

Abstract
G protein-gated, inwardly-rectifying, potassium (GIRK) channels are important regulators of cellular excitability throughout the body. GIRK channels are heterotetrameric and homotetrameric combinations of the Kir3.1-4 (GIRK1-4) subunits. Different subunit combinations are expressed throughout the central nervous system (CNS) and the periphery, and most of these combinations contain a GIRK1 subunit. For example, the predominance of GIRK channels in the CNS are comprised of GIRK1 and GIRK2 subunits, while the GIRK channels in cardiac atrial myocytes are made up mostly of GIRK1 and GIRK4 subunits. Although the vast majority of GIRK channels contain a GIRK1 subunit, discrete populations of cells that express non-GIRK1-containing GIRK (non-GIRK1/X) channels do exist. For instance, dopaminergic neurons in the ventral tegmental area of the brain, associated with addiction and reward, do not express the GIRK1 subunit. Targeting these non-GIRK1/X channels with subunit-selective pharmacological probes could lead to important insights into how GIRK channels are involved in reward and addiction. Such insights may, in turn, reveal therapeutic opportunities for the treatment or prevention of addiction. Previously, our laboratory discovered small molecules that can specifically modulate the activity of GIRK1-containing GIRK channels. However, efforts to generate compounds active on non-GIRK1/X channels from these scaffolds have been unsuccessful. Recently, ivermectin was shown to modulate non-GIRK1/X channels, and historically, ivermectin is known to modulate a wide variety of neuronal channels and receptors. Further, ivermectin is a complex natural product, which makes it a challenging starting point for development of more selective, effective, and potent compounds. Thus, while ivermectin provides proof-of-concept as a non-GIRK1/X channel activator, it is of limited utility. Therefore, we sought to discover a synthetic small molecule that would serve as a starting point for the development of non-GIRK1/X channel modulators. To accomplish this, we used a high-throughput thallium flux assay to screen a 100,000-compound library in search of activators of homomeric GIRK2 channels. Using this approach, we discovered VU0529331, the first synthetic small molecule reported to activate non-GIRK1/X channels, to our knowledge. This discovery represents the first step towards developing potent and selective non-GIRK1/x channel probes. Such molecules will help elucidate the role of GIRK channels in addiction, potentially establishing a foundation for future development of therapies utilizing targeted GIRK channel modulation.
Keywords: GIRK channel, Kir3, GIRK2, ion channel, modulator, activator, small molecule, high-throughput screening, thallium flux assay, whole-cell patch-clamp electrophysiology
Graphical Abstract

INTRODUCTION
Opioids are some of the most effective pain medications, but they are also extremely addictive. The nationwide opioid epidemic claims tens of thousands of lives each year as people addicted to opioids overdose.1 The difficulty in combating this issue is demonstrated by the increasing number of overdose deaths, which are tied to a dramatic increase in opioid prescriptions for treatment of acute and chronic pain over the last decade.2 Research into understanding the underlying mechanisms of opioid addiction has also become increasingly intense, aiming to illuminate new paths for the prevention and treatment of opioid addiction. One such area of focus involves a family of inwardly-rectifying potassium (K+) channels, called GIRKs.3–5
The G protein-gated, inwardly-rectifying, K+ channel (GIRK) family, also known as the Kir3 family, is part of the larger inwardly-rectifying potassium channel (Kir) family. GIRK channels function by inhibiting cellular excitability though membrane hyperpolarization. Numerous studies have revealed the importance of GIRK channel function in neural processes6 such as analgesia,7 reward,8–13 anxiety,14 memory,15 respiration,16 and seizures17 as well as in diverse functions including embryonic development,18,19 regulation of heart rate,20,21 and hormone secretion.22,23 The GIRK channel family is comprised of GIRK1, 2, 3, and 4 (Kir3.1, 3.2, 3.3, and 3.4) subunits that are encoded by the KCNJ3, 6, 9, and 5 genes, respectively. These individual subunits form homotetrameric and heterotetrameric channels. Differential GIRK channel expression between various human organs and cell types has been identified.4 GIRK1 or GIRK3 homotetrameric channels are not believed to be expressed in vivo,6,24,25 while GIRK1-containing (GIRK1/X) heterotetrameric channels are believed to be most commonly found GIRK channels throughout the body. GIRK1, GIRK2, and GIRK3 subunits are widely expressed throughout the central nervous system25,26 (CNS) while GIRK1 and GIRK4 subunits are expressed predominantly throughout peripheral organs, i.e. in the heart24,27 and pancreas.28 Wherever the GIRK1 subunit is expressed, GIRK1/X channels are therefore expressed; however, non-GIRK1/X channels have been reported in a few discrete brain regions.26 One example of specific GIRK expression is in dopaminergic (DA) neurons of the ventral tegmental area (VTA), which express only GIRK2 and GIRK3 subunits.29 Here, GIRK2 homotetrameric and GIRK2/3 heterotetrameric channels may play a critical role in regulating addiction and reward circuitry.8–13,30 To date, research has shown that the activity of addictive substances like ethanol9 and methamphetamine8 is affected by GIRK3 expression in DA neurons of the VTA. These data raise the intriguing possibility that modulation of non-GIRK1/X channels in the VTA may decrease DA neuron activity and provide a mechanism for decreasing drug abuse. While our knowledge of GIRK channel involvement in regulating key neuronal circuitry in reward and addiction is still in its infancy, improving our understanding of the role of GIRK channels in drug abuse may provide new targets for development of medicines to help combat the increasingly deadly opioid epidemic.
Modulation of GIRK channel activity by a number of broadly-active ligands has been studied for many years.31 Phosphatidylinositol 4,5-bisphosphate (PIP2) is necessary for channels to open while βγ-subunits of inhibitory G proteins (Gi/o) are major endogenous intracellular channel activators. Gi/oβγ-subunits of Gi/o proteins bound to Gi/o-coupled G protein-coupled receptors (GPCR) are activated after ligand binding, and these Gi/oβγ-subunits can activate GIRK channels.32,33 Gi/o protein-coupled GPCRs include an array of receptors that are important in normal physiology and are the targets of a large number of drug-based therapies, namely subtypes of opioid, muscarinic, serotonergic, and dopaminergic receptors. GIRK channels have been shown to be targets of alcohol action34, and a discrete alcohol binding pocket on these channels has been discovered and characterized.35–37 Intracellular sodium ions (Na+) have also been shown to modulate GIRK channel activity.38 A crystal structure, solved by Wharton et al., 39 with PIP2, Gi/oβγ-subunit, and Na+ bound to homomeric GIRK2 revealed the binding pockets of these ligands. The activity of GIRK channels has also been demonstrated to be sensitive to cholesterol,40–42 inhibited by Gα subunits of Gi/o-coupled GPCRs,43 decreased due to PIP2 degradation by phospholipase C (PLC) after Gq-coupled GPCR activation,44 increased with phosphorylation by protein kinase A (PKA),45,46 and decreased by phosphorylation by protein kinase C (PKC).47 While GIRK channel modulation by this broad array of ligands and mechanisms has been thoroughly studied, none of these entities are selective modulators of GIRK channels.
Recently, our lab has discovered and characterized a number of small-molecule activators of GIRK channels. While these modulators are proving highly useful as probes to investigate a variety of GIRK-expressing systems, i.e. the CNS,14,17,48–50 they show extraordinary selectivity for GIRK1/X channels.51,52 Two critical amino acids in GIRK1 govern the ability of these molecules to activate only GIRK1/X channels,14 and all efforts to generate analogs that can modulate non-GIRK1/X channels have failed. To date, we have generated and tested nearly 800 compounds based on scaffolds discovered by screening >250,000 compounds on GIRK1/2-expressing HEK293 cells, and the only non-GIRK1/X-active compound we identified was abamectin, a compound closely related to ivermectin, recently described to activate GIRK channels by Su et al.53 and Chen et al.54 Abamectin and ivermectin belong to the avermectin family of compounds and have been utilized as insecticides55 and antihelminthic medications56 for decades. Although avermectins provide proof-of-concept for pharmacologic probes that activate non-GIRK1/X channels, they are far from ideal. Perhaps most damaging, avermectins are active on a wide variety of ion channels, including many Cys-loop, ligand-gated, ion channels, namely the glycine receptor chloride channel (GlyR),59 the γ-aminobutyric acid A receptor (GABAAR),60,61 the nicotinic acetylcholine receptors (nAChR),62 and the insect glutamate-gated chloride channels (GluR).63,64 Further, ivermectin has been demonstrated to activate P2X purinoceptor 4 (P2X4R).57,58 Several of these avermectin targets are co-expressed in the same neuronal populations as GIRK channels. This severely limits avermectins’ utility as probes to study the role of GIRK channels in the VTA and other brain and peripheral tissues. These issues of poor selectivity are compounded by the compounds’ high molecular weight (MW > 850 gmol−1) and relatively low potency. Further, the avermectins are complex natural products,65 making synthesis and derivatization considerably more difficult than typical drug-like small molecules. Even in light of the failures of ourselves and others53,54 to discover synthetically tractable small-molecule modulators of non-GIRK1/X channels, we conducted a ~100,000 compound screen exclusively targeting homomeric GIRK2 channels. Herein, we describe the discovery and characterization of VU0529331, a synthetic small molecule capable of activating non-GIRK1/X channels.
RESULTS AND DISCUSSION
In an attempt to discover small-molecule modulators of non-GIRK1/X channels, we performed a high-throughput screen (HTS) of a collection of ~100,000 compounds using a fluorescence-based thallium (Tl+) flux assay (Figure 1). This screen was conducted on HEK293 cells engineered to express GIRK2 and the neuropeptide Y receptor type 4 (NPY4R), a Gi/o-coupled GPCR capable of increasing the activity of GIRK2 channels in the presence of its agonist, human pancreatic polypeptide (hPP). Henceforth, this HEK293 cell line expressing GIRK2 and NPY4R will be referred to as G2Y4 cells. We confirmed GIRK2 expression by western blot (Figure 2, full membrane shown in Supporting Information, Figure S1) and NPY4R expression using a functional assay, through which we confirmed hPP activity in G2Y4 cells using Tl+ flux, similar to the experiment reported in Figure 4b. During the HTS, we screened a partially-activated GIRK2 channel using an EC30 concentration of hPP. We chose this approach in order to enable discovery of inhibitors, activators, or potentiators of GIRK2 channels.
Figure 1.
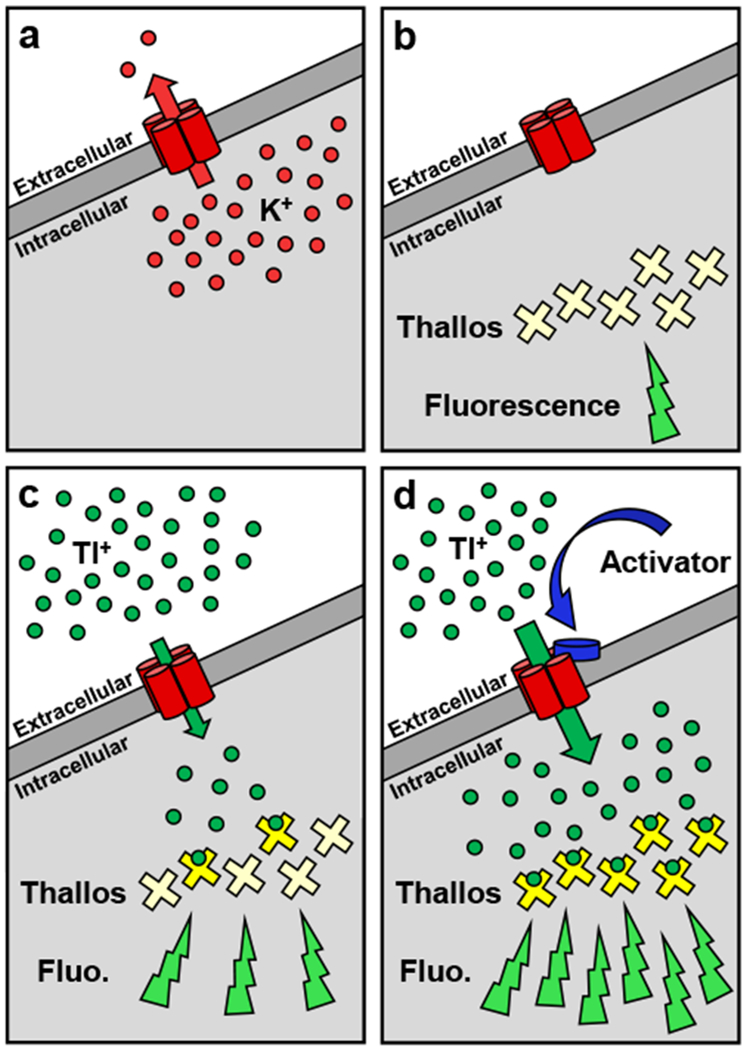
The thallium (Tl+) flux assay enabled identification of compounds that modulated Tl+ entry pathways on the cellular membrane. (a) When GIRK homotetrameric channels are expressed in a cell, they efflux potassium (K+) under normal physiological conditions. During a Tl+ flux assay, (b) an intracellularly-loaded Tl+-sensitive dye, Thallos, enables (c) measurement of the influx of Tl+ via fluorescence (Fluo.). (d) Channel activators increase Tl+ influx, which increases fluorescence and enables compound identification.
Figure 2.
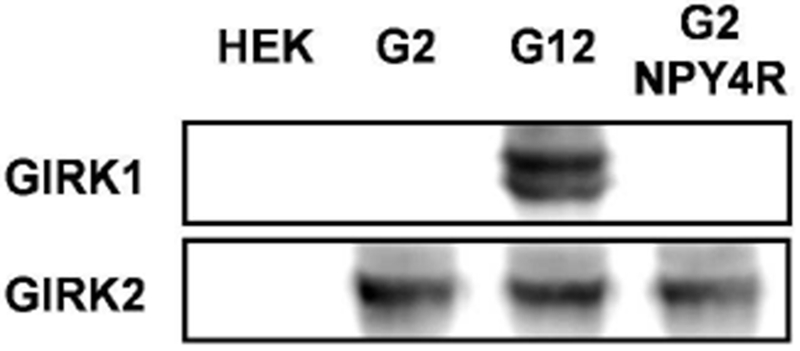
GIRK1 and GIRK2 protein expression levels shown in untransfected HEK293 cells or cells engineered to express GIRK2, GIRK1/2, or GIRK2 and NPY4R. The full gel is shown in Supporting Information, Figure 1S.
Figure 4.
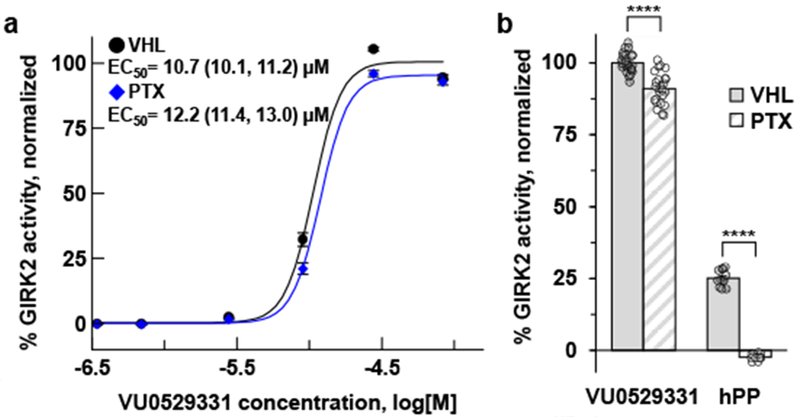
(a) Pertussis toxin (PTX) does not affect the activity of VU0529331 on G2Y4 cells while (b) activation of GIRK2 through the Gβγ-subunit after GPCR activation is fully inhibited. The G2Y4 cells express neuropeptide Y receptor type 4 (NPY4R) in addition to any GPCRs native to HEK293 cells. NPY4R was activated using a maximally-effective concentration, 200 nM, of human pancreatic polypeptide (hPP). All values in (a) were normalized to the predicted maximum activity of VU0529331 under VHL conditions. VU0529331 activity in (b) is normalized data from the 30 μM value in (a). 95% confidence intervals for the EC50 measurements were calculated using a 2-tailed Mann-Whitney test. **** indicates p<0.0001. Error bars represent the standard error of the mean (SEM).
Using this screening approach, we discovered VU0529331 (Figure 3a). We characterized the ability of VU0529331 to activate GIRK channels using Tl+ flux assays and whole-cell patch-clamp electrophysiology. Because the screen was conducted in HEK293 cells that express a variety of Tl+ influx pathways that may be responsible for false-positive hits, we conducted studies to determine which proteins were responsible for VU0529331’s ability to increase Tl+ influx. First, we observed that VU0529331 does not increase Tl+ influx in untransfected HEK293 cells (Figure 3d), suggesting that VU0529331 was dependent on the expression of GIRK channels and/or NPY4R for activity. To investigate whether VU0529331 was dependent on NPY4R expression for activity, we conducted Tl+ flux assays using HEK293 cells engineered to express GIRK2 channels but not NPY4R. We observed VU0529331 activity in the absence of NPY4R, demonstrating that this phenomenon is dependent on GIRK2 expression (Figure 3b). Representative examples of the raw Tl+ flux assay fluorescence traces used to generate concentration series graphs in Figure 3 are provided in the Supporting Information, Figure S2. Next, we sought to identify whether a natively expressed Gi/o-coupled GPCR in HEK293 cell lines was the target of VU0529331 and, thus, indirectly responsible for the increase in GIRK activity downstream of the GPCR. To investigate this possibility, we tested VU0529331 in G2Y4 cells treated with pertussis toxin (PTX), a bacterial exotoxin that inhibits Gi/o-coupled GPCR signaling by catalyzing ADP-ribosylation of the Gα subunit of the G proteins. Through this mechanism, PTX inhibits Gi/oβγ protein signaling to GIRK channels from any native Gi/o-coupled GPCR expressed in HEK293 cells. We conducted Tl+ flux experiments using PTX-treated G2Y4 cells and found that PTX treatment induced little to no difference in the activity of VU0529331 (Figure 4a). In contrast, PTX treatment dramatically inhibited a maximally-effective dose of hPP from activating GIRK channels indirectly (Figure 4b). Because VU0529331 was insensitive to PTX, we concluded that the activity of VU0529331 was independent of Gi/o protein signaling through any Gi/o-coupled GPCR that may be expressed in HEK293 cells. Together, these data suggest that VU0529331 is active on GIRK channels engineered into HEK293 cells. We sought to explore how broadly active this compound was on closely and distantly related K+ channels.
Figure 3.
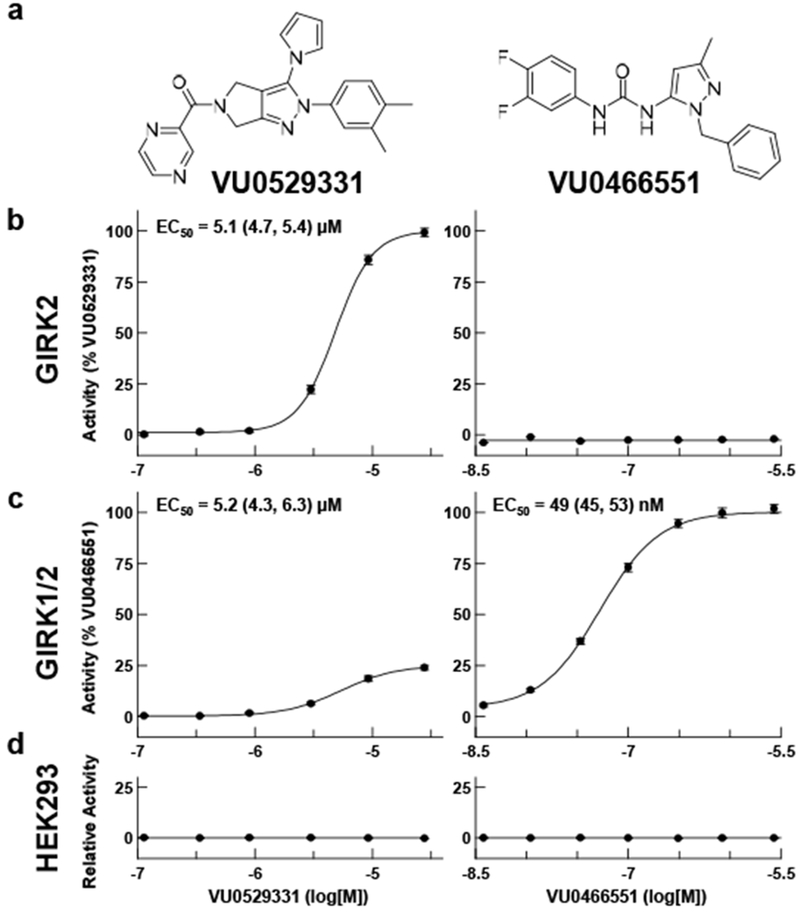
Characterization of VU0529331 efficacy and potency using Tl+ flux. VU0529331 activated GIRK2 and GIRK21/2 channels in a concentration-dependent manner. In contrast, VU0466551 only activated GIRK1-containing channels. Potency (EC50) values were provided for active compounds. Both compounds were inactive in untransfected HEK293 cells. GIRK2 channel activity was normalized to a maximally-effective concentration of VU0529331, while GIRK1/2 channel activity was normalized to a maximally-effective concentration of VU0466551. All data shown are representative of, at minimum, 3 independent experiments. Error bars represent the standard error of the mean (SEM).
We tested whether VU0529331 activity was specific to homomeric GIRK2 channels or whether VU0529331 was capable of activating other GIRK channels as well. Using Tl+ flux assays, we observed that VU0529331 was able to activate GIRK1/2 (Figure 3c), GIRK1/4, and GIRK4 channels (Table 1) expressed in HEK293 cells. This finding is not unexpected considering that GIRK subunits are highly homologous, GIRK4 being the most homologous member of the Kir3 family with respect to GIRK2. However, this finding was exciting, since no other small molecules that activate homomeric GIRK4 channels have been reported, to our knowledge. To study VU0529331 on GIRK1/X channels, we compared the activity of VU0529331 against VU0466551, an efficacious, potent, and selective activator of GIRK1/X channels that was previously discovered by our laboratory.50 We verified that VU0466551 was active on GIRK1/2 channels with a potency of ~50 nM and inactive on both untransfected and GIRK2-expressing HEK293 cells. We found that the efficacy of VU0529331 on GIRK1/2 and GIRK1/4 was ~25% and ~20% of the maximum efficacy of VU0466551, respectively. The raw Tl+ flux traces (Supporting Information, Figure S2) demonstrated that VU0529331 was capable of generating a much greater total fluorescence in cells expressing just GIRK2 as compared to cells expressing GIRK1/2 channels. These data suggest that VU0529331 may be more effective at opening homotetrameric GIRK2 channels than GIRK1/2 or GIRK1/4 heterotetrameric channels. Overall, these results suggest that VU0529331 is a modestly selective, non-GIRK1/X channel activator in addition to being the first synthetic small molecule known to activate both GIRK2 and GIRK4 homomeric channels.
Table 1.
VU0529331 was tested on an array of targets expressed in HEK293 cells. VU0529331 activated GIRK2, GIRK1/2, GIRK1/4, GIRK4, Kir6.1/SUR2a, and Kir6.1/SUR2b channels. The compound was inactive on all other listed targets.
| Target | Efficacy, % (95% CI) | Potency, μM (95% CI) |
|---|---|---|
| GIRK2 | AD100 | 5.1 (4.7, 5.4) |
| GIRK1/2 | B25.9 (+/− 2.4) | 5.2 (4.3, 6.3) |
| GIRK1/4 | B18.1 (+/− 1.1) | 4.8 (4.2, 5.4) |
| GIRK4 | AD100 | 22.3 (21.1, 23.7) |
| Kir6.1/SUR2a | C1418 (+/− 108) | 15.7 (13.8, 17.8) |
| Kir6.1/SUR2b | C127(+/− 2) | 1.23 (1.21, 1.25) |
| Kir6.2/SUR1 | Inactive | Inactive |
| Kir4.1 | Inactive | Inactive |
| Kir2.1 | Inactive | Inactive |
| Slack | Inactive | Inactive |
| Kv2.1 | Inactive | Inactive |
| α1β2 MaxiK | Inactive | Inactive |
| α1β4 MaxiK | Inactive | Inactive |
| α1 GlyR | Inactive | Inactive |
| Untransfected | Inactive | Inactive |
Normalized to maximum VU0529331 activity;
Normalized to maximum VU0466551 activity;
Normalized to 250 μM pinacidil activity;
Confidence interval (95% CI) cannot be calculated.
To further investigate the selectivity of VU0529331 within several families of K+ channels, we tested the ability of VU0529331 to modulate the activity of a variety of Kir channels as well as more distantly related K+ channels (Supporting Information, Figure S4). Using Tl+ flux assays on HEK293 cells engineered to express the channels of interest, we found that VU0529331 was inactive on Kir2.1, Kir4.1, α1β2 MaxiK, α1β4 MaxiK, Kv2.1, and Slack channels. We did discover that VU0529331 activated Kir6.1/SUR2a and Kir6.1/SUR2b channels. This finding suggests that Kir6.1 and the Kir3 channel family share some, thus-far, unappreciated pharmacological homology. Kir6 and Kir3 are closely related Kir families, and future experiments involving site-directed mutagenesis and chimeric channels will aim to elucidate the amino acids involved in VU0529331 binding and activity. In addition to testing VU0529331 selectivity amongst K+ channels, we also investigated whether VU0529331 modulated homomeric α1 GlyR,59 one of the many Cys-loop receptors activated and potentiated by ivermectin. To test the activity of VU0529331 on α1 GlyR, we expressed α1 GlyR in a HEK293 cell line engineered to express both SuperClomeleon, a genetically encoded chloride ion (Cl−) sensor,68 and the K+-Cl− transporter member 5 (KCC2), which is essential for the generation of a Cl− gradient that favors inward Cl− flux.69 While ivermectin demonstrated both activation of α1 GlyR and potentiation of glycine on α1 GlyR, VU0529331 neither activated α1 GlyR nor potentiated the activity of glycine on α1 GlyR (Supporting Information, Figure S3). The selectivity results for VU0529331, including efficacies and potencies, are listed in Table 1.
After establishing that the ability of VU0529331 to increase Tl+ flux in GIRK-expressing HEK293 cells was mediated by GIRK channels, we then explored the GIRK-activating properties of VU0529331 using whole-cell patch-clamp electrophysiology. First, we investigated the effect of VU0529331 on the current-voltage relationship (IV) of GIRK channels. Here, we utilized the same engineered HEK293 cells expressing GIRK2 and GIRK1/2 channels used for Tl+ flux assays. Currents were recorded in 12 increments of 10 mV, from −100 mV to 10 mV, in the absence or presence of 80 μM VU0529331, which was a maximally-effective concentration. Representative traces of the recordings used to generate these IVs are shown in Supporting Information, Figure S5. IVs generated in GIRK2 (Figure 5a) and GIRK1/2 (Figure 5b) cell lines were consistent with inwardly-rectified K+ channels. Recordings demonstrated that inward and outward GIRK2 and GIRK1/2 currents were increased in the presence of VU0529331, while we did not record any changes in the currents of untransfected HEK293 cells (Figure 5c). The magnitude of activation was greater for GIRK2 channels than GIRK1/2 channels when comparing the physiologically-relevant outward currents (Figure 5d), further supporting the view that VU0529331 is a better activator of GIRK2 channels than GIRK1/2 channels. For both channels, inward rectification was maintained in the presence of VU0529331, and the rectification ratio (RR = (−1)*Current Density−100mV/Current Density−30mV) was not significantly altered by VU0529331 (Figure 6). Additionally, no significant change in the reversal potential was observed (GIRK295%CI: (−43.9, −40.8) mVVHL vs (−44.4, −42.8) mVVU0529331; GIRK1/295%CI: (−42.9, −42.0) mVVHL vs (−44.1, −41.9) mVVU0529331) indicating that K+ selectivity was unaffected. Together, these data indicate that VU0529331 does not affect the inward rectification or the K+ selectivity of GIRK channels.
Figure 5.
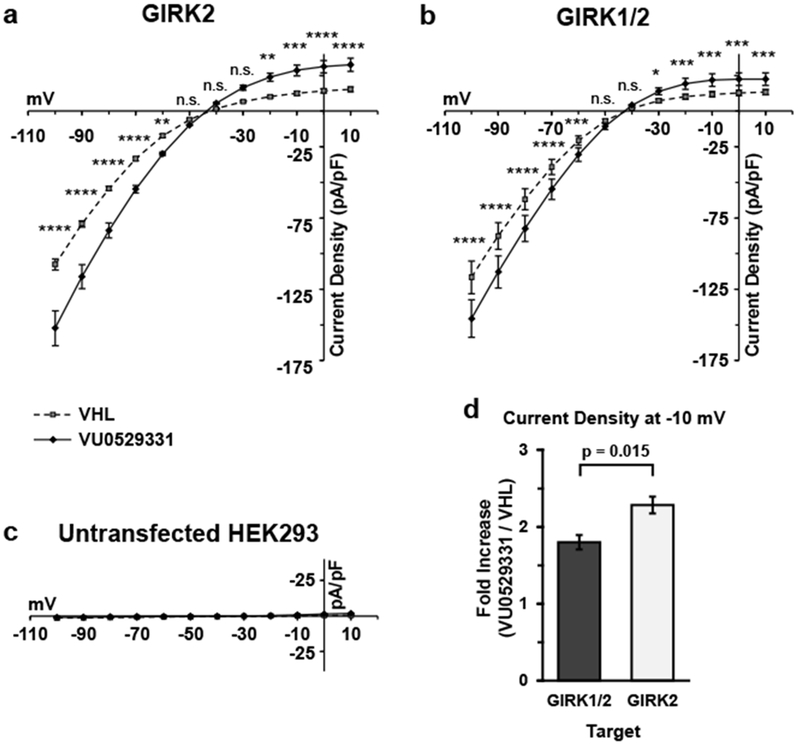
80 μM VU0529331 significantly increased both inward and outward currents in HEK293 cells expressing (a) GIRK2 and (b) GIRK1/2 channels. VU0529331 did not evoke currents in (c) untransfected HEK293 cells. (d) The increase in current density for GIRK1/2 and GIRK2 cells at −10 mV was significantly different. For each cell type in (a), (b), and (c), the conditions were compared using a 2-way ANOVA with Bonferroni’s multiple comparisons test. * p<0.05, ** p<0.01, *** p<0.001, and **** p<0.0001. The fold differences in (d) were compared using a Welch’s t-test. Recordings were generated in 20 mM K+ external buffer solution containing 0.25% (v/v) DMSO (VHL). Error bars indicate standard error around the mean (SEM).
Figure 6.
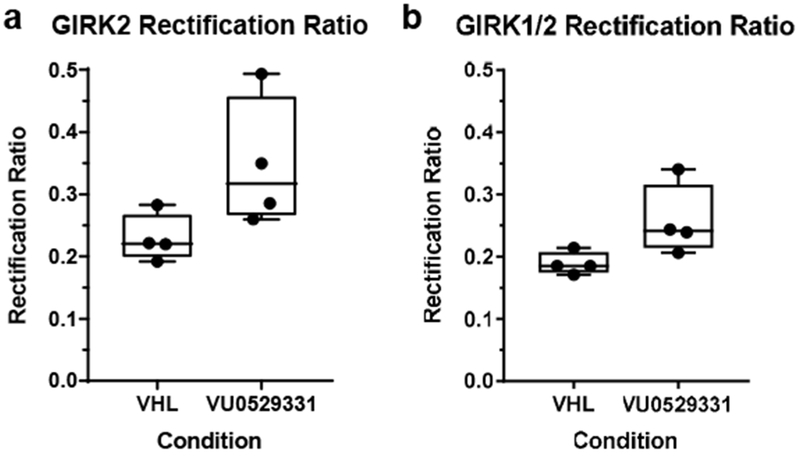
80 μM VU0529331 had no effect on the rectification ratio of HEK293 cells expressing (a) GIRK2 and (b) GIRK1/2 channels. Whole-cell patch-clamp electrophysiology recordings were generated under 20 mM K+ external buffer solution containing 0.25% (v/v) DMSO (VHL). A Wilcoxon matched-pairs signed rank test analysis indicated that there was not a statistically significant difference between the two conditions in (a) or (b).
To further investigate the activity of VU0529331, we recorded currents with the membrane potential clamped at −60 mV. Increasing concentrations of VU0529331 incrementally increased inward GIRK2 and GIRK1/2 currents in engineered HEK293 cells (Figure 7a,d) while failing to induce changes in currents in untransfected HEK293 cells (Figure 7b). Application of an extracellular solution containing 2 mM barium (Ba2+) blocked nearly all basal and VU0529331-evoked GIRK currents (Figure 7c,e). These data corroborated the results of the Tl+ flux data and demonstrated that VU0529331 does act on and increase currents through homotetrameric and heterotetrameric GIRK channels. Overall, these electrophysiology experiments demonstrated that VU0529331 is capable of increasing the activity of GIRK channels while maintaining the K+ selectivity and inward rectification inherent to these channels.
Figure 7.
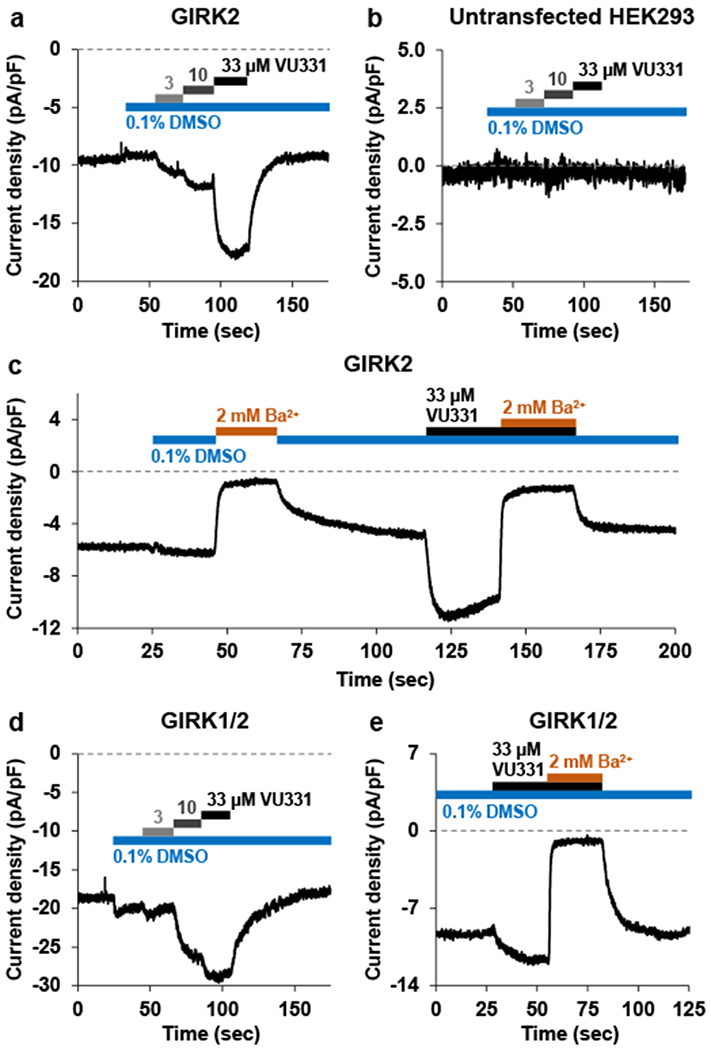
Whole-cell patch-clamp electrophysiology using either untransfected HEK293 cells or cells engineered to express GIRK channels. Currents were held at −60 mV while cells were bathed in 20 mM K+ external buffer. Exposure of (a) GIRK2 cells or (d) GIRK1/2 cells to escalating VU0529331 (VU331) concentrations showed concentration-dependent increases in inward current density. (b) Untransfected HEK293 cells did not show any changes in currents after VU331 exposure. Both (c) GIRK2 and (e) GIRK1/2 currents evoked by maximally-active VU331 concentrations were blocked by exposure to extracellular solutions containing 2 mM barium (Ba2+), the non-selective inwardly-rectifying K+ channel inhibitor.
Although VU0529331 represents an exciting opportunity for small-molecule-based modulation of non-GlRK1/X channels, the measured potency at GIRK2 channels of ~5 μM is not ideal. We sought to identify analogs of VU0529331 with improved potency by studying the structure-activity relationship (SAR) between analogs and GIRK channels. To this end, we purchased 43 analogs (Supporting Information, Figure S6) and synthesized 4 additional analogs with substitutions that were not available for purchase (Supporting Information, Figure S7). The synthetic strategy is illustrated in Supporting Information, Scheme S1. By testing this limited analog library on GIRK2 and GlRK1/2 channels, we found that none of the modifications to the parent structure of VU0529331 were tolerated. Although this limited number of analogs did not yield any improvement in the activity of VU0529331, further medicinal chemistry efforts may result in improvements in both potency and efficacy. However, these efforts are beyond the scope of the present study.
While GIRK physiology has been extensively studied for the past two decades, development of molecular probes to study non-GIRK1/X channels has proven a difficult endeavor. To date, two other screens independently identified ivermectin as an activator of non-GIRK1/X channels. While an exciting discovery, ivermectin is a large, macrocyclic natural product with multiple known important CNS targets that evoke a variety of known in vivo effects. More specifically, ivermectin’s activation and potentiation of Cys-loop receptors, such as GlyR and GABAAR receptors, severely limit ivermectin’s utility, especially since GABAAR and GIRK channels are often co-localized in the same neurons throughout the CNS. Thus, efforts to utilize ivermectin as a GIRK probe are quite limited. Therefore, as a GIRK channel probe, VU0529331 fills an important and unique pharmacologic gap. Although the activity of VU0529331 on Kir6.1/SUR2a and Kir6.1/SUR2b is not ideal, the lack of Kir6.1 expression in neurons mitigates some of the concern. Additionally, the Kir6.2/SUR1 channel, which is expressed in neurons,70,71 was not activated by VU0529331, further mitigating concerns. However, Kir6.1 is expressed in astrocytes70 and, even though SUR expression in astrocytes has not been explored,72 this target should be considered when VU0529331 is used ex vivo (e.g. brain slice preparations). Nevertheless, we are eager to capitalize on the similarity between Kir3 and Kir6 to explore the amino acids important in VU0529331 activity on these families. Such work may reveal opportunities to develop more potent and selective compounds for both Kir3 and Kir6.1 channels, alike.
CONCLUSIONS
In summary, we have discovered and characterized VU0529331, the first small-molecule GIRK-channel activator capable of activating non-GIRK1/X channels. Although this compound activated all the GIRK channels we tested, we demonstrated that its activity was greater on non-GIRK1/X channels. Further, VU0529331 did not change the K+ selectivity or rectification of GIRK channels. Neither VU0529331 nor a limited number of its analogs possessed sufficient potency for ex vivo brain slice or in vivo studies; however, VU0529331 provides evidence that a synthetic small molecule can activate non-GIRK1/X channels. This molecule presents a starting point for the development of analogs with improved potency, selectivity, and favorable ADME properties. Such improved analogs will enable ex vivo brain slice and in vivo studies, which have the potential to help better our understanding of the role of GIRK channels in addiction and reward. Additionally, such molecules may indicate that modulation of non-GIRK1/X channels with pharmaceuticals could be an important new mechanism through which to help combat the nationwide opioid epidemic and other forms of addiction.
METHODS
Cell Line Generation and Cell Culture
HEK293 cells expressing both human GIRK1 and human GIRK2 subunits were generated by transfecting a pCMV6-A-BSD vector (Origene, Rockville, MD) containing KCNJ3 and a pCMV6-A-puro vector (Origene, Rockville, MD) with KCNJ6 into low-passage HEK293 cells (CRL-1573, ATCC, Manassas, VA). The FuGENE 6 (Promega, Madison, WI) transfection reagent and protocol, together with Opti-MEM (Thermo Fisher Scientific, Waltham, MA), were utilized in all transfections herein. Similarly, HEK293 cells expressing the human GIRK2 subunit were generated by transfecting only the KCNJ6-containing vector. Further, GIRK2 cells were transfected with a vector encoding human NPY4R, generously provided by the Dr. Annette Beck-Sickinger laboratory,73,74 to create the G2Y4 cells used for HTS and PTX experiments. To study GIRK4 channels, HEK293 cells were transiently-transfected with a pCMV6-A-Neo vector (Origene, Rockville, MD) containing human KCNJ5. HEK293 cells stably-expressing human α1β2 MaxiK and α1β4 MaxiK channels were generated by transfecting a pCMV6-A-puro vector (Origene, Rockville, MD) with human KCNMA1 and a pcDNA3.1(+)/zeo vector (Genscript, Piscataway, NJ) with either human KCNMB2 or human KCNMB4, respectively. HEK293 cells expressing human Slack channels were generated by transfecting a pCMV6-Entry vector (Origene, Rockville, MD) containing the KCNT1 gene. HEK293 cells expressing human Kv2.1 channels were generated by transfecting a vector encoding the KCNB1 gene, generously provided by the Dr. Jennifer Kearney laboratory.76 HEK293 cells expressing both human Kir6.1 and rat SUR2a were generated by transfecting a pBUDCE4.1 vector (Invitrogen, Carlsbad, CA) containing KCNJ8 and ABCC9 isoform A. HEK293 cells expressing both human Kir6.1 and rat SUR2b were generated by transfecting a pcDNA5/TO vector (Origene, Rockville, MD) containing KCNJ8 and a pcDNA3.1 vector, generously provided by the Dr. Colin Nichols laboratory, containing ABCC9 isoform B. Stably-transfected monoclonal T-REx-HEK293 cell lines expressing human Kir2.1,77 Kir4.1,78 or Kir6.2/SUR179 under a tetracycline-inducible promoter were previously generated and graciously provided by the laboratory of Dr. Jerod S. Denton. HEK293 cells stably-expressing SuperClomeleon and KCC2 were generated in a T-REx-HEK293 background by transducing a pLenti CMV Puro DEST vector containing SuperClomeleon and transfecting a pcDNA4/TO vector containing human KCC2 isoform B. To study α1 GlyR, this cell line was then transiently-transfected with a pCMV6-A-hygro vector containing human GLRA1. All stably-transfected cells were maintained under antibiotic selection over their lifetime.
Monoclonal cell lines were generated by limiting-dilution cloning, where polyclonal populations of cells were distributed in 384-well amine-coated plates at <0.8 cells per well and cultured till colonies grew to encompass at least a quarter of a well. These colonies were further tested and selected for expansion as described previously.80 Selected clones were expanded for use in experiments and for cryopreservation. Cell culture medium consisted of Minimal Essential Medium, Alpha Medium (Corning, Corning, NY) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) and GlutaGro (Corning, Corning, NY). Medium was supplemented with antibiotics, as appropriate, to maintain selection pressure.
For use in Tl+ flux experiments, monoclonal HEK293 cells engineered to express proteins of interest were grown in cell culture medium on tissue-culture-treated and vented T75, T150, or T300 flasks (TPP, Trasadingen, Switzerland) in a humidified incubator at 37°C and 5% CO2. These cells were grown to reach 90% confluence on the day before experimentation. Experiments with GIRK4 and α1 GlyR channels were conducted in transiently-transfected cells, which were not processed for monoclonal selection. To generate transiently-expressing cell lines for experiments, cells were transfected when flasks were 40% confluent, and the flasks were incubated overnight to reach 90% confluence in a humidified incubator at 37°C and 5% CO2. To prepare cells for transfer into 384-well plates, culture medium was removed from flasks, flasks were treated using TrypLE Express (Thermo Fisher Scientific, Waltham, MA) for 5 min to dislodge cells, and single-cell suspensions were created through trituration. Cells were resuspended in antibiotic-containing medium at 1,000 cells/μL. 1 μg/mL of tetracycline (Sigma-Aldrich, St. Louis, MO) was added to suspensions where cells expressed a protein under a tetracycline-inducible promoter. 20 μL of cellular suspensions were plated in Corning® PureCoat™, amine-coated, 384-well microtiter plates (cell plates; cat#354719, Corning, Corning, NY) to achieve a cell density of 20,000 cells/well. Cell plates were incubated overnight in a humidified incubator at 37°C and 5% CO2.
Thallium (Tl+) Flux Experiments
The Tl+ flux assays were conducted largely as previously described,81,82 and below we detail the specifics. Compounds for primary screening were managed by the Vanderbilt High-throughput Screening Facility and were received on the day of screening as 80 nL aliquots of 10 mM compound stocks dissolved in dimethyl sulfoxide (DMSO). With 1 compound/well, 320 compounds were formatted in 384-well, round-bottom, polypropylene microplates (compound plates; Greiner, Monroe, NC) and diluted to 40 μL with assay buffer comprising Hanks Balanced Salt Solution (Thermo Fisher Scientific, Waltham, MA) and 20 mM HEPES pH 7.3 (Corning, Corning, NY). This generated compound solutions at concentrations twice the final concentration because solutions were diluted 2-fold during the assay to a final concentration of 10 μM. For commercially purchased VU0529331 and its available analogs, dry compounds were ordered through AldrichMarketSelect (Sigma-Aldrich, St. Louis, MO) and dissolved in DMSO to generate 10-100 mM stocks. Specifically, VU0529331 was stored in DMSO at 33.3 mM. For an experiment, 10 μL aliquots in 384-well, Echo qualified, low dead volume microplates (Labcyte, Sunnyvale, CA) were reformatted using an Echo555 plate reformatter (Labcyte, Sunnyvale, CA) to generate 10-concentration, 3-fold dilution series in compound plates. Compounds were similarly diluted with assay buffer to twice the final concentration. To help solubilize compounds, compound plates with diluted compounds were sealed using a PlateLoc Thermal Microplate Sealer (Agilent, Santa Clara, CA), briefly ultrasonicated in a bath sonicator (Cole-Parmer, Vernon Hills, IL), and shaken vigorously using a Teleshake (Inheco, Martinsried, Germany) for at least 30 min. Compounds were used within an hour of preparation, and the final DMSO concentrations in all assays were ≤0.25% (v/v).
Cells for experiments were prepared 1 day before screening, as described in the Methods section above. For experiments involving PTX treatment, medium in cell plates was replaced with fresh medium containing 800 ng/mL PTX (Tocris, Bristol, United Kingdom) and incubated in a humidified incubator at 37°C and 5% CO2 for 8 hours prior to dye-loading. Dye was loaded into cells for 1 hour, directly prior to imaging. For this, culture medium was replaced with 20 μL/well of dye-loading buffer comprising assay buffer with 0.6 μM Tl+-sensitive dye Thallos-AM. Thallos-AM was stored dry in 20 μg aliquots and resuspended with 30 μL of DMSO (Sigma-Aldrich, St. Louis, MO) containing 6.7% (w/v) Pluronic F-127 (Sigma-Aldrich, St. Louis, MO) immediately prior to dilution. After incubating cells in dye-loading buffer for 1 hour at room temperature (RT), the buffer was replaced with assay buffer and loaded onto the WaveFront Biosciences Panoptic (Franklin, TN). Data were acquired at 5 Hz (excitation 482 ± 35 nm, emission 536 ± 40 nm) for 10 s, at which time 20 μL/well of test compounds was added and allowed to equilibrate for 120 s. Next, 10 μL/well of Tl+ stimulus buffer (125 mM NaHCO3, 1.8 mM CaSO4, 1 MgSO4, 5 mM glucose, 4 mM Tl2SO4, and 20 mM HEPES pH 7.3) was added. Imaging was concluded after an additional 120 s. When preparing hPP for testing, 100 μM hPP stock solutions were diluted to 5-fold over the desired final concentration in Tl+ stimulus buffer additionally containing 0.0025% fatty acid free, low endotoxin, lyophilized, powdered bovine serum albumin (Sigma-Aldrich, St. Louis, MO). During the screen, a final concentration of 0.7 nM hPP was used to achieve approximately 30% GIRK channel activity through the NPY4R, and 200 nM hPP was a maximally-effective concentration. To better observe activity of certain channels, assay parameters were varied as follows. MaxiK-expressing cells were tested using a Tl+ stimulus buffer containing 3 μM ionomycin, which elevated intracellular Ca2+ and promoted channel activity to approximately EC30. Kv2.1-expressing cells were tested using a modified Tl+ stimulus buffer (87.5 mM NaHCO3, 37.5 mM KHCO3, 1.8 mM CaSO4, 1 MgSO4, 5 mM glucose, 4 mM Tl2SO4, and 20 mM HEPES pH 7.3) containing an elevated K+ concentration to promote channel activity at approximately EC30.
Tl+ flux assay data was collected using WaveGuide (WaveFront Biosciences, Franklin, TN) and analyzed using Excel (Microsoft, Redmond, WA). Data reported are the averages of at least 3 independent experiments. Compound activity was extracted from normalized and control-subtracted fluorescence waves generated over 250 s of imaging. The wave for each well was normalized by dividing by the average fluorescence from the first 8 s of imaging (F/F0), which accounted for differences in cell number and dye loading. Next, each wave was control-subtracted, and the change in wave amplitude (ΔA) due to stimulus buffer addition was calculated. At the time of stimulus buffer addition (Ts), the ΔA of active wells increased dramatically, as demonstrated in Supporting Information, Figure S2. ΔA was calculated as the difference in amplitude between time points Ts–5 s and Ts+X s, where X ranges between 2 and 15 s depending on the particular cell line. For each cell line, this time point was chosen to provide a high signal-to-noise ratio while measuring nearest the linear portion of the initial Tl+ influx. These values are inversely proportional to the overall measured activity of a specific channel. Specifically, X=15 for untransfected HEK293, GIRK2, Kir6.1/SUR2a, Kir6.1/SUR2b, Kir6.2/SUR1, α1β2 MaxiK, and α1β4 MaxiK cells, X=5 for GIRK1/4, Slack, Kv2.1, Kir2.1, and Kir4.1 cells, and X=2 for GIRK1/2 cells. ΔA values for each compound at different concentrations were used to generate fits to a four-parameter logistic equation in XLfit (IDBS, Guildford, Surrey, United Kingdom). Statistical analyses were performed using Prism 7 (GraphPad, La Jolla, CA).
As described in Results and Discussion, we utilized the Tl+ flux assay in the HTS format to screen partially-activated G2Y4 cells for activators of GIRK2 channels. Potential activators, or “hits”, were chosen if the compound ranked in the top 0.5% of molecules that displayed the greatest ΔA relative to vehicle controls. Compounds were then characterized through a series of assays against a variety of engineered cell types, as described in the Results and Discussion section, and led to the discovery of VU0529331. Although here we have reported our efforts towards identifying a novel activator from this HTS, our screen did also yield hundreds of hits that inhibited Tl+ influx in our system. However, due to our focus on activators, we have yet to further explore the activity and selectivity of these compounds.
Screened Compound Library
The compound collection that we screened for this work was composed of the following libraries: Vanderbilt Discovery Collection, which is selected from the Life Chemicals collection for HTS. The compounds in this collection were chosen by Vanderbilt medicinal and computational chemists to provide lead-like motifs, minimum pan-assay interference, and maximum diversity; NIH Clinical Collection I and II, which are small molecules that have history of use in human clinical trials; NCI Focused Natural Product Collection, which is comprised of pure compounds acquired by the NCI from Analytical and MerLion; Cayman Lipid Library, which is a broad variety of bioactive lipids; Ion Channel Library, which is a collection from Life Chemicals targeted to ion channels compiled using 2D fingerprint similarity methodology; Epigenetics Collection, which is a group of small molecule modulators with biological activity for use in epigenetic research; Marnett Collection, which contains NSAID derivatives that contain cyclooxygenase inhibitors, PPARγ activators, and apoptosis inducers; and the Enzo Kinase Inhibitor Library, which is the Screen-Well™ Kinase Inhibitor Library containing 80 known kinase inhibitors of well-defined activity.
Western Blot
Untransfected and engineered GIRK1/2, GIRK2, and G2Y4-expressing HEK293 cell lines were grown to 100% confluence in tissue culture treated 150 cm2 dishes. The growth medium was aspirated and plates were twice washed with 5 mL phosphate-buffered saline (PBS) pH 7.4 (Thermo Fisher Scientific, Waltham, MA). Cells were scraped from the dish and triturated. Cell suspensions were then centrifuged at 500 g for 5 min at RT. Next, the supernatant solution was removed, and the pellet was resuspended in 1 mL of PBS and centrifuged once more. After removal of the supernatant solution, cells were resuspended in 1 mL of RIPA buffer (Sigma-Aldrich, St. Louis, MO) containing Halt™ Protease Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA) and stored on ice. Suspensions were then ultrasonicated in 4 brief 5 s pulses and allowed to cool on ice for 15 min before centrifugation at 6,000 g for 10 min in a 4°C room. The supernatant was recovered, and the protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA) with a SpectraMax Plus 384 Microplate Reader (Molecular Devices, San Jose, CA). Sample were diluted to contain 30 μg of protein in 25 μL of Laemmli Sample Buffer (Bio-Rad, Hercules, CA) containing 355 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO). Samples were heated at 55 °C for 10 min before loading, and the gel was run according to the NuPage Bis-Tris/MOPS protocol using NuPage buffers and gel (Invitrogen, Carlsbad, CA). 25 μL/well of each sample was loaded onto a precast NuPAGE 4-12% Bis-Tris Gel and separated for 50 min at 200 V in NuPAGE MOPS SDS Running Buffer. The gel was transferred onto Odyssey Nitrocellulose Membrane (LI-COR Biosciences, Lincoln, NE) for 1 h at 30 V in NuPAGE Transfer Buffer. The membrane was rinsed using Tris-buffered Saline (TBS; Corning, Corning, NY) before blocking with 25 mL of TBS containing 5% (w/v) Blotting Grade Blocker Non-fat Dry Milk (Bio-Rad, Hercules, CA) at RT for 1 hour. For the remainder of the membrane treatments, 0.05% Tween 20 was added to the milk-containing TBS buffer and will be referred to as TT20. Next, the membrane was incubated overnight at 4°C in TT20 containing a 1:2,000 dilution of a monoclonal IgG2b mouse anti-GIRK1 antibody (cat#TA504152, Origene, Rockville, MD) and a 1:1,000 dilution of monoclonal IgG rabbit anti-GIRK2 antibody (cat#25797, Cell Signaling Technology, Danvers, MA). On the next day, the membrane was washed 3 times with 5 mL of TT20 for 5 min each before incubating for 1 h at RT in TT20 containing a 1:20,000 dilution of fluorescent IRDye 800CW donkey anti-rabbit antibody (LI-COR Biosciences, Lincoln, NE) and a 1:20,000 dilution of fluorescent IRDye 680LT donkey antimouse antibody (LI-COR Biosciences, Lincoln, NE). Afterwards, the membrane was washed 3 times in 5 mL of TT20 for 5 minutes before a final rinse with TBS. Fluorescence from secondary antibodies was detected using the Odyssey CLx (LI-COR Biosciences, Lincoln, NE).
SuperClomeleon Chloride (Cl−) Influx Assay of Cys-Loop Receptor Activity
Two days before an experiment, T-REx-HEK293 cells stably expressing SuperClomeleon and KCC2 were grown to 40% confluency in multiple T25 flasks (Corning, Corning, NY). Half of these flasks were transfected with α1 GlyR using FuGENE 6, and all the flasks were incubated overnight in a humidified incubator at 37°C and 5% CO2. One day before an experiment, cell medium was treated with 1 μg/mL tetracycline to enable KCC2 expression and cells were transferred onto 384-well cell plates as described above at 20,000 cells/well. Each cell plate contained at least 1 transfected and 1 untransfected cell type. On the day of an experiment, one compound plate containing VU0529331 and ivermectin (Sigma-Aldrich, St. Louis, MO) at 2-fold the desired final compound concentrations was generated as described above. A second compound plate containing 5-fold the desired final glycine concentrations was prepared by hand. Compound plates were used within an hour of preparation, and the final DMSO concentrations in all conditions assayed was 0.25% (v/v). At 20 minutes prior to imaging, culture medium on cell plates was replaced with 20 μL/well of assay buffer. At the time of imaging, the experiment was loaded into the WaveFront Biosciences Panoptic, and data were acquired at 1 Hz with alternating filters set to capture both the Cl’-insensitive cyan fluorescent protein (CFP) signal (440 ± 40 nm excitation; 480 ± 17 nm emission filters) and the Cl−-sensing yellow fluorescent protein (YFP) signal (440 ± 40 nm excitation; 536 ± 40 nm emission). After 10 s of imaging, 20 μL/well from the first compound plate was added to the cell plate and imaged for 120 s. Next, 10 μL/well from the second compound plate was added, and imaging was concluded after an additional 120 s. Fluorescence data was collected using WaveFront Biosciences WaveGuide and analyzed using Microsoft Excel. Statistical analyses were performed using GraphPad Prism 7. Data reported are the averages of at least 3 independent experiments. The potential of a compound to affect the movement of Cl− across the cell membrane was calculated from normalized and control-subtracted measurements of the area under the curve (AUC) of the FRET ratio (FCFP/FYFP) generated throughout imaging. In this way, increased signal correlated with increased intracellular Cl− concentrations.
Whole-cell Patch-clamp Electrophysiology
Serial dilutions of compounds were done by hand in DMSO, and DMSO stocks were diluted into a buffer consisting of 20 mM KCl, 140 mM, NaCl, 0.5 mM CaCl2, 2 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4; 315 mOsm), named the 20K extracellular solution (KES). Cells were prepared by triturating flasks of engineered cells (cultured and dissociated as described above) to create single-cell suspensions. Cell suspensions were sparsely plated on 35 mm cell culture dishes (cat#83.3900, SARSTEDT, Nümbrecht, Germany) and incubated overnight in a humidified incubator at 37°C and 5% CO2. Before patching, culture medium was removed and each dish was washed twice with 1 mL of assay buffer (Hanks Balanced Salt Solution and 20 mM HEPES pH 7.3) before being filled with 1 mL of KES. Borosilicate glass electrodes (3-7 MΩ resistance) were filled with intracellular solution containing 130 mM KCl, 20 mM NaCl, 5 mM EGTA, 5.46 mM MgCl2, and 10 mM HEPES (pH 7.4; 305 mOsm). Whole-cell current recordings were conducted in KES using a system comprised of an EPC10 amplifier (HEKA Elektronik, Ludwigshafen am Rhein, Germany), an MP-285 micromanipulator (Sutter Instruments, Novato, CA), and an Axiovert 200 microscope (Zeiss, Oberkochen, Germany). HEKA PatchMaster software was used to control the amplifier, enabling semiautomatic signal compensation for electrode capacitance, cellular capacitance, and series resistance. Currents were filtered using the 6-pole Bessel prefilter with 10 kHz bandwidth, and signals were digitized at 20 kHz. Cells were exposed to solutions using a homemade, 8-channel, local superfusion manifold with manual solution switching. Sensitivity to Ba2+ was measured by applying KES, +/− compound, containing 2 mM BaCl2 during recordings. Current-voltage (IV) relationships were measured with cells held at −40 mV before switching between −100 mV and 10 mV in 10 mV steps lasting 250 ms and separated by 500 ms. Current density at ~200 ms into each pulse for each step was extracted and averaged for 3 consecutive pulse protocols conducted on an individual cell at each condition. Current density was calculated by dividing current amplitudes by each cell’s cellular capacitance. Data presented are averages of IVs generated for at least 3 different cells per channel. Data was analyzed using Microsoft Excel. Statistical analyses were performed using GraphPad Prism 7.
Compound Synthesis
N-(tert-butoxycarbonyl)-N-(prop-2-yn-1-yl)glycine (2):
NaH (2.8 g, 2.5 equiv., 60% in mineral oil) was added to a solution of Boc-glycine (1, 5 g, 1.0 eq.) in DMF (100 mL) at 0°C. The suspension was stirred for 1 hr, followed by addition of propargyl bromide (4.95 mL, 1.8 eq., 80% in toluene) at 0°C. The reaction was allowed to react up to RT overtime and stirred overnight. After completion product was extracted between ethyl acetate and cold HCl-water (pH 4). Organic layer was separated, dried over sodium sulfate, filtered, concentrated and used as such. (Yield-4.8 g, 80%). 1H NMR (499 MHz, Chloroform-d) δ 4.29 – 4.09 (m, 4H), 2.29 (t, J = 2.5 Hz, 1H), 1.48 (d, J = 20.4 Hz, 9H).
N-(3-bromoprop-2-yn-1-yl)-N-(tert-butoxycarbonyl)glycine (3):
To a solution of KOH (2.2 g, 7 eq.) in water (40 mL) at 0 °C was added bromine (0.319 mL, 1.1 eq.). After 30 min, a solution of 2 (1.2 g) in MeOH (15 mL) was added dropwise over 2 min and reaction was stirred for 1 h at 0°C. The solution was diluted with water and washed with ethyl acetate. Aqueous layer was acidified with HCl (pH 4) and product was extracted with ethyl acetate. Organic layer was separated, dried over sodium sulfate, filtered, concentrated and used as such. (Yield = 1.24 g, 75%). 1H NMR (499 MHz, Chloroform-d) δ 4.29 – 4.08 (m, 4H), 1.56 – 1.41 (m, 9H).
tert-butyl(3-bromoprop-2-yn-1-yl)(2-(2-(3,4-dimethylphenyl)hydrazmeyl)-2-oxoethyl)carbamate (4):
To a stirred solution of 3 (1 g, 1 eq.), (3,4-dimethylphenyl)hydrazine hydrochloride (1.2 g, 1 eq.), TEA (1.2 mL, 2.5 eq.) in DCM (10 mL) at 0°C was added propylphosphonic anhydride solution ≥50 wt. % in ethyl acetate (2.5 mL, 1.2 eq.). The reaction was stirred at RT for 12 h. Product formed was partitioned between DCM and water. Organic layer was separated, dried over sodium sulfate, filtered, concentrated, and used as such. (Yield = 1.4 g crude). Analytical LCMS, single peak (254 nm), RT = 2.69 min; MS (ESI+) m/z = 432.1 [M + Na]+.
tert-butyl(EH3-bromoprop-2-yn-1-yl)(2-chloro-2-(2-(3,4-dimethylphenyl)hydrazineylidene)ethyl)carbamate (5):
Resin bound tripehnyl phosphine (6.0 g, 3 eq.) was added to a stirred solution of 4 (1 g, 1 eq.) and CCl4 (2 mL, 3 eq.) in CAN (20 mL). Reaction was stirred overnight. Reaction was filtered, filtrate was evaporated and purified by Normal phase flash chromatography (0–50% Ethyl acetate:Hexane). (Yield = 0.31 g, 30%). Analytical LCMS, single peak (254 nm), RT = 3.38 min; MS (ESI+) m/z = 450.0 [M + Na]+. 1H NMR (499 MHz, DMSO-d6) δ 9.27 (s, 1H), 7.00 – 6.93 (m, 2H), 6.88 (dd, J = 8.2, 2.3 Hz, 1H), 4.28 (s, 2H), 4.12 (dd, J = 14.5, 6.1 Hz, 2H), 2.16 (s, 3H), 2.12 (s, 3H), 1.45 – 1.36 (m, 9H).
tert-butyl3-bromo-2-(3,4-dimethylphenyl)-2,6-dihydropyrrolo[3,4-c]pyrazole-5(4H)-carboxylate (6):
A solution of compound 5 (0.78 g, 1 eq.) and TEA (0.76 mL, 3 eq.) in toluene (15 mL) was refluxed at 110 °C for 3 hr. Water was added to the reaction mixture, and extracted with ethyl acetate. Organic layer was separated, dried over sodium sulfate, filtered, concentrated and purified by flash chromatography (0-50% Ethyl acetate:Hexane). (Yield = 0.49 g, 70%). Analytical LCMS, single peak (254 nm), RT = 3.27 min; MS (ESI+) m/z = 392.0 [M + H]+. 1H NMR (499 MHz, DMSO-d6) δ 7.30 (d, J = 8.1 Hz, 2H), 7.22 (dt, J = 8.2, 2.0 Hz, 1H), 4.46 (d, J = 11.3 Hz, 2H), 4.38 (d, J = 17.0 Hz, 2H), 2.29 (s, 6H), 1.47 (d, J = 1.5 Hz, 9H).
3-bromo-2-(3,4-dimethylphenyl)-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole (7):
A solution of compound 6 (0.45 g) and TFA (1 mL) in DCM 5 mL was stirred overnight and concentrated. The residue was dissolved in DCM and washed with saturated aqueous NaHCO3. The organic phase was dried over MgSO4, filtered and concentrated to afford desired product. (Yield = 0.32 g, 95%). Analytical LCMS, single peak (254 nm), RT = 1.88 min; MS (ESI+) m/z = 292.0 [M + H]+. 1H NMR (499 MHz, DMSO-d6) δ 7.28 (dd, J = 5.3, 2.8 Hz, 2H), 7.21 (dd, J = 8.0, 2.3 Hz, 1H), 3.88 (s, 2H), 3.83 (s, 2H), 2.29 (s, 6H).
(3-bromo-2-(3,4-dimethylphenyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl)(pyrazin-2-yl)methanone (8):
pyrazine-2-carbonyl chloride (0.17 g, 1.1 eq.) was added to a stirred solution of compound 7 (0.32 g, 1 eq.) and TEA (0.45 mL, 3 eq.) in DCM (8 mL) at 0°C. Reaction was stirred at RT for 2 hr. Product was partitioned between water and ethyl acetate. Organic layer was dried over sodium sulfate, filtered, concentrated, and purified by flash chromatography 0–80% Ethyl acetate:Hexane. (Yield = 0.38 g, 87%). Analytical LCMS, single peak (254 nm), RT = 2.6 min; MS (ESI+) m/z = 398.0 [M + H]+. 1H NMR (499 MHz, Chloroform-d) δ 9.30 (d, J = 6.4 Hz, 1H), 8.73 (dd, J = 5.0, 2.4 Hz, 1H), 8.64 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 6.5 Hz, 1H), 7.26 (s, 2H), 5.18 (s, 1H), 5.08 (s, 1H), 4.97 (s, 1H), 4.87 (s, 1H), 2.35 (s, 6H). 13C NMR (126 MHz, CDCl3) chemical shifts of the major rotamer are reported δ 164.46, 155.84, 153.87, 146.42, 145.93, 142.24, 137.67, 137.49, 129.96, 126.76, 123.03, 121.86, 119.70, 48.83, 47.27, 19.84, 19.56.
(2-(3,4-dimethylphenyl)-3-(pyridin-4-yl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl)(pyrazin-2-yl)methanone (9):
A solution of compound 8 (50 mg, 1 eq.), pyridin-4-ylboronic acid (30 mg, 2 eq.), K3PO4 (80 mg, 3 eq.) in dioxane (1.5 mL) was degassed with argon for 10 min. Tetrakis (7.21 mg, 0.05 eq.) was added to the mixture and reaction was subjected to microwave irradiation at 140 °C for 2 hours. Water was added to crude, and product was extracted with ethyl acetate. Organic layer was dried over sodium sulfate, filtered, concentrated, and purified by reverse phase Prep-HPLC (0–100% ACN:water). Analytical LCMS, single peak (254 nm), RT = 2.08 min; MS (ESI+) m/z = 397.1 [M + H]+. 1H NMR (499 MHz, DMSO-d6) δ 9.08 (dd, J = 17.3, Hz, 1H), 8.83 (dd, J = 6.1, 2.5 Hz, 1H), 8.77 (ddd, J = 8.2, 2.6, 1.4 Hz, 1H), 8.59 – 8.56 (m, 1H), 8.54 – 8.51 (m, 1H), 7.20 (dt, J = 8.3, 3.3 Hz, 3H), 7.13 – 7.11 (m, 1H), 6.96 (ddd, J = 8.4, 6.5, 2.2 Hz, 1H), 5.07 (d, J = 23.8 Hz, 2H), 4.96 (s, 1H), 4.84 (s, 1H), 2.27 – 2.21 (m, 6H). 13C NMR (126 MHz, DMSO) chemical shifts of the major rotamer are reported δ 164.86, 155.48, 150.59, 150.56, 148.94, 146.62, 145.66, 143.51, 138.23, 137.93, 133.70, 130.56, 126.78, 123.40, 122.69, 122.64, 122.20, 120.04, 48.32, 46.95, 19.78, 19.50.
(2-(3,4-dimethylphenyl)-3-(pyridin-3-yl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl)(pyrazin-2-yl)methanone (10):
Same procedure as compound 9. Analytical LCMS, single peak (254 nm), RT = 2.2 min; MS (ESI+) m/z = 397.1 [M + H]+. 1H NMR (499 MHz, DMSO-d6) δ 9.08 (dd, J = 15.6, 1.5 Hz, 1H), 8.83 (dd, J = 10.4, 2.5 Hz, 1H), 8.77 (dt, J = 14.6, 1.9 Hz, 1H), 8.57 – 8.49 (m, 1H), 8.44 (dd, J = 27.6, 2.2 Hz, 1H), 7.60 (ddt, J = 53.3, 8.2, 2.0 Hz, 1H), 7.40 (ddd, J = 25.3, 8.0, 4.8 Hz, 1H), 7.22 – 7.14 (m, 2H), 6.94 (dt, J = 8.1, 2.3 Hz, 1H), 5.06 (d, J = 15.8 Hz, 2H), 4.93 (s, 1H), 4.84 (s, 1H), 2.23 (d, J = 17.8 Hz, 7H). 13C NMR (126 MHz, DMSO) chemical shifts of the major rotamer are reported δ 165.08, 153.36, 149.62, 149.02, 146.55, 145.64, 143.48, 138.12, 137.06, 136.04, 133.11, 130.47, 126.84, 125.98, 124.12, 123.24, 121.52, 119.25, 48.24, 46.76, 19.78, 19.45.
2-(3,4-dimethylphenyl)-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole (11):
To a solution of compound 8 (60 mg, 1 eq.) in THF (4 mL) at −78°C was added n-BuLi (0.15 mL, 2.0 eq., 2.5 M in hexane), the reaction was stirred at −78°C for 30 min, and quenched with water. Product was extracted with ethyl acetate. Organic layer was dried over sodium sulfate, filtered, concentrated and used as such. Analytical LCMS, single peak (254 nm), RT = 2.08 min; MS (ESI+) m/z = 214.1 [M + H]+.
(2-(3,4-dimethylphenyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl)(pyrazin-2-yl)methanone (12):
pyrazine-2-carbonyl chloride (0.22 mg, 1.1 eq.) was added to a stirred solution of compound 11 (30 mg, 1 eq.) and TEA (58 μL, 3 eq.) in DCM (1 mL) at 0°C. Reaction was stirred at RT for 2 hours. Product was partitioned between water and ethyl acetate. Organic layer was dried over sodium sulfate, filtered, concentrated and purified by reverse phase Prep-HPLC. (Yield = 25 mg, 55%). Analytical LCMS, single peak (254 nm), RT = 2.08 min; MS (ESI+) m/z = 214.1 [M + H]+. 1H NMR (499 MHz, Chloroform-d) δ 9.29 (dd, J = 4.8, 1.5 Hz, 1H), 8.74 – 8.70 (m, 1H), 8.63 (dt, J = 8.0, 1.8 Hz, 1H), 7.69 (d, J = 43.4 Hz, 1H), 7.48 (dd, J = 8.2, 2.3 Hz, 1H), 7.36 (dd, J = 8.2, 2.3 Hz, 1H), 7.22 (d, J = 8.1 Hz, 1H), 5.14 (d, J = 22.7 Hz, 2H), 4.95 (d, J = 21.7 Hz, 2H), 2.33 (d, J = 17.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) chemical shifts of the major rotamer are reported δ 164.53, 156.22, 154.18, 146.40, 145.85, 142.28, 138.44, 138.02, 135.21, 130.44, 121.05, 120.61, 120.21, 118.77, 116.52, 48.60, 46.84, 19.97, 19.31.
Supplementary Material
ACKNOWLEDGMENTS
We thank the laboratory of Prof. Dr. Annette G. Beck-Sickinger for their generous supply of the NPY4R plasmid and the human pancreatic polypeptide. We thank the laboratory of Dr. Colin G. Nichols for their generous supply of the SUR2b plasmid. We thank the laboratory of Dr. Jennifer A. Kearney for their generous supply of the KV2.1 plasmid. We thank Dr. Connie Cepko for her generous gift of pCAG-GFP (Addgene plasmid # 11150). We are grateful to Dr. Brendan F. Dutter for synthesizing Thallos-AM for our laboratory and for his comments on the manuscript. The NIH Clinical Collection is provided through the National Institutes of Health Molecular Libraries Roadmap Initiative, and the Marnett library was donated by Dr. Lawrence J. Marnett. We thank Dr. Paige N. Vinson for her descriptions of the HTS libraries. All the libraries utilized in this work were distributed by the Vanderbilt High-throughput Screening (HTS) Core Facility. Many experiments were performed in the HTS Core with assistance provided by Corbin Whitwell. The HTS Core receives support from the Vanderbilt Institute of Chemical Biology and the Vanderbilt Ingram Cancer Center (P30 CA68485).
Funding
Funding for this project was provided by the National Institute of Mental Health (NIMH) of the National Institutes of Health under the award number 5R21MH099363. This research was supported by funds from the Vanderbilt University Pharmacology Department and Vanderbilt Institute of Chemical Biology (C.D.W.). Additional funds were available through University of Nebraska Medical Center start-up (C.R.H.). Research funding was also provided through the PhRMA Foundation’s Paul Calabresi Medical Student Research Fellowship (K.A.K.). Funding for the WaveFront Biosciences Panoptic kinetic imaging plate reader was provided by the Office of The Director (OD) of the National Institutes of Health under the award number 1S10OD021734. The Panoptic is housed and managed within the Vanderbilt High-throughput Screening Core Facility, an institutionally supported core. Research reported in this publication was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award numbers T32GM007347 (K.A.K.), T32GM07628 (B.D.S.), and T32GM065086 (F.J.P.). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS USED
- AUC
area under the curve
- Ba2+
barium ion
- CFP
cyan fluorescent protein
- CNS
central nervous system
- DA
dopaminergic
- DCM
dichloromethane
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EC30
30% of maximum efficacy
- EC50
potency
- FRET
fluorescence resonance energy transfer
- G2Y4
GIRK2 and NPY4R-expressing HEK293 cells
- GABAAR
γ-aminobutyric acid A receptor
- Gi/o
inhibitory G protein
- GIRK
G proteingated inwardly-rectifying potassium channel
- GIRK1/X
GIRK1-subunit-containing
- GlyR
glycine receptor chloride channel
- GPCR
G protein-coupled receptor
- hPP
human pancreatic polypeptide
- HTS
high-throughput screen
- IV
current-voltage
- K+
potassium ion
- KCC2
potassium-chloride transporter member 5
- kDa
kilodalton
- KES
20 mM potassium ion extracellular solution
- Kir
inwardly-rectifying potassium channel family
- LCMS
liquid chromatography-mass spectrometry
- MS
mass spectrometry
- Na+
sodium ion
- NMR
nuclear magnetic resonance
- NPY4R
neuropeptide Y4 receptor
- PBS
phosphate-buffered saline
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PLC
phospholipase C
- PKA
protein kinase A
- PKC
protein kinase C
- RT
room temperature
- TEA
triethanolamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- Tl+
thallium ion
- VTA
ventral tegmental area
- VU331
VU0529331
- YFP
yellow fluorescent protein
Footnotes
Supporting Information
Additional figures illustrating the complete western blot, thallium flux fluorescence traces, raw electrophysiology traces, potassium channel dendrogram, FRET assay results, VU0529331 analog structures, and synthetic strategy scheme are provided in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
C.D.W. is an owner of WaveFront Biosciences (Franklin, TN), the manufacturer of Panoptic plate readers. C.D.W. also receives royalties from the sale of Thallos.
REFERENCES
- (1).Manchikanti L; Helm S; Fellows B; Janata JW; Pampati V; Grider JS; Boswell MV Opioid Epidemic in the United States. Pain Physician 2012, 15 (3 Suppl), ES9–38. [PubMed] [Google Scholar]
- (2).Frenk SM; Porter KS; Paulozzi LJ Prescription Opioid Analgesic Use Among Adults: United States, 1999–2012. NCHS Data Brief 2015, February (189), 1–8. [PubMed] [Google Scholar]
- (3).Marker CL; Cintora SC; Roman MI; Stoffel M; Wickman K Hyperalgesia and Blunted Morphine Analgesia in G Protein-Gated Potassium Channel Subunit Knockout Mice. Neuroreport 2002, 13 (18), 2509–2513. [DOI] [PubMed] [Google Scholar]
- (4).Luján R; Marron Fernandez de Velasco, E.; Aguado, C.; Wickman, K. New Insights into the Therapeutic Potential of Girk Channels. Trends Neurosci. 2014, 37 (1), 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tipps ME; Buck KJ GIRK Channels: A Potential Link Between Learning and Addiction, 1st ed.; Elsevier Inc, 2015; Vol. 123. [DOI] [PubMed] [Google Scholar]
- (6).Lüscher C; Slesinger PA Emerging Roles for G Protein-Gated Inwardly Rectifying Potassium (GIRK) Channels in Health and Disease. Nat. Rev. Neurosci 2010, 11 (5), 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tsantoulas C; McMahon SB Opening Paths to Novel Analgesics: The Role of Potassium Channels in Chronic Pain. Trends Neurosci. 2014, 37 (3), 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Munoz MB; Padgett CL; Rifkin R; Terunuma M; Wickman K; Contet C; Moss SJ; Slesinger PA A Role for the GIRK3 Subunit in Methamphetamine-Induced Attenuation of GABAB Receptor-Activated GIRK Currents in VTA Dopamine Neurons. J. Neurosci 2016, 36 (11), 3106–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Herman MA; Sidhu H; Stouffer DG; Kreifeldt M; Le D; Cates-Gatto C; Munoz MB; Roberts AJ; Parsons LH; Roberto M; et al. GIRK3 Gates Activation of the Mesolimbic Dopaminergic Pathway by Ethanol. Proc. Natl. Acad. Sci 2015, 112 (22), 7091–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lomazzi M; Slesinger PA; Lüscher C Addictive Drugs Modulate GIRK-Channel Signaling by Regulating RGS Proteins. Trends Pharmacol. Sci 2008, 29 (11), 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Munoz MB; Slesinger PA Sorting Nexin 27 Regulation of G Protein-Gated Inwardly Rectifying K+ Channels Attenuates InVivo Cocaine Response. Neuron 2014, 82 (3), 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kotecki L; Hearing M; McCall NM; Marron Fernandez de Velasco E; Pravetoni M; Arora D; Victoria NC; Munoz MB; Xia Z; Slesinger PA; et al. GIRK Channels Modulate Opioid-Induced Motor Activity in a Cell Type- and Subunit-Dependent Manner. J. Neurosci 2015, 35 (18), 7131–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pravetoni M; Wickman K Behavioral Characterization of Mice Lacking GIRK/Kir3 Channel Subunits. Genes, Brain Behav. 2008, 7 (5), 523–531. [DOI] [PubMed] [Google Scholar]
- (14).Wydeven N; Marron Fernandez de Velasco E; Du Y.; Benneyworth MA; Hearing MC; Fischer RA; Thomas MJ; Weaver CD; Wickman K Mechanisms Underlying the Activation of G-Protein-Gated Inwardly Rectifying K+ (GIRK) Channels by the Novel Anxiolytic Drug, ML297. Proc. Natl. Acad. Sci 2014, 111 (29), 10755–10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ostrovskaya O; Xie K; Masuho I; Fajardo-Serrano A; Lujan R; Wickman K; Martemyanov KA RGS7/Gbeta5/R7BP Complex Regulates Synaptic Plasticity and Memory by Modulating Hippocampal GABABR-GIRK Signaling. Elife 2014, 3, e02053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Montandon G; Ren J; Victoria NC; Liu H; Wickman K; Greer JJ; Horner RL G Protein-Gated Inwardly-Rectifying Potassium Channels Modulate Respiratory Depression by Opioids. Anesthesiology 2015, 124 (3), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kaufmann K; Romaine I; Days E; Pascual C; Malik A; Yang L; Zou B; Du Y; Sliwoski G; Morrison RD; et al. ML297 (VU0456810), the First Potent and Selective Activator of the GiRK Potassium Channel, Displays Antiepileptic Properties in Mice. ACS Chem. Neurosci 2013, 4 (9), 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Masotti A; Uva P; Davis-Keppen L; Basel-Vanagaite L; Cohen L; Pisaneschi E; Celluzzi A; Bencivenga P; Fang M; Tian M; et al. Keppen-Lubinsky Syndrome Is Caused by Mutations in the Inwardly Rectifying K+ channel Encoded by KCNJ6. Am. J. Hum. Genet 2015, 96 (2), 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Best TK; Siarey RJ; Galdzicki Z Ts65Dn, a Mouse Model of Down Syndrome, Exhibits Increased GABAB-Induced Potassium Current. J. Neurophysiol 2007, 97 (1), 892–900. [DOI] [PubMed] [Google Scholar]
- (20).Mesirca P; Alig J; Torrente AG; Müller JC; Marger L; Rollin A; Marquilly C; Vincent A; Dubel S; Bidaud I; et al. Cardiac Arrhythmia Induced by Genetic Silencing of “funny” (f) Channels Is Rescued by GIRK4 Inactivation. Nat. Commun 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wickman K; Nemec J; Gendler SJ; Clapham DE Abnormal Heart Rate Regulation in GIRK4 Knockout Mice. Neuron 1998, 20 (1), 103–114. [DOI] [PubMed] [Google Scholar]
- (22).Hu L; Wada K; Mores N; Krsmanovic LZ; Catt KJ Essential Role of G Protein-Gated Inwardly Rectifying Potassium Channels in Gonadotropin-Induced Regulation of GnRH Neuronal Firing and Pulsatile Neurosecretion. J. Biol. Chem 2006, 281 (35), 25231–25240. [DOI] [PubMed] [Google Scholar]
- (23).Oki K; Plonczynski MW; Lam ML; Gomez-Sanchez EP; Gomez-Sanchez CE The Potassium Channel, Kir3.4 Participates in Angiotensin II-Stimulated Aldosterone Production by a Human Adrenocortical Cell Line. Endocrinology 2012, 153 (9), 4328–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Krapivinsky G; Gordon E. a; Wickman K; Velimirović B; Krapivinsky L; Clapham DE The G-Protein-Gated Atrial K+ Channel IKACh Is a Heteromultimer of Two Inwardly Rectifying K(+)-Channel Proteins. Nature. 1995, pp 135–141. [DOI] [PubMed] [Google Scholar]
- (25).Liao YJ; Jan YN; Jan LY Heteromultimerization of G-Protein-Gated Inwardly Rectifying K+ Channel Proteins GIRK1 and GIRK2 and Their Altered Expression in Weaver Brain. J. Neurosci 1996, 16 (22), 7137–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Karschin C; Dißmann E; Stu W; Karschin A. IRK (1–3) and GIRK (1–4) Inwardly Rectifying K+ Channel MRNAs Are Differentially Expressed in the Adult Rat Brain. J. Neurosci 1996, 16 (11), 3559–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Anderson A; Kulkarni K; Marron Fernandez De Velasco E; Carlblom N; Xia Z; Nakano A; Martemyanov KA; Tolkacheva EG; Wickman K Expression and Relevance of the G Protein-Gated K+ Channel in the Mouse Ventricle. Sci. Rep 2018, 8 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Iwanir S; Reuveny E Adrenaline-Induced Hyperpolarization of Mouse Pancreatic Islet Cells Is Mediated by G Protein-Gated Inwardly Rectifying Potassium (GIRK) Channels. Pflugers Arch. Eur. J. Physiol 2008, 456 (6), 1097–1108. [DOI] [PubMed] [Google Scholar]
- (29).Cruz HG; Ivanova T; Lunn M-L; Stoffel M; Slesinger PA; Lüscher C Bi-Directional Effects of GABAB Receptor Agonists on the Mesolimbic Dopamine System. Nat. Neurosci 2004, 7 (2), 153–159. [DOI] [PubMed] [Google Scholar]
- (30).Jelacic TM; Kennedy ME; Wickman K; Clapham DE Functional and Biochemical Evidence for G-Protein-Gated Inwardly Rectifying K+(GIRK) Channels Composed of GIRK2 and GIRK3. J. Biol. Chem 2000, 275 (46), 36211–36216. [DOI] [PubMed] [Google Scholar]
- (31).Huang CL; Feng SY; Hilgemann DW Direct Activation of Inward Rectifier Potassium Channels by PIP2 and Its Stabilization by Gβγ. Nature 1998, 391 (6669), 803–806. [DOI] [PubMed] [Google Scholar]
- (32).Logothetis DE; Kurachi Y; Galper J; Neer EJ; Clapham DE The Βγ Subunits of GTP-Binding Proteins Activate the Muscarinic K+ Channel in Heart. Nature. 1987, pp 321–326. [DOI] [PubMed] [Google Scholar]
- (33).Riven I; Iwanir S; Reuveny E GIRK Channel Activation Involves a Local Rearrangement of a Preformed G Protein Channel Complex. Neuron 2006, 51 (5), 561–573. [DOI] [PubMed] [Google Scholar]
- (34).Lewohl JM; Wilson WR; Mayfield RD; Brozowski SJ; Morrisett RA; Harris RA G-Protein-Coupled Inwardly Rectifying Potassium Channels Are Target of Alcohol Action. Nat. Neurosci 1999, 2 (12), 1084–1090. [DOI] [PubMed] [Google Scholar]
- (35).Aryal P; Dvir H; Choe S; Slesinger PA A Discrete Alcohol Pocket Involved in GIRK Channel Activation. Nat. Neurosci 2009, 12 (8), 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bodhinathan K; Slesinger PA Molecular Mechanism Underlying Ethanol Activation of G-Protein-Gated Inwardly Rectifying Potassium Channels. Proc. Natl. Acad. Sci 2013, 110 (45), 18309–18314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Toyama Y; Kano H; Mase Y; Yokogawa M; Osawa M; Shimada I Structural Basis for the Ethanol Action on G-Protein–activated Inwardly Rectifying Potassium Channel 1 Revealed by NMR Spectroscopy. Proc. Natl. Acad. Sci 2018, 115 (15), 3858–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ho IHM Molecular Determinants for Sodium-Dependent Activation of G Protein-Gated K+ Channels. 1999, 274 (13), 8639–8648. [DOI] [PubMed] [Google Scholar]
- (39).Whorton MR; MacKinnon R Crystal Structure of the Mammalian GIRK2 K + Channel and Gating Regulation by G Proteins, PIP 2, and Sodium. Cell 2011, 147 (1), 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Glaaser IW; Slesinger PA Dual Activation of Neuronal G Protein-Gated Inwardly Rectifying Potassium (GIRK) Channels by Cholesterol and Alcohol. Sci. Rep 2017, 7 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rosenhouse-dantsker A Cholesterol-Binding Sites in GIRK Channels: The Devil Is in the Details. 2018, 5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bukiya AN; Durdagim S; Noskov S; Rosenhouse-Dantske A Cholesterol Up-Regulates Neuronal G Protein-Gated Inwardly Rectifying Potassium (GIRK) Channel Activity in the Hippocampus. J. Biol. Chem 2017, 292 (15), 6135–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Clancy SM; Fowler CE; Finley M; Suen KF; Arrabit C; Berton F; Kosaza T; Casey PJ; Slesinger PA Pertussis-Toxin-Sensitive Gαsubunits Selectively Bind to C-Terminal Domain of Neuronal GIRK Channels: Evidence for a Heterotrimeric G-Protein-Channel Complex. Mol. Cell. Neurosci 2005, 28 (2), 375–389. [DOI] [PubMed] [Google Scholar]
- (44).Lei Q; Talley EM; Bayliss DA Receptor-Mediated Inhibition of G Protein-Coupled Inwardly Rectifying Potassium Channels Involves G??Q Family Subunits, Phospholipase C, and a Readily Diffusible Messenger. J. Biol. Chem 2001, 276 (20), 16720–16730. [DOI] [PubMed] [Google Scholar]
- (45).Müllner C; Vorobiov D; Bera AK; Uezono Y; Yakubovich D; Frohnwieser-Steinecker B; Dascal N; Schreibmayer W Heterologous Facilitation of G Protein-Activated K(+) Channels by Beta-Adrenergic Stimulation via CAMP-Dependent Protein Kinase. J. Gen. Physiol 2000, 115 (5), 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Medina I; Krapivinsky G; Arnold S; Kovoor P; Krapivinsky L; Clapham DE A Switch Mechanism for Gβγ Activation of IKACh. J. Biol. Chem 2000, 275 (38), 29709–29716. [DOI] [PubMed] [Google Scholar]
- (47).Stevens EB; Shah BS; Pinnock RD; Lee K Bombesin Receptors Inhibit G Protein-Coupled Inwardly Rectifying K+ Channels Expressed in Xenopus Oocytes through a Protein Kinase C-Dependent Pathway. Mol Pharmacol 1999, 55(6), 1020–1027. [PubMed] [Google Scholar]
- (48).Days E; Kaufmann K; Romaine I; Niswender C; Lewis M; Utley T; Du Y; Sliwoski G; Morrison R; Dawson ES; et al. Discovery and Characterization of a Selective Activator of the G-Protein Activated Inward-Rectifying Potassium (GIRK) Channel. Probe Reports from NIH Mol. Libr. Progr 2013, No. Md. [PubMed] [Google Scholar]
- (49).Barber DM; Schönberger M; Burgstaller J; Levitz J; Weaver CD; Isacoff EY; Baier H; Trauner D Optical Control of Neuronal Activity Using a Light-Operated GIRK Channel Opener (LOGO). Chem. Sci 2016, 7(3), 2347–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wen W; Wu W; Romaine IM; Kaufmann K; Du Y; Sulikowski GA; Weaver CD; Lindsley CW Discovery of “molecular Switches” within a GIRK Activator Scaffold That Afford Selective GIRK Inhibitors. Bioorganic Med. Chem. Lett 2013, 23 (16), 4562–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ramos-Hunter SJ; Engers DW; Kaufmann K; Du Y; Lindsley CW; Weaver CD; Sulikowski GA Discovery and SAR of a Novel Series of GIRK1/2 and GIRK1/4 Activators. Bioorganic Med. Chem. Lett 2013, 23(18), 5195–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wen W; Wu W; Weaver CD; Lindsley CW Discovery of Potent and Selective GIRK1/2 Modulators via “molecular Switches” within a Series of 1-(3-Cyclopropyl-1-Phenyl-1H-Pyrazol-5-YI)Ureas. Bioorganic Med. Chem. Lett 2014, 24 (21), 5102–5106. [DOI] [PubMed] [Google Scholar]
- (53).Su Z; Btrn EC; Wang W; MacKinnon R Novel Cell-Free High-Throughput Screening Method for Pharmacological Tools Targeting K+ Channels. Proc. Natl. Acad. Sci 2016, 113 (20), 5748–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Chen IS; Tateyama M; Fukata Y; Uesugi M; Kubo Y Ivermectin Activates GIRK Channels in a PIP2-Dependent, Gβγ-lndependent Manner and an Amino Acid Residue at the Slide Helix Governs the Activation. J. Physiol 2017, 595(17), 5895–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jackson HCC Ivermectin as a Systemic Insecticide. Parasitol. Today 1989, 5 (1988), 146–156. [DOI] [PubMed] [Google Scholar]
- (56).Barragry TB A Review of the Pharmacology and Clinical Uses of Ivermectin. Can. Vet. J 1987, 28 (8), 512–517. [PMC free article] [PubMed] [Google Scholar]
- (57).Khakh BS; Proctor WR; Dunwiddie TV; Labarca C; Lester HA Allosteric Control of Gating and Kinetics at P2X(4) Receptor Channels. J. Neurosci 1999, 19 (17), 7289–7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Priel A; Silberberg SD Mechanism of Ivermectin Facilitation of Human P2X 4 Receptor Channels. J. Gen. Physiol 2004, 123 (3), 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Shan Q; Haddrill JL; Lynch JW Ivermectin, an Unconventional Agonist of the Glycine Receptor Chloride Channel. J. Biol. Chem 2001, 276 (16), 12556–12564. [DOI] [PubMed] [Google Scholar]
- (60).Robertson B Actions of Anaesthetics and Avermectin on GABA(A) Chloride Channels in Mammalian Dorsal Root Ganglion Neurones. Br. J. Pharmacol 1989, 98 (1), 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Dawson GR; Wafford KA; Smith A; Marshall GR; Bayley PJ; Schaeffer JM; Meinke PT; Mckernan RM Anticonvulsant and Adverse Effects of Avermectin Analogs in Mice Are Mediated through the ⋅ -Aminobutyric Acid A Receptor. 2000, 295 (3), 1051–1060. [PubMed] [Google Scholar]
- (62).Krause RM; Buisson B; Bertrand S; Corringer PJ; Galzi JL; Changeux JP; Bertrand D Ivermectin: A Positive Allosteric Effector of the Alpha7 Neuronal Nicotinic Acetylcholine Receptor. Mol. Pharmacol 1998, 53 (2), 283–294. [DOI] [PubMed] [Google Scholar]
- (63).Scott RH; Duce IR Effects of 22,23-Dihydroavermectin B1a on Locust (Schistocerca Gregaria) Muscles May Involve Several Sites of Action. Pestic. Sci 1985, 16 (April), 599–604. [Google Scholar]
- (64).Arena P; Cully DF Expression of a Glutamate-Activated Chloride Current in Xenopus Oocytes Injected with Caenorhabditis Elegans RNA: Evidence for Modulation by Avermectin. 2000, 15 (1992), 339–348. [DOI] [PubMed] [Google Scholar]
- (65).Danishefsky SJ; Armistead DM; Wincott FE; Selnick HG; Hungate R The Total Synthesis of Avermectin Ala. J. Am. Chem. Soc 1989, No. 111, 2967–2980. [Google Scholar]
- (66).Ma D; Zerangue N; Raab-Graham K; Fried SR; Jan YN; Jan LY Diverse Trafficking Patterns Due to Multiple Traffic Motifs in G Protein-Activated Inwardly Rectifying Potassium Channels from Brain and Heart. Neuron 2002, 33 (5), 715–729. [DOI] [PubMed] [Google Scholar]
- (67).Kofuji P; Davidson N; Lester HA Evidence That Neuronal G-Protein-Gated Inwardly Rectifying K+ Channels Are Activated by GI3y Subunits and Function as Heteromultimers. Neurobiology 1995, 92 (July), 6542–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Grimley JS; Li L; Wang W; Wen L; Beese LS; Hellinga HW; Augustine GJ Visualization of Synaptic Inhibition with an Optogenetic Sensor Developed by Cell-Free Protein Engineering Automation. J. Neurosci 2013, 33 (41), 16297–16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Boffi JC; Knabbe J; Kaiser M; Kuner T KCC2-Dependent Steady-State Intracellular Chloride Concentration and PH in Cortical Layer 2/3 Neurons of Anesthetized and Awake Mice. Front. Cell. Neurosci 2018, 12 (January), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Thomzig A; Laube G; Prüss H; Veh RW Pore-Forming Subunits of K-ATP Channels, Kir6.1 and Kir6.2, Display Prominent Differences in Regional and Cellular Distribution in the Rat Brain. J. Comp. Neurol 2005, 484 (3), 313–330. [DOI] [PubMed] [Google Scholar]
- (71).Zhang C; Bosch MA; Levine JE; Ronnekleiv OK; Kelly MJ Gonadotropin-Releasing Hormone Neurons Express KATP Channels That Are Regulated by Estrogen and Responsive to Glucose and Metabolic Inhibition. J. Neurosci 2007, 27 (38), 10153–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Medsker B; Forno E; Simhan H; Juan C; Sciences R ABCC9/SUR2 in the Brain: Implications for Hippocampal Sclerosis of Aging and a Potential Therapeutic Target Peter. 2016, 70 (12), 773–779. [Google Scholar]
- (73).Pedragosa-Badia X; Sliwoski GR; Nguyen ED; Lindner D; Stichel J; Kaufmann KW; Meiler J; Beck-Sickinger AG Pancreatic Polypeptide Is Recognized by Two Hydrophobic Domains of the Human Y4 Receptor Binding Pocket. J. Biol. Chem 2014, 289 (9), 5846–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Mäde V; Bellmann-Sickert K; Kaiser A; Meiler J; Beck-Sickinger AG Position and Length of Fatty Acids Strongly Affect Receptor Selectivity Pattern of Human Pancreatic Polypeptide Analogues. ChemMedChem 2014, 9 (11), 2463–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Matsuda T; Cepko CL Electroporation and RNA Interference in the Rodent Retina in Vivo and in Vitro. Proc. Natl. Acad. Sci 2004, 101 (1), 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Torkamani A; Bersell K; Jorge BS; Bjork RL; Friedman JR; Bloss CS; Cohen J; Gupta S; Naidu S; Vanoye CG; et al. De Novo KCNB1 Mutations in Epileptic Encephalopathy. Ann. Neurol 2014, 76 (4), 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Lewis LM; Bhave G; Chauder BA; Banerjee S; Lornsen KA; Redha R; Fallen K; Lindsley CW; Weaver CD; Denton JS High-Throughput Screening Reveals a Small-Molecule Inhibitor of the Renal Outer Medullary Potassium Channel and Kir7.1. Mol. Pharmacol 2009, 76 (5), 1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Raphemot R; Kadakia RJ; Olsen ML; Banerjee S; Days E; Smith SS; Weaver CD; Denton JS Development and Validation of Fluorescence-Based and Automated Patch Clamp–Based Functional Assays for the Inward Rectifier Potassium Channel Kir4.1. Assay Drug Dev. Technol 2013, 11 (9–10), 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Raphemot R; Swale DR; Dadi PK; Jacobson DA; Cooper P; Wojtovich AP; Banerjee S; Nichols CG; Denton JS Direct Activation of Beta-Cell KATP Channels with a Novel Xanthine Derivative. Mol. Pharmacol 2014, 85 (6), 858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Du Y; Days E; Romaine I; Abney KK; Kaufmann K; Sulikowski G; Stauffer S; Lindsley CW; Weaver CD Development and Validation of a Thallium Flux-Based Functional Assay for the Sodium Channel NaV1.7 and Its Utility for Lead Discovery and Compound Profiling. ACS Chem. Neurosci 2015, 6 (6), 871–878. [DOI] [PubMed] [Google Scholar]
- (81).Weaver CD; Harden D; Dworetzky SI; Robertson B; Knox RJ A Thallium-Sensitive, Fluorescence-Based Assay for Detecting and Characterizing Potassium Channel Modulators in Mammalian Cells. J. Biomol. Screen 2004, 9 (8), 671–677. [DOI] [PubMed] [Google Scholar]
- (82).Weaver CD Thallium Flux Assay for Measuring the Activity of Monovalent Cation Channels and Transporters In Potassium Channels: Methods and Protocols; Shyng S-L, Valiyaveetil FI, Whorton M, Eds.; Springer New York: New York, NY, 2018; pp 105–114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.