been published in independent studies testing other cellulose‐producing strains (Keshk & Sameshima, 2005; Mikkelsen, Flanagan, Dykes, & Gidley, 2009; Nguyen, Flanagan, Gidley, & Dykes, 2008). The most explicit differences between all tested strains were pronounced when grown on glucose. Specifically, K. hansenii ATCC 53582 and K. xylinus E26 strains productivity was four and three times higher as compared to the other two K. xylinus strains, respectively. Interestingly, we noticed exceptionally high cellulose productivity for the K. xylinus E26 strain grown on the medium with glycerol as the main carbon source and K. xylinus BCRC 12334 strain grown on sucrose (Figure 1b). Another important difference between the phenotypes of the investigated strains is stability of cellulose production. Among the four in‐house strains compared here, K. hansenii ATCC 53582 strain was the most productive in submerged cultures (data not shown).
Figure 1.
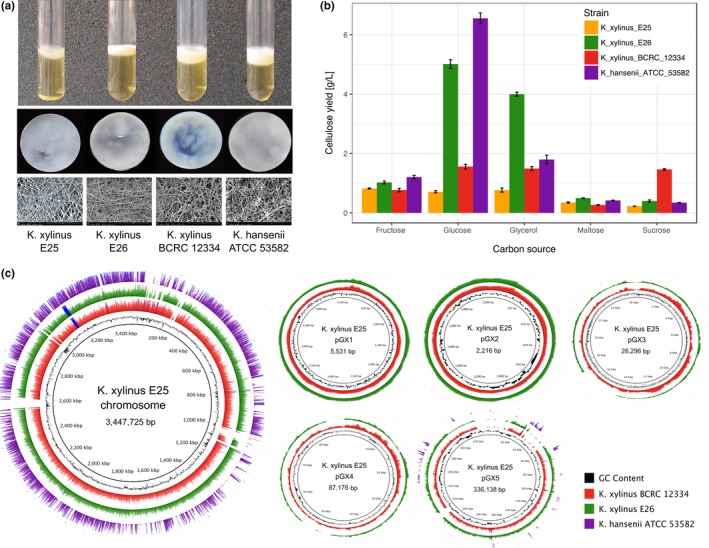
Phenotypic and genomic characteristics of the in‐house strains. (a) Comparison of cellulose pellicles. Liquid culture (top panel), macroscopic (middle panel), and SEM pictures (bottom panel) of membranes. (b) Cellulose yield in media containing five different carbon sources (fructose, glucose, glycerol, maltose, and sucrose). Error bar represents standard deviation calculated from six replicated cultures. (c) Mapping coverage of sequencing reads from three NGS libraries (color rings: K. xylinus E26—green, K. xylinus BCRC 12334—red, K. hansenii ATCC 53582—purple) to the genome of K. xylinus E25 (chromosome and five plasmids)
To understand the genetic basis of the observed phenotypic differences between the three K. xylinus in‐house strains and the K. hansenii ATCC 53582 strain, their genomes were sequenced using the NGS technology. Since the complete genome of K. xylinus E25 strain was already available (Kubiak et al., 2014), the remaining three genomes were sequenced. The sequencing was conducted in a cost‐effective manner, at low coverage of approximately 20×. The assembled draft genomes consist of 300 contigs, on average (Table 1). The assembly of K. hansenii ATCC 53582 genome is of the highest quality (the highest contiguity) and has the smallest GC content among the sequenced genomes (Table 1). Moreover, mapped read sequences of K. xylinus E26 and K. xylinus BCRC 12334 strains, unlike those of K. hansenii ATCC 53582 strain, covered the genome of K. xylinus E25 strain almost completely (Figure 1c).
Next, we estimated genomic distances between these four genomes using the MUM index, which is based on the number of maximal unique and exact matches shared by two genomes and takes values between 0 and 1, for very similar and very diverged genomes, respectively (Deloger, El Karoui, & Petit, 2009). This analysis confirmed that the genomes of K. xylinus E25, K. xylinus E26, and K. xylinus BCRC 12334 strains are much more similar to one another (MUMi values close to 0), whereas, for K. hansenii ATCC 53582 strain, MUMi values were of approximately 0.5, suggesting higher sequence divergence with the other three strains. Further comparative genome analysis produced a list of SNPs, InDels and missing or unique genes of mostly unknown function (data not shown). Finding which of these differences affect cellulose phenotype would be challenging without carrying out more experimental studies. What is more, other genomic differences, such as genome rearrangements, cannot be accurately investigated at the draft level of genome assembly. Therefore, we decided to include more of Komagataeibacter genomes and to perform a wide comparative study to extract consistent genomic patterns characterizing the genus and highlighting the most meaningful differences.
We annotated de novo our and publicly available genome sequences of Komagataeibacter strains using Prokka (Seemann, 2014). Additionally, one complete genome of Gluconacetobacter diazotrophicus PAl 5, a well‐studied free‐living strain, was included as a reference. Table 1 presents the general characteristics of the twenty analyzed genomes. Only three genomes of this set are completely assembled, whereas the others are in a draft state, with a varying degree of quality. The genome length of Komagataeibacter strains is in the range of 3.13–4.52 Mbp. The DNA G+C content is rather similar and in the range of 59%–62%, with the K. hansenii strains displaying the lowest values (Supporting Information Figure S1). Next, we generated a cladogram based on the MUM index to gain a rough overview of the sequence similarity among the twenty genome sequences. This analysis grouped genomes into several clades according to reciprocally low values (0–0.2; Figure 2a). Three main clades can be distinguished based on this cladogram. One clade groups the K. europaeus, the K. xylinus, K. oboediens, the K. intermedius, and K. rhaeticus strains. In general, the K. hansenii strains scored much higher MUMi values with other genomes suggesting higher sequence divergence. On the other hand, K. medellinensis NBRC 3288 and K. kakiaceti JCM 25156 genomes localize in between of the K. xylinus and the K. hansenii clades.
Figure 2.
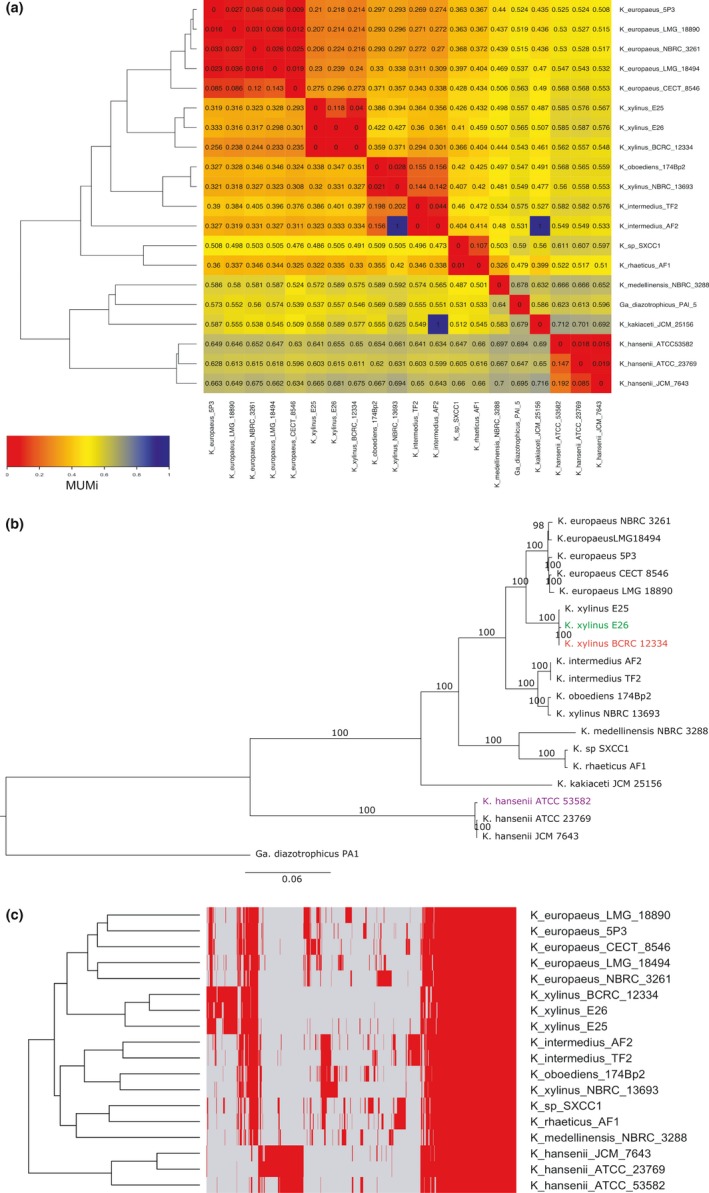
Relationship between Komagataeibacter genomes. (a) Reciprocal MUMi values between each of the 20 analyzed Komagataeibacter and a Gluconacetobacter diazotrophicus PAl 5 genome. Color scale from red to blue corresponds to lowest (highest similarity) and highest (lowest similarity) MUMi values, respectively. Dendrogram was generated based on hierarchical clustering analysis. (b) Maximum Likelihood phylogenetic tree calculated based on sequences of 868 orthologous proteins. Colored are the newly sequenced strains of K. xylinus E26 (green), K. xylinus BCRC 12334 (red), and K. hansenii ATCC 53582 (purple). The bar represents 6% sequence divergence. The numbers above branches represent bootstrap values. The tree was generated in RAxML and drawn in FigTree. (c) Clustering of Komagataeibacter strains based on presence (red)/absence (gray) of orthologs pattern. Dendrogram was generated based on hierarchical clustering analysis. Y‐axis: strains clustering; x‐axis: protein clustering (dendrogram not shown). K. kakiaceti JCM 25156 is not included due to poor quality of its genome
3.2. Phylogenetic analysis and the core genome of the Komagataeibacter genus
In order to verify the cladding pattern based on MUMi values, we performed the phylogenetic analysis based on the 16S rRNA sequence. This yielded a tree of low confidence, as tested by bootstrap test (Supporting Information Figure S2A). Moreover, on this tree, many of the K. xylinus strains clustered together with the K. europaeus strains. To perform a more accurate phylogenetic classification, we searched for a stronger taxonomic signal than a single gene sequence. We decided to use all available genomic data. For this purpose, orthologs were searched among their proteomes, which resulted in generation of 6,724 clusters holding proteins from at least two strains. Out of these, 1,578 orthologous gene clusters are present in every Komagataeibacter genome (constituting the core genome). After further filtering of the core genome set to select those clusters, which hold only single copy genes, we chose a set of 868 ortholog clusters. The phylogenetic analysis based on this group yielded a tree separating the genomes into species‐specific clades (Figure 2b). The tree topology was further confirmed with a previously proposed method, which employed dnaK‐groEL‐rpoB genes only (Supporting Information Figure S2B; Cleenwerck, De Vos, & De Vuyst, 2010). The advantage of using genomic data is manifested by a higher reliability, as measured by the bootstrap test. At this stage, it is clear that two distinct clades divide the genus, one that groups the K. europaeus, the K. xylinus, the K. intermedius, K. oboediens 174Bp2, K. medellinensis NBRC 3288, K. rhaeticus AF1, and K. kakiaceti JCM 25156 strains (from now on referred to as the K. xylinus clade in this work); the second clade constitutes of the three K. hansenii strains (now on called in this work the K. hansenii clade).
Additionally, we tested the sharing pattern of the entire orthologs set (Supporting Information Figure S3). Here, again the fewest orthologs were shared between the K. hansenii and the K. xylinus clade. Next, we focused on the groups of orthologs, which are not shared by all Komagataeibacter strains by investigating the cladding of their presence/absence pattern (Figure 2c). Roughly three major orthologs’ clusters were formed, one, the biggest, grouping genes present in every Komagataeibacter genome (genes clustered at the right side of the Figure 2c); one characteristic of the K. xylinus species; and another one distinguishing the K. hansenii strains. In general, the distinction of the K. hansenii strains from the rest of the Komagataeibacter spp. presented here is in accordance with previous studies (Andrés‐Barrao et al., 2013; Cleenwerck et al., 2010).
Based on our results, the two strains sequenced in this work, K. xylinus E26 and K. xylinus BCRC 12334, clustered together with K. xylinus E25, whereas the reference strain K. hansenii ATCC 53582 clearly grouped with other K. hansenii strains. The other K. xylinus strain, NBRC 13693, clustered rather far from K. xylinus E25, which was unexpected. To clarify these taxonomy differences, we decided to include the partial sequences for dnaK, groEL, and rpoB genes of the type strains from the work of Cleenwerck et al. (2010). The resulting tree showed that the majority of the strains are correctly classified at the species level (Supporting Information Figure S4). However, the K. xylinus genomes, contributed by our work (K. xylinus E25, K. xylinus E26, K. xylinus BCRC 12334) as well as K. xylinus NBRC 13693, cluster far from K. xylinus LMG 1515, the K. xylinus type strain (Yamada, 2016). Although, the K. xylinus E25, K. xylinus E26, and K. xylinus BCRC 12334 strains position close to the K. swingsii LMG 22125 (isolated from apple juice in South Tyrol region in Italy), they form a distinctive clade in this tree. These results suggest that K. xylinus NBRC 13693 probably belongs to K. oboediens; K. sp. SXCC1 is likely a K. rhaeticus strain, whereas the K. xylinus strains contributed by this work formed a group, which could be separated as species or subspecies, when more data would accumulate.
3.3. The mobile genome elements in Komagataeibacter strains
Sequence‐based and phylogenetic analysis conducted so far defined the core genome of the tested Komagataeibacter strains and enabled their classification, suggesting the clear separateness of the K. hansenii clade. Next, we focused on the mobile part of Komagataeibacter genomes (namely plasmid DNA, genomic islands GIs, insertion sequences IS, and prophages) because phenotypic diversity among bacterial strains of the same species is typically connected with this portion of DNA (Darmon & Leach, 2014).
3.3.1. Plasmids
The diversity among plasmid DNA has already been connected with phenotypic changes for other AAB strains (Akasaka et al., 2015; Azuma et al., 2009). Conjugative plasmid DNA transfer, in cellulose‐producing and cellulose nonproducing derivatives of K. xylinus ATCC 10245 (in the original paper: Acetobacter xylinum ATCC 10245) strain, has been shown in pioneering studies of Jackson, Vinatzer, Arnold, Dorus, and Murillo (2011). The complete genomic sequences of Komagataeibacter strains (K. xylinus E25 (Kubiak et al., 2014), K. medellinensis NBRC 3288 (Ogino et al., 2011), and the two strains deposited during the time of preparation of this manuscript—K. nataicola RZS01—(Zhang et al., 2017) and K. europaeus SRCM 101446; unpublished) have shown the presence of five, seven, six, and three plasmids in these strains, accordingly. In case of the three in‐house strains sequenced here, the results of mapping of sequencing reads to the K. xylinus E25 genome suggested the presence of at least two small plasmids (~5 kbp and ~2 kbp) in K. xylinus E26 and K. xylinus BCRC 12334 strains (Figure 1c). However, the depth of sequencing was not sufficient for proper plasmid sequence reconstruction, and therefore, we verified this prediction by identification of plasmid replication protein RepA in the whole‐genome assemblies. RepA homologs were detected on two separate contigs in K. xylinus E26 and K. xylinus BCRC 12334 genomes and one on one contig of K. hansenii ATCC 53582 genome (Supporting Information Table S1). Together with the published results, our data suggest that the variability in plasmid DNA content may contribute to the phenotypic diversity of the Komagataeibacter strains.
3.3.2. Short mobile genetic elements prediction
Two best‐studied groups of short mobile elements commonly related to bacterial strains phenotypic diversity are insertion sequences (ISs) and prophages. There are well‐established bioinformatics tools enabling prediction of these mobile sequences available. In order to assure accuracy, complete assemblies of genomes are required. Therefore, four chromosome sequences from the completed or closed genomes (Table 1) were used. Results showed a tendency of lower number of both, IS and prophage sequences, in K. hansenii ATCC 23769 strain when compared to K. xylinus E25 and K. medellinensis NBRC 3288 strains (Supporting Information Figure S5). The last‐mentioned strain shows a similar level of short mobile sequences as compared to the free‐living G. diazotrophicus PAI 5 strain used as a reference. The only mobile elements reported before in Komagataeibacter strains (namely K. hansenii ATCC 23770 and K. hansenii ATCC 23769) are insertion sequence IS 1031 and its derivatives (Coucheron, 1991, 1993; Standal et al., 1994). In contrast, our predictions showed quite complex composition of the IS families; for example, fifteen of them were identified in K. xylinus E25 genome (Supporting Information Figure S6). Absence of prophage sequences is very unusual in bacterial genomes but has been reported previously, for example, for Acetobacter pasteurianus 386B strain—a stable and industrially exploited AAB representative (Illeghems, De Vuyst, & Weckx, 2013). On the other hand, the presence of numerous diverse prophages is often linked with short‐term strain variation, for example, in Streptococcus pneumoniae lineages (Croucher et al., 2014). Therefore, the relatively low number of insertion sequences and lack of intact prophages in K. hansenii ATCC 23769 strain genome may be interpreted as an indication of the highest stability of this genome among the compared three representatives of the Komagataeibacter genus.
3.3.3. Genomic islands prediction
In contrast to prophages and ISs, genomic islands (GIs) are linked with more ancient evolutionary events and frequently are composed of genes of horizontal transfer origin, which appeared to be beneficial for a bacterial cell (Croucher et al., 2014; Gyles & Boerlin, 2014). Interestingly, genes encoded on GIs may take part in attenuation of other genetic elements’ mobility (Darmon & Leach, 2014). The occurrence of genomic islands in all nineteen analyzed genomes was predicted using the closest complete (or closed) sequence as reference (the chromosome of K. hansenii ATCC 23769 for the K. hansenii strains and K. xylinus E25 for the remaining draft genomes). It should be stated here, that, inconsistently with Island Viewer's settings, a GI should have length of at least 10 kbp, according to the most frequently used definition (Bellanger, Payot, Leblond‐Bourget, & Guédon, 2014). The pattern of predicted genomic islands (in the meaning of size and number) varied for the two main intragenus clades. The lowest number of GIs was found in the K. hansenii strains, whereas the opposite result was obtained in all of the K. europaeus strains (Figure 3a). Large number of diverse mobile genetic elements are of the key importance, for example, in the survival in extreme environments for the acidophilic Acidithiobacillus caldus ATCC 51756 strain (Acuña et al., 2013); and in maintaining virulence of numerous pathogens, including enterotoxigenic Escherichia coli, Salmonella typhimurium, Streptococcus pyogenes, and Clostridium perfringens (Gyles & Boerlin, 2014). It was expected to find the source of phenotypic divergence among the in‐house K. xylinus strains in the sequences or orthologs’ content of the identified genomic islands (Supporting Information Figure S7A). It appeared that only a few rearrangements on GI sequences could be found and that in each of the three analyzed strains, only 13‐20% of GI‐localized genes were unique (Supporting Information Figure S7B). Among unique sequences, genes connected with DNA rearrangements, transcriptional regulation, and cellular stress response (e.g., error‐prone polymerases, HTH‐transcriptional regulators, type IV secretion system genes and chaperons) were found, suggesting indirect influence on cellulose production process. There was a huge predominance of CDSs annotated as hypothetical proteins among them (namely: 68% in K. xylinus E25, 69% in K. xylinus E26, and 80% in K. xylinus BCRC 12334 strain), causing difficulty in posing any hypothesis about the functional pathways adopted on these islands.
Figure 3.
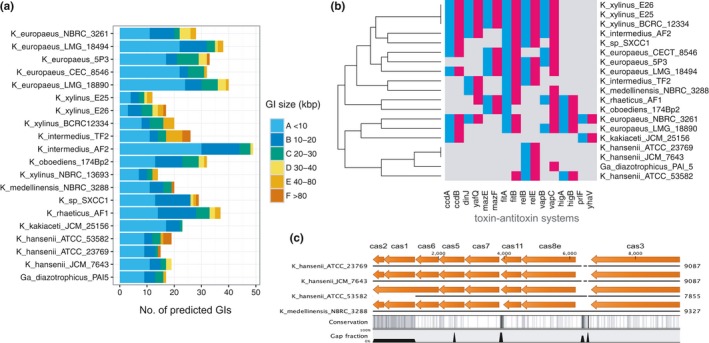
The flexible genome characteristics. (a) Number of predicted GIs of various sizes (kbp) in all analyzed 20 genomes of Komagataeibacter and a Gluconacetobacter diazotrophicus PAl 5 genome. (b) Presence (colored)/absence (gray) map of toxin–antitoxin systems in Komagataeibacter and a Gluconacetobacter strains. Antitoxins are marked in blue, toxins in red and arranged consecutively in pairs. Dendrogram (y‐axis: strains clustering) was generated based on hierarchical clustering analysis. (c) CRISPR‐Cas gene cluster in K. hansenii strains and K. medellinensis NBRC 3288 constituting of eight cas genes. In K. hansenii ATCC 53582, cas1 is incomplete, as it is located at a contig border; cas2 is present on another contig. Figure produced using SnapGene and modified
3.3.4. Predicted mechanisms regulating flexibility of Komagataeibacter genomes
Mobile genetic elements are under control of protective molecular mechanisms, like toxin antitoxin systems (TA) found on GIs or CRISPR‐Cas systems, which sustain a balance between genome flexibility and stability assuring cell survival (Darmon & Leach, 2014). First, we tested for the presence of TA systems in the studied genomes, using our orthologous groups identified previously. Only one toxin‐antitoxin system (namely fitA/B) was found as common in all Komagataeibacter strains tested with the exception of K. kakiaceti JCM 25156 and the whole K. hansenii clade, in which relB/E system is primarily found (Figure 3b).
Surprisingly, we found that only the K. hansenii clade and K. medellinensis NBRC 3288 strain harbors a CRISPR‐Cas system, in particular, of the Type I‐E, as predicted by the presence of cas3 signature gene and sequence similarity to E. coli K12 proteins (Figure 3c; Choi & Lee, 2016; Makarova, Zhang, & Koonin, 2017). These genomes contain one loci of the cas gene cluster consisting of 4‐20 palindromic repeats of 29 bp, separated by 32‐bp spacers (Supporting Information Table S2). We searched the NCBI nucleotide database with spacer sequences of the K. hansenii strains but were unable to find any valid match with a viral, prophage or plasmid sequences. Only one spacer from K. medellinensis NBRC 3288 shared a moderate sequence similarity with the pAC58‐29 plasmid from Acetobacter pasteurianus AC2‐58 (data not shown). Therefore, speculation about foreign DNA invasion events, “remembered” by CRISPR‐Cas system, is impossible at this stage of knowledge. Higher representation of TA systems in the strains possessing numerous IS and/or prophage sequences (e.g., K. medellinensis NBRC 3288 or K. xylinus E25 strains) seems reasonable. Moreover, the K. hansenii clade again showed to be clearly separated from all the rest of the analyzed strains with the smallest number of toxin–antitoxin systems and unique presence of CRISPR‐Cas system. The importance of the CRISPR‐Cas system in maintaining genome stability in other AAB representatives, Acetobacter pasteurianus CICC 20001 and CGMCC 1.41 strains and their derivatives, was proved recently (Wang, Shao, Chen, Chen, & Chen, 2016), but no such studies have been done for the Komagataeibacter genus so far.
3.4. Functional diversity of the Komagataeibacter genus
Ortholog‐conservation pattern displayed distinctive clusters, which may group proteins responsible for species‐specific features (Figure 2c). Investigation of these clusters, however, did not expose any particular functional groups among them, likely due to poor annotation (data not shown). In the next steps, we focused on functional analyses of the core genome. For this purpose, we investigated functions represented by the core genes. To do this, we performed functional enrichment, based on the COG and RAST functional categories, in respect to K. xylinus E25 genome annotation only (Supporting Information Figure S8A,8B). This analysis revealed a few overrepresented housekeeping COG categories, such as nucleotide transport and metabolism (F); coenzyme transport and metabolism (H); translation, ribosomal structure and biogenesis (J); posttranslational modification, protein turnover, chaperones (O). This could suggest some divergence among other functional groups and thus higher than expected intragenus functional diversity of the Komagataeibacter strains. Nevertheless, a meaningful analysis is not yet possible for these species, since functional categories, either COG or RAST, were assigned to only ca. 30% of the proteins in K. xylinus E25 genome. Therefore, we further concentrated on the most characteristic features, such as carbon source preference and exopolysaccharides production, including cellulose.
3.4.1. Predicted carbohydrate uptake mechanisms
One of the surprising results revealed by functional enrichment was the lack of “Carbohydrates” category (Supporting Information Figure S8A,S8B) since soluble exopolysaccharides and cellulose secretion are regarded as shared features of the genus. In our four in‐house strains, we observed variability in the utilization of the two carbon sources that promote the highest cellulose yield, that is, glucose and glycerol (Figure 1b). Comparative analysis between K. xylinus E25, K. xylinus E26, and K. xylinus BCRC 12334 strains showed a very high sequence conservation of the proteins involved in glucose metabolism and cellulose biosynthesis, such as periplasmic PQQ‐dependent glucose dehydrogenase, glucokinase, glucose‐6‐phosphate dehydrogenase (and its isoforms), phosphoglucomutase, UDP‐glucose pyrophosphorylase, cellulose synthase subunits (BcsA, BcsB, BcsC, BcsD) (Chawla, Bajaj, Survase, & Singhal, 2009; Kuo, Teng, & Lee, 2015) (data not shown). Therefore, it seemed that genetic basis contributing to the differences in cellulose yield in these strains is likely not related to the direct catalytic machinery. For this reason, we decided to further investigate carbohydrate uptake and exopolysaccharides production pathways. Although a variety of carbohydrate transport systems have been characterized in bacteria, none has been experimentally investigated in this genus. The availability of Komagataeibacter genomes allows now making predictions based on sequence similarity to known channels and transporters.
3.4.2. Predicted glucose uptake mechanism
In gram‐negative bacteria, glucose must pass through two membranes before it enters the cytosol. The permeability of the outer membrane is often dependent on the channels formed by porins. Based on Prokka annotation and InterProScan search, one candidate for a glucose transporter in Komagataeibacter strains may be the homolog of OprB porin from Pseudomonas aeruginosa (Wylie & Worobec, 1995). OprBs of Komagataeibacter are also likely beta‐barrel proteins (predictions made using Boctopus2). The number of oprB gene copies varies depending on genome, with the highest number of fifteen genes predicted for K. sp. SXCC1 strain (Supporting Information Figure S9). Due to the high number of oprB gene copies in each Komagataeibacter genome, it is possible that these proteins play an important role in carbohydrates transport (potentially, other than glucose as well; Wylie & Worobec, 1995). The conservation pattern of OprB orthologs highlights their diversity in the Komagataeibacter genus (Figure 4a).
Figure 4.
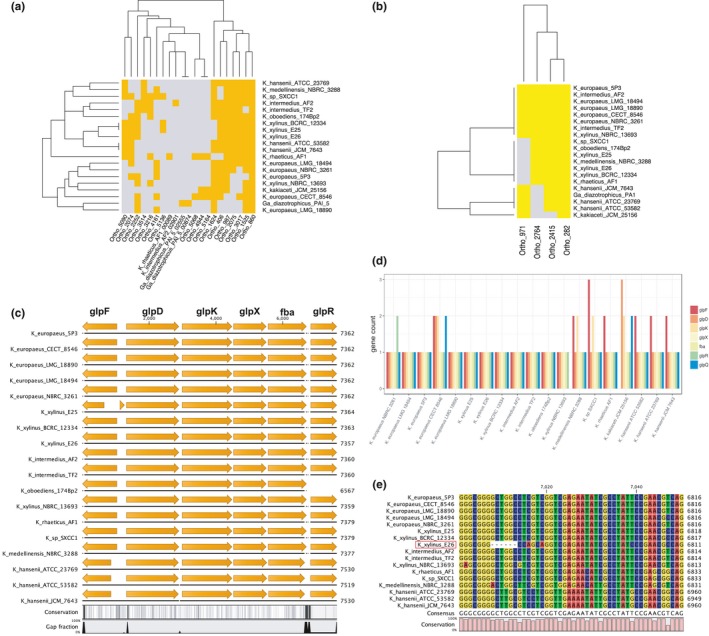
Conservation of glucose and glycerol transporters, and metabolic enzymes. (a) Presence (gold)/absence (gray) map of OprB proteins. (b) Presence (yellow)/absence (gray) map of GalP proteins. In both panels, (a and b), dendrograms were generated based on hierarchical clustering analysis. Y‐axis: strains clustering; x‐axis: protein clustering. (c) Conservation of glycerol metabolism operon. Aligned are only operon sequences contained within intact contigs. The gene symbols relate to putative functions: glpF—membrane diffusion facilitator for glycerol; glpD—aerobic G3P dehydrogenase; glpK—cytoplasmic glycerol kinase; glpX—fructose‐1,6‐bisphosphatase; fba—fructose‐bisphosphate aldolase; glpR—repressor of the glp regulon. (d) Number of glp genes in Komagataeibacter genomes. (e) Multiple sequence alignment of GlpR protein. Panels c and e generated using CLC Sequence Viewer
Transport of glucose through the cytoplasmic membrane in bacteria most commonly include phosphoenolpyruvate‐dependent sugar phosphotransferase system (PTS), transporters dependent on proton or cation gradient, diffusion facilitators, or ATP‐driven ABC transporters (Jahreis, Pimentel‐Schmitt, Brückner, & Titgemeyer, 2008). Blastp search did not result in finding of homologs of E. coli and V. furnissii glucose‐PTS and mannose‐PTS systems’ components in any of the Komagataeibacter strains. Based on Prokka annotation and InterProScan search, we detected only the EIIA component of fructose‐specific PTS system. This gene is present in all the K. xylinus strains and is clustered together with several other components of PTS system (Supporting Information Figure S10). However, the genomes lacked other domains of the enzyme II complex (such as IIB, IIC, IID; Saier & Reizer, 1992), which may suggest that this system does not function in fructose transport. We continued blastp‐based search using sequences of known glucose transporters of E. coli (the methyl‐galactoside permease MglBAC and the galactose permease GalP of E. coli; the sodium‐coupled glucose permease GLT of V. parahaemolyticus; the glucose facilitator, Glf of Z. mobilis; glucose transport protein GlcP of S. scabiei) and found only homologs of the galactose permease GalP (Hernández‐Montalvo et al., 2003). Up to four copies of galP gene are present in the Komagataeibacter genomes and their products are well conserved (Figure 4b). Summarizing, our findings suggest, that glucose is not phosphorylated during transport through the inner membrane, but in the cytosol (by glucokinase).
The conservation pattern of both, OprB and GalP proteins, is almost exactly the same in K. xylinus E25, K. xylinus E26, K. xylinus BCRC 12334 strains. Furthermore, direct genome sequence comparison between these three strains did not identify a putative transporter present uniquely in K. xylinus E26 strain. Therefore, we cannot explain, at this stage, why K. xylinus E26 strain is more efficient in glucose‐based cellulose synthesis. It is therefore possible that the observed differences in cellulose synthesis yield between the three strains are not due to differences in glucose transport, but due to distinctive modes of glucose metabolism regulation.
3.4.3. Predicted glycerol metabolic enzymes
It has been shown in Escherichia coli that the proteins encoded by the glp regulon mediate the utilization of glycerol and sn‐glycerol‐3‐phosphate (G3P) (Cozzarelli, Freedberg, & Lin, 1968). This regulon comprises of five operons which are located in three different regions of the genome (Zhao et al., 1994). We found homologs of glp proteins, which form one putative operon composed of glpD, glpK, glpX, fba genes in the tested Komagataeibacter strains (Figure 4c). The components of this operon encode: aerobic G3P dehydrogenase, a cytoplasmic glycerol kinase, fructose‐1,6‐bisphosphatase, fructose‐bisphosphate aldolase, respectively. The sequence upstream of the glp operon encodes the glpF gene, whose product is a cytoplasmic membrane protein facilitating diffusion of glycerol into the cell (Weissenborn, Wittekindtn, & Larsonsii, 1992). Adjacent to the glp operon is located the glpR gene, which encodes the repressor of the glp regulon of the deoR family of the transcriptional regulators (Zeng, Ye, & Larson, 1996). The repression of the glp regulon is relived in the presence of glycerol‐P (Zeng et al., 1996). The majority of Komagataeibacter genomes also contain a homolog of the glpQ gene (located outside of the glp operon), whose product is a periplasmic glycerophosphodiester phosphodiesterase. Moreover, several of the genomes carry additional copies of the predicted glp genes (Figure 4d). The structure of the glp operon is well conserved in the Komagataeibacter genomes, with the highest divergence displayed by the K. hansenii strains (Figure 4c). It can be noticed that, the glpF gene is much shorter in K. xylinus E25 strain than in other K. xylinus strains (Figure 4c). It is due to a single‐nucleotide insertion in its sequence, resulting in a frameshift and a premature termination (data not shown). Since GlpF is a putative glycerol diffusion facilitator, its disruption may negatively influence the glycerol uptake. This could partially explain, why K. xylinus E25 strain has the worst cellulose productivity out of the four in‐house strains, in the medium containing glycerol as carbon source (Figure 1b). On the other hand, a good performance of K. hansenii ATCC 53582 in this medium may be due to the presence of an additional copy of glpF gene in its genome (Figure 4d). A further inspection of the nucleotide sequence of the glp regulon allowed the detection of a polymorphism in the otherwise well‐conserved region of the glpR gene (Figure 4e). The glpR sequence of K. xylinus E26 strain contained a six‐nucleotide deletion and several mutations, which translated to the deletion of two amino acids (Leu61 and Ala62 in the consensus sequence; Supporting Information Figure S11) and four amino acid substitutions (Ser63Gln, Ser64Gln, Ser71Pro, and Gly156Arg in the consensus sequence; Supporting Information Figure S11). The deletion and the adjacent mutations are located at the C‐terminus of the DeoR‐like helix‐turn‐helix DNA binding domain predicted in this protein (InterProScan analysis). It is possible that binding of the GlpR repressor to the operator of the glp operon is impaired in K. xylinus E26 strain. Therefore, the expression of the glp operon may be active even in the absence of glycerol. This may explain why K. xylinus E26 strain has the highest cellulose yield in the medium containing glycerol among the tested strains (Figure 1b). From a more general point of view, this result shows that such an important phenotypic difference may be due to discrete changes in a single gene sequence, which influences indirectly the metabolic pathways via regulatory mechanisms.
3.4.4. Acetan biosynthesis gene cluster
Komagataeibacter species harbor the acetan biosynthesis cluster. These genes are homologous to gum‐like heteropolysaccharide genes of Xanthomonas campestris responsible for xanthan synthesis, which has been recently discovered in Kozakia baliensis (Brandt, Jakob, Behr, Geissler, & Vogel, 2016). In K. xylinus E25 strain, this cluster consists of seventeen genes (Figure 5a). It is completely absent in K. europaeus LMG 18494, whereas the majority of these genes is missing in the K. hansenii strains, which was additionally verified using tblastn (Figure 5b). The most important seems to be the absence of aceA (gumD), which is thought to initiate acetan biosynthesis by transferring a glucosyl‐1‐phosphate residue from UDP‐glucose to an undecaprenyl‐phosphate lipid carrier anchored in the inner membrane (Brandt et al., 2016; Ishida, Sugano, & Shoda, 2002). This would implicate that the K. hansenii strains and K. europaeus LMG 18494 should not produce acetan. However, it has been shown that some strains of these species do secrete EPS of similar monosaccharides composition as in acetan (Fang & Catchmark, 2014, 2015; Valepyn, Berezina, & Paquot, 2012). It has been also observed that the presence of acetan in a culture medium influences its viscosity, thus enhancing cellulose dispersion (Ishida, Sugano, Nakai, et al., 2002). Moreover, it has been shown that EPS can modulate bundling and width of cellulose ribbons, and thus influencing cellulose porosity (Fang & Catchmark, 2014, 2015). Therefore, the differences in cellulose membrane structure observed for K. hansenii and the other three in‐house strains may be due to the divergence in soluble EPS synthesizing enzymes.
Figure 5.
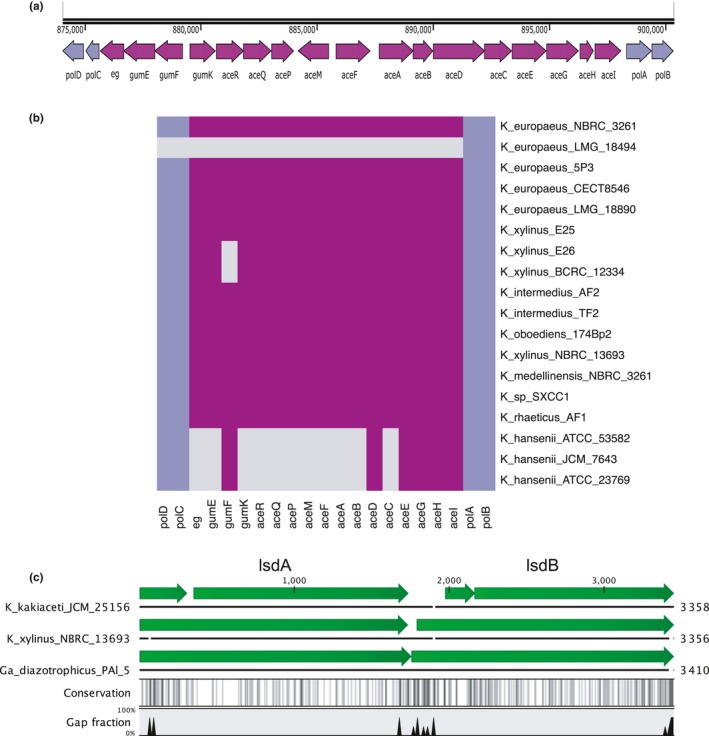
Conservation and characteristics of the soluble EPS gene clusters. (a) Organization of the acetan gene cluster in K. xylinus E25. Figure produced using SnapGene and modified. (b) Presence (magenta)/absence (gray) map of the acetan gene cluster and pol genes (navy‐blue). The gene symbols relate to putative functions: polD (rmlD)—dTDP‐4‐dehydrorhamnose reductase; polC (rmlC)—dTDP‐4‐dehydrorhamnose 3,5‐epimerase; e.g.,—endoglucanase; gumE—polymerization or export protein; gumF—acyltransferase; gumK—glucuronosyltransferase; aceR—rhamnosyl transferase; aceQ—glucosyl transferase; aceP—glucosyl transferase; aceM (ugd)—UDP‐glucose 6‐dehydrogenase; aceF (mpg)—mannose‐phosphate‐guanyl transferase; aceA (gumD)—UDP‐glucose:undecaprenyl‐phosphate glucose‐1‐phosphate transferase; aceB (gumM)—glycosyltransferase; aceD (gumC)—putative tyrosine‐protein kinase; aceC (gumH)—glycosyltransferase; aceE—polysaccharide transporter or flippase; aceG—putative beta‐barrel porin 2 family protein; aceH (gumB)—polymerization and export protein; aceI—acyltransferase; polA (aceJ; rmlB)—dTDP‐glucose 4,6‐dehydratase; polB (rmlA)—glucose‐1‐phosphate thymidylyltransferase. Annotation is based on homology with gum‐like gene cluster in Kozakia baliensis and acetan biosynthesis cluster from K. sucrofermentans DSM 15973 (Brandt et al., 2016; Ishida, Sugano, & Shoda, 2002). (c) Organization and conservation of the levan operon in K. kakiaceti JCM 25156, K. xylinus NBRC 13693, and Ga. diazotrophicus PAl 5. The gene symbols relate to putative functions: lsdA—levansucrase precursor; lsdB—levanase precursor. Annotation based on sequence homology with Ga. diazotrophicus SRT4's genes. Figure generated using CLC Sequence Viewer
The polymerization and export of acetan in the K. xylinus species seems to follow the Wzx/Wzy pathway due to the presence of aceD, aceE, aceG, and aceH genes, which putatively code for polysaccharide copolymerase (PCP), flippase, beta‐barrel porin, and outer membrane transport protein (OPX), respectively (Schmid, Sieber, & Rehm, 2015). Interestingly, these genes are among the few conserved in the K. hansenii strains. Genomic analysis of the neighborhood of these genes, in the K. hansenii strains, revealed presence of several putative and unknown glycosyltransferases (data not shown). It is possible that the K. hansenii strains synthesize other than acetan, yet an uncharacterized EPS.
The acetan gene cluster is flanked by pol genes (colored in navy blue in the Figure 5a,b). These genes are organized in a cluster in Acetobacter tropicalis and Kozakia baliensis (polABCDE; Deeraksa et al., 2005; Brandt et al., 2016). This cluster is responsible for the biosynthesis of capsular polysaccharide (CPS), which consists of galactose, glucose, and rhamnose. It has been shown that polE plays an important role in this process, since it is likely responsible for anchoring of the polysaccharide to the cell surface (Deeraksa et al., 2005). CPS is thought to serve as a protective barrier from acetic acid in AAB. However, it has been observed that Komagataeibacter species do not produce this membrane polysaccharide (Andrés‐Barrao et al., 2013; Barja et al., 2016). Our genomic analysis supports this finding as we observed that in all analyzed strains, polABCD operon is disrupted by the acetan cluster and, moreover, lacks the crucial polE gene (Figure 5a,b). Komagataeibacter species must clearly have a distinctive, from other AAB, mechanism for acetic acid resistance.
3.4.5. Levan biosynthesis
Apart from acetan, K. xylinus was also reported to synthesize levan, an EPS produced from extracellular sucrose (Kornmann et al., 2003; Limoli, Jones, & Wozniak, 2015). Activity of levansucrase, which is the main enzyme involved in the levan biosynthesis process, has been reported in K. xylinus I‐2281 strain (Kornmann et al., 2003). By screening Komagataeibacter genomes (using tblastn with the levansucrase (LsdA) protein sequence of Ga. diazotrophicus SRT4; Arrieta et al., 2004), we were unable to find levansucrase gene in all but K. xylinus NBRC 13693 and K. kakiaceti JCM 25156 strains. In K. xylinus NBRC 13693 strain, levansucrase gene is arranged in a cluster with levanase (lsdB) gene (Figure 5c) and the components of the type II secretion system (data not shown), similarly as it has been reported in Ga. diazotrophicus (Arrieta et al., 2004). The levanase is responsible for hydrolysis of levan to free fructose. Therefore, these genes are necessary for the bacterium to feed on sucrose. It has been reported that in Gluconacetobacter representatives, sucrose cannot be transported through cell membrane and has to be hydrolyzed into glucose and fructose in the periplasm (Velasco‐Bedrán & López‐Isunza, 2007). However, the obtained results suggest that the majority of Komagataeibacter strains should not synthesize levan. Other systems of sucrose transport and hydrolysis were not reported for this genus, and we were unable to discover them in the sequenced genomes. We have shown that culturing of the in‐house strains in the medium with sucrose results in a low cellulose yield, except for K. xylinus BCRC 12334 strain (Figure 1b). Since the genome of K. xylinus BCRC 12334 strain does not encode the lsdA‐lsdB operon, this would suggest that there should exist another, yet undefined, system for sucrose metabolism in Komagataeibacter strains.
3.4.6. Conservation of cellulose synthase operons’ structure
In Komagataeibacter, cellulose synthase enzyme is encoded by two types of operons (Matsutani et al., 2015). The type I bcs operon consists of four genes: bcsAI, bcsBI, bcsCI, and bcsDI (Matsutani et al., 2015). BcsA is a β‐glycosyltransferase, an inner membrane protein, which catalyzes the synthesis of β‐1,4‐glucan from UDP‐glucose (Jedrzejczak‐Krzepkowska et al., 2016; Morgan et al., 2016). Furthermore, PilZ domain of this subunit has been shown as c‐di‐GMP binding site (Fujiwara et al., 2013; Morgan, Strumillo, & Zimmer, 2012). BcsB is a periplasmic protein, which participates in β‐glucan chain synthesis and translocation (Jedrzejczak‐Krzepkowska et al., 2016). Role of BcsC and BcsD is unclear, and however, they are required for cellulose biosynthesis in vivo (Jedrzejczak‐Krzepkowska et al., 2016; Römling, 2002). The type I cellulose synthase operon has on average size of 9 kbp (Figure 6a). When the structure of this operon is compared inside of the Komagataeibacter genus, several differences can be observed. First, in the K. hansenii strains, genes coding for subunits bcsAI and bcsBI are fused, unlike in the K. xylinus strains, as it was reported before (Saxena et al., 1994). What is more, some diversity in the structure of bcs genes can be observed. The subunit A of K. hansenii ATCC 53582 strain is much longer than in any of the Komagataeibacter strains. For K. hansenii ATCC 23769 and K. hansenii JCM 7643 strains, bcsCI is split into two, not similar parts. In case of other strains, for K. medellinensis, bcsBI is disrupted due to a frameshift mutation, as previously reported for this cellulose‐negative strain (Matsutani et al., 2015). The alignment shown in the Figure 6a highlights a large deletion in the sequence of bcsCI in two K. europaeus strains, LMG 18494 and NBRC 3261. Overall, the bcs operons of the K. xylinus E25, E26, and BCRC 12334 strains are very similar to those of the K. europaeus strains. Interestingly, one subunit that is very well conserved among Komagataeibacter strains is bcsDI, which function is up to date the most speculative. It was suggested that bcsDI has originally evolved in AAB since it lacks homology with any other bacteria (Matsutani et al., 2015).
Figure 6.
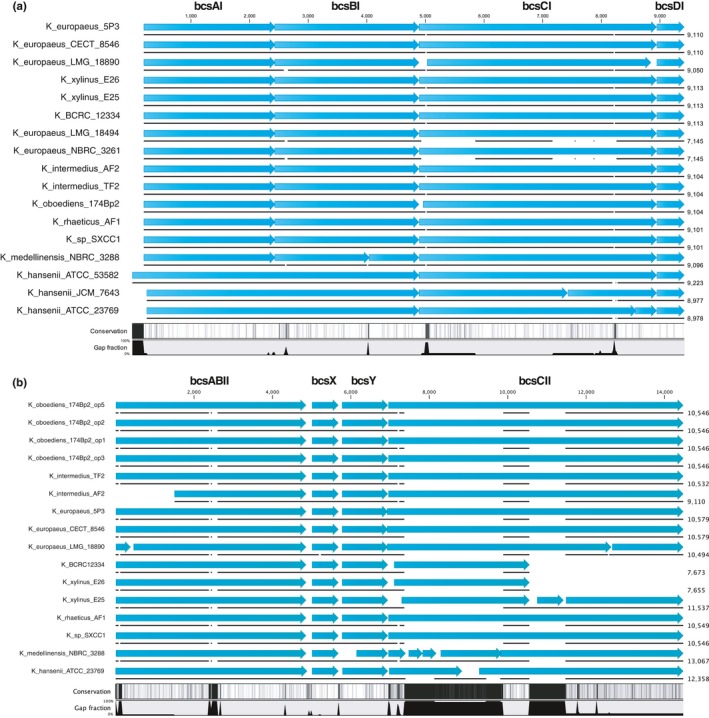
Comparison of cellulose synthase operons’ structure and nucleotide sequence among the Komagataeibacter strains. Operons contained within a single contig or a chromosome are only shown. (a) Operon type I. (b) Operon type II. Figures generated using CLC Sequence Viewer and modified
The type II bcs operon consists of four genes, bcsABII, bcX, bcsY, and bcsCII. It is believed that bcsII synthesizes the acylated, amorphous cellulose due to homology of bcsY to transacylase (Chawla et al., 2009; Umeda et al., 1999). This operon is absent in K. europaeus LMG 18494 and K. europaeus NBRC 3261 strains, as well as in Ga. diazotrophicus PAl 5 strain (Figure 6b), which was additionally checked using tblastn. K. oboediens 174Bp2 is unusual, as its genome contains four copies of bcsII operon (Figure 6b). In other Komagataeibacter strains, bcsCII is often disrupted, as in the case of K. europaeus LMG 18890, K. xylinus E25, K. medellinensis NBRC 3288, and K. hansenii ATCC 23769 strains. In K. xylinus E25 and K. medellinensis NBRC 3288 strains, this disruption is caused by an insertion sequence, as it was previously reported (Matsutani et al., 2015). Additionally, there are many insertions or deletions (InDels) present at the start of the bcsCII across the compared strains, likely causing frameshift mutations in some of them.
Sequenced‐based predictions suggest that bcsCII, like bcsCI, is a beta‐barrel protein, likely forming a channel in the outer membrane (predictions made using Boctopus2; Hayat, Peters, Shu, Tsirigos, & Elofsson, 2016). Since the putative role of bcsC is export of cellulose, disturbance of bcsCII may influence secretion of the acylated polymer. Furthermore, sequence variations, in the both subunits, across Komagataeibacter strains may be responsible for differences in cellulose structure. Generally, the often‐seen disruption of mainly the bcsC subunits suggests that cellulose export may be the first target of evolutionary forces.
3.4.7. Diversity in the c‐di‐GMP‐based regulatory network
Cyclic di‐GMP ubiquitous second messenger, until recently regarded as bacteria‐specific (Hengge, Gründling, Jenal, Ryan, & Yildiz, 2016), crucial for life style and cell cycle regulation was discovered in K. xylinus in late 1990s, but elucidation of complexity of its signaling network was done in other species (Tal et al., 1998). Most frequently c‐di‐GMP signaling is being proved to be involved in the regulation of soluble and insoluble EPS components production during biofilm formation and dispersion (Römling & Galperin, 2015 and references therein). Typically, c‐di‐GMP regulatory networks are composed of numerous enzymes catalyzing its synthesis (diguanylate synthases with DGGEF domains, DGCs) and hydrolyzing it into linear pGpG (phosphodiesterases with EAL domains, PDEs) or directly into two molecules of GTP (phosphodiesterases with HD‐GYP domains). Complexity of this network may be illustrated by the number of genes involved: from around 20 in, for example, Escherichia coli K‐12 (29 genes; Povolotsky & Hengge, 2016) or alpha‐proteobacterium Sinorhizobium meliloti (22 genes; Schäper et al., 2015), up to dozens, for example, in Pseudomonas aeruginosa (42 genes; Valentini & Filloux, 2016) or in alpha‐proteobacteria Bradyrhizobium japonicum (55 genes; Schäper et al., 2015). Besides enzymatic activity, GGDEF/EAL proteins frequently serve as signal receiving proteins via their sensory domains (e.g., PAS, GAF, CHASE or BLUF; Römling & Galperin, 2015; Hengge et al., 2016). Complete understanding of biological effect regulated by c‐di‐GMP includes identification of effector molecules containing riboswitches and diverse proteins capable of c‐di‐GMP molecule binding (most frequently but not exclusively via PilZ domains; Hengge et al., 2016). Since pioneering work of Prof. Benziman's group resulting in discovery of c‐di‐GMP and three operons composed of pairs of DGC and PDEs (cdg1‐3; Tal et al., 1998), no further elucidation of this signaling network in any Komagataeibacter strain has been published. Therefore, we decided to take advantage of the available genomic data to shed some light into level of complexity of this important regulatory network in the tested genus. Furthermore, analysis presented here suggests that source of phenotypic diversity observed among in‐house strains does not stem from any large changes in the genome content, but rather from discrete changes, outside of the main metabolic pathways (e.g., the glycerol repressor in K. xylinus E26 strain).
By running InterProScan on the entire predicted proteomes of Komagataeibacter strains, we selected proteins, which matched EAL domain or/and GGDEF domain pattern. In the majority of proteins, both domains were present in one protein. Number of EAL/GGDEF proteins varied, with the K. xylinus clade genomes having up to seventeen and the K. hansenii clade harboring consistently eight proteins (Figure 7a). Already at this point, it appears a unique feature of the Komagataeibacter genus to have both domains present in tandem in the majority of c‐di‐GMP‐metabolizing proteins (Figure 7a). In Gluconacetobacter diazotrophicus PAl 5 and in strains of genera Acetobacter and Gluconobacter, it appears that this distribution is less skewed and there are many proteins harboring only a single, mostly a GGDEF, domain (Figure 7a,b). These numbers are nevertheless much higher than previously proven by Prof. Moshe Benziman (three cdg operons mentioned above; Tal et al., 1998).
Figure 7.
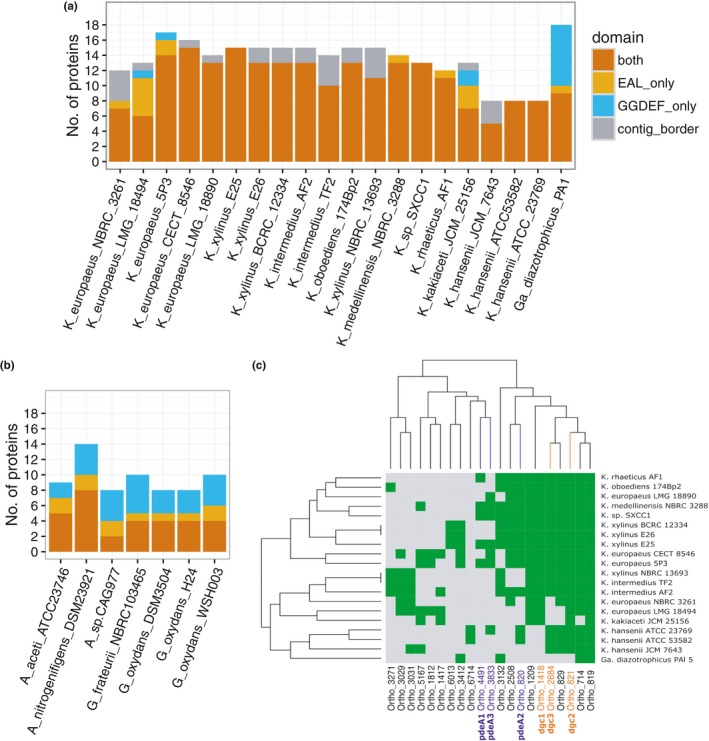
Characteristics and conservation of c‐di‐GMP‐metabolizing proteins. (a) Number of GGDEF/EAL domain‐containing proteins in genomes of Komagataeibacter spp. and Gluconacetobacter diazotrophicus PAl 5. Distinguished are the proteins containing both domains (in red) or only one (EAL in yellow; GGDEF—in blue). In gray are given single‐domain proteins encoded by genes that are located at contigs borders and therefore are of lower reliability (see Methods). (b). Genomes of the Acetobacter (A) and the Gluconobacter (G) genus. The strains were selected based on the completeness of their genome sequence (full genome, or less than 50 contigs). (c) Presence (green)/absence (gray) map of GGDEF/EAL domain‐containing proteins in the analyzed genomes. Orthologs of diguanylate cyclases (dgc) and phosphodiesterases (pde) identified earlier (Tal et al., 1998) are colored in orange and dark blue respectively. Dendrogram was generated based on hierarchical clustering analysis. Y‐axis: strains clustering; x‐axis: protein clustering
After observing diversity in the number of c‐di‐GMP turnover proteins across the Komagataeibacter genomes, we next investigated how well these proteins are conserved. We observed a very complicated presence/absence pattern of these important proteins in the twenty analyzed genomes (Figure 7c). Furthermore, even the three canonical operons identified by Tal et al. (1998) turned out not to be well conserved (highlighted in blue [PDEs] and in orange [DGCs]). Surprisingly, PdeA1 and Dgc1, the most highly expressed proteins in K. xylinus strains tested in pioneering studies (in original paper: A. xylinum strains: 1306‐3, an isolate from strain B42; North Regional Research Laboratories, Peoria, Ill; Tal et al., 1998), are both absent in the genomes of the K. hansenii strains. Moreover, diguanylate cyclases (dgc1‐3) seem to be better conserved than phosphodiesterases (pdeA1, pdeA3). These observations are drawn from the analysis based on mostly draft genomes and therefore have to be treated cautiously. For example, for both, the K. xylinus BCRC 12334 and K. xylinus E26 genome, we checked, using tblastn, that these genomes may indeed contain the pdeA1 and pdeA3 genes, but at contig borders. This may likely be the explanation of this surprising incompleteness of these important operons, since the two complete genomes of K. xylinus E25 and K. medellinensis NBRC 3288 harbor them. On the other hand, the only closed K. hansenii genome sequence, ATCC 23769, is missing the cdg1 operon, which would support the hypothesis that the c‐di‐GMP regulatory network have diverged in the K. hansenii strains, with respect to the K. xylinus strains. Furthermore, none of the cdg operons is conserved in Ga. diazotrophicus PAl 5 strain. Additionally, searching of orthologs of these genes in other AAB (as those shown in the Figure 7b) was similarly unsuccessful.
Fast evolution of c‐di‐GMP signaling network was previously observed in genomic comparison study of 61 pathogenic and commensal Escherichia coli strains (Povolotsky & Hengge, 2016). The authors identified numerous losses of DGCs besides many mutational changes in pathogenic strains when compared to nonviral strains. Therefore, we expect that the observed by us diversity in the atypical (as composed of only several and mainly two‐domain GGDEF/EAL proteins) c‐di‐GMP signaling network in Komagataeibacter species is of great importance for phenotypes of cellulose producers. Elucidation of biochemical role of the identified genes in Komagataeibacter strains and precise analysis of their possible interactions with EPS synthesis and secretion machineries as well as with other cellular processes should bring a valuable impact on productive strain engineering.
4. CONCLUSIONS
This first comparative genomic approach conducted by us in the Komagataeibacter genus brought several genomic evidences for a clear separation of two main clades, namely: of the K. hansenii strains and larger, of the K. xylinus‐related strains. This conclusion is based on whole genome sequence similarity comparisons measured using MUM index as well as through phylogenetic analysis employing 868 ortholog gene clusters. The distinctiveness of the K. hansenii strains was further supported by the pattern of intragenus distribution of the predicted mobile elements and the presence of different genome defense systems, that is, variable toxin‐antitoxin systems in the K. xylinus group and CRISPR‐Cas loci in the K. hansenii strains. Moreover, functional diversity analysis showed that genetic diversity between the two main Komagataeibacter clades is beyond the well‐known differences in cellulose operon organization and is manifested by, for example, lack of cluster of genes responsible for acetan synthesis and changes in the structure of the putative glycerol diffusion facilitator gene, as well as differences in size and composition of c‐di‐GMP signaling network (including absence of PdeA1 and Dgc1) in the K. hansenii strains. All these results taken together suggest that K. hansenii strains seem to form a subgenus, separated from all other strains tested here (named the K. xylinus clade).
Genomic diversity among Komagataeibacter strains, shown here on numerous examples, is particularly interesting from the practical point of view for two functional categories: EPS synthesis and c‐di‐GMP signaling network. Better characterization of soluble EPS synthesized, in parallel or alternatively to cellulose, in different Komagataeibacter strains may aid optimization of BNC productivity. Moreover, cellulose pellicle formation is a very precisely controlled process and involves molecular regulatory mechanisms such as c‐di‐GMP signaling. We predicted presence of several proteins with GGDEF and EAL domains in every Komagataeibacter strain. Such predominant presence of dual‐domain proteins is not usual in c‐di‐GMP signaling networks in AAB and may suggest primitive stage of these pathways organization in the Komagataeibacter genus. There is not enough data to speculate about specialization of the discovered genes in c‐di‐GMP metabolism or signaling by allosteric interactions. Nevertheless, cellulose producers seem to be a very poorly studied group of bacterial strains, which, when explored further, can bring interesting molecular discoveries in nucleotide‐regulated signaling field.
Importantly, this work contributed the first description of gene clusters important for carbohydrate metabolism (glucose and glycerol transport, EPS synthesis and secretion). Furthermore, comparative analysis enabled finding of Komagataeibacter features, believed to be genus‐typical, which appeared to be strain‐specific, for example, acetan and levan synthesis, or presence of three cdg operons. Therefore, many metabolic features, revealed by the previously reported production process optimizations, may appear not common for the whole genus. Moreover, great phenotypic differences may not be clearly distinguishable on genome level as it was exemplified here by our K. xylinus in‐house strains, which were overall similar in DNA sequence. Most probably phenotypic diversity among the compared strains was due to discrete sequence changes (glpR polymorphism in K. xylinus E26 strain) or fine‐tuning of gene expression and protein activities via yet to‐be‐discovered signaling pathways (the poorly understood c‐di‐GMP signaling network, hypothetical proteins identified as unique on genomic islands and in ortholog groups). Taken together, our findings showed that the genomic approach is sensible for elucidation of the numerous misleading results found in the literature. Precise description of any new strain, preferably together with genomic sequence, is of great importance for utility of published results concerning Komagataeibacter representatives. Phylogenetic studies, performed in this work, brought evidence for usefulness of Cleenwerck et al. method for intragenus classification of the Komagataeibacter strains and should be chosen for taxonomy verification of any new strain.
In the future, together with the growing number of genomic sequences and biochemical studies, new genome engineering strategies should become available for cellulose production optimization.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHORS CONTRIBUTION
MR, KK, and SB conceived and designed the study. KK, MJ‐K, and PJ cultured bacteria and isolated DNA. MR assembled and annotated the genomes. KK performed the bioinformatics analyses regarding IS, prophages, and GIs. MR performed the remaining bioinformatics analyses. MR and KK drafted the manuscript. All authors read and approved the final manuscript.
ETHICAL STATEMENT
The presented research did not involve studies with human or animal subjects or recombinant DNA.
Supporting information
ACKNOWLEDGMENTS
We are grateful to OpenExome s.c. and Department of Genetics, World Hearing Center, Institute of Physiology and Pathology of Hearing, Kajetany/Warsaw, Poland, for providing the training and facilities for genome sequencing. We would like to thank Teresa Pankiewicz for her valuable and multilevel help throughout the project's time. We thank Jolanta Płoszyńska for her help with media and strain preparation. We would like to further acknowledge Aleksandra Placzyńska (nee Kot) for her contribution to CRISPR‐Cas loci finding. Furthermore, we would like to acknowledge Ganga Jena for her support. We are grateful to Nova Do and Bartłomiej Jan Blus for proofreading of the manuscript.
Ryngajłło M, Kubiak K, Jędrzejczak‐Krzepkowska M, Jacek P, Bielecki S. Comparative genomics of the Komagataeibacter strains—Efficient bionanocellulose producers. MicrobiologyOpen. 2019;8:e731 10.1002/mbo3.731
DATA ACCESSIBILITY
The DNA sequences of the assembled genomes are available at NCBI under the BioProject accession: PRJNA339514, PRJNA339679, PRJNA339678. Additional data sets generated in this work are provided in the Supporting Information Data S1 of this article.
REFERENCES
- Acuña, L. G. , Cárdenas, J. P. , Covarrubias, P. C. , Haristoy, J. J. , Flores, R. , Nuñez, H. , … Quatrini, R. (2013). Architecture and gene repertoire of the flexible genome of the extreme acidophile Acidithiobacillus caldus . PLoS ONE, 8(11), e78237 10.1371/journal.pone.0078237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akasaka, N. , Astuti, W. , Ishii, Y. , Hidese, R. , Sakoda, H. , & Fujiwara, S. (2015). Change in the plasmid copy number in acetic acid bacteria in response to growth phase and acetic acid concentration. Journal of Bioscience and Bioengineering, 119(6), 661–668. 10.1016/j.jbiosc.2014.11.003 [DOI] [PubMed] [Google Scholar]
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Andrés‐Barrao, C. , Benagli, C. , Chappuis, M. , Ortega Pérez, R. , Tonolla, M. , & Barja, F. (2013). Rapid identification of acetic acid bacteria using MALDI‐TOF mass spectrometry fingerprinting. Systematic and Applied Microbiology, 36(2), 75–81. 10.1016/j.syapm.2012.09.002 [DOI] [PubMed] [Google Scholar]
- Andrés‐Barrao, C. , Falquet, L. , Calderon‐Copete, S. P. , Descombes, P. , Ortega Pérez, R. , & Barja, F. (2011). Genome sequences of the high‐acetic acid‐resistant bacteria Gluconacetobacter europaeus LMG 18890T and G. europaeus LMG 18494 (reference strains), G. europaeus 5P3, and Gluconacetobacter oboediens 174Bp2 (isolated from vinegar). Journal of Bacteriology, 193(10), 2670–2671. 10.1128/JB.00229-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt, D. , Grant, J. R. , Marcu, A. , Sajed, T. , Pon, A. , Liang, Y. , & Wishart, D. S. (2016). PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Research, 44(W1), W16–W21. 10.1093/nar/gkw387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieta, J. G. , Sotolongo, M. , Menéndez, C. , Alfonso, D. , Trujillo, L. E. , Soto, M. , … Hernández, L. (2004). A type II protein secretory pathway required for levansucrase secretion by Gluconacetobacter diazotrophicus . Journal of Bacteriology, 186(15), 5031–5039. 10.1128/JB.186.15.5031-5039.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausmees, N. , Mayer, R. , Weinhouse, H. , Volman, G. , Amikam, D. , Benziman, M. , & Lindberg, M. (2001). Genetic data indicate that proteins containing the GGDEF domain possess diguanylate cyclase activity. FEMS Microbiology Letters, 204(1), 163–167. 10.1111/j.1574-6968.2001.tb10880.x [DOI] [PubMed] [Google Scholar]
- Ausubel M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., & Struhl K. (Eds.) (1992). Current protocols in molecular biology. New York, NY: John Wiley & Sons Incorporated. [Google Scholar]
- Aziz, R. K. , Bartels, D. , Best, A. A. , DeJongh, M. , Disz, T. , Edwards, R. A. , … Zagnitko, O. (2008). The RAST Server: Rapid annotations using subsystems technology. BMC Genomics, 9(1), 75 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma, Y. , Hosoyama, A. , Matsutani, M. , Furuya, N. , Horikawa, H. , Harada, T. , … Shirai, M. (2009). Whole‐genome analyses reveal genetic instability of Acetobacter pasteurianus . Nucleic Acids Research, 37(17), 5768–5783. 10.1093/nar/gkp612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja, F. , Andrés‐Barrao, C. , Ortega Pérez, R. , María Cabello, E. , & Chappuis, M.‐L. (2016). Physiology of Komagataeibacter spp. during acetic acid fermentation In Matsushita K., Toyama H., Tonouchi N. & Okamoto‐Kainuma A. (Eds.), Acetic acid bacteria: Ecology and physiology (pp. 201–221). Tokyo, Japan: Springer. [Google Scholar]
- Bellanger, X. , Payot, S. , Leblond‐Bourget, N. , & Guédon, G. (2014). Conjugative and mobilizable genomic islands in bacteria: Evolution and diversity. FEMS Microbiology Reviews, 38(4), 720–760. 10.1111/1574-6976.12058 [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological), 57, 289–300. [Google Scholar]
- Beppu, T. (1993). Genetic organization of Acetobacter for acetic acid fermentation. Antonie van Leeuwenhoek, 64(2), 121–135. [DOI] [PubMed] [Google Scholar]
- Bertalan, M. , Albano, R. , de Pádua, V. , Rouws, L. , Rojas, C. , Hemerly, A. , … Ferreira, P. C. (2009). Complete genome sequence of the sugarcane nitrogen‐fixing endophyte Gluconacetobacter diazotrophicus Pal5. BMC Genomics, 10(1), 450 10.1186/1471-2164-10-450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt, J. U. , Jakob, F. , Behr, J. , Geissler, A. J. , & Vogel, R. F. (2016). Dissection of exopolysaccharide biosynthesis in Kozakia baliensis . Microbial Cell Factories, 15(1), 170 10.1186/s12934-016-0572-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzuszkiewicz, E. , Gottschalk, G. , Ron, E. , Hacker, J. , & Dobrindt, U. (2009). Adaptation of pathogenic E. coli to various niches: Genome flexibility is the key. Genome Dynamics, 6, 110–125. 10.1159/000235766 [DOI] [PubMed] [Google Scholar]
- Cacicedo, M. L. , Castro, M. C. , Servetas, I. , Bosnea, L. , Boura, K. , Tsafrakidou, P. , … Castro, G. R. (2016). Progress in bacterial cellulose matrices for biotechnological applications. Bioresource Technology, 213, 172–180. 10.1016/j.biortech.2016.02.071 [DOI] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10(1), 421 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, A. P. , Sutton, G. , DePew, J. , Krishnakumar, R. , Choi, Y. , Huang, X. Z. , … Fouts, D. E. (2015). A novel method of consensus pan‐chromosome assembly and large‐scale comparative analysis reveal the highly flexible pan‐genome of Acinetobacter baumannii . Genome Biology, 16(1), 143 10.1186/s13059-015-0701-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla, P. R. , Bajaj, I. B. , Survase, S. A. , & Singhal, R. S. (2009). Microbial cellulose: Fermentative production and applications. Food Technology and Biotechnology, 47(2), 107–124. [Google Scholar]
- Choi, K. R. , & Lee, S. Y. (2016). CRISPR technologies for bacterial systems: Current achievements and future directions. Biotechnology Advances, 34(7), 1180–1209. 10.1016/j.biotechadv.2016.08.002 [DOI] [PubMed] [Google Scholar]
- Cleenwerck, I. , De Vos, P. , & De Vuyst, L. (2010). Phylogeny and differentiation of species of the genus Gluconacetobacter and related taxa based on multilocus sequence analyses of housekeeping genes and reclassification of Acetobacter xylinus subsp. sucrofermentans as Gluconacetobacter sucrofermentans (T. International Journal of Systematic and Evolutionary Microbiology, 60(10), 2277–2283. 10.1099/ijs.0.018465-0 [DOI] [PubMed] [Google Scholar]
- Coucheron, D. H. (1991). An Acetobacter xylinum insertion sequence element associated with inactivation of cellulose production. Journal of Bacteriology, 173(18), 5723–5731. 10.1128/jb.173.18.5723-5731.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coucheron, D. H. (1993). A family of IS1031 elements in the genome of Acetobacter xylinum: Nucleotide sequences and strain distribution. Molecular Microbiology, 9(1), 211–218. 10.1111/j.1365-2958.1993.tb01682.x [DOI] [PubMed] [Google Scholar]
- Couso, R. O. , Ielpi, L. , & Dankert, M. A. (1987). A Xanthan‐gum‐like polysaccharide from Acetobacter xylinum . Journal of General Microbiology, 133(8), 2123–2135. [Google Scholar]
- Cozzarelli, N. R. , Freedberg, W. B. , & Lin, E. C. C. (1968). Genetic control of the l‐α‐glycerophosphate system in Escherichia coli . Journal of Molecular Biology, 31(3), 371–387. 10.1016/0022-2836(68)90415-4 [DOI] [PubMed] [Google Scholar]
- Croucher, N. J. , Coupland, P. G. , Stevenson, A. E. , Callendrello, A. , Bentley, S. D. , & Hanage, W. P. (2014). Diversification of bacterial genome content through distinct mechanisms over different timescales. Nature Communications, 5, 5471 10.1038/ncomms6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja, W. K. , Young, D. J. , Kawecki, M. , & Brown, R. M. Jr (2007). The future prospects of microbial cellulose in biomedical applications. Biomacromolecules, 8(1), 1–12. 10.1021/bm060620d [DOI] [PubMed] [Google Scholar]
- Darmon, E. , & Leach, D. R. F. (2014). Bacterial genome instability. Microbiology and Molecular Biology Reviews, 78(1), 1–39. 10.1128/MMBR.00035-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeraksa, A. , Moonmangmee, S. , Toyama, H. , Yamada, M. , Adachi, O. , & Matsushita, K. (2005). Characterization and spontaneous mutation of a novel gene, polE, involved in pellicle formation in Acetobacter tropicalis SKU1100. Microbiology, 151(12), 4111–4120. 10.1099/mic.0.28350-0 [DOI] [PubMed] [Google Scholar]
- Deloger, M. , El Karoui, M. , & Petit, M.‐A. (2009). A genomic distance based on MUM indicates discontinuity between most bacterial species and genera. Journal of Bacteriology, 191(1), 91–99. 10.1128/JB.01202-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon, B. K. , Laird, M. R. , Shay, J. A. , Winsor, G. L. , Lo, R. , Nizam, F. , … Brinkman, F. S. (2015). IslandViewer 3: More flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Research, 43(W1), W104–W108. 10.1093/nar/gkv401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos, R. A. , Berretta, A. A. , Barud Hda, S. , Ribeiro, S. J. , González‐García, L. N. , Zucchi, T. D. , … Riaño‐Pachón, D. M. (2014). Draft genome sequence of Komagataeibacter rhaeticus strain AF1, a high producer of cellulose, isolated from Kombucha tea. Genome Announcements, 2(4), e00731‐14 10.1128/genomeA.00731-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos, R. A. , Berretta, A. A. , Barud Hda, S. , Ribeiro, S. J. , González‐García, L. N. , Zucchi, T. D. , … Riaño‐Pachón, D. M. (2015). Draft genome sequence of Komagataeibacter intermedius strain AF2, a producer of cellulose, isolated from Kombucha tea. Genome Announcements, 3(6), e01404–e01415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, X. J. , Jia, S. R. , Yang, Y. , & Wang, S. (2011). Genome sequence of Gluconacetobacter sp. strain SXCC‐1, isolated from Chinese vinegar fermentation starter. Journal of Bacteriology, 193(13), 3395–3396. 10.1128/JB.05147-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duardo, F. , Ryngajllo, M. , Jedrzejczak‐Krzepkowska, M. , Bielecki, S. , & Gama, F. M. (2016). Chapter 1 – Taxonomic Review and Microbial Ecology in Bacterial NanoCellulose Fermentation In Gama M., Dourado F., & Bielecki S. (Eds.), Bacterial nanocellulose: From biotechnology to bio‐economy (pp. 1–17). New York, NY: Elsevier. [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, L. , & Catchmark, J. M. (2014). Characterization of water‐soluble exopolysaccharides from Gluconacetobacter xylinus and their impacts on bacterial cellulose crystallization and ribbon assembly. Cellulose, 21(6), 3965–3978. 10.1007/s10570-014-0443-8 [DOI] [Google Scholar]
- Fang, L. , & Catchmark, J. M. (2015). Characterization of cellulose and other exopolysaccharides produced from Gluconacetobacter strains. Carbohydrate Polymers, 115, 663–669. 10.1016/j.carbpol.2014.09.028 [DOI] [PubMed] [Google Scholar]
- Florea, M. , Reeve, B. , Abbott, J. , Freemont, P. S. , & Ellis, T. (2016). Genome sequence and plasmid transformation of the model high‐yield bacterial cellulose producer Gluconacetobacter hansenii ATCC 53582. Scientific Reports, 6, 23635 10.1038/srep23635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara, T. , Komoda, K. , Sakurai, N. , Tajima, K. , Tanaka, I. , & Yao, M. (2013). The c‐di‐GMP recognition mechanism of the PilZ domain of bacterial cellulose synthase subunit A. Biochemical and Biophysical Research Communications, 431(4), 802–807. 10.1016/j.bbrc.2012.12.103 [DOI] [PubMed] [Google Scholar]
- Gama, M. , Dourado, F. , & Bielecki, S. (2016). Bacterial nanocellulose: From biotechnology to bio‐economy. New York, NY: Elsevier; 10.1201/b12936 [DOI] [Google Scholar]
- Grissa, I. , Vergnaud, G. , & Pourcel, C. (2007). CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Research, 35(Web Server issue), W52–W57. 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich, A. , Saveliev, V. , Vyahhi, N. , & Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics, 29(8), 1072–1075. 10.1093/bioinformatics/btt086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyles, C. , & Boerlin, P. (2014). Horizontally transferred genetic elements and their role in pathogenesis of bacterial disease. Veterinary Pathology, 51(2), 328–340. 10.1177/0300985813511131 [DOI] [PubMed] [Google Scholar]
- Hayat, S. , Peters, C. , Shu, N. , Tsirigos, K. D. , & Elofsson, A. (2016). Inclusion of dyad‐repeat pattern improves topology prediction of transmembrane B‐barrel proteins. Bioinformatics, 32(10), 1571–1573. 10.1093/bioinformatics/btw025 [DOI] [PubMed] [Google Scholar]
- Hengge, R. , Gründling, A. , Jenal, U. , Ryan, R. , & Yildiz, F. (2016). Bacterial signal transduction by cyclic di‐GMP and other nucleotide second messengers. Journal of Bacteriology, 198(1), 15–26. 10.1128/JB.00331-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández‐Montalvo, V. , Martínez, A. , Hernández‐Chavez, G. , Bolivar, F. , Valle, F. , & Gosset, G. (2003). Expression of galP and glk in a Escherichia coli PTS mutant restores glucose transport and increases glycolytic flux to fermentation products. Biotechnology and Bioengineering, 83(6), 687–694. 10.1002/bit.10702 [DOI] [PubMed] [Google Scholar]
- Hestrin, S. , & Schramm, M. (1954). Synthesis of cellulose by Acetobacter xylinum. II. Preparation of freeze‐dried cells capable of polymerizing glucose to cellulose. The Biochemical Journal, 58(2), 345–352. 10.1042/bj0580345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, S. Q. , Gao, Y. G. , Tajima, K. , Sunagawa, N. , Zhou, Y. , Kawano, S. , … Yao, M. (2010). Structure of bacterial cellulose synthase subunit D octamer with four inner passageways. Proceedings of the National Academy of Sciences of the United States of America, 107(42), 17957–17961. 10.1073/pnas.1000601107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G. L. , Locascio, P. F. , Land, M. L. , Larimer, F. W. , & Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11(1), 119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino, T. , Suzuki, R. , Tanaka, N. , Kosako, Y. , Ohkuma, M. , Komagata, K. , & Uchimura, T. (2012). Gluconacetobacter kakiaceti sp. nov., an acetic acid bacterium isolated from a traditional Japanese fruit vinegar. International Journal of Systematic and Evolutionary Microbiology, 62(7), 1465–1469. 10.1099/ijs.0.031773-0 [DOI] [PubMed] [Google Scholar]
- Illeghems, K. , De Vuyst, L. , & Weckx, S. (2013). Complete genome sequence and comparative analysis of Acetobacter pasteurianus 386B, a strain well‐adapted to the cocoa bean fermentation ecosystem. BMC Genomics, 14(1), 526 10.1186/1471-2164-14-526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida, T. , Sugano, Y. , Nakai, T. , & Shoda, M. (2002). Effects of acetan on production of bacterial cellulose by Acetobacter xylinum . Bioscience, Biotechnology, and Biochemistry, 66(8), 1677–1681. 10.1271/bbb.66.1677 [DOI] [PubMed] [Google Scholar]
- Ishida, T. , Sugano, Y. , & Shoda, M. (2002). Novel glycosyltransferase genes involved in the acetan biosynthesis of Acetobacter xylinum . Biochemical and Biophysical Research Communications, 295(2), 230–235. 10.1016/S0006-291X(02)00663-0 [DOI] [PubMed] [Google Scholar]
- Iversen, T. , Standal, R. , Pedersen, T. , & Coucheron, D. H. (1994). IS1032 from Acetobacter xylinum, a new mobile insertion sequence. Plasmid, 32(1), 46–54. 10.1006/plas.1994.1043 [DOI] [PubMed] [Google Scholar]
- Iyer, P. R. , Catchmark, J. , Brown, N. R. , & Tien, M. (2011). Biochemical localization of a protein involved in synthesis of Gluconacetobacter hansenii cellulose. Cellulose, 18(3), 739–747. 10.1007/s10570-011-9504-4 [DOI] [Google Scholar]
- Iyer, P. R. , Geib, S. M. , Catchmark, J. , Kao, T. H. , & Tien, M. (2010). Genome sequence of a cellulose‐producing bacterium, Gluconacetobacter hansenii ATCC 23769. Journal of Bacteriology, 192(16), 4256 10.1128/JB.00588-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, R. W. , Vinatzer, B. , Arnold, D. L. , Dorus, S. , & Murillo, J. (2011). The influence of the accessory genome on bacterial pathogen evolution. Mobile Genetic Elements, 1(1), 55–65. 10.4161/mge.1.1.16432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahreis, K. , Pimentel‐Schmitt, E. F. , Brückner, R. , & Titgemeyer, F. (2008). Ins and outs of glucose transport systems in eubacteria. FEMS Microbiology Reviews, 32(6), 891–907. 10.1111/j.1574-6976.2008.00125.x [DOI] [PubMed] [Google Scholar]
- Jedrzejczak‐Krzepkowska, M. , Kubiak, K. , Ludwicka, K. , & Bielecki, S. (2016). Chapter 2 – Bacterial NanoCellulose Synthesis, Recent Findings In Gama M., Dourado F., & Bielecki S. (Eds.), Bacterial nanocellulose: From biotechnology to bio‐economy (pp. 19–46). New York, NY: Elsevier; 10.1016/B978-0-444-63458-0.00002-0 [DOI] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H. Y. , Fraser, M. , Li, W. , McAnulla, C. , … Hunter, S. (2014). InterProScan 5: Genome‐scale protein function classification. Bioinformatics, 30(9), 1236–1240. 10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K. , & Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshk, S. , & Sameshima, K. (2005). Evaluation of different carbon sources for bacterial cellulose production. African Journal of Biotechnology, 4(6), 478–482. [Google Scholar]
- Kolbe, D. L. , & Eddy, S. R. (2011). Fast filtering for RNA homology search. Bioinformatics, 27(22), 3102–3109. 10.1093/bioinformatics/btr545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann, H. , Duboc, P. , Marison, I. , & von Stockar, U. (2003). Influence of nutritional factors on the nature, yield, and composition of exopolysaccharides produced by Gluconacetobacter xylinus I‐2281. Applied and Environmental Microbiology, 69(10), 6091–6098. 10.1128/AEM.69.10.6091-6098.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystynowicz, A. , Czaja, W. , Wiktorowska‐Jezierska, A. , Gonçalves‐Miśkiewicz, M. , Turkiewicz, M. , & Bielecki, S. (2002). Factors affecting the yield and properties of bacterial cellulose. Journal of Industrial Microbiology & Biotechnology, 29(4), 189–195. 10.1038/sj.jim.7000303 [DOI] [PubMed] [Google Scholar]
- Krystynowicz, A. , Koziołkiewicz, M. , Wiktorowska‐Jezierska, A. , Bielecki, S. , Klemenska, E. , Masny, A. , & Płucienniczak, A. (2005). Molecular basis of cellulose biosynthesis disappearance in submerged culture of Acetobacter xylinum . Acta Biochimica Polonica, 52(3), 691. [PubMed] [Google Scholar]
- Kubiak, K. , Jedrzejczak‐Krzepkowska, M. , Ludwicka, K. , & Bielecki, S. (2016). Chapter 3 – Molecular Control Over BNC Biosynthesis In Gama M., Dourado F., & Bielecki S. (Eds.), Bacterial nanocellulose: From biotechnology to bio‐economy (pp. 47–58). New York, NY: Elsevier; 10.1016/B978-0-444-63458-0.00003-2 [DOI] [Google Scholar]
- Kubiak, K. , Kurzawa, M. , Jędrzejczak‐Krzepkowska, M. , Ludwicka, K. , Krawczyk, M. , Migdalski, A. , … Bielecki, S. (2014). Complete genome sequence of Gluconacetobacter xylinus E25 strain‐Valuable and effective producer of bacterial nanocellulose. Journal of Biotechnology, 176(1), 18–19. 10.1016/j.jbiotec.2014.02.006 [DOI] [PubMed] [Google Scholar]
- Kung, V. L. , Ozer, E. A. , & Hauser, A. R. (2010). The accessory genome of Pseudomonas aeruginosa . Microbiology and Molecular Biology Reviews, 74(4), 621–641. 10.1128/MMBR.00027-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, C. H. , Chen, J.‐H. , Liou, B.‐K. , & Lee, C.‐K. (2016). Utilization of acetate buffer to improve bacterial cellulose production by Gluconacetobacter xylinus . Food Hydrocolloids, 53, 98–103. 10.1016/j.foodhyd.2014.12.034 [DOI] [Google Scholar]
- Kuo, C. H. , Teng, H. Y. , & Lee, C. K. (2015). Knock‐out of glucose dehydrogenase gene in Gluconacetobacter xylinus for bacterial cellulose production enhancement. Biotechnology and Bioprocess Engineering, 20(1), 18–25. 10.1007/s12257-014-0316-x [DOI] [Google Scholar]
- Laslett, D. , & Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Research, 32(1), 11–16. 10.1093/nar/gkh152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner, M. , Findeiss, S. , Steiner, L. , Marz, M. , Stadler, P. F. , & Prohaska, S. J. (2011). Proteinortho: Detection of (co‐)orthologs in large‐scale analysis. BMC Bioinformatics, 12, 124 10.1186/1471-2105-12-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limoli, D. H. , Jones, C. J. , & Wozniak, D. J. (2015). Bacterial extracellular polysaccharides in biofilm formation and function. Microbiology Spectrum, 3(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.‐P. , Loira Calvar, I. , Catchmark, J. M. , Liu, J.‐R. , Demirci, A. , & Cheng, K.‐C. (2013). Biosynthesis, production and applications of bacterial cellulose. Cellulose, 20(5), 2191–2219. 10.1007/s10570-013-9994-3 [DOI] [Google Scholar]
- MacCormick, C. A. , Harris, J. E. , Gunning, A. P. , & Morris, V. J. (1993). Characterization of a variant of the polysaccharide acetan produced by a mutant of Acetobacter xylinum strain CR1/4. Journal of Applied Microbiology, 74(2), 196–199. [DOI] [PubMed] [Google Scholar]
- Makarova, K. S. , Zhang, F. , & Koonin, E. V. (2017). SnapShot: Class 1 CRISPR‐Cas systems. Cell, 168(5), 946.e1. [DOI] [PubMed] [Google Scholar]
- Masaoka, S. , Ohe, T. , & Sakota, N. (1993). Production of cellulose from glucose by Acetobacter xylinum . Journal of Fermentation and Bioengineering, 75(1), 18–22. 10.1016/0922-338X(93)90171-4 [DOI] [Google Scholar]
- Matsutani, M. , Ito, K. , Azuma, Y. , Ogino, H. , Shirai, M. , Yakushi, T. , & Matsushita, K. (2015). Adaptive mutation related to cellulose producibility in Komagataeibacter medellinensis (Gluconacetobacter xylinus) NBRC 3288. Applied Microbiology and Biotechnology, 99(17), 7229–7240. 10.1007/s00253-015-6598-x [DOI] [PubMed] [Google Scholar]
- Mikkelsen, D. , Flanagan, B. M. , Dykes, G. A. , & Gidley, M. J. (2009). Influence of different carbon sources on bacterial cellulose production by Gluconacetobacter xylinus strain ATCC 53524. Journal of Applied Microbiology, 107(2), 576–583. 10.1111/j.1365-2672.2009.04226.x [DOI] [PubMed] [Google Scholar]
- Morgan, J. L. , McNamara, J. T. , Fischer, M. , Rich, J. , Chen, H. M. , Withers, S. G. , & Zimmer, J. (2016). Observing cellulose biosynthesis and membrane translocation in crystallo. Nature, 531(7594), 329–334. 10.1038/nature16966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, J. L. W. , Strumillo, J. , & Zimmer, J. (2012). Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature, 493(7431), 181–186. 10.1038/nature11744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, V. T. , Flanagan, B. , Gidley, M. J. , & Dykes, G. A. (2008). Characterization of cellulose production by a Gluconacetobacter xylinus strain from Kombucha. Current Microbiology, 57(5), 449–453. 10.1007/s00284-008-9228-3 [DOI] [PubMed] [Google Scholar]
- Nurk, S. , Bankevich, A. , Antipov, D. , Gurevich, A. A. , Korobeynikov, A. , Lapidus, A. , … Pevzner, P. A. (2013). Assembling single‐cell genomes and mini‐metagenomes from chimeric MDA products. Journal of Computational Biology, 20(10), 714–737. 10.1089/cmb.2013.0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino, H. , Azuma, Y. , Hosoyama, A. , Nakazawa, H. , Matsutani, M. , Hasegawa, A. , … Shirai, M. (2011). Complete genome sequence of NBRC 3288, a unique cellulose‐nonproducing strain of Gluconacetobacter xylinus isolated from vinegar. Journal of Bacteriology, 193(24), 6997–6998. 10.1128/JB.06158-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, S. , Mehta, K. , & Brown, R. M. (2016a). Complete genome sequence of a Gluconacetobacter hansenii ATCC 23769 isolate, AY201, producer of bacterial cellulose and important model organism for the study of cellulose biosynthesis. Genome Announcements, 4(4), 15–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, S. , Mehta, K. , & Brown, R. M. (2016b). Complete genome sequence of Gluconacetobacter hansenii strain NQ5 (ATCC 53582), an efficient producer of bacterial cellulose. Genome Announcements, 4(4), e00785‐16 10.1128/genomeA.00785-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, S. , Santos, R. , Ebels, M. , Bordbar, D. , & Brown, R. M. Jr (2017a). Complete genome sequence of Komagataeibacter hansenii LMG 23726(T). Genome Announcements, 5(15), e00168‐17 10.1128/genomeA.00168-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, S. , Santos, R. , Ebels, M. , Bordbar, D. , & Brown, R. M. Jr (2017b). Complete genome sequence of Komagataeibacter hansenii Strain HUM‐1. Genome Announcements, 5(15), e00167‐17 10.1128/genomeA.00167-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, S. , Santos, R. , Ebels, M. , Bordbar, D. , & Brown, R. M. Jr (2017c). Complete genome sequence of Komagataeibacter hansenii strain SC‐3B. Genome Announcements, 5(15), e00169‐17 10.1128/genomeA.00169-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povolotsky, T. L. , & Hengge, R. (2016). Genome‐based comparison of cyclic di‐GMP signaling in pathogenic and commensal Escherichia coli strains. Journal of Bacteriology, 198(1), 111–126. 10.1128/JB.00520-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling, U. (2002). Molecular biology of cellulose production in bacteria. Research in Microbiology, 153(4), 205–212. 10.1016/S0923-2508(02)01316-5 [DOI] [PubMed] [Google Scholar]
- Römling, U. (2012). Cyclic di‐GMP, an established secondary messenger still speeding up. Environmental Microbiology, 14(8), 1817–1829. 10.1111/j.1462-2920.2011.02617.x [DOI] [PubMed] [Google Scholar]
- Römling, U. , & Galperin, M. Y. (2015). Bacterial cellulose biosynthesis: Diversity of operons, subunits, products, and functions. Trends in Microbiology, 23(9), 545–557. 10.1016/j.tim.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling, U. , Galperin, M. Y. , & Gomelsky, M. (2013). Cyclic di‐GMP: The first 25 years of a universal bacterial second messenger. Microbiology and Molecular Biology Reviews, 77(1), 1–52. 10.1128/MMBR.00043-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, P. , Aloni, Y. , Weinhouse, C. , Michaeli, D. , Weinberger‐Ohana, P. , Meyer, R. , & Benziman, M. (1985). An unusual guanyl oligonucleotide regulates cellulose synthesis in Acetobacter xylinum . FEBS Letters, 186(2), 191–196. 10.1016/0014-5793(85)80706-7 [DOI] [PubMed] [Google Scholar]
- Ross, P. , Weinhouse, H. , Aloni, Y. , Michaeli, D. , Weinberger‐Ohana, P. , Mayer, R. , … Benziman, M. (1987). Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature, 325(6101), 279–281. 10.1038/325279a0 [DOI] [PubMed] [Google Scholar]
- Saier, M. H. , & Reizer, J. (1992). Proposed uniform nomenclature for the proteins and protein domains of the bacterial phosphoenolpyruvate: Sugar phosphotransferase system. Journal of Bacteriology, 174(5), 1433–1438. 10.1128/jb.174.5.1433-1438.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, I. M. , & Brown, R. M. (1995). Identification of a second cellulose synthase gene (acsAII) in Acetobacter xylinum . Journal of Bacteriology, 177(18), 5276–5283. 10.1128/jb.177.18.5276-5283.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, I. M. , Kudlicka, K. , Okuda, K. , & Brown, R. M. Jr (1994). Characterization of genes in the cellulose‐synthesizing operon (acs operon) of Acetobacter xylinum: Implications for cellulose crystallization. Journal of Bacteriology, 176(18), 5735–5752. 10.1128/jb.176.18.5735-5752.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäper, S. , Krol, E. , Skotnicka, D. , Kaever, V. , Hilker, R. , Søgaard‐Andersen, L. , & Becker, A. (2015). Cyclic Di‐GMP regulates multiple cellular functions in the symbiotic Alphaproteobacterium Sinorhizobium meliloti . Journal of Bacteriology, 198(3), 521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, J. , Sieber, V. , & Rehm, B. (2015). Bacterial exopolysaccharides: Biosynthesis pathways and engineering strategies. Frontiers in Microbiology, 6(MAY). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics, 30(14), 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Simm, R. , Morr, M. , Kader, A. , Nimtz, M. , & Römling, U. (2004). GGDEF and EAL domains inversely regulate cyclic di‐GMP levels and transition from sessility to motility. Molecular Microbiology, 53(4), 1123–1134. 10.1111/j.1365-2958.2004.04206.x [DOI] [PubMed] [Google Scholar]
- Sokollek, S. J. , Hertel, C. , & Hammes, W. P. (1998). Cultivation and preservation of vinegar bacteria. Journal of Biotechnology, 60(3), 195–206. 10.1016/S0168-1656(98)00014-5 [DOI] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30(9), 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standal, R. , Iversen, T. G. , Coucheron, D. H. , Fjaervik, E. , Blatny, J. M. , & Valla, S. (1994). A new gene required for cellulose production and a gene encoding cellulolytic activity in Acetobacter xylinum are colocalized with the bcs operon. Journal of Bacteriology, 176(3), 665–672. 10.1128/jb.176.3.665-672.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwanposri, A. , Yukphan, P. , Yamada, Y. , & Ochaikul, D. (2013). Identification and biocellulose production of Gluconacetobacter strains isolated from tropical fruits in Thailand. Maejo International Journal of Science and Technology, 7(1), 70–82. [Google Scholar]
- Takemura, H. , Horinouchi, S. , & Beppu, T. (1991). Novel insertion sequence IS1380 from Acetobacter pasteurianus is involved in loss of ethanol‐oxidizing ability. Journal of Bacteriology, 173(22), 7070–7076. 10.1128/jb.173.22.7070-7076.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal, R. , Wong, H. C. , Calhoon, R. , Gelfand, D. , Fear, A. L. , Volman, G. , … Benziman, M. (1998). Three cdg operons control cellular turnover of cyclic di‐GMP in Acetobacter xylinum: Genetic organization and occurrence of conserved domains in isoenzymes. Journal of Bacteriology, 180(17), 4416–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30(12), 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiboni, M. , Grzybowski, A. , Passos, M. , Barison, A. , Morais Lião, L. , Ramos Campos, F. , … Domingos Fontana, J. (2012). The use of dyed bacterial cellulose to monitor cellulase complex activity. Cellulose, 19(6), 1867–1877. 10.1007/s10570-012-9787-0 [DOI] [Google Scholar]
- Toyosaki, H. , Naritomi, T. , Seto, A. , Matsuoka, M. , Tsuchida, T. , & Yoshinaga, F. (1995). Screening of bacterial cellulose‐producing Acetobacter strains suitable for agitated culture. Bioscience, Biotechnology, and Biochemistry, 59(8), 1498–1502. 10.1271/bbb.59.1498 [DOI] [Google Scholar]
- Treangen, T. J. , Ondov, B. D. , Koren, S. , & Phillippy, A. M. (2014). Rapid core‐genome alignment and visualization for thousands of microbial genomes. Genome Biology, 15, 524 10.1186/s13059-014-0524-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda, Y. , Hirano, A. , Ishibashi, M. , Akiyama, H. , Onizuka, T. , Ikeuchi, M. , & Inoue, Y. (1999). Cloning of cellulose synthase genes from Acetobacter xylinum JCM 7664: Implication of a novel set of cellulose synthase genes. DNA Research, 6(2), 109–115. 10.1093/dnares/6.2.109 [DOI] [PubMed] [Google Scholar]
- Valentini, M. , & Filloux, A. (2016). Biofilms and cyclic di‐GMP (c‐di‐GMP) signaling: Lessons from Pseudomonas aeruginosa and other bacteria. Journal of Biological Chemistry, 291(24), 12547–12555. 10.1074/jbc.R115.711507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valepyn, E. , Berezina, N. , & Paquot, M. (2012). Optimization of production and preliminary characterization of new exopolysaccharides from Gluconacetobacter hansenii LMG1524. Advances in Microbiology, 2, 488–496. 10.4236/aim.2012.24062 [DOI] [Google Scholar]
- Valera, M. J. , Poehlein, A. , Torija, M. J. , Haack, F. S. , Daniel, R. , Streit, W. R. , … Mas, A. (2015). Draft genome sequence of Komagataeibacter europaeus CECT 8546, a cellulose‐producing strain of vinegar elaborated by the traditional method. Genome Announcements, 3(5), e01231–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera, M. J. , Torija, M. J. , Mas, A. , & Mateo, E. (2015). Cellulose production and cellulose synthase gene detection in acetic acid bacteria. Applied Microbiology and Biotechnology, 99(3), 1349–1361. 10.1007/s00253-014-6198-1 [DOI] [PubMed] [Google Scholar]
- Varani, A. M. , Siguier, P. , Gourbeyre, E. , Charneau, V. , & Chandler, M. (2011). ISsaga is an ensemble of web‐based methods for high throughput identification and semi‐automatic annotation of insertion sequences in prokaryotic genomes. Genome Biology, 12(3), R30 10.1186/gb-2011-12-3-r30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco‐Bedrán, H. , & López‐Isunza, F. (2007). The unified metabolism of Gluconacetobacter entanii in continuous and batch processes. Process Biochemistry, 42(8), 1180–1190. 10.1016/j.procbio.2007.05.017 [DOI] [Google Scholar]
- Wang, S.‐S. , Han, Y.‐H. , Ye, Y.‐X. , Shi, X.‐X. , Xiang, P. , Chen, D.‐L. , & Li, M. (2017). Physicochemical characterization of high‐quality bacterial cellulose produced by Komagataeibacter sp. strain W1 and identification of the associated genes in bacterial cellulose production. RSC Advances, 7(71), 45145–45155. 10.1039/C7RA08391B [DOI] [Google Scholar]
- Wang, B. , Shao, Y. , Chen, T. , Chen, W. , & Chen, F. (2016). Global insights into acetic acid resistance mechanisms and genetic stability of Acetobacter pasteurianus strains by comparative genomics. Scientific Reports, 5(1), 18330 10.1038/srep18330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenborn, D. L. , Wittekindtn, N. , & Larsonsii, T. J. (1992). Structure and regulation of the gZpFK operon encoding glycerol. Biochemistry, 267(9), 6122–6131. [PubMed] [Google Scholar]
- Williams, W. S. , & Cannon, R. E. (1989). Alternative environmental roles for cellulose produced by Acetobacter xylinum . Applied and Environmental Microbiology, 55(10), 2448–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, H. C. , Fear, A. L. , Calhoon, R. D. , Eichinger, G. H. , Mayer, R. , Amikam, D. , … Emerick, A. W. (1990). Genetic organization of the cellulose synthase operon in Acetobacter xylinum . Proceedings of the National Academy of Sciences of the United States of America, 87(20), 8130–8134. 10.1073/pnas.87.20.8130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. M. , & Liu, R. H. (2012). Thin stillage supplementation greatly enhances bacterial cellulose production by Gluconacetobacter xylinus . Carbohydrate Polymers, 90(1), 116–121. 10.1016/j.carbpol.2012.05.003 [DOI] [PubMed] [Google Scholar]
- Wylie, J. L. , & Worobec, E. A. (1995). The OprB porin plays a central role in carbohydrate uptake in Pseudomonas aeruginosa . Journal of Bacteriology, 177(11), 3021–3026. 10.1128/jb.177.11.3021-3026.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, Y. (2014). Transfer of Gluconacetobacter kakiaceti, Gluconacetobacter medellinensis and Gluconacetobacter maltaceti to the genus Komagataeibacter as Komagataeibacter kakiaceti comb. nov., Komagataeibacter medellinensis comb. nov. and Komagataeibacter maltaceti comb. International Journal of Systematic and Evolutionary Microbiology, 64(5), 1670–1672. 10.1099/ijs.0.054494-0 [DOI] [PubMed] [Google Scholar]
- Yamada, Y. (2016). Systematics of acetic acid bacteria In Matsushita K., Toyama H., Tonouchi N., & Okamoto‐Kainuma A. (Eds.), Acetic acid bacteria: Ecology and physiology (pp. 1–50). Tokyo, Japan: Springer. [Google Scholar]
- Yamada, Y. , Yukphan, P. , Lan Vu, H. T. , Muramatsu, Y. , Ochaikul, D. , Tanasupawat, S. , & Nakagawa, Y. (2012). Description of Komagataeibacter gen. nov., with proposals of new combinations (Acetobacteraceae). The Journal of General and Applied Microbiology, 58(5), 397–404. 10.2323/jgam.58.397 [DOI] [PubMed] [Google Scholar]
- Yamada, Y. , Yukphan, P. , Vu, H. T. L. , Yuki, M. , Duangjai, O. , & Yasuyoshi, N. (2012). Subdivision of the genus Gluconacetobacter Yamada, Hoshino and Ishikawa 1998: The proposal of Komagatabacter gen. nov., for strains accommodated to the Gluconacetobacter xylinus group in the α‐Proteobacteria. Annals of Microbiology, 62(2), 849–859. 10.1007/s13213-011-0288-4 [DOI] [Google Scholar]
- Yoshinaga, F. , Tonouchi, N. , & Watanabe, K. (1997). Research progress in production of bacterial cellulose by aeration and agitation culture and its application as a new industrial material. Bioscience, Biotechnology, and Biochemistry, 61(2), 219–224. 10.1271/bbb.61.219 [DOI] [Google Scholar]
- Zeng, M. , Laromaine, A. , & Roig, A. (2014). Bacterial cellulose films: Influence of bacterial strain and drying route on film properties. Cellulose, 21(6), 4455–4469. 10.1007/s10570-014-0408-y [DOI] [Google Scholar]
- Zeng, G. , Ye, S. , & Larson, T. J. (1996). Repressor for the sn‐glycerol 3‐phosphate regulon of Escherichia coli K‐12: Primary structure and identification of the DNA‐binding domain. Journal of Bacteriology, 178(24), 7080–7089. 10.1128/jb.178.24.7080-7089.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Poehlein, A. , Hollensteiner, J. , & Daniel, R. (2018). Draft genome sequence of Komagataeibacter maltaceti LMG 1529T, a vinegar‐producing acetic acid bacterium isolated from malt vinegar brewery acetifiers. Genome Announcements, 6(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Xu, X. , Chen, X. , Yuan, F. , Sun, B. , Xu, Y. , … Sun, D. (2017). Complete genome sequence of the cellulose‐producing strain Komagataeibacter nataicola RZS01. Scientific Reports, 7(1), 4431 10.1038/s41598-017-04589-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, N. , Oh, W. , Trybul, D. , Thrasher, K. S. , Kingsbury, T. J. , & Larson, T. J. (1994). Characterization of the interaction of the glp repressor of Escherichia coli K‐12 with single and tandem glp operator variants. Journal of Bacteriology, 176(8), 2393–2397. 10.1128/jb.176.8.2393-2397.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DNA sequences of the assembled genomes are available at NCBI under the BioProject accession: PRJNA339514, PRJNA339679, PRJNA339678. Additional data sets generated in this work are provided in the Supporting Information Data S1 of this article.