

SUMMARY
Super-enhancers are clusters of enhancers, which activate gene expression. Broad trimethyl histone H3 lysine 4 (H3K4me3) often defines active tumor suppressor genes. However, how these epigenomic signatures are regulated for tumor suppression is little understood. Here, we show that brain-specific knockout of the H3K4 methyltransferase MLL4 (a COMPASS-like enzyme; alias KMT2D) in mice spontaneously induces medulloblastoma. Mll4 loss upregulates oncogenic Ras and Notch pathways while downregulating neuronal gene expression programs. MLL4 enhances DNMT3A-catalyzed DNA methylation and SIRT1/BCL6-mediated H4K16 deacetylation, which antagonize expression of Ras activators and Notch pathway components, respectively. Notably, Mll4 loss downregulates tumor suppressor genes (e.g., Dnmt3a and Bcl6) by diminishing broad H3K4me3 and super-enhancers and also causes widespread impairment of these epigenomic signatures during medulloblastoma genesis. These findings suggest an anti-tumor role for super-enhancers and provide a unique tumor-suppressive mechanism in which MLL4 is necessary to maintain broad H3K4me3 and super-enhancers at tumor suppressor genes.
Keywords: Epigenetics, Histone methylation, Histone methyltransferase, MLL4, DNA methylation, H4K16 deacetylation, Super-enhancers, Broad H3K4me3, tumor suppressor
Graphical Abstarct
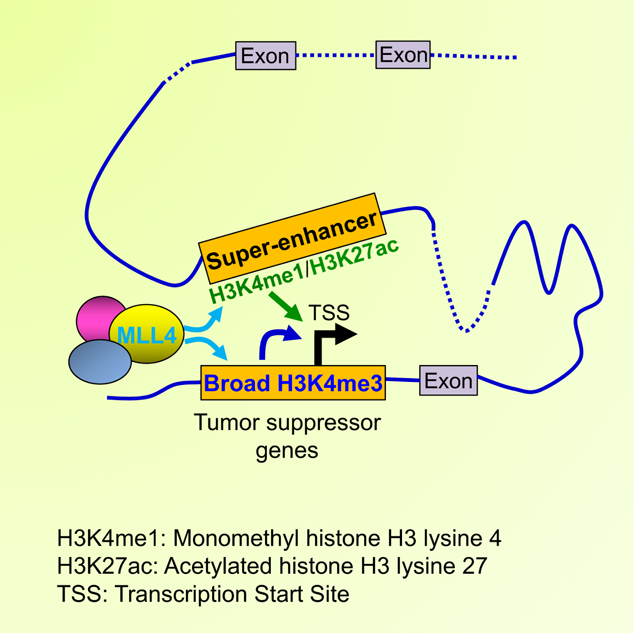
eTOC Blurb
Dhar et al. show that MLL4 suppreses medulloblastoma by establishing super-enhancers and broad H3K4me3 to activate multiple mechanisms that lead to activation of tumor suppressor genes and repression of oncogenes.
INTRODUCTION
Histone methylation, a type of histone posttranslational modification, is a hallmark of epigenetic and transcriptional regulation of gene expression. In particular, methylation at histone H3 lysine 4 (H3K4) is associated with active expression or poised (activatable but repressed) bivalent states of genes at the genome-wide level (Barski et al., 2007; Dhar et al., 2016; Guenther et al., 2007). Like other lysine methylation sites within histones, H3K4 methylation can take place at three different levels, i.e., mono-, di-, or trimethyl (me1, me2, or me3). The genomic distribution profiles of monomethyl H3K4 (H3K4me1), H3K4me2, and H3K4me3 often overlap but are distinct. H3K4 methylation is catalyzed by H3K4 methyltransferases, such as mixed lineage leukemia 1‒4 (MLL1‒4). It can be reversed by the H3K4 demethylases LSD1 and JARID1A‒D (KDM5A‒D) (Gu and Lee, 2013).
Notably, H3K4me3 decorates as much as 75% of all human gene-regulatory regions around the transcription start sites (TSSs) (Guenther et al., 2007). Recently, we and others have identified an epigenomic signature called broad H3K4me3 (also known as H3K4me3 breadth), which has a skewed distribution of H3K4me3 signals covering at least -500 bp to +3,500 bp and is associated with high gene expression (Benayoun et al., 2014; Chen et al., 2015). Broad H3K4me3 peaks are different from sharp H3K4me3 peaks, which are narrower and higher. Interestingly, broad H3K4me3 in normal cells denotes tumor suppressor genes and cell identity genes, is shortened in cancer, and anti-correlates with DNA methylation and H3K27me3 (Chen et al., 2015; Dahl et al., 2016; Liu et al., 2016). It was suggested that JARID1A and JARID1B decrease broad H3K4me3 peaks during early embryo development (Dahl et al., 2016). However, how broad H3K4me3 peaks for tumor suppression are established remains unknown.
Enhancers are a key epigenetic regulatory module that mediates the spatial and temporal activation of genes. H3K4me1, together with acetylated H3K27 (H3K27ac), signifies enhancers (Smith and Shilatifard, 2014). Interestingly, clusters of enhancers that are much larger than typical enhancers with a median size of 0.7–1.3 kb have been described as super-enhancers (Whyte et al., 2013). Super-enhancers strongly activate gene expression and are known to be associated with the regulation of cell identity and diseases (Hnisz et al., 2013; Whyte et al., 2013). For instance, oncogenic super-enhancers can be newly formed in cancer (Hnisz et al., 2013) and are linked to cancer subtypes (Lin et al., 2016). However, little is known about whether super-enhancers contribute to tumor suppression in normal cells or how super- enhancers are established for tumor suppression.
Medulloblastoma (MB), a type of cerebellar cancer, is the most common brain tumor in children and adolescents younger than age 15 years (up to 30% of all childhood brain tumors). MB can spread to the spinal cord or to other parts of the brain. MB is categorized as a high-grade malignant primary tumor because of its rapid growth and can be classified into four major molecular subtypes: wingless (WNT), sonic hedgehog (SHH), Group 3, and Group 4 (Kool et al., 2012; Taylor et al., 2012). Interestingly, several histone methyltransferases and demethylases, including MLL4 and UTX, are frequently mutated in MB (Northcott et al., 2012a).
We have previously shown that MLL4 (a COMPASS-like enzyme; also called KMT2D, MLL2, and ALR) induces expression of many differentiation-specific genes by depositing H3K4me3 during retinoic acid (RA)-mediated neuronal differentiation of human neuron-lineage NT2/D1 stem cells (Dhar et al., 2012). MLL4 belongs to the COMPASS (COMplex of Proteins ASsociated with Set1) family of histone H3K4 methyltransferases, which are conserved from yeast to human and include SET1A, SET1B, and MLL1‒3 in mammals (Shilatifard, 2012). In the present study, we generated an Mll4 brain-specific knockout (Mll4 BSKO) mouse model to understand the role of MLL4-mediated H3K4 methylation in neural differentiation in an in vivo context. Using this mouse model, we found that brain-specific loss of Mll4 induced spontaneous MB and activates oncogenic programs, such as Ras activators and Notch pathway components (e.g., Hes1 and Jag1). To our surprise, MLL4 positively regulates both super-enhancers and broad H3K4me3 to directly activate tumor-suppressor genes, such as the DNA methyltransferase gene Dnmt3a and the transcriptional repressor gene Bcl6. Mechanistically, we provided evidence that expression of Ras activators and Notch pathway components was derepressed by the downregulation of DNMT3A-catalyzed DNA methylation and SIRT1/BCL6-mediated H4K16 deacetylation, respectively, by Mll4 loss. Our results reveal an unexpected epigenetic antitumor mechanism in which MLL4 establishes both super-enhancers and broad H3K4me3 signals to install MB-suppressive programs.
RESULTS
Brain-Specific Knockout of Mll4 in Mouse Induces Spontaneous Medulloblastoma
To establish an Mll4 brain-specific knockout (Mll4 BSKO) mouse model, we generated Mll4flox/+ (=Mll4f/+) mice that harbor the Mll4 gene with a region (exons 16–19) encompassed by two loxP sites (i.e., floxed Mll4) in one allele in chromosome 15 (Figures S1A–S1C). To inactivate Mll4 in mouse brain, we used Nestin (Nes)-Cre+/- mice, which express Cre recombinase (as early as E8.5) under the Nestin promoter and enhancer that are specific largely for both neural stem cells and neural progenitor cells in the central nervous system (Dubois et al., 2006). This resulted in Nes-Cre Mll4f/f (i.e., Mll4 BSKO) mice in which the homozygous deletion of exons 16– 19 occurred mainly in the central nervous system, including the cerebellum (Figures S1A and S1D).
To determine whether Mll4 loss affects mouse brain development or structure, we analyzed Mll4f/f, Nes-Cre Mll4f/+, and Mll4 BSKO brains using magnetic resonance imaging (MRI). Our MRI data showed that Mll4 BSKO mice, but not the other mouse groups (Mll4f/f and Nes-Cre Mll4f/+), were likely to develop spontaneous tumors in the cerebellum (Figures 1A, 1B, S2A, and S2B). In support of this, brains that were isolated from 7-month-old Mll4 BSKO mice displayed tumors in the cerebella (Figure 1C). Histological analysis confirmed that MBs were in the cerebella of 7-month-old Mll4 BSKO mice but not in the cerebella of control mice (Figures 1D, S2C, and S2D). These tumors were classic MB, as the neoplastic cells were small and poorly differentiated and had uniform nuclei without an obvious nuclear size pleomorphism (Figure 1D). There were neither prominent nucleoli nor areas of tumor cells engulfing other tumor cells, which are features of anaplastic and large cell MB. In line with Mll4 loss in Mll4 BSKO cerebella, immunohistochemical (IHC) analyses showed that MLL4 levels were greatly reduced in 4- and 7-month-old Mll4 BSKO cerebella compared with control cerebella (Figure S2E).
Figure 1. Mll4 Loss in Mouse Brain Results in Spontaneous MB.

(A) T2-weighted horizontal magnetic resonance images of control (Mll4f/f), Nes-Cre Mll4f/+, and Nes-Cre Mll4f/f (Mll4 brain-specific knockout, i.e., Mll4 BSKO) mice (4- and 7-month-old). Yellow-dotted lines denote tumors in the cerebellum in Mll4 BSKO mice. White scale bars, 0.25 mm.
(B) Quantification of tumor volume in Mll4 BSKO mice (7-month-old) using MRI data. Data are presented as the mean ± SEM (error bars) (n ≥ 3).
(C) Representative pictures for Mll4f/f and Mll4 BSKO Mll4f/f mice and their perfused brains. Arrows indicate cerebellum and tumor-bearing Mll4 BSKO cerebellum.
(D) H&E staining of sagittal brain sections from Mll4f/f, Nes-Cre Mll4f/+, and Mll4 BSKO (1-, 4- and 7-month-old). Whole brains and cerebellar regions are shown. The green-boxed regions are also presented in a higher magnification. Representative image for Mll4f/f (n=3), Nes-Cre Mll4f/+ (n=3), and Mll4 BSKO (n=5) cerebella are presented. Black scale bars, 200 μm; yellow scale bars, 40 μm.
(E) Tumor-free percentages in Mll4f/f, Nes-Cre Mll4f/+, and Mll4 BSKO mice. Tumors in Mll4 BSKO mice (18 males and 8 females) were confirmed using H&E staining or MRI.
(F – H) Transplantation experiments of Mll4 BSKO MB. Four weeks after cell injection, tumor nodules were formed on the right flank (F). Tumor volume analysis (G) and H&E staining (H) were performed. Yellow scale bars, 50 μm.
See also Figures S1 and S2.
Our analysis of tumor incidence showed that 9 male mice of 26 Mll4 BSKO mice (34.6%) had tumors in the cerebella, consistent with male preference of human MB (Taylor et al., 2012) (Figure 1E). Interestingly, our analysis of MB genomics databases in cBioPortal showed that MLL4 (about 8.2%) was among the most frequently mutated genes in MB and that its mutation rates were similar to those of CTNNB1 and PTCH1, two highly mutated genes in MB (Figure S2F). To confirm tumorigenicity of Mll4 BSKO MB cells, we performed tumor transplantation experiments. Specifically, we isolated cells from 7-month-old wild type cerebellum and tumor-bearing Mll4 BSKO cerebellum and then injected control cells and Mll4 BSKO MB cells into left flanks and right flanks of nude mice, respectively. Our results showed that all 4 mice injected with Mll4 BSKO MB cells generated tumors whereas none of mice injected with normal cerebellar cells produced a tumor in the same mice (Figures 1F and 1G). Hematoxylin and eosin (H&E) staining showed that these tumor cells were similar to those in the Mll4 BSKO MB (Figure 1H). These results indicate that MLL4 has a tumor-suppressive function in the cerebellum.
Mll4 Loss Increases Cell Proliferation in the Cerebellum but Negatively Impacts Cerebellar Neurons
Because tumorigenesis requires dysregulated cell proliferation (Hanahan and Weinberg, 2011), we assessed the effect of Mll4 loss on cell proliferation in the cerebellum by performing IHC analysis for the cell proliferation marker Ki-67. Ki-67 levels were highly increased in 4-month-old Mll4 BSKO cerebellum and in 7-month-old tumor-bearing Mll4 BSKO cerebella but not in 1-month-old Mll4 BSKO cerebella, compared with control Mll4f/f cerebella (Figure 2A). Increased Ki-67 levels in the neoplastic cells suggest that Mll4 BSKO MB is a highly proliferative type of neoplasm. It is worth noting that 4-month-old Mll4 BSKO mice might not have full blown MB but have increased cell proliferation in the cerebellum. In contrast, cellular levels of the apoptosis marker caspase-3 were generally low in both Mll4f/f and Mll4 BSKO cerebella and were not changed between Mll4f/f and Mll4 BSKO cerebella (Figure 2B). These results indicate that Mll4 loss increases cell proliferation without obviously affecting cell apoptosis.
Figure 2. Mll4 Loss Augments Cell Proliferation in the Cerebellum and Impairs Cerebellar Neurons.

(A and B) IHC staining of Ki-67 (cell proliferation marker; brown nuclear stain; A) and caspase-3 (apoptotic marker; brown nuclear stain; B) in Mll4f/f and Mll4 BSKO cerebella (n=3 for each group).
(C and D) IHC analysis of NeuN (neuronal cell marker; brown nuclear stain; C) and calbindin (Purkinje cell marker; red or brown nuclear and cytoplasmic stain; D) in Mll4f/f and tumor-bearing Mll4 BSKO cerebella (1-, 4- and 7-month-old). NeuN- and calbindin-positive cells were quantified. Blue color was from blue nuclei staining by hematoxylin, suggesting the lack of marker staining. Black scale bars, 200 μm; yellow scale bars, 40 μm.
In the cerebellum, Purkinje cells and granule cells are among the major types of neurons, with granule cells being the most abundant cell type (White and Sillitoe, 2013) (Figure S2C). Calbindin is a neuronal marker for Purkinje cells. NeuN is a marker for most mature neuronal cell types, although in the cerebellum it labels only granule cells. To determine whether Mll4 loss affects Purkinje and granule neurons, we compared control Mll4f/f cerebella with Mll4 BSKO cerebella by performing IHC staining for NeuN and Calbindin. NeuN levels were decreased in 1- and 4-month-old Mll4 BSKO cerebella and in 7-month-old Mll4 BSKO cerebella bearing tumors, compared with control Mll4f/f cerebella. NeuN staining results suggest the lack of neuronal maturation or differentiation in the neoplastic cells (Figure 2C). The number of Calbindin-stained Purkinje cells was reduced in Mll4 BSKO cerebella compared with Mll4f/f cerebella (Figure 2D). These results indicate that Mll4 loss negatively impacts cerebellar neurons.
Mll4 Loss Enhances Oncogenic Signaling Programs, Including Ras Activators and Notch Pathway Components, but Downregulates Neuronal Gene Expression Programs
To understand the molecular mechanism by which Mll4 loss induce MB, we compared transcriptomic profiles between Mll4f/f and Mll4 BSKO cerebella using RNA-seq. Gene ontology analysis showed that multiple genes associated with oncogenic signaling programs, including guanine nucleotide exchange factors (GEFs) for the Ras superfamily (hereinafter referred as to RasGEFs) and Notch signaling components, were upregulated by Mll4 loss (Figure 3A). In contrast, neuronal gene expression programs, such as cerebellar granule cell differentiation and postsynaptic membrane genes, were downregulated by Mll4 loss (Figure 3B; Table S1), consistent with our histological analysis (Figure 2).
Figure 3. Mll4 Loss Downregulates Tumor Suppressor Genes and Neuronal Genes While Upregulating Oncogenic Ras and Notch Pathway Components.

(A and B) Ontology analysis of genes that were at least 2-fold upregulated or downregulated by Mll4 loss. Gene ontology analysis of RNA-seq data of 1-month and 4-month-old cerebella was determined using DAVID.
(C–F) Comparison of expression levels of RasGEFs (C and D) and Notch pathway components (E and F) between Mll4f/f and Mll4 BSKO cerebella (4- and 7-month-old) using quantitative RT-PCR.
(G, H, and I) Expression of neuronal genes (G), tumor suppressors/transcriptional repressor genes (H), Dnmt1, Dnmt3a, and Dnmt3b (I) in Mll4f/f and Mll4 BSKO cerebella (4-month-old). Quantitative RT-PCR was performed. Dnmt3a expression data are shown in both H and I for direct comparison. Data are presented as the mean ± SEM (error bars) of at least three independent experiments. Green scale bars, 40 μm; *p < 0.05, **p < 0.01, and ***p<0.001.
RasGEF genes upregulated by Mll4 loss include Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1, which encode activators for the Ras superfamily (Ras activators) (Vigil et al., 2010). Rasgrp1, Rasgrf1, and Rasgrf2 activate Ras GTPases, such as HRas, NRas, and KRas, by mediating the exchange of GDP for GTP in Ras GTPases (Vigil et al., 2010). In their GTP-bound state, Ras GTPases bind their effectors to stimulate subsequent Ras signaling processes. Rgl1 is a GEF for the Ras homologue Ral (Vigil et al., 2010). Rasgrp1 and Rasgrf1 were shown to promote tumorigenesis (Sharma et al., 2014; Tarnowski et al., 2012). In line with RNA-Seq results, quantitative reverse transcription-polymerase chain reaction (RT-PCR) experiments showed that expression of these RasGEF genes in 4-month-old and 7-month-old mice was up to 200-fold increased by Mll4 loss (Figures 3C and 3D). Upregulated expression of Notch signaling components (e.g., the ligands Jag1 and Jag2 as well as the transcriptional effectors Hes1 and Hes5) by Mll4 loss was confirmed by quantitative RT-PCR results (Figures 3E and 3F). Increased Notch signaling has been shown to promote neural stem cell self-renewal and MB formation (Natarajan et al., 2013). Decreased expression of several neuronal program genes by Mll4 loss were also confirmed by quantitative RT-PCR results (Figure 3G).
To examine whether Mll4 loss changes expression patterns in signature genes for four major molecular MB subtypes (Genovesi et al., 2013; Northcott et al., 2012b), we analyzed our RNA-Seq data of 4-month-old Mll4f/f and Mll4 BSKO cerebellum. Expression of multiple signature genes for the most devastating but poorly characterized MB subtype Group 3 was elevated in Mll4 BSKO cerebella compared with control cerebella (Figure S3A). In addition, some signature genes for other three subtypes (albeit fewer than those for Group 3) were upregulated. To confirm these results, we compared expression patterns of signature genes between 7-month-old Mll4 BSKO cerebella bearing MB and 7-month-old control cerebella using quantitative RT-PCR analysis. Results from this analysis were similar to those from RNA-Seq analysis (Figure S3B). We next performed IHC analysis of 7-month-old Mll4 BSKO MB using antibodies against well-known molecular markers for each MB subtypes (CTNNB1 and DKK2 for WNT; GAB1 for SHH; NPR3 and Myc for Group 3; and KCNA1 for Group 4) (Taylor et al., 2012). In line with the above RNA-seq results, IHC analysis showed that the Group 3 markers NPR3 and Myc were increased in Mll4 BSKO MB compared with control cerebellum whereas CTNNB1, DKK2, GAB1, and KCNA1 levels were not changed by Mll4 loss (Figure S3C). Thus, Mll4 BSKO MB might be close to Group 3. Because MB is classified as an embryonal brain tumor type according to the world health organization, we analyzed expression patterns of signature genes for other embryonal brain tumor types. However, Mll4 loss did not obviously change expression of signature genes for several CNS-PNET subtypes (Sturm et al., 2016) (Figure S3D). These results indicate that Mll4 BSKO MB has at least in part human MB characteristics.
Tumor-Suppressive Transcriptional Co-repressors, Such as DNMT3A, SIRT1, and BCL6, Are Downregulated by Mll4 Loss
To further understand the molecular basis underlying MB driven by Mll4 loss, we sought to assess how oncogenic RasGEFs and Notch signaling components are highly upregulated by Mll4 loss. Because MLL4 generally acts as a transcriptional co-activator and MLL4’s target genes are likely downregulated by Mll4 loss, expression of these oncogenic factors may not be directly downregulated by MLL4. Therefore, we reasoned that expression of RasGEFs and Notch signaling components may be downregulated by tumor-suppressive transcriptional repressors (or co-repressors) that are directly activated by MLL4. We searched transcriptional co-repressor/repressor genes among tumor suppressor genes that are downregulated by Mll4 loss. Such transcription-repressive tumor suppressor genes included Dnmt3a, Sirt1, Bcl6, Pax6, Foxo3, and Cbfa2t3 (Table S1).
DNMT3A is considered to be a tumor suppressor protein, because deletion of Dnmt3a promotes K-Ras–induced lung tumorigenesis (Gao et al., 2011) and DNMT3A mutations correlate with poor survival (Ley et al., 2010). It has been shown that the well-known lymphoma oncoprotein BCL6, together with SIRT1, suppresses MB (Tiberi et al., 2014). Our quantitative RT-PCR confirmed the downregulation of Dnmt3a, Bcl6, Sirt1, and other genes (Pax6, Foxo3, and Cbfa2t3) by Mll4 loss (Figure 3H). In contrast, expression of other DNA methyltransferases, Dnmt1 and Dnmt3b, was not affected by Mll4 loss (Figure 3I).
MLL4 Indirectly Downregulates Ras Activators by Enhancing DNMT3A-Mediated DNA Methylation
Several promoters of RasGEF genes, including Rasgrp1, Rasgrf1, and Rasgrf2, belong to H3K4me3-low/CpG-poor group (data not shown). Interestingly, DNMT3A is a de novo DNA methyltransferase that can repress gene expression by methylating CpG sites at H3K4me3-low/CpG-poor promoters (Wu et al., 2010). Therefore, we tested the possibility that low DNMT3A levels in Mll4 BSKO decrease DNA methylation levels at Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1 genes to derepress the same genes. We compared CpG methylation states at these RasGEF genes between Mll4f/f and Mll4 BSKO cerebella. DNA methylation analysis involving sodium bisulphite treatment showed that CpG methylation levels at the gene-regulatory regions in Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1 genes were decreased by Mll4 loss (Figures 4A–4F). Our quantitative ChIP data demonstrated that DNMT3A occupied the same regions in these RasGEF genes (Figure 4G). Consistent with global reduction of its levels (Figure 4H), DNMT3A levels at the gene-regulatory regions of RasGEF genes were decreased by Mll4 loss (Figure 4I). These results indicate that DNMT3A directly methylates CpG sites in Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1 genes to repress gene expression.
Figure 4. Mll4 Loss Reduces DNMT3A-mediated CpG Methylation at the Gene-regulatory Regions of Ras Activators to Increase Their Expression.

(A – E) DNA methylation states at Rasgrp1 (A), Rasgrf1 (B), Rasgrf2 (C), Rapgef5 (D), and Rgl1 (E) genes in Mll4f/f and Mll4 BSKO cerebella. Cerebella from 4-month-old mice were used for bisulfite treatment. Green lines and each circle represent PCR amplicons for DNA methylation assay and a CG dinucleotide, respectively. Red line above the schematic representation of each gene indicates PCR amplicons for ChIP. Open circles, unmethylated; filled circles, methylated; arrows, transcription start sites.
(F) Quantification of methylation levels at CpG sites marked by the red rectangles in A–E.
(G and I) DNMT3A occupancy at Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1 genes in 4-month-old Mll4f/f (G and I) and Mll4 BSKO cerebella (I). Chromatin levels for DNMT3A were analyzed by quantitative ChIP (qChIP) assay.
(H) IHC analysis of DNMT3A in Mll4f/f and tumor-bearing Mll4 BSKO cerebella. Green scale bars, 40 μm.
(J and K) The effect of acute Dnmt3a knockdown on Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1 mRNA levels in primary neurospheres (J) and the effect of ectopic Dnmt3a expression on Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1 mRNA levels in Mll4-deleted neurospheres (K). Quantitative RT-PCR assays were used to measure mRNA levels.
(L) Repression of RasGEF genes (i.e., Ras activators) by MLL4-mediated upregulation of the DNMT3A levels.
To vigorously determine whether DNMT3A represses Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1 genes, we examined the effect of acute Dnmt3a knockdown on expression of these RasGEF genes using siRNAs against Dnmt3a. For acute knockdown experiment, we used neurospheres isolated from the cerebella, because neurospheres known as non-adherent spherical clusters of cells containing neural stem and progenitor cells are often used to model neural development and differentiation (Reynolds and Rietze, 2005). Our RNA interference experiments with use of neurospheres isolated from 5-day-old mouse cerebella demonstrated that DNMT3A knockdown increased expression of Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1 genes in neurospheres (Figure 4J). In addition, in Mll4-deleted neurospheres with reduced Dnmt3a expression, ectopic Dnmt3a expression repressed expression of these RasGEF genes upregulated by Mll4 loss (Figure 4K). These results confirm that MLL4 upregulates Dnmt3a expression to repress multiple RasGEF genes via DNMT3A-mediated DNA methylation in the cerebellum (Figure 4L).
MLL4 Indirectly Antagonizes Notch Pathway Components (e.g., Hes1 and Jag1) by Upregulating SIRT1/BCL6-Mediated H4K16 Deacetylation
As mentioned above, Mll4 loss increased expression of the Notch signaling pathway genes Hes1, Hes5, Jag1, and Jag2. Canonical Notch signaling is activated by the interaction of the receptors Notch 1‒4 with their ligands (e.g., JAG1 and JAG2). JAG1 and JAG2 were found to be critical pro-survival factors in MB (Fiaschetti et al., 2014). The transcription factors HES1 and HES5 act as essential Notch effectors for Notch-mediated inhibition of neuron differentiation (Ohtsuka et al., 1999). In addition, expression levels of Hes1 and Hes5 are frequently upregulated in MB (Hallahan et al., 2004). Therefore, we examined how MLL4 regulates Hes1, Hes5, Jag1, and Jag2.
SIRT1 has been reported to suppress the proliferation of neural stem cells by repressing Hes5 (Ma et al., 2014). This report prompted us to analyze whether SIRT1 is recruited to Hes1, Hes5, Jag1, and Jag2 genes using publicly available SIRT1 data from chromatin immunoprecipitation (ChIP) with DNA sequencing (ChIP-seq) of the cerebellum. SIRT1 occupied all of these genes (Figure 5A). Because SIRT1 and BCL6 cooperate for the MB-suppressive function (Tiberi et al., 2014) and Mll4 loss decreased expression of both Bcl6 and Sirt1 (Figure 3H), we examined the possibility that BCL6 is also recruited to Hes1, Hes5, Jag1, and Jag2 genes. Our ChIP data suggested that BCL6 and SIRT1 co-occupied Hes1, Hes5, Jag1, and Jag2 genes (Figures 5B–E). In line with global reduction of BCL6 and SIRT1 (Figure 5F), Mll4 loss decreased BCL6 and SIRT1 levels at Hes1, Hes5, Jag1, and Jag2 genes (Figures 5B–5E). Because SIRT1 preferentially deacetylates the gene-activating mark acetylated H4K16 (H4K16ac) (Vaquero et al., 2004), we assessed the effect of Mll4 loss on H4K16ac levels in Hes1, Hes5, Jag1, and Jag2 genes. Our ChIP data showed that Mll4 loss increased H4K16ac levels at these genes (Figures 5B–5E). To further determine whether BCL6 and SIRT1 downregulates Hes1, Hes5, Jag1, and Jag2 genes, we examined the effect of acute BCL6 or SIRT1 knockdown on expression of these genes in primary cerebellar neurospheres. Our results demonstrated that BCL6 or SIRT1 knockdown increased expression of Hes1, Hes5, Jag1, and Jag2 genes (Figures 5G & 5H). In addition, in Mll4-deleted neurospheres with decreased Bcl6 and Sirt1 expression, ectopic Bcl6 or Sirt1 expression downregulated expression of these Notch pathway genes upregulated by Mll4 loss (Figures 5I & 5J). These results indicate that SIRT1 and BCL6 co-repress expression of Hes1, Hes5, Jag1, and Jag2 by downregulating H4K16ac levels and that MLL4 increases SIRT1/BCL6-mediated deacetylation of H4K16ac to indirectly repress expression of these Notch signaling components (Figure 5K).
Figure 5. Mll4 Loss Decreases SIRT1/BCL6-Mediated H4K16 Deacetylation at the Gene-regulatory Regions of Notch Pathway Components to Increase Their Expression.

(A) ChIP-seq profiles for SIRT1 at Hes1, Hes5, Jag1, and Jag2 genes in mouse cerebellum (from the publicly available data GSM427095).
(B – E) The effect of Mll4 loss on chromatin levels of BCL6, SIRT1, and H4K16ac at the promoters of Hes1 (B), Hes5 (C), Jag1 (D), and Jag2 (E) genes. Quantitative ChIP assays was performed using samples prepared from 4-month-old mouse cerebella.
(F) IHC analysis of SIRT1 and BCL6 in Mll4f/f and tumor-bearing Mll4 BSKO cerebella. Green scale bars, 40 μm.
(G and H) The effect of SIRT1 (G) or BCL6 (H) knockdown on Hes1, Hes5, Jag1, and Jag2 mRNA levels in primary neurospheres.
(I and J) The effect of ectopic SIRT1 (I) or BCL6 (J) expression on Hes1, Hes5, Jag1, and Jag2 mRNA levels in Mll4-deleted neurospheres. Quantitative RT-PCR assays were used to measure mRNA levels.
(K) Regulation of Notch pathway components by MLL4.
Data are presented as the mean ± SEM (error bars) of at least three independent experiments. *p < 0.05, **p < 0.01, and ***p<0.001.
MLL4 Is Indispensable for Genome-wide Establishment of Super-enhancers and Broad H3K4me3 peaks
To comprehensively assess the epigenetic basis underlying MB genesis driven by Mll4 loss, we sought to examine the effect of Mll4 loss on genome-wide landscapes of MLL4-regulated epigenetic marks using ChIP-seq. It has been shown that MLL4 deposits H3K4me1 in enhancers in Drosophila and mammalian cells (Herz et al., 2012; Hu et al., 2013; Lee et al., 2013). Moreover, we and others have shown that MLL4 is able to catalyze trimethylation besides mono-methylation at H3K4 (Dhar et al., 2012; Lee et al., 2007; Zhang et al., 2015b). We first examined the effect of Mll4 loss on global H3K4me1 and H3K4me3 levels in mouse cerebella using Western blot analysis H3K27ac was also included because H3K4me1 in the presence of H3K27ac denotes active enhancers (Smith and Shilatifard, 2014). Interestingly, 4-month-old Mll4 BSKO cerebella, which showed high cell proliferation as a sign of cellular transformation, still had low H3K4me1, H3K27ac, and H3K4me3 levels that resulted from Mll4 loss (Figures S4A and S4B). However, these histone marks were substantially recovered in 7-month-old tumor-bearing Mll4 BSKO cerebella, unlike in 4-month-old Mll4 BSKO cerebella, suggesting that other events, such as altered H3K4 methyltransferases and demethylases, may cause to recover H3K4me3 and H3K4me1 levels during tumorigenesis (Figure S4C). Because this recovery in 7-month-old tumor-bearing Mll4 BSKO cerebella is likely an indirect consequence of Mll4 loss during tumorigenesis, we used 4-month-old cerebellar tissues rather than 7-month-old cerebellar tissues to perform ChIP-seq.
Enhancers can be classified into super-enhancers and typical enhancers (Whyte et al., 2013). Our ChIP-seq analysis of super-enhancers and typical enhancers on the basis of H3K27ac signals showed that Mll4 loss decreased average H3K4me1 and H3K27ac levels more in super-enhancers (top 1000 enhancers) than in typical enhancers (Figures 6A–6D and S4D–S4H). Similar results were obtained from additional enhancer analysis on the basis of H3K4me1 signals (Figures 6A–6D vs. S5A–S5E). We also determined whether Mll4 loss affects broad H3K4me3, an epigenomic signature associated with tumor suppression (Chen et al., 2015). Our analysis showed that average H3K4me3 levels in broad H3K4me3 (top 1000 broad H3K4me3 peaks) were decreased by Mll4 loss (Figures 6E and S5F). In addition, Mll4 loss reduced H3K4me1 and H3K27ac levels in broad H3K4me3 (Figures 6F and 6G). In contrast, Mll4 loss had no obvious effect on average densities of sharp H3K4me3 peaks (Figure 6H). Interestingly, MLL4 ChIP-seq analysis showed that MLL4 appeared to be enriched in super-enhancers and broad H3K4me3 (Figure S5G). Substantial percentages of 1,000 super-enhancers and 1,000 broad H3K4me3 peaks had MLL4 signals in Mll4f/f cerebella, and most of the signal intensities of MLL4-positive super-enhancers and MLL4-positive broad H3K4me3 peaks were decreased by Mll4 loss (Figures S5H–S5N). To further address the role of MLL4 in regulating super-enhancers and broad H3K4me3, we examined the effect of acute Mll4 deletion on super-enhancers and broad H3K4me3. Specifically, we infected primary neurospheres isolated from Mll4f/f cerebella using Ad-Cre viruses and then performed ChIP-seq for H3K4me1, H3K4me3, and H3K27ac. Consistent with ChIP-seq results from use of Mll4f/f and Mll4 BSKO cerebella, our results showed that Mll4 loss in neurospheres reduced average H3K4me1 and H3K27ac levels more in super-enhancers than in typical enhancers (Figures 6I–6L and S5O–S5R). In addition, Mll4 loss in neurospheres decreased average H3K4me3, H3K27ac, and H3K4me1 levels in broad H3K4me3 but did not have an obvious effect on average densities of sharp H3K4me3 peaks (Figures 6M–6P). These results suggest that during MB genesis in Mll4 BSKO mice, super-enhancers and broad H3K4me3 at the genome-wide levels are severely impaired by Mll4 loss.
Figure 6. Mll4 Loss Results in Genome-wide Impairment of Broad H3K4me3 and Super-enhancers.

(A ‒ D) Average ChIP-seq read densities for H3K27ac (A and B) and H3K4me1 (C and D) at super-enhancers (A and C) and all typical enhancers (B and D) in 4-month-old Mll4f/f and Mll4 BSKO cerebella. Enhancers were defined using H3K27ac signals.
(E ‒ H) Average ChIP-seq read densities for H3K4me3 (E), H3K4me1 (F), and H3K27ac (G) at broad H3K4me3 regions and for H3K4me3 (H) at sharp H3K4me3 regions in 4-month-old Mll4f/f and Mll4 BSKO cerebella.
(I ‒ L) Average ChIP-seq read densities for H3K27ac (I and J) and H3K4me1 (K and L) at super-enhancers (I and K) and all typical enhancers (J and L) in cerebellar neurospheres treated with Adeno-empty or Adeno-Cre. Enhancers were defined using H3K27ac signals.
(M ‒ P) Average ChIP-seq read densities for H3K4me3 (M), H3K4me1 (N), and H3K27ac (O) at broad H3K4me3 regions and for H3K4me3 (P) at sharp H3K4me3 regions in cerebellar neurospheres treated with Adeno-Empty or Adeno-Cre.
(Q) Boxplots showing eRNA levels (Fragments Per Kilobase of transcript per Million mapped reads; FPKM) from super-enhancers and typical enhancers in Mll4f/f and Mll4 BSKO cerebella (4-month-old).
(R and S) Comparison of mRNA levels (FPKM) of genes associated with super-enhancers (R), typical enhancers (R), broad H3K4me3 (S), and sharp H3K4me3 (S) between Mll4f/f and Mll4 BSKO cerebella (4-month-old).
For A‒H and I‒P, average profiles of two ChIP-seq data was used to generate individual density plots, and horizontal axis represents a region from −10 kb to +10 kb with the respect to the center. For comparison in Q‒S, 1,000 genes were used for typical enhancers and sharp H3K4me3. The p values were calculated using the Wilcoxon signed rank test.
See also Figures S4 and S5.
Enhancer regions are transcribed by RNA Polymerase II in a bidirectional manner and express their own RNAs called enhancer RNAs (eRNAs) (Kim and Shiekhattar, 2015). Enhancer activities can be measured by eRNA expression levels (Kim and Shiekhattar, 2015). Interestingly, analysis of our RNA-seq data of Mll4f/f and Mll4 BSKO cerebella showed that Mll4 loss decreased eRNA levels more in super-enhancers than in typical enhancers (Figure 6Q). We also analyzed whether alterations of super-enhancers and broad H3K4me3 by Mll4 loss are linked to changes in gene expression. Decreases in super-enhancer and broad H3K4me3 signals by Mll4 loss were accompanied with significantly reduced levels in gene expression (Figures 6R and 6S). In contrast, decreases of typical enhancers and sharp H3K4me3 signals by Mll4 loss had no substantial effect on gene expression (Figures 6R and 6S). Consistent with the high gene-activating function of super-enhancers and broad H3K4me3, genes associated with super-enhancer and broad H3K4me3 overall had higher expression than did those associated with typical enhancers and sharp H3K4me3 (Figures 6R and 6S). These results indicate that activities of super-enhancers and broad H3K4me3 are reduced during cellular transformation driven by Mll4 loss. They also suggest that Mll4 loss preferentially downregulates genes associated with super-enhancers and broad H3K4me3 rather than those associated with typical enhancers and sharp H3K4me3 peaks.
Mll4 Loss Diminishes Both Super-enhancers and Broad H3K4me3 Peaks in Tumor Suppressor Genes (e.g., Dnmt3a and Bcl6) and Neuronal Genes (e.g., Neurod2)
To understand how MLL4 epigenetically activates tumor suppressor genes, we examined ChIP-seq profiles of H3K4me1 and H3K27ac at Dnmt3a and Bcl6 genes. Interestingly, Dnmt3a and Bcl6 were marked by both super-enhancer and broad H3K4me3 in mouse cerebella and mouse cerebellar neurospheres (Figures 7A, 7B, S6A and S6B). MLL4 was localized at both super-enhancers and broad H3K4me3 peaks in Dnmt3a and Bcl6 in mouse cerebella (Figures 7A–7C). Quantitative ChIP showed that MLL4 occupancy was decreased in Mll4 BSKO cerebella compared with Mll4f/f cerebella (Figure 7C). ChIP-seq results showed that in Dnmt3a and Bcl6 genes in mouse cerebella and mouse cerebellar neurospheres, Mll4 loss decreased H3K4me1 and H3K27ac levels in the super-enhancers and slightly reduced H3K4me3 levels in the broad H3K4me3 peaks (Figures 7A, 7B, S6C, and S6D). To complement ChIP-seq data, we performed quantitative ChIP assays. Results showed that Mll4 loss clearly decreased not only H3K4me1 levels but also H3K4me3 levels at Dnmt3a and Bcl6 in mouse cerebella (Figures 7D and 7E). These results indicate that MLL4 directly and positively regulates expression of Dnmt3a and Bcl6 by establishing broad H3K4me3 peaks and super-enhancers at these genes in mouse cerebellum.
Figure 7. Mll4 Loss Weakens Both Super-enhancers and Broad H3K4me3 in the Dnmt3a and Bcl6 Genes.

(A and B) ChIP-seq profiles for H3K27ac (green), H3K4me1 (gray), and H3K4me3 (blue) at the Dnmt3a (A) and Bcl6 (B) genes in 4-month-old mouse Mll4f/f and Mll4 BSKO cerebella. ChIP-seq profiles for MLL4 (pink) in Mll4f/f cerebellum are also shown. ChIP-Seq signals in the tracks were generated by subtracting input signals from their original signals. Peak tracks in the BigBed format are shown as black lines under the bottom of ChIP-Seq tracks.
(C) The effect of Mll4 loss on MLL4 occupancy at Dnmt3a and Bcl6.
(D and E) The effect of Mll4 loss on chromatin levels of H3K4me3 (D) and H3K4me1 (E) at Dnmt3a and Bcl6.
(F) Chromatin interactions (3C) between the promoter and the super-enhancer’s regions at Dnmt3a and Bcl6 in Mll4f/f and Mll4 BSKO cerebella.
(G) Chromatin interactions (3C) among the various regions in the super-enhancers at Dnmt3a and Bcl6 in Mll4f/f and Mll4 BSKO cerebella.
(H) The effect of Mll4 loss on eRNA levels expressed from various regions in the super-enhancers in Dnmt3a and Bcl6. eRNA levels were measured using quantitative RT-PCR.
(I) The effect of Mll4 loss on chromatin levels of UTX and P300 at Dnmt3a and Bcl6 enhancers. Quantitative ChIP assay (C, D, E, and I) was performed using 4-month-old Mll4f/f and Mll4 BSKO cerebella.
(J) A hypothetical model showing a tumor suppressive role for MLL4. MLL4 may activate expression of tumor suppressor genes by establishing super-enhancers and broad H3K4me3 peaks. *p < 0.05, **p < 0.01, and ***p<0.001.
See also Figures S6 and S7 and Table S1.
Enhancers activate gene expression by interacting with the promoter regions (Kim and Shiekhattar, 2015). Because H3K4me1 and H3K27ac levels at super-enhancers in Dnmt3a and Bcl6 were decreased by Mll4 loss, we determined whether decreased H3K4me1 and H3K27ac levels affect the interactions between the super-enhancer and the promoter in Dnmt3a as well as in Bcl6 using a chromatin conformation capture (3C) assay. Our 3C results showed that chromatin interactions between the promoter with broad H3K4me3 peak and several regions in the super-enhancer in Dnmt3a as well as in Bcl6 were highly reduced by Mll4 loss (Figure 7F). In addition, the interactions among the various regions in the super-enhancers in Dnmt3a and Bcl6 were lowered by Mll4 loss (Figure 7G). We then examined the effect of Mll4 loss on eRNA expression from the super-enhancers of Dnmt3a and Bcl6. Because these super-enhancers are located in the gene body, we measured anti-sense eRNAs from the various regions in the super-enhancers. Our quantitative RT-PCR results showed that eRNA levels in the various regions in the super-enhancers in Dnmt3a and Bcl6 were decreased by Mll4 loss (Figure 7H). These results suggest that activities of the super-enhancers and broad H3K4me3 regions in Dnmt3a and Bcl6 are downregulated by Mll4 loss.
Because it has been reported that a crosstalk between MLL4-UTX complex and the acetyltransferase p300 is critical for enhancer establishment (Wang et al., 2017), we examined the effect of Mll4 loss on chromatin levels of the H3K27 demethylase UTX and p300 in Dnmt3a and Bcl6 genes. Mll4 loss decreased the occupancy of UTX and p300 in Dnmt3a and Bcl6 (Figure 7I), suggesting inter-dependency between MLL4, UTX and p300 in establishing Dnmt3a and Bcl6 super-enhancers.
Other MLL4-activated tumor suppressor genes with both super-enhancer and broad H3K4me3 included Foxo3 and Pax6 (Figures S6E–S6H; Table S1). However, Sirt1 had a sharp H3K4me3 and regular enhancer (data not shown). MLL4-activated neuronal genes with both super-enhancer and broad H3K4me3 included Neurod2, which is necessary for the survival of cerebellar neurons (Olson et al., 2001) (Figure S7A and S7B; Table S1).
We comprehensively analyzed the effect of Mll4 loss on expression profiles of genes associated with super-enhancers or broad H3K4me3 peaks. A significant number of genes associated with super-enhancers (348 genes; P = 3.05 × 10−144) or broad H3K4me3 (332 genes; P = 2.29 × 10−130) were downregulated by Mll4 loss (Figures S7C and S7D). Interestingly, of such genes downregulated by Mll4 loss, genes associated with super-enhancers highly overlapped with those associated with broad H3K4me3 (Figure S7E). Consistent with strongly reduced NeuN and Calbindin staining of Mll4 BSKO cerebella, many neuron differentiation genes associated with super-enhancers and broad H3K4me3 were downregulated by Mll4 loss (Figures S7F and S7G; Table S1). In addition, our analysis using the tumor suppressor list from a publicly available database (Zhao et al., 2013) showed that a significant number of tumor suppressor genes associated with super-enhancers and broad H3K4me3 were downregulated by Mll4 loss (Figures S7H and S7I; Table S1). These results further support a role for MLL4 in activating tumor suppressor genes and neuronal genes by positively regulating super-enhancers and broad H3K4me3 peaks.
DISCUSSION
Broad H3K4me3 is an epigenomic signature that occupies the TSSs in tumor suppressor genes and cell identity genes. Super-enhancers activate gene expression by interacting with the TSSs via their associated factors. Our findings showed that brain-specific Mll4 loss caused genome-wide decreases of broad H3K4me3 and super-enhancer signals while frequently inducing cerebellar tumors. Notably, Mll4 loss diminished broad H3K4me3 and super-enhancer signals at tumor suppressor genes while decreasing the interactions between broad H3K4me3 and super-enhancer at the same genes. Among tumor suppressors with broad H3K4me3 and super-enhancer are DNMT3A and BCL6, which repress expression of oncogenic programs, such as Ras activators and Notch signaling components, respectively. Therefore, our findings uncover MLL4 as the first-identified histone modifier that establishes both broad H3K4me3 and super-enhancers to directly activate tumor suppressor genes (e.g., Dnmt3a and Bcl6) (Figure 7J). In addition, our results indicate that MLL4 suppresses oncogenic pathways for MB by directly activating expression of tumor suppressor genes.
Super-enhancers have been linked to cancer (Hnisz et al., 2013; Lin et al., 2016; Loven et al., 2013). Specifically, this epigenomic signature is often established to activate oncogenes in cancer cells through several mechanisms (Sur and Taipale, 2016). One such mechanism is super-enhancers that are aberrantly established by overexpressed trans-acting factors. For example, overexpression of the transcription factor TAL1 in T-cell acute lymphoblastic leukemia is associated with newly formed super-enhancer in the MYC locus (Hnisz et al., 2013). In addition, alterations in noncoding DNA sequences, such as mutations, translocations, and focal amplification, can cause the de novo formation of super-enhancers in oncogene loci and in turn activate oncogene expression. For instance, somatic mutations in T-cell acute lymphoblastic leukemia can generate DNA-binding motifs for the transcription factor MYB, which leads to super-enhancer formation for activation of the oncogene TAL1 (Mansour et al., 2014). For these reasons, it has been suggested that oncogenes can be selectively inhibited by disruption of super-enhancers via BET-bromodomain inhibitors (e.g., JQ1) (Loven et al., 2013). In the current study, we unexpectedly found that Mll4 loss in normal cerebellar cells decreased signals and activities of super-enhancers associated with tumor suppressor genes. Therefore, although the de novo formation of oncogenic super-enhancers during cellular transformation promotes tumorigenesis, it is likely that MLL4-established super-enhancers play an important role in tumor suppression in normal cells. This bifunctional view of super-enhancers is consistent with the Cao et al. study, which suggests that regions occupied by super-enhancers are related to both oncogenes and tumor suppressor genes (Cao et al., 2017).
Our results showed that Mll4 loss reduced more super-enhancer signals than typical enhancer signals (Figure 6). Why may Mll4 loss have a greater effect on super-enhancer signals than on typical enhancer signals? It has been shown that multiple transcription factors are more abundantly enriched at super-enhancers than at typical enhancers (Hnisz et al., 2013). Because of cooperative interaction of multiple factors in super-enhancers, activities of super-enhancers are decreased more than are those of typical enhancers if enhancer-associated factors are inhibited or reduced (Loven et al., 2013). In this regard, the inhibition of the enhancer-associated BET protein BRD4 by JQ1 reduced more the gene transcription associated with super-enhancers than that associated with typical enhancers (Loven et al., 2013). Interestingly, MLL4 may cooperate with UTX and p300 for enhancer establishment for MLL4 target genes (Figure 7I) (Lai et al., 2017; Wang et al., 2017). Therefore, MLL4 may contribute directly to the concerted action of multiple factors in super-enhancers, although MLL4 protein, rather than MLL4-catalyzed H3K4 mono-methylation, is relevant to enhancer function (Dorighi et al., 2017; Rickels et al., 2017).
Epigenetic modifiers, including MLL4, are altered in MB. In contrast to our advanced understanding of sonic hedgehog and wingless pathways, little was known about the roles of altered epigenetic modifiers in MB development. Surprisingly, our results showed that the loss of Mll4 encoding a H3K4 methyltransferase alone caused MB formation. To our knowledge, our Mll4 BSKO mouse model is the first to show that the knockout of an epigenetic modifier can cause MB. We also showed that MLL4 is a tumor-suppressive epigenetic modifier that antagonizes oncogenic programs, e.g., Ras activators and Notch pathway components. Therefore, our findings would provide previously unknown molecular insights into how an epigenetic modifier suppresses MB. In line with its tumor-suppressive role, MLL4 has also been shown to have an antitumor function against B-cell lymphoma genesis (Ortega-Molina et al., 2015; Zhang et al., 2015a). On the contrary, MLL4 may play a role in oncogenesis. For example, reduced MLL4 levels correlate with good prognosis in pancreatic cancer (Dawkins et al., 2016) and MLL4 knockdown decreased breast cancer cell proliferation (Kim et al., 2014). Thus, the function of MLL4 may be dependent on the cellular context, and MLL4 may regulate a different set of genes in a tissue-dependent manner. MLL4 is considered a transcriptional coactivator that may cooperate with a transcription factor to be recruited to its target genes. For instance, MLL4 associates with the adipogenic transcription factors PPARγ and C/EBPβ and regulates adipocyte gene expression (Lee et al., 2013). PPARγ and C/EBPβ are linked to tumor suppression (Lourenco and Coffer, 2017; Yu et al., 2010). Therefore, it is possible that MLL4 may interact with a MB-suppressive transcription factor to suppress MB. It would be interesting to identify such MLL4-interacting transcription factor and study its role in MB in the future.
MB predominantly occurs in children and adolescents younger than age 15 years. However, our data showed that MB lesions were visible in adult Mll4 BSKO mice (e.g., 7-month-old). This suggests that additional genetic or epigenetic alterations besides Mll4 loss may be required for early development of MB in mice. In fact, several mouse models for MB have been developed on the basis of heterozygous or homozygous knockout of two genes (Northcott et al., 2012a). In this regard, it is likely that Mll4 loss cooperates with other oncogenic hits to promote MB formation. For these reasons, our Mll4 BSKO model may be useful for future studies of MB.
DNA methylation is known to be positively and directly associated with gene-repressive histone methylation, such as H3K9 methylation. In contrast, DNA methylation has an antagonistic relationship with H3K4 methylation in regulating gene expression (Cedar and Bergman, 2009). Distinct from this, our results indicate that MLL4-mediated H3K4 methylation upregulates expression of the Dnmt3a gene to increase DNA methylation levels at the promoters of RasGEFs (e.g., Rasgrp1 and Rasgrf1) and to consequently repress RasGEF genes. Thus, our findings reveal that H3K4 methylation positively regulates DNA methylation to indirectly repress gene expression during MB genesis. In addition, MLL4 counteracts Notch signaling programs by increasing BCL6/SIRT1-mediated H4K16 deacetylation at Notch pathway genes, such as Hes1 and Jag1. Therefore, MLL4-mediated H3K4 methylation acts as a regulatory mode that enhances both DNMT3A-mediated DNA methylation and SIRT1/BCL6-mediated H4K16 deacetylation. This mode of MLL4-mediated regulation is unique because a histone methylation modifier indirectly antagonizes oncogenic signaling programs by enhancing two other types of epigenetic modifications, i.e., DNA methylation and H4K16 deacetylation. Moreover, although we cannot exclude the possibility that other yet-to-be-identified pathways may also contribute to MB genesis in Mll4 BSKO cerebella, our findings reveal an unexpected tumor-suppressive mechanism in which MLL4 suppresses oncogenic Ras and Notch pathways, at least in part, by directly activating tumor suppressor genes via the establishment of super-enhancers and broad H3K4me3 peaks. Our findings are likely relevant to a better understanding of human MB with MLL4 mutations.
STAR ★ METHODS
Detailed methods are provided in the online version of this paper and including the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for further information or reagents should be directed to the Lead Contact and corresponding author, Min Gyu Lee (mglee@mdanderson.org)
EXPERIMENTAL MODEL AND METHODS
Mll4 Brain-Specific Knockout (Mll4 BSKO) Mice
To generate an Mll4 brain-specific knockout (Mll4 BSKO) mouse model, an Mll4 targeting vector was transfected into mouse embryonic stem (ES) cells. The Mll4 vector contains the Mll4 exons 16–19 surrounded by the two loxP sites, a neomycin resistance gene, and a thymidine kinase gene. G418-resistant mouse ES cells were injected into the inner cell mass of fresh blastocysts, which were implanted into a pseudo-pregnant female. This approach produced Mll4flox/+ (=Mll4f/+) mice that harbored the Mll4 gene with a region (exons 16–19) encompassed by two loxP sites (i.e., floxed Mll4) in one allele in chromosome 15.
To inactivate Mll4 in mouse brain, Mll4f/+ mice were crossed with Nestin (Nes)-Cre (Jackson no. 003771; B6.Cg-Tg(Nes-Cre)1Kln/J), which expresses Cre recombinase (as early as E8.5) under the Nestin promoter and enhancer specific for both neural stem cells and neural progenitor cells in the central nervous system (Dubois et al., 2006). Mll4f/f mice were crossed with Nes-Cre Mll4f/+ mice. This breeding strategy resulted in Nes-Cre Mll4f/f (i.e., Mll4 BSKO) mice in which the homozygous deletion of the region surrounded by two loxP sites occurred mainly in the central nervous system, including the cerebellum. Cre deleted exons 16–19 and caused open reading frame shift to create a stop codon in exon 20. A truncated MLL4 protein lacking the C-terminal ~4200aa is expected to be produced from Mll4 knockout allele. For genotyping the Mll4 alleles, allelic PCR primers were developed as follows: F1: 5′-AGAATGGACACTGGAGCTCC -3′; R1: 5′-GAAATCCCCAACCACAGC -3’; F2: 5’- TCTCACAGAAGGGACAAGGC -3′; R2: 5′- ACAAGCAGTGACAGACAAGTCC -3’. Genotyping of Nes-Cre was performed by PCR analysis using the following primers: 5’- GCGGTCTGGCAG TAAAAACTATC-3’, 5’- GTGAAACAGCATTGCTGTCACTT -3’, 5’- GAGACTCTGGCTACTCATCC -3’, and 5’- CCTTCAGCAAGAGCTGGGGAC -3’. Humane end-points were implemented in the mouse experiments.
Mouse Study Approval
All animals used and studied were approved by the Institutional Animal Care and Use Committee (IACUC) of The University of Texas MD Anderson Cancer Center.
Magnetic Resonance Imaging (MRI)
To monitor tumor development and progression, MRI was frequently performed for 1- to 12-month-old mice using a BioSpec 70/30 MRI system (Bruker Corp., Billerica, MA). Tumor volumes in mice bearing cerebellar tumors were calculated on the basis of MRI data and ranged from 2.5 mm3 to 15 mm3.
Although tumor formation in both male and female Mll4 BSKO mice were monitored using MRI, female mice did not develop any obvious sign of brain tumors until they were 18‒24 months old. Therefore, male mice were used for subsequent experiments, including ChIP, ChIP-seq, RT-PCR, RNA-seq, 3C, and bisulfite sequencing.
Tumor Transplantation in Subcutaneous Xenograft Mouse Model
Medulloblastoma in the cerebellum was dissected out from 7-month-old Mll4 BSKO mouse. The tumor tissue was finely chopped into small pieces in a sterile petri dish containing Neurobasal medium with penicillin/streptomycin (1 % v/v) (ThermoFisher Scientific, Waltham, MA). Tumor pieces were further mixed with equal volume of Matrigel. Nude mice were anesthetized with isoflurane. The tumor cell /Matrigel mixture (100 μl per mouse) was injected into the right flank of the mouse using 1ml syringe. Similar to medulloblastoma sample, normal cerebellum from Mll4f/f 7-month-old mouse was processed to be used as a control, and cell /Matrigel mixture was injected into the left flank in the same mouse. The treated mice were placed in a clean cage and were observed until complete recovery from anesthesia. Tumor growth was weekly monitored by regular measurement of the length and the width using a Vernier caliper. Tumor volume was calculated by the most commonly used ellipsoid formula: 0.5 × length × width2. Four weeks later, mice were sacrificed. Tumors were excised and the final tumor volume was calculated.
Immunohistochemical Analysis
Mice (4–7 months old) were perfused with 4% paraformaldehyde (PFA), and serial 40-μm-thick coronal sections were cut on a cryostat and collected as free-floating sections. For histological analysis of paraffin sections (cut at 8 μm), a standard hematoxylin/eosin staining procedure and Immunohistochemical (IHC) analysis were used. Briefly, sections were subjected to antigen retrieval (antigen retrieval solution, Vector Laboratories, Burlingame, CA) followed by blocking in 3% BSA with 0.1% Triton X-100 for 1 h. The primary antibodies were used as listed in key resources table. Anti-caspase-3 detects the cleaved large fragment (17 kDa) for caspase-3. IHC staining was quantified with the Aperio Nuclear Algorithm software after images were scanned on Aperio Imagescope from Leica Biosystems.
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ki-67 (IHC) | BD Biosciences | Cat # 550609; RRID:AB_393778 |
| Caspase-3, Cleaved (Asp175) (D3E9) Rabbit mAb (IHC) | Cell Signaling | Cat # 9579; RRID:AB_1520863 |
| NeuN (IHC) | Chemicon | Cat # MAB377; RRID:AB_2298772 |
| Calbindin D-28k (IHC) | Swant | Cat # CB38; RRID:AB_2721225 |
| MLL4 (Western and IHC) | Sigma-Aldrich | Cat # HPA035977 |
| MLL4 (ChIP, Western) | Dr. Ali Shilatiffard | Homemade |
| MLL4-C (ChIP, Western) | Abgent | Cat # AP6183a; RRID:AB_353335 |
| H3K4me1 (ChIP, Western, and IHC) | Abcam | Cat # ab8895; RRID:AB_306847 |
| H3K4me1 (ChIP) | Dr. Ali Shilatiffard | Homemade |
| H3K4me3 (ChIP, Western and IHC) | Millipore | Cat # 07–473; RRID:AB_1977252 |
| H3K27ac (ChIP, Western and IHC) | Abcam | Cat # ab4729; RRID:AB_2118291 |
| H4K16ac (ChIP) | Active Motif | Cat # 39168; RRID:AB_2636968 |
| DNMT3A (ChIP and IHC) | Active Motif | Cat # 39206; RRID:AB_2722512 |
| SIRT1 (ChIP and IHC) | Santa cruz | Cat # sc-19857, RRID:AB_2301809 Cat # sc-74504; RRID:AB_2188348 |
| BCL6 (ChIP and IHC) | Santa cruz Active Motif |
Cat # sc-858, RRID:AB_2063450 Cat # 61194; RRID:AB_2063450 |
| UTX (ChIP) | Bethyl | Cat# A302–374A; RRID:AB_1907257 |
| P300 (ChIP) | Novus Biologics | Cat # NB100–616, RRID:AB_10002598 |
| CTNNB1 (IHC) | Life Span | Cat# LS-C45628–50, RRID:AB_1508636 |
| DKK2 (IHC) | Thermo Fisher Scientific | Cat# PIPA547318; RRID:AB_2576803 |
| GAB1 (IHC) | Abcam | Cat# 1626–1, RRID:AB_562118 |
| NPR3 (IHC) | Abcam | Cat# ab37617, RRID:AB_776650 |
| MYC (IHC) | Bethyl | Cat# A301–833A-T; RRID:AB_1264336 |
| KCNA1 (IHC) | Thermo Fisher Scientific | Cat# PIPA519593; RRID:AB_10978519 |
| Plasmids | ||
| pcDNA3/Myc-DNMT3A | Addgene | Cat# 35521 |
| Flag-SIRT1 | Addgene | Cat#1791 |
| PCXN2-BCL6 | Addgene | Cat#40346 |
| siRNA Sequences | IDT | |
| mm.Ri.Dnmt3a.13.1 | AAUGUUGAUGUGGAAUCUUACUUTC | GAAAGUAAGAUUCCACAUCAACAUUGA |
| mm.Ri.Dnmt3a.13.2 | AUCCUUACAAGGAAGUUUACACCGA | UCGGUGUAAACUUCCUUGUAAGGAUUC |
| mm.Ri.Sirt1.13.1 | ACCAAAGAAUGGUAUUUCACACUTT | AAAGUGUGAAAUACCAUUCUUUGGUCU |
| mm.Ri.Sirt1.13.2 | GACUCUUCUGUGAUUGCUACACUTG | CAAGUGUAGCAAUCACAGAAGAGUCUU |
| mm.Ri.Bcl6.13.1 | GCUAGUGAUGUUCUUCUCAACCUTA | UAAGGUUGAGAAGAACAUCACUAGCGU |
| mm.Ri.Bcl6.13.4 | CCAGUUGAAAUGCAACCUUAGUGTA | UACACUAAGGUUGCAUUUCAACUGGUC |
| Commercial Assay Kits | ||
| Quick-RNA MiniPrep | Zymo research | Cat # 11–328 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat # 1708840 |
| QIAamp DNA Mini kit | Qiagen | Cat # 51304 |
| Epitect Fast DNA Bisulfite kit | Qiagen | Cat # 59802 |
| Experimental Models | ||
| Mouse Nestin Cre | The Jackson Laboratory | Stock No. 003771 |
| Mouse Mll4flox/flox | This paper | N/A |
| Deposited Data | ||
| ChIP-seq | GEO Datasets | GSE95626 |
| RNA-seq | GEO Datasets | GSE95626 |
| Primer List | ||
| Gene | 5'-3' Forward Sequence | 5'-3' Reverse Sequence |
| RT-PCR | ||
| Rasgrp1 | GCCAGCTCCATCTATTCCAAG | TTCATCCCGCAGTCTTTACAG |
| Rasgrf1 | GTGACAGAGTGGAGTCGAAG | CATAAGACAGTTAAAGCAGGCG |
| Rasgrf2 | ACGTTCTTGACCTACCCAATG | AGCGAACTCCAGACTTTTCC |
| Rapgef5 | AAACTCAGATGTCCCTTGTCG | CCAGCACGTTCCTGTATATGG |
| Rgl1 | AGAGGAAGAGGAACTGGAGAG | GAGACCAAATACAGCCCAGG |
| Hes1 | CCAGCCAGTGTCAACACGA | AATGCCGGGAGCTATCTTTCT |
| Hes5 | AGTCCCAAGGAGAAAAACCGA | GCTGTGTTTCAGGTAGCTGAC |
| Jag1 | CCTCGGGTCAGTTTGAGCTG | CCTTGAGGCACACTTTGAAGTA |
| Jag5 | CAATGACACCACTCCAGATGAG | GGCCAAAGAAGTCGTTGCG |
| Dnmt3a | GGACTTTATGAGGGTACTGGC | GATGTCCCTCTTGTCACTAACG |
| Dnmt1 | GACCTACTTGAGAGCATCCAG | TTCCCTTTCCCTTTGTTCCC |
| Dnmt3b | GATGTCCCTCTTGTCACTAACG | TTGATCTTTCCCCACACGAG |
| Bcl6 | CCGGCACGCTAGTGATGTT | TGTCTTATGGGCTCTAAACTGCT |
| Sirt1 | GCTGACGACTTCGACGACG | TCGGTCAACAGGAGGTTGTCT |
| Neurod1 | ATGACCAAATCATACAGCGAGAG | TCTGCCTCGTGTTCCTCGT |
| Pax6 | TACCAGTGTCTACCAGCCAAT | TGCACGAGTATGAGGAGGTCT |
| Foxo3 | CTGGGGGAACCTGTCCTATG | TCATTCTGAACGCGCATGAAG |
| Cbfa2t3 | CACCCTCCTTCACACCTCAC | TGAGATGTCACTGCCAAACTG |
| ChIP | ||
| Dnmt3a | ||
| b1 | GCAACTGTCCCTTTGATCCT | ACCCACGGCACACTTATTT |
| b2 | AGAACTGCAGGGCGAAG | CATCCAGCACTGGTCGTAG |
| b3 | CTCATGCCAATCCAGGTAGTC | CTCCTCCTGTTCCTCTCCTT |
| Hes1 | CCTTTCTGCCCAGTAGGAAAT | TGGATGCTTGTCTCCCTCTA |
| Hes5 | TACATGAGTCCCTCTCTCATCC | ACAGGCACAGTGGGTTAAAG |
| Jag1 | ATGGATTTGGGAAGGGATGG | AGACTCTGCTGGGAACTTTG |
| Jag2 | CTCTTGACATGGTCCACTATCC | GGCCATCGCTACATTCTCTTAT |
| Rasgrp1 | CTTACGCAGTGCCCTCTTT | GGAACATGGCCACGGTAATA |
| Rasgrf1 | GAGCGAGAGAGAGAGAGAGAG | GAGACTGCTACGCACTTACAT |
| Rasgrf2 | CAGAATTCTAGGAAACCGAGGAG | AGGGTGGTGGTGATGATAATG |
| Rapgef5 | CACAGGACCAACTTCTGACTT | TCTCTCTCTCTCTCTCTCTCTCT |
| Rgl1 | ATGCCTGTAATCTCAGCACTC | CCTACTGCCCACTCTTGTTT |
| ERNA | ||
| Dnmt3a | ||
| E1 | CATAGACCCTGATGTCAAGCC | AGGGCTTCAGAGAAGGAATTG |
| E2 | GTTCACTCCGCTTCTCCAAG | ACAGCTCTGGAGGCTTCT |
| E3 | AGCCACTCATCCCGTTTC | CAGAGGCCTGGTTCTCTTC |
| E4 | GTAGAACTCAAAGAAGAGGCGG | CATGGCCACCACATTCTCA |
| Bcl6 | ||
| E1 | GACTGTGAAGCAAGGCACT | ACACACACACACACACACA |
| E2 | TGTGTCACAGCAACATCTACTC | GGGAAGTATGGAGCATTCCG |
| E3 | ATGGGACCTTCTTCTGCAAC | GATCACATTTGTATGGTTTGTCACT |
| 3C-primers | Primers spanning around Bgl II site | |
| Dnmt3a | ||
| P1 | GGAGAGGTGAGCACTACAGA | CAGTAAGCTTTCCGTACCTTGA |
| E0 | AGCTAGGGTTACTGGGTTACT | TTCTCCAACCATGTGGGTTC |
| E1 | AGTTGTTCCCTGGGTGTTAAA | CAGCAAGGTGACCAGGAAA |
| E2 | AGATGGCTCAGTGGGTAAGA | GCCTTCACTGCTGGACTTT |
| E3 | CAAGCCTGAGAGAGCTGAAA | GGAACTTTCTCCAGGACCAG |
| E4 | TTGCAGGCTTCCAAGACAC | GGCTTGGTGCTGGGTTAAA |
| Bcl6 | ||
| P1 | CAAGATCAGGAGGGAAGAAG | ATCACTCACAAAGATCTCCCTATC |
| E1 | CAACGCCGATCTCAGTCC | AGAAGGTCTGCTGACAATGC |
| E2 | CAAATGTATAGGCCATTCTCTATGT | TTCCAAAGGTATGTTCGTTCAA |
| E3 | GTGATCGGTTACAGGCAATAGA | GACTGCTGCCCTAGTTGAAG |
| E4 | CAGAGCTGCTTGCTCCA | CACACTGAGACCTGGGATTG |
RNA-seq and Quantitative RT-PCR
Total RNA was isolated from cerebellar tissues using Trizol reagent (ThermoFisher Scientific, Waltham, MA). RNA samples were sequenced by using the Illumina HiSeq 2000. The RNA-seq reads were mapped to the mouse reference genome version mm9. The mouse reference gene set (UCSC knownGene) was downloaded from https://genome.ucsc.edu, and the function Cuffdiff in the software CuffLinks version 2.2.1 (Trapnell et al., 2013) was used to calculate expression level (fragments per kilobase per million, or FPKM) for each reference gene. Gene expression values between samples were normalized using the geometric method in Cuffdiff. DAVID (version 6.8) was used for Gene Ontology (GO) analysis. Each GO term with a p value less than 1 × 10−2 was considered to be significantly enriched.
Reverse transcription (RT)-PCR was performed as previously described (Dhar et al., 2012). In brief, total RNA was reverse-transcribed using the iScript cDNA Synthesis Kit (BioRad). For quantitative PCR, iQ SYBR Green Supermix (BioRad, Hercules, CA) was used during PCR amplification in the CFX384 real-time PCR detection system (BioRad). GAPDH mRNA or 18s rRNA levels were used as internal normalization controls for mRNA quantification. Each experiment was performed in triplicate.
Bisulfite Sequencing
Genomic DNA was extracted from the cerebella using the QIAamp DNA Mini Kit (Qiagen, Venlo, Netherlands). DNA was chemically modified by bisulfite treatment using Epitect Fast DNA Bisulfite Kit (Qiagen). Bisulfite-converted DNA serves as a template in PCR. Specific PCR primers were designed using MethPrimer program, which identifies predicted CpG sites for methylation. The bisulfite-converted DNA was amplified by PCR (annealing temperature between 55°C and 60°C; 40 cycles). Amplicon sizes w ere between 150 and 300 bp. The PCR products were cloned using the TOPO TA Cloning Kit, and the clones were subjected to DNA sequencing.
Neurosphere Culture
Cerebella from Mll4f/f mice (postnatal day 5) were used for the generation of neurospheres. The cerebella were gently washed with PBS and then placed in a 60 mm dish with 2ml of the Neurobasal medium. Cerebellar tissues were finely chopped using sterile scalpels. The pieces of cerebellar tissues were placed and slowly mashed in sterile 70 μm cell strainer and collected in 50 ml tube containing the Neurobasal medium. The dissociated cells were plated in 100 mm dishes at a density of 5 × 105 cells per dish in Neurobasal medium supplemented with B27 supplement, 20ng/ml Epidermal Growth Factor (EGF), 20 ng/mL basic Fibroblast Growth Factor (bFGF) and 2 μg /mL heparin (ThermoFisher Scientific, Waltham, MA). Three to five days later, the cells propagated as free-floating neurospheres.
RNA Interference and Gene Expression in Neurospheres
For knockdown experiments, siRNAs were purchased from Integrated DNA technologies (IDT; See key resources table for siRNA sequences). Neurospheres were resuspended and cells (5 × 103) were seeded in a 6-well plate. These cells were transfected with siRNAs at a final concentration of 100 nM using Lipofectamine RNAiMAX (ThermoFisher Scientific, Waltham, MA). Following 48 hours incubation, cells were harvested for RNA isolation.
Cells resuspended from neurospheres were plated in 60 mm dishes at a density of 2 × 104 cells per dish. Adeno-Cre virus particles at 1x 104 pfu were used to delete Mll4 in neurospheres. Neurospheres infected with Adeno-Empty virus particles were used as a control. Forty eight hours after infection of either Adeno Empty or Adeno-Cre, cells were collected, and RNA was isolated to confirm Mll4 deletion by RT-PCR.
To ectopically express DNMT3A, SIRT1, and BCL6 gene in cerebellar neurospheres treated with either Adeno Empty or Adeno-Cre, their cDNA constructs (See key resources table) were transfected into neurospheres using Lipofectamine 3000 (ThermoFisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Following incubation for 48 hours, cells were harvested, and total RNA was isolated for further analysis.
Enhancer RNA Analysis
Because enhancers in the Dnmt3a and Bcl6 genes are located in the gene body, we measured anti-sense eRNAs from enhancer regions using quantitative RT-PCR. In our RNA-seq data analysis for eRNAs, RSeQC (Wang et al., 2012) was used to confirm strand specificity of the RNA-seq data. After mapping RNA-seq reads to the mouse genome, we used BEDTools (Quinlan and Hall, 2010) to assign the reads to enhancer regions. For enhancers that overlapped with reference genes, the RNA-seq reads were removed on the basis of strand specificity, and the remaining reads were used to calculate FPKM value of each enhancer region.
ChIP Assays and ChIP-seq
ChIP assays of mouse cerebellar tissues or neurospheres were performed using a modified version of the previously described methods (Dhar et al., 2016; Dhar et al., 2012). Cerebellar tissues (about 100 mg per ChIP antibody) isolated from 4-month-old mice were crosslinked and sonicated using Ren lab’s protocol from Roadmap Epigenome (https://www.encodeproject.org). For neurospheres, cerebellar cells were plated in 100 mm dishes at a density of 1 × 106 cells per dish. Cells were cultured for 7 days after infection of either Adeno Empty or Adeno-Cre virus and were crosslinked using 1% formaldehyde for 10 min at room temperature. The crosslinking reaction was stopped with 125 mM glycine. ChIP assay was performed using ChIP-IT High Sensitivity (HS) Kit from Active Motif.
In brief, DNA was purified from chromatin immuneprecipitates for H3K4me1, H3K4me3, H3K27ac, MLL4, UTX, p300 or IgG using phenol/chloroform extraction. IgG was used as a negative control for ChIP. Then, DNA was amplified by quantitative PCR and normalized to input. To compare H3K4me1, H3K27ac, and H3K4me3 levels between Mll4f/f and Mll4-deleted samples, ChIP with reference exogenous genome (ChIP-Rx) was carried out as previously described (Orlando et al., 2014). In brief, two biological replicates were used for ChIP-Rx experiments. Drosophila melanogaster chromatin and Drosophila-specific H2Av antibody were added to each ChIP assay as a minor fraction. The following components for ChIP-Rx were purchased from Active Motif: the spike-in chromatin (53083), Drosophila-specific H2Av antibody (61686), spike-in ChIP positive control primer set (71037), and negative control primer set (71028). The immunoprecipitated DNAs were used for ChIP-seq library preparation, followed by next-generation sequencing (UCI GHTF).
ChIP-seq Analysis
All ChIP-seq sequencing reads were mapped to the mouse genome version mm9 or hg19 using Bowtie version 1.1.2 (Langmead et al., 2009). The Dregion function in DANPOS (version 2.2.3) (Chen et al., 2013) was used to calculate read density and define enrichment peaks. Briefly, we extended each read at the 3′ end to be 200 bp long, which is approximately the nucleosome unit size in mouse or human, and then calculated read density as the number of reads covering each base pair in the genome. For each sample, the total number of mapped reads was normalized to 10 million. For H3K4me1, H3K27ac, and MLL4 data, we used Poisson test P value 1 × 10−10 for read density cutoff to define seed peaks. For H3K4me3, we used Poisson test P value 1 × 10−5 for read density cutoff to define seed peaks. We then used Poisson test P value 1 × 10−3 for read density cutoff to extend each seed peak. The function profile in DANPOS version 2.2.3 was used to calculate the average read density flanking a group of TSSs or peak regions. The software MEV version 4.8.1 (Howe et al., 2011) was used to plot heatmaps.
Chromosome Confirmation Capture
For chromosome confirmation capture (3C) experiments, cerebellar (4-month-old) tissues were cross-linked (1% formaldehyde, 10 mM NaCl, 100 μM EDTA, 50 μM EGTA, 5 mM HEPES, pH 8.0) for 15 min at room temperature. Cross-linking was terminated by the addition of 125 mM glycine, and cells were dissociated by using a Dounce homogenizer. Nuclei was isolated by incubating cells in a lysis buffer (10 mM Tris, pH 8.0, 10 mM NaCl, 0.2% NP-40) on ice with agitation for 30 min. Chromatin was subsequently released by treating isolated nuclei with 0.3% SDS and was digested with 400 units of Bgl II overnight. Thereafter, the restriction enzyme was heat-inactivated, and chromatin was diluted to a low genomic DNA concentration of 2.5 ng/μl in T4 ligation buffer. Chromatin re-ligation was performed by incubating diluted chromatin with 100 Weiss units of T4 ligase for 4 h at 16°C. After RNA se A digestion, the 3C chromatin was purified by phenol:chloroform extraction. The interactions between the promoter P1 and several regions in the enhancers of the Dnmt3a and Bcl6 genes were determined using quantitative PCR and normalized to an internal control such as 18S or β-actin gene. In addition, the interaction among several regions (E1‒E4) in the enhancers of both Dnmt3a and Bcl6 genes were also determined using quantitative PCR.
Statistical Analysis
In general, statistical analyses were performed using Prism GraphPad unless otherwise indicated. Data were presented as the mean ± SEM (error bars) of at least three independent experiments. Statistical significance was tested by the two-tailed Student t test unless otherwise indicated. * (p <0.05), ** (p <0.01), and *** (p <0.001) indicated statistically significant differences. Tumor-free curves were plotted with the Kaplan-Meier method and compared by using a two-sided log-rank test.
Supplementary Material
Highlights.
Brain-specific Mll4 loss in mice results in spontaneous medulloblastoma
MLL4 upregulates DNMT3A-catalyzed DNA methylation to repress Ras activators
MLL4 enhances SIRT1/BCL6-mediated H4K16 deacetylation to downregulate Notch pathways
MLL4 establishes broad H3K4me3 and super-enhancers to activate tumor suppressor genes
ACKNOWLEDGMENTS.
We are thankful to Dr. M. McArthur for histopathological analysis, to S. Zhang and Dr. Z. Han for technical assistance, and to K. Michel, K. Maldonado, and C. Kingsley in the Small Animal Imaging Facility (supported by the NIH/NCI under Core grant award number P30CA16672) at The University of Texas MD Anderson Cancer Center for technical advice. We also thank the IDDRC neuropathology core (Core grant award number U54HD083092) and RNA In Situ Hybridization core at The Baylor College of Medicine for histology and image scan services. This work was supported in part by grants to M.G.L. from the Cancer Prevention and Research Institute of Texas (CPRIT; RP140271), the NIH (R01CA157919, R01CA207098, and R01CA207109), and the Center for Cancer Epigenetics at MD Anderson, by a grant A.S. from the NIH (R35CA197569), by grants to W.L. from the CPRIT (RP150292) and the NIH (R01HG007538 and R01CA193466), by grants to V.G. from the CPRIT (RP150301) and the NIH (R01NS079715 and R03NS077021), by a grant to R.V.S. from the NIH (R01NS089664), by a grant to E.R.F. from the NIH (R35CA197452), and by a fellowship to S.S.D. from the Center for Cancer Epigenetics at MD Anderson. K. C. was supported by startup from The Methodist Hospital Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS. The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures and one table and can be found with this article online.
REFERENCES
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, and Zhao K (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, et al. (2014). H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Fang Y, Tan HK, Goh Y, Choy JYH, Koh BTH, Hao Tan J, Bertin N, Ramadass A, Hunter E, et al. (2017). Super-Enhancers and Broad H3K4me3 Domains Form Complex Gene Regulatory Circuits Involving Chromatin Interactions. Sci Rep 7, 2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, and Bergman Y (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10, 295–304. [DOI] [PubMed] [Google Scholar]
- Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, et al. (2015). Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet 47, 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, and Li W (2013). DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome research 23, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, Li G, Kuan S, Li B, Lee AY, et al. (2016). Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawkins JB, Wang J, Maniati E, Heward JA, Koniali L, Kocher HM, Martin SA, Chelala C, Balkwill FR, Fitzgibbon J, et al. (2016). Reduced Expression of Histone Methyltransferases KMT2C and KMT2D Correlates with Improved Outcome in Pancreatic Ductal Adenocarcinoma. Cancer Res 76, 4861–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Lee SH, Chen K, Zhu G, Oh W, Allton K, Gafni O, Kim YZ, Tomoiga AS, Barton MC, et al. (2016). An essential role for UTX in resolution and activation of bivalent promoters. Nucleic acids research 44, 3659–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, Reinberg D, and Lee MG (2012). Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev 26, 2749–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD 2nd, Garcia BA, Adelman K, and Wysocka J (2017). Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Molecular cell 66, 568–576 e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois NC, Hofmann D, Kaloulis K, Bishop JM, and Trumpp A (2006). Nestin-Cre transgenic mouse line Nes-Cre1 mediates highly efficient Cre/loxP mediated recombination in the nervous system, kidney, and somite-derived tissues. Genesis 44, 355–360. [DOI] [PubMed] [Google Scholar]
- Fiaschetti G, Schroeder C, Castelletti D, Arcaro A, Westermann F, Baumgartner M, Shalaby T, and Grotzer MA (2014). NOTCH ligands JAG1 and JAG2 as critical pro-survival factors in childhood medulloblastoma. Acta neuropathologica communications 2, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Steine EJ, Barrasa MI, Hockemeyer D, Pawlak M, Fu D, Reddy S, Bell GW, and Jaenisch R (2011). Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proceedings of the National Academy of Sciences of the United States of America 108, 18061–18066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovesi LA, Ng CG, Davis MJ, Remke M, Taylor MD, Adams DJ, Rust AG, Ward JM, Ban KH, Jenkins NA, et al. (2013). Sleeping Beauty mutagenesis in a mouse medulloblastoma model defines networks that discriminate between human molecular subgroups. Proceedings of the National Academy of Sciences of the United States of America 110, E4325–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, and Lee MG (2013). Histone H3 lysine 4 methyltransferases and demethylases in self-renewal and differentiation of stem cells. Cell Biosci 3, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, and Young RA (2007). A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, et al. (2004). The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 64, 7794–7800. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, and Shilatifard A (2012). Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26, 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe EA, Sinha R, Schlauch D, and Quackenbush J (2011). RNA-Seq analysis in MeV. Bioinformatics 27, 3209–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Gao X, Morgan MA, Herz HM, Smith ER, and Shilatifard A (2013). The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol 33, 4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Sharma A, Dhar SS, Lee SH, Gu B, Chan CH, Lin HK, and Lee MG (2014). UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res 74, 1705–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, and Shiekhattar R (2015). Architectural and Functional Commonalities between Enhancers and Promoters. Cell 162, 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D, et al. (2012). Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123, 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai B, Lee JE, Jang Y, Wang L, Peng W, and Ge K (2017). MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic acids research 45, 6388–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, et al. (2013). H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2, e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, and Shiekhattar R (2007). Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. (2010). DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, et al. (2016). Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang C, Liu W, Li J, Li C, Kou X, Chen J, Zhao Y, Gao H, Wang H, et al. (2016). Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537, 558–562. [DOI] [PubMed] [Google Scholar]
- Lourenco AR, and Coffer PJ (2017). A tumor suppressor role for C/EBPalpha in solid tumors: more than fat and blood. Oncogene 36, 5221–5230. [DOI] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CY, Yao MJ, Zhai QW, Jiao JW, Yuan XB, and Poo MM (2014). SIRT1 suppresses self-renewal of adult hippocampal neural stem cells. Development 141, 4697–4709. [DOI] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. (2014). An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan S, Li Y, Miller EE, Shih DJ, Taylor MD, Stearns TM, Bronson RT, Ackerman SL, Yoon JK, and Yun K (2013). Notch1-induced brain tumor models the sonic hedgehog subgroup of human medulloblastoma. Cancer Res 73, 5381–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD, et al. (2012a). Medulloblastomics: the end of the beginning. Nature reviews Cancer 12, 818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Shih DJ, Remke M, Cho YJ, Kool M, Hawkins C, Eberhart CG, Dubuc A, Guettouche T, Cardentey Y, et al. (2012b). Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol 123, 615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, and Kageyama R (1999). Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. The EMBO journal 18, 2196–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JM, Asakura A, Snider L, Hawkes R, Strand A, Stoeck J, Hallahan A, Pritchard J, and Tapscott SJ (2001). NeuroD2 is necessary for development and survival of central nervous system neurons. Developmental biology 234, 174–187. [DOI] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell reports 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. (2015). The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, and Rietze RL (2005). Neural stem cells and neurospheres--re-evaluating the relationship. Nat Methods 2, 333–336. [DOI] [PubMed] [Google Scholar]
- Rickels R, Herz HM, Sze CC, Cao K, Morgan MA, Collings CK, Gause M, Takahashi YH, Wang L, Rendleman EJ, et al. (2017). Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat Genet 49, 1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Fonseca LL, Rajani C, Yanagida JK, Endo Y, Cline JM, Stone JC, Ji J, Ramos JW, and Lorenzo PS (2014). Targeted deletion of RasGRP1 impairs skin tumorigenesis. Carcinogenesis 35, 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A (2012). The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81, 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, and Shilatifard A (2014). Enhancer biology and enhanceropathies. Nature structural & molecular biology 21, 210–219. [DOI] [PubMed] [Google Scholar]
- Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I, et al. (2016). New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur I, and Taipale J (2016). The role of enhancers in cancer. Nature reviews Cancer 16, 483–493. [DOI] [PubMed] [Google Scholar]
- Tarnowski M, Schneider G, Amann G, Clark G, Houghton P, Barr FG, Kenner L, Ratajczak MZ, and Kucia M (2012). RasGRF1 regulates proliferation and metastatic behavior of human alveolar rhabdomyosarcomas. Int J Oncol 41, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, et al. (2012). Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberi L, Bonnefont J, van den Ameele J, Le Bon SD, Herpoel A, Bilheu A, Baron BW, and Vanderhaeghen P (2014). A BCL6/BCOR/SIRT1 complex triggers neurogenesis and suppresses medulloblastoma by repressing Sonic Hedgehog signaling. Cancer Cell 26, 797–812. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature biotechnology 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, and Reinberg D (2004). Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Molecular cell 16, 93–105. [DOI] [PubMed] [Google Scholar]
- Vigil D, Cherfils J, Rossman KL, and Der CJ (2010). Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nature reviews Cancer 10, 842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH, Nakadai T, Chramiec A, Krivtsov AV, Armstrong SA, et al. (2017). A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Molecular cell 67, 308–321 e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, and Sillitoe RV (2013). Development of the cerebellum: from gene expression patterns to circuit maps. Wiley Interdiscip Rev Dev Biol 2, 149–164. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, and Sun YE (2010). Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Shen B, Chu ES, Teoh N, Cheung KF, Wu CW, Wang S, Lam CN, Feng H, Zhao J, et al. (2010). Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro. Hepatology 51, 2008–2019. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. (2015a). Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mittal A, Reid J, Reich S, Gamblin SJ, and Wilson JR (2015b). Evolving Catalytic Properties of the MLL Family SET Domain. Structure 23, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Sun J, and Zhao Z (2013). TSGene: a web resource for tumor suppressor genes. Nucleic acids research 41, D970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.