

Abstract
Aldo–keto reductase 1C3 (AKR1C3) catalyzes the synthesis of 9α,11β-prostaglandin (PG) F2α and PGF2α prostanoids that sustain the growth of myeloid precursors in the bone marrow. The enzyme is overexpressed in acute myeloid leukemia (AML) and T-cell acute lymphoblastic leukemia (T-ALL). Moreover, AKR1C3 confers chemother-apeutic resistance to the anthracyclines: first-line agents for the treatment of leukemias. The highly homologous isoforms AKR1C1 and AKR1C2 inactivate 5α-dihydrotestosterone, and their inhibition would be undesirable. We report herein the identification of AKR1C3 inhibitors that demonstrate exquisite isoform selectivity for AKR1C3 over the other closely related isoforms to the order of >2800-fold. Biological evaluation of our isoform-selective inhibitors revealed a high degree of synergistic drug action in combination with the clinical leukemia therapeutics daunorubicin and cytarabine in in vitro cellular models of AML and primary patient-derived T-ALL cells. Our developed compounds exhibited >100-fold dose reduction index that results in complete resensitization of a daunorubicin-resistant AML cell line to the chemotherapeutic and >100-fold dose reduction of cytarabine in both AML cell lines and primary T-ALL cells.
Graphical Abstract

INTRODUCTION
Acute myeloid leukemia (AML) is a debilitating condition that affects both pediatric and elderly populations. With an estimated incidence rate of more than 19520, it is the second most common type of hematologic malignancy.1 The underlying pathophysiology for AML is marked by a differentiation block and clonal expansion of immature blast cells in the bone marrow that can be ascribed to various chromosomal translocations offering a proliferative advantage.2,3 Clinically, AML is classified into seven distinct subtypes (M0–M7) depending on the type of cells affected within the myelocytic lineage during hematopoiesis. The M3 subtype or acute promyelocytic leukemia stems from a reciprocal chromosomal translocation of promyelocytic leukemia/retinoic acid receptor α oncogene that prevents cellular differentiation.4 Treatment with retinoic acid is a primary therapeutic intervention that is employed along with arsenic trioxide or chemotherapy to induce remission.5 However, the use of retinoic acid is limited to only the M3 subtype, due to adverse effects.6 Standard treatment of care for the other types of AML relies heavily on the use of anthracyclines and the antimetabolite cytarabine as first-line chemotherapeutics for remission induction as well as for maintenance therapy. Continuous infusion of cytarabine for 7 days in combination with daunorubicin for 3 days is employed as induction chemotherapy (7 + 3 regimen) followed by cytarabine administration for maintenance.2 Since AML predominantly affects the elderly with higher incidence rates among patients >65 years of age, the tolerability of intensive chemotherapy remains a grave concern.7 Moreover, the disease prognosis among older patients is poor due to higher relapse rates and unfavorable cytogenetics.8
Acute lymphocytic leukemia (ALL) stems from a malignant transformation of the lymphoid precursors in the bone marrow and is estimated to affect 5960 individuals per year in the U.S., predominantly children and adolescents.1 Approximately 25% of all ALL cases are classified as T-cell ALL (T-ALL) that affects the T lymphocytes of the white blood cell lineage.9 Owing to chromosomal translocation of transcription factors that are then placed under the control of strong T-cell receptor gene promoters, immature precursor T-cells gain a proliferative advantage. The clinical outcome for patients suffering from primary refractory or relapsed T-ALL remains bleak.10,11 Chemotherapeutics used to treat AML are commonly used to treat T-ALL and encounter similar resistance profiles in relapse.11 Development of nontoxic chemical agents that can increase the efficacy of currently employed chemotherapeutics and prevent the development of resistance represents an attractive therapeutic strategy to manage leukemic disease progression.
Aldo–keto reductase 1C3 (AKR1C3) is overexpressed in a range of leukemic cell lines spanning various AML subtypes and in high-risk T-ALL.12,13 By virtue of the enzyme’s prostaglandin (PG) F synthase activity, AKR1C3 converts the precursor prostanoid PGD2 to 11β-PGF2α, which induces proliferation of leukemic blasts. The proliferative activity of 11β-PGF2α is attributed to the activation of prostaglandin F2α receptor and the downstream mitogen-activated protein kinase cascade.14 In the absence of AKR1C3, PGD2 nonenzymatically converts to PGJ2 and eventually to 15-deoxy-Δ12,14-prostaglandin J2 (15Δ-PGJ2). 15Δ-PGJ2 serves as a ligand for the peroxisome proliferator-activated receptor-γ (PPARγ) and promotes cellular differentiation.15 Prior reports have validated the role AKR1C3 plays in inducing myeloid cell proliferation and demonstrated that upon AKR1C3 upregulation, the cells become resistant to differentiation by all-trans retinoic acid (ATRA), whereas the loss of AKR1C3 transgene expression confers sensitivity to ATRA.12 Studies have shown that PGD2 synergizes with ATRA in HL-60 promyelocytic cells to induce differentiation and apoptosis, whereas, in contrast, incubation with 11β-PGF2α significantly increased cell proliferation.12 Since direct administration of prodifferentiation prostanoids PGJ2 and 5Δ-PGJ2 is severely limited by high toxicity and low bioavailability, a superior therapeutic strategy will be to inhibit the activity of AKR1C3 to divert the prostanoid metabolism toward the production of PGJ2 series prostanoids to gain a beneficial therapeutic outcome.16 The related AKR1C3 isoforms AKR1C1 and 1C2 share >84% sequence homology to 1C3 and are responsible for the inactivation of the potent androgen (e.g., 5α-dihydrotestosterone); thus, the inhibition of these isoforms is undesirable since it would cause androgen excess.17,18 Importantly, AKR1C3 acts as a phase I biotransformation enzyme, transforming anthracyclines (doxorubicin, daunorubicin, and idarubicin) into their less-potent C-13 hydroxy metabolites, which results in chemotherapeutic resistance.19,20 Apart from AML and T-ALL, AKR1C3 is overexpressed in a variety of neoplasms of the breast,21 colon,22 prostate,23,24 endometrium,25,26 and lungs.27
Significant efforts have been made to discover and develop different classes of AKR1C3 inhibitors from diverse structural classes [steroids,26 flavones,28 jasmonates,29 and nonsteroidal anti-inflammatory drugs (NSAIDs)]21,24,26,30 (Figure 1). Among steroids, medroxyprogesterone acetate (MPA) and an estrogen lactone (EM1404) are potent inhibitors of AKR1C3 but are limited in application due to the lack of isoform selectivity. Jasmonic acids suffer from the same fate and lack inhibition potency and selectivity toward AKR1C3. Among flavonoids, 2-hydroxyflavone exhibits moderate potency while inhibiting AKR1C3 with 157-fold isoform selectivity. Repurposed NSAIDs possess high inhibitory potency for AKR1C3 and selectivity but have not progressed into the clinic. For example, derivatization of the indomethacin scaffold to the des-methyl indomethacin analogue, modification of naproxen to (R)-ethyl naproxen, and the structural optimization of flufenamic acid to a meta isomer abrogated cyclooxygenase inhibition activities while retaining inhibitory potency for AKR1C3. The N-naphthylamino benzoate analogue was found to be a bifunctional AKR1C3 inhibitor that also antagonizes the androgen receptor (AR) functions.21,26,31–35 The inhibitor SN33638 demonstrated only a moderate inhibition of testosterone production and a modest reduction in cell viabilities evaluated in AKR1C3-dependent castration-resistant prostate cancer (CRPC) cell lines, whereas ASP9521, taken through a phase I/II clinical trial in metastatic CRPC patients, established no observable clinical activity.36–38 Another bifunctional inhibitor GTX-560 has been shown to inhibit the AR coactivator functions of AKR1C3 in addition to being an AKR1C3 inhibitor.39 Recently reported, a benzisoxazole derivative designed by applying a scaffold hopping approach to flufenamic acid displayed a greater isoform selectivity for AKR1C3 inhibition as compared to AKR1C1. However, the inhibition potency still remained in the mid-nanomolar range (Figure 1).40 There may be multiple reasons for AKR1C3 inhibitor failure, which have prevented their clinical use.
Figure 1.
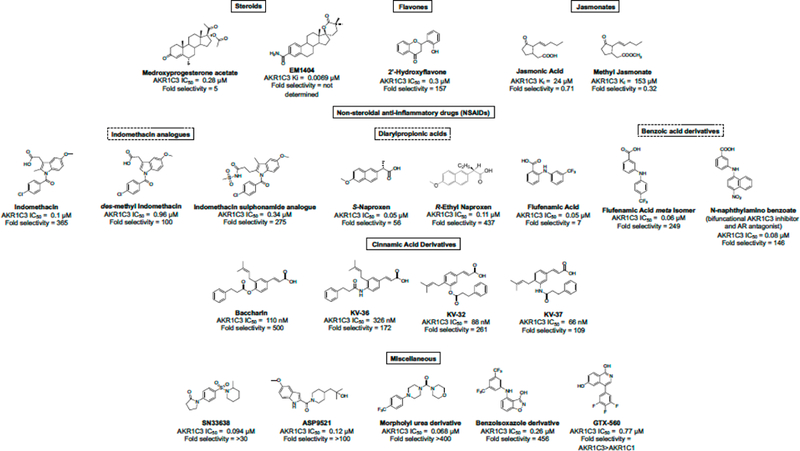
Structures of known AKR1C3 inhibitors.
Even though indomethacin yielded an IC50 value = 0.1 μM and exhibited a 356-fold selectivity for AKR1C3, it displayed a weak antiproliferative effect on AML cells.12,41 A study combining the pan-AKR1C inhibitor medroxyprogesterone acetate (MPA) (AKR1C3 IC50 = 2.7 μM, 0.66-fold selectivity for AKR1C3) and bezafibrate (a PPAR α/γ agonist) provided an approximate 2-fold potentiation of bezafibrate activity, demonstrating a reduction in cell proliferation with increased differentiation and apoptosis in AML cells. However, further studies on the mechanism of action of MPA attributed this activity to targets other than the AKR1C family. A recent study reported that the specific AKR1C3 inhibitor 4-methyl(de-dimethylamine)-tetracycline (IC50 = 0.51 μM) does not give the adjuvant effect at concentrations up to 50 μM that a pan-AKR1C inhibitor does, thus requiring greater credentialing of the AKR1C3 target in leukemia.42 Although inhibitors with nanomolar potency and relatively high selectivity to AKR1C3 (up to 500-fold) over the other isoforms have been identified from prior studies, the discovery of new compounds for preclinical development is still required.
Derivatives of cinnamic acids have been shown to exhibit inhibition activities for AKR isozymes bearing selectivity toward AKR1C3. Among these compounds, baccharin (Figure 1), a natural product extracted from honeybee propolis, has been shown to potently and selectively inhibit AKR1C3 with an IC50 of 0.11 μM and a 500-fold selectivity over AKR1C2.43 This discovery has made baccharin a promising hit to develop a new series of potent and specific inhibitors against AKR1C3. We adopted this natural product scaffold and have previously reported the synthesis and a preliminary structure–activity relationship (SAR) for AKR1C3 inhibition.44 The dihydro-cinnamoyloxy moiety of baccharin is reported as a structural prerequisite for AKR1C3 inhibition, and other groups have synthesized potent AKR1C3 inhibitors based on this scaffold.45 However, the presence of an ester linkage introduces a hydrolytic liability into baccharin, making these analogues unsuitable for drug discovery. Hydrolysis of the ester results in formation of the known phenol drupanin, which leads to a complete abrogation of AKR1C3 inhibitory potency.46 Our prior studies have reported the design, synthesis, and evaluation of potent AKR1C3 inhibitors bearing a more stable amide bioisostere (Figure 1). These AKR1C3 inhibitors exhibit a 6-fold potentiation of etoposide and a 10-fold potentiation of daunorubicin cytotoxicity in AML cell lines.47 However, the selectivity over AKR1C1 and AKR1C2, although significantly improved above that of MPA (0.66-fold), remained relatively low (109-fold). Continuing our efforts to identify highly isoform-selective AKR1C3 inhibitors, we herein report the discovery of a library of optimized compounds possessing >2800-fold selectivity for AKR1C3 inhibition, with retention of inhibitory potency in the nanomolar range. Furthermore, we demonstrate that our highly isoform-selective AKR1C3 inhibitors provide up to 100-fold potentiation of the clinical chemotherapeutics daunorubicin and cytarabine across a panel of AML cell lines and in primary patient-derived T-ALL cells.
RESULTS AND DISCUSSION
Chemistry.
Based on the baccharin structural scaffold, three different classes of analogues were synthesized and evaluated for AKR1C3 inhibitory potency and selectivity toward the other highly homologous AKR isoforms (Figure 2). To explore modifications of the dihydrocinnamoyloxy moiety, class I compounds possess an ester or amide link to the terminal phenyl B ring. The spacer is of varied length and features a diversity of substituents on the phenyl ring. Class II compounds have the acrylate side chain moved to the meta position and substitution on the terminal phenyl ring is varied. In addition, class II compounds also explore 1,3,5 substituents on the central A ring. Class III compounds constitute varying substitution patterns on the central A ring and also explore boronic acid bioisosteres of the cinnamic acid.
Figure 2.
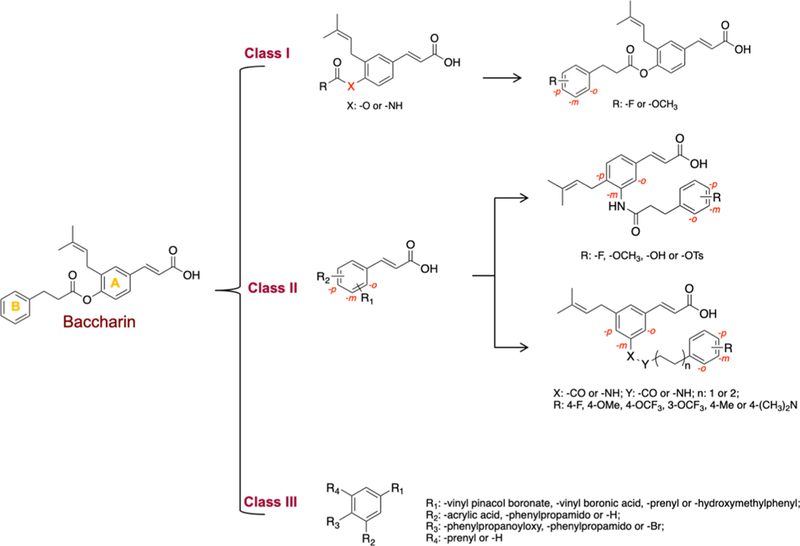
SAR strategy for the design of baccharin derivatives.
Baccharin (1) and compounds 7 and 7a (class III) and 8b–n (class I) were synthesized using a modified literature procedure.48 Nucleophilic substitution of commercially available 4-iodophenol (2) with prenyl bromide yielded the alkylated intermediate 3,49 which was subsequently esterified with an appropriately substituted acid chloride (4a–n) to yield ester intermediates (5a–n). Mizoroki–Heck50,51 coupling of the aryl iodide intermediates (5a–n) with tert-butylacrylate or vinyl boronic acid pinacol ester using Pd(OAc)2 and PPh3 as catalyst–ligand complex produced α,β-unsaturated olefins 6a–n and 7, respectively. Chemoselective hydrolysis of the tert-butyl ester52 of 6a–n afforded baccharin (1) and derivatives 8b–n, whereas hydrolysis of the pinacol ester53,54 afforded boronic acid 7a (Scheme 1).
Scheme 1. Synthesis of Baccharin (1),44 Substituted Ester Derivatives (8b–g, 8h,44 8i–n) of Class I, and Boronic Acid Analogues (7 and 7a) of Class IIIa.
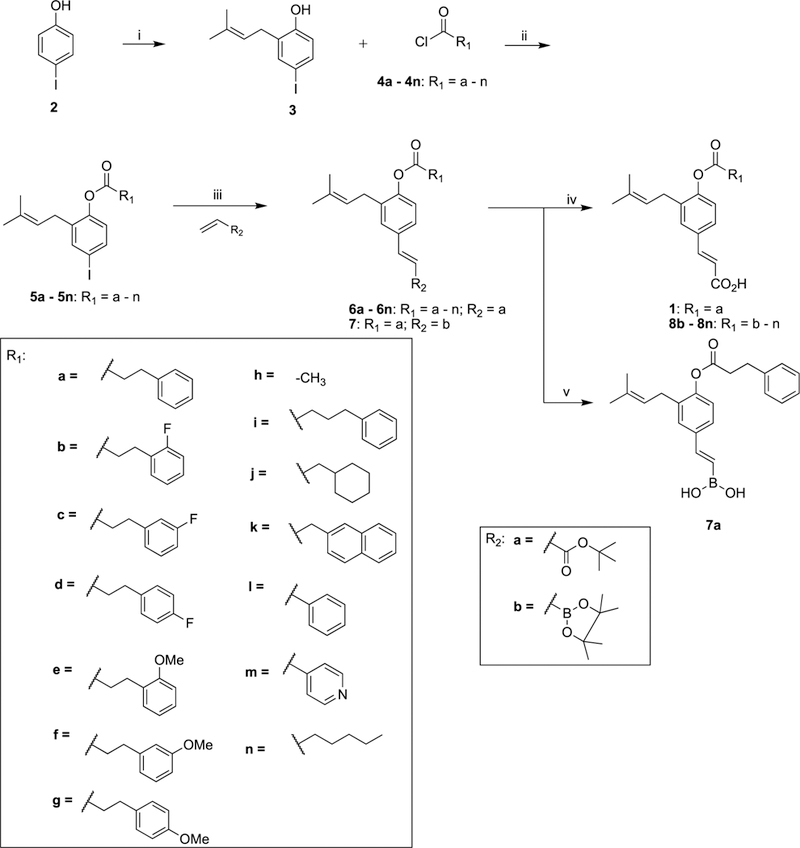
a Reagents and conditions: (i) Prenyl bromide, NaH, PhMe, 55%; (ii) 4-dimethylaminopyridine (DMAP), NEt3, dichloromethane (DCM); (iii) NEt3, P(Ph)3, Pd(OAc)2, PhMe, reflux; (iv) SiO2, PhMe, reflux; (v) MeOH/H2O, 80 °C.
The reaction sequence was then modified where p-iodoaniline (9) was regioselectively brominated using a modified literature procedure that afforded dihalide 10.55 Exploiting the greater reactivity of iodide over bromide56 in Mizoroki–Heck51 couplings, 10 was selectively coupled with tert-butylacrylate yielding the intermediate aniline 11. Coupling with the corresponding acid chloride gave amide intermediates (12a–e). A palladium-catalyzed Suzuki- Miyaura57,58 cross-coupling reaction installed the prenyl side chain that yielded intermediates (13a–e), which underwent chemoselective hydrolysis of the tert-butyl ester to afford the final compounds (14a47 and 14b–e) of class I (Scheme 2).
Scheme 2. Synthesis of Amide Bioisosteres (14a47 and 14b–e) of Class I Derivativesa.
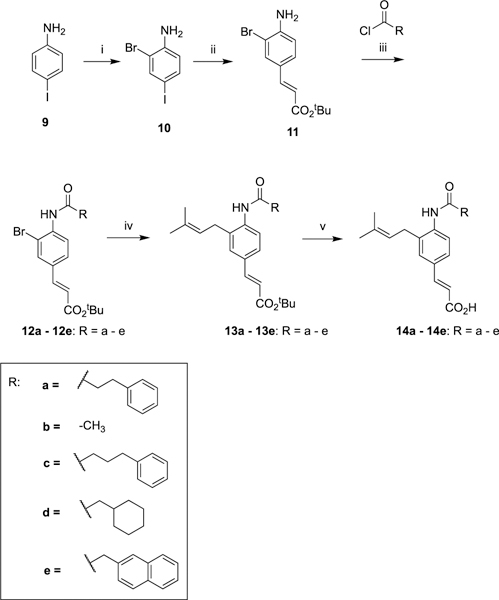
aReagents and conditions: (i) Br2, AcOH, room temperature (RT), 44%; (ii) tert-butylacrylate, NEt3, P(Ph)3, Pd(OAc)2, PhMe, reflux, 64%; (iii) NEt3, DMAP, DCM, 40 °C; (iv) prenylboronic acid pinacol ester, Pd(dppf)Cl2, Cs2CO3, dimethylformamide (DMF), 90 °C; (v) SiO2, PhMe, reflux.
Class II compounds in which the substitution pattern on the A ring of baccharin is altered, wherein the ester chain was moved to the meta position relative to the acrylate side chain were synthesized as follows. Commercially available m-coumaric acid (15) was protected as the methyl ester and subsequently brominated, with column chromatography yielding the desired regioisomer in 35% yield. The brominated intermediate (17) was deprotected under basic conditions to yield compound 17a, which was followed by esterification of the phenol using phenylpropionyl acid chloride to yield ester derivative 18. Suzuki–Miyaura57 reaction with the appropriately substituted boronic acid pinacol ester afforded prenyl and allyl derivatives (19a47 and 19b) of class II analogues (Scheme 3).
Scheme 3. Synthesis of meta-Substituted Ester Derivatives (19a47 and 19b) of Class IIa.
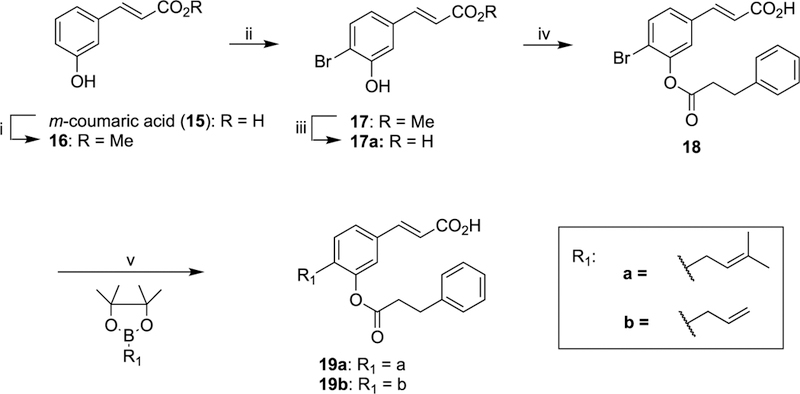
aReagents and conditions: (i) MeOH, H2SO4, reflux, 93%; (ii) Br2, AcOH, RT, 35%; (iii) NaOH, H2O, reflux, 90%; (iv) 3-phenylpropanoyl chloride, DMAP, NEt3, DCM, RT, 36%; (v) Pd(dppf)Cl2, Cs2CO3, DMF, 90 °C.
The amide bioisosteres 26a–1, 28 (class II), and 24 (class III) were accessed through Mizoroki–Heck51 reaction of commercially available 4-bromo-2-iodoaniline (20) with tert-butylacrylate or vinyl boronic acid pinacol ester to afford 21a and 21b, respectively. Amide formation with the appropriate acid chloride as previously described yielded amides 23a–j and 24. Subsequent Suzuki-Miyaura57 cross-coupling afforded intermediates 25a–j and 27, which were chemoselectively hydrolyzed to yield final compounds 26a47 and 26b–j and 28. Furthermore, hydrolysis of the tosyl moieties in 26i and 26j afforded phenols 26k and 26l, respectively (Scheme 4).
Scheme 4. Synthesis of meta-Substituted Amide Derivatives (26a,47 26b–1, and 28) of Class II and Boronic Ester Derivative 24 of Class IIIa.
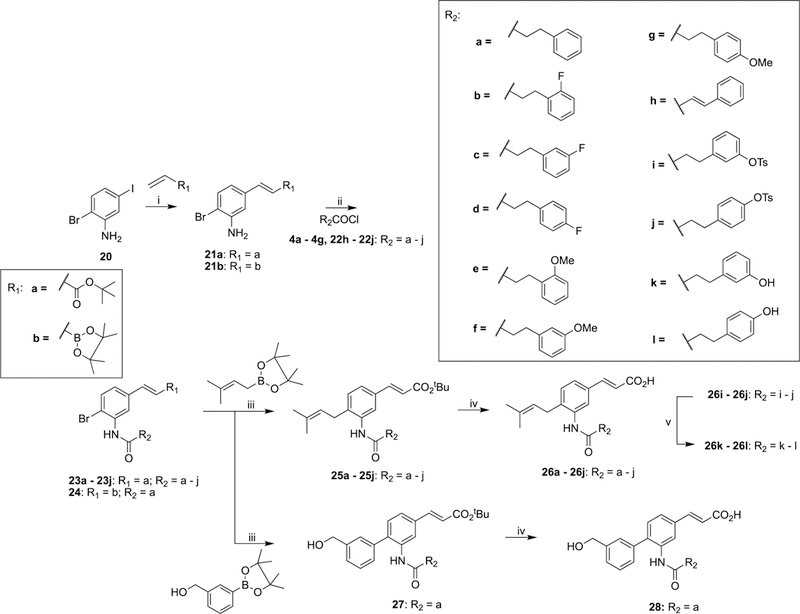
aReagents and conditions: (i) Pd(OAc)2, P(Ph)3, NEt3, PhMe, 110 °C; (ii) DMAP, NEt3, DCM, 40 °C; (iii) Pd(dppf)Cl2, Cs2CO3, DMF, 90 °C; (iv) SO PhMe, reflux; (v) MeOH, 1 M NaOH, H2O, reflux.
Class III analogues (33a and 33b) were prepared from the commercially available 4-bromo-2-iodoaniline (29) as a starting material. After installation of the amide chain by reaction with acid chloride as previously described, Mizoroki–Heck cross-coupling chemoselectively afforded intermediate 31 due to the increased reactivity of iodide over bromide for the reaction.51 Suzuki–Miyaura57 cross-coupling with substituted pinacol boronate followed by tert-butyl ester deprotection yielded compounds 33a and 33b (Scheme 5).
Scheme 5. Synthesis of 33a and 33b as Amide Derivatives of Class IIIa.
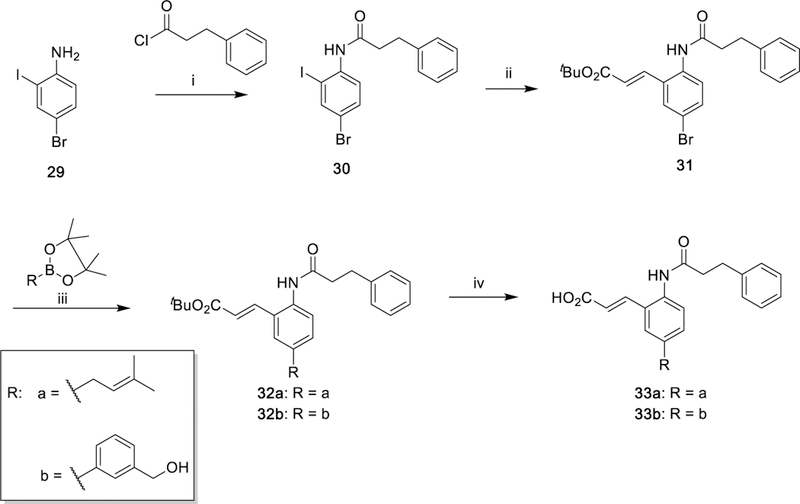
aReagents and conditions: (i) Net3 DMAP, DCM, 40 °C, 69%; (ii) tert-butylacrylate, NEt3, P(Ph)3, Pd(OAc)2, PhMe, reflux, 70%; (iii) Pd(dppf)Cl2, Cs2CO3, DMF, 90 °C; (iv) SiO2, PhMe, reflux.
Furthermore, to modify the side chain substitution pattern of the parent scaffold A ring to the 1,3,5-meta-substituted pattern, 3-bromo-5-iodobenzoic acid (34) was used as the common starting material for the synthesis of amide, ester, or retroinverse amide analogues. Conversion of 34 to 3-bromo-5- iodoaniline (35) was achieved by a modified Curtius rearrangement,59 followed by tert-butyl deprotection. A reaction sequence of amide synthesis using acid chloride, followed by Pd-catalyzed cross-couplings in the order of Mizoroki–Heck reaction followed by Suzuki–Miyaura reaction, afforded tert-butyl-protected intermediates that were subsequently hydrolyzed to afford compounds 39a and 39b. Conversion of 34 to its acid chloride followed by esterification or amide formation using phenylethyl alcohol, or the substituted primary amine, yielded the ester and amide intermediates (42 and 41a–h), respectively. The reaction sequences of Mizoroki–Heck, Suzuki–Miyaura, and tert-butyl ester hydrolysis were used to obtain compounds 49a–h and 50–52 of class III (Scheme 6).
Scheme 6. Synthesis of 1,3,5-meta-Substituted Derivatives (39a and 39b, 49a–h and 50–52) of Class IIa.
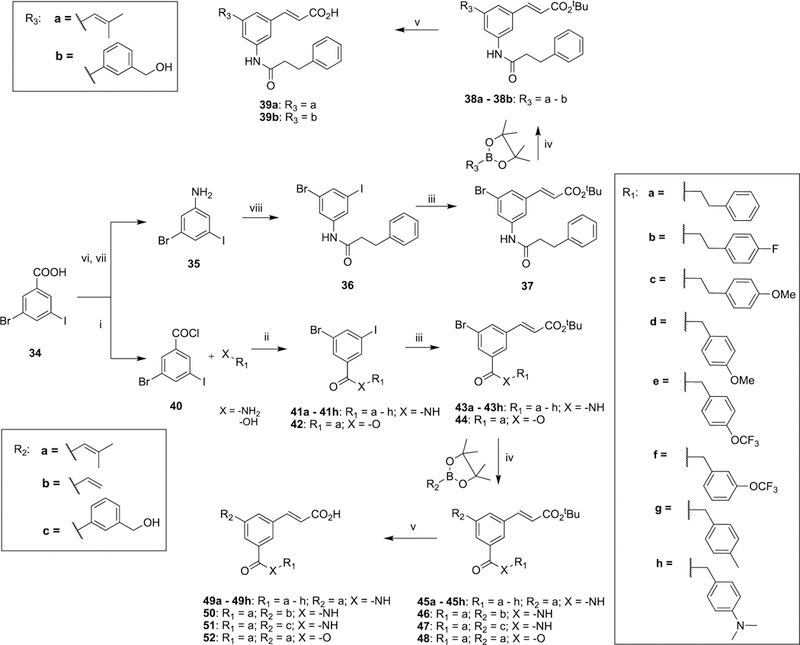
aReagents and conditions: (i) SOCl2, PhMe, reflux; (ii) DMAP, NEt3, DCM, 40 °C; (iii) tert-butylacrylate, Pd(OAc)2, P(Ph)3, NEt3, PhMe, 110 °C; (iv) Pd(dppf)Cl2, Cs2CO3, DMF, 90 °C; (v) SiO2, PhMe, reflux; (vi) diphenyl phosphoryl azide, NEt3, tBuOH; (vii) 4 M HCl, dioxane, 40%; (viii) 3-phenylpropanoyl chloride, DMAP, NEt3, DCM, 40 °C, 33%.
Ether analogues 59a and 59b were synthesized using 3-bromo-4-hydroxybenzoic acid (53) as the starting material, which was reacted with benzyl bromide or 3-methoxybenzyl bromide to install the ether side chain. After activating the carboxylic acid to an acid chloride, a reaction sequence of amide formation followed by Mizoroki–Heck reaction and tert-butyl ester hydrolysis afforded final compounds (59a and 59b) of class II (Scheme 7).
Scheme 7. Synthesis of 1,3,5-meta-Substituted Ether Derivatives (59a and 59b) of Class IIa.
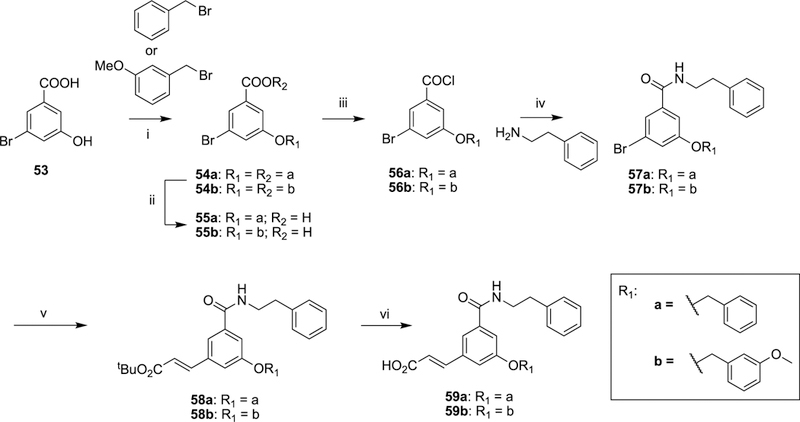
aReagents and conditions: (i) N,N-diisopropylethylamine, 150 °C; (ii) 1 M KOH, MeOH, reflux; (iii) SOCl2, PhMe, reflux; (iv) DMAP, NEt3, DCM, 40 °C; (v) tert-butylacrylate, Pd(OAc)2, DABCO, K2CO3, DMF, 110 °C; (vi) SiO2, PhMe, reflux.
Structure–Activity Relationship.
Class I Analogues.
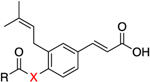
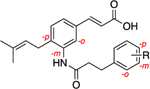
Consistent with previous findings,43,44,47 baccharin exhibited an IC50 of 105 nM for AKR1C3 inhibition with 510-fold selectivity over AKR1C2. Removal of the dihydrocinnamoyloxy group to afford 1a (drupanin) resulted in complete abrogation of AKR1C3 inhibitory potency and selectivity. Replacement of the dihydrocinnamoyloxy group (compounds 8h–n) resulted in low or submicromolar inhibition potency for AKR1C3, with a significant reduction of selectivity. Introduction of an acetoxy group (8h) and a cyclohexylacetoxy group (8j) decreased both AKR1C3 inhibitory potency and selectivity by 4-fold. As compared to 1, an increase in the carbon spacer in 8i decreased AKR1C3 inhibitory potency and selectivity by 2-fold and 4-fold, respectively. Excision of the ethyl linker to afford the benzoyloxy moiety (8l) reduced inhibitory potency by 3-fold and isoform selectivity to AKR1C3 by 8-fold. Replacement of the terminal phenyl moiety of 1 with a bulky napthyl (8k) or a simple hexane chain (8n) resulted in a decrease of the inhibitory potency by 3-fold with loss of selectivity being 12-fold (8k) and 6-fold (8n) as compared to 1. Replacement of the phenethyl moiety in 1 with a pyridine ring (8m) significantly reduced potency and selectivity. Comparison with the phenyl derivative (8l) indicates that the pyridine ring is largely responsible for this drop-in potency, potentially due to its ability to ionize under the assay conditions. Replacement of the ester linkage with an amide linkage (14a–e) led to less-potent inhibitors than their bioisosteric counterparts (1, 8h–n). Differences in potency varied from 3-fold reduction (compare 1 to 14a and 8i to 14c) to an 8-fold reduction in AKR1C3 inhibitory potency (compare 8h to 14b). Likewise, the amide derivatives, with the exception of 14a, were also significantly more promiscuous between AKR1C isoforms with only moderate to minimal selectivity for the AKR1C3 isoform over AKR1C2. Fortuitously, the amide derivative of baccharin (14a) retained potency and high selectivity, IC50 = 0.420 μM with 93-fold selectivity, for AKR1C3 over the closely related AKR1C2 isozyme. Additionally, ligand-lipophilicity efficiency (LLE), which is a measure of “drug-likeliness” and takes into account the potency and lipophilicity of compounds, was also calculated. Even though the amide bioisosteres (14a–e) are less-potent inhibitors of AKR1C3, they exhibit a more favorable LLE profile (except 14b) as compared to the ester derivatives (1, 8i–k) due to their greater lipophilicity (Table 1).
Table 1.
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | X | IC50 AKR1C3 (μM) |
IC50 AKR1C2 (μM) |
Selectivity 1C2:1C3 |
LLEb |
| 144 |  |
O | 0.105 | 51 | 510 | 1.29 |
|
1a44 (Drupanin) |
- | OH | 15 | 107 | 7 | 1.30 |
| 8h44 | -CH3 | O | 0.440 | 45 | 102 | 2.76 |
| 8i |  |
O | 0.233 | 29 | 124 | 0.56 |
| 8j | 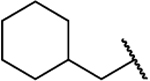 |
O | 0.453 | 44 | 97 | 0.10 |
| 8k | 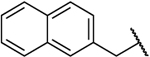 |
O | 0.370 | 16 | 43 | −0.17 |
| 8l | 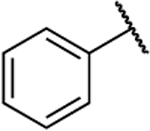 |
O | 0.348 | 21 | 60 | 0.74 |
| 8m | 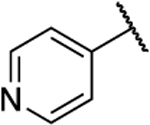 |
O | 1.8 | 14 | 7.8 | 1.52 |
| 8n | O | 0.300 | 25 | 83 | 0.81 | |
| 14a | 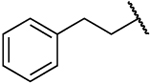 |
NH | 0.420 | 39 | 93 | 1.89 |
| 14b | −CH3 | NH | 3.6 | 30 | 8 | 2.15 |
| 14c | 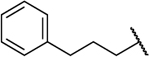 |
NH | 0.866 | 30 | 35 | 1.19 |
| 14d | 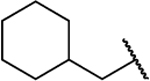 |
NH | 2.8 | 38 | 14 | 0.51 |
| 14e | 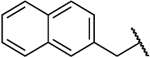 |
NH | 1.8 | 42 | 23 | 0.64 |
IC50 values were measured in quadruplicate, and technical replicates gave mean + standard error (SE). The SE is less than 10%, and the mean value is reported. AKR1C3 IC50 and fold selectivity over AKR1C2 highlighted in red.
LLE: ligand-lipophilicity efficiency (calculated as pIC50 − c log P
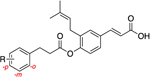
Introduction of electron-withdrawing groups onto the phenyl B ring (8b–d) resulted in compounds with inhibitory profiles similar to those of 1 with only marginally decreased selectivity. Introduction of an electron-donating methoxy group on the B ring gave compounds equipotent with the parent baccharin scaffold at all substitution positions. However, a clear correlation between selectivity and substitution position was observed. When the substitution occurs at the para (8g) or ortho (8e) positions, a 2-fold loss of selectivity is apparent. When the substitution occurs at the meta position (8f), selectivity is retained. The derivatives 8e–g display an LLE profile similar to that of 1 (Table 2).
Table 2.
Inhibitory Properties of Class I Analogues on AKR1C3 and AKR1C2a
 | |||||
|---|---|---|---|---|---|
| compound | R | IC50 AKR1C3 (μM) |
IC50 AKR1C2 (μM) |
selectivity 1C2:1C3 |
LLEb |
| 8b | 2-F | 0.125 | 45 | 360 | 1.07 |
| 8c | 3-F | 0.156 | 62 | 397 | 0.97 |
| 8d | 4-F | 0.135 | 67 | 496 | 1.04 |
| 8e | 2-OMe | 0.135 | 37 | 274 | 1.26 |
| 8f | 3-OMe | 0.126 | 57 | 452 | 1.29 |
| 8g | 4-OMe | 0.132 | 34 | 258 | 1.27 |
IC50 values were measured in quadruplicate, and technical replicates gave mean + SE. The SE is less than 10%, and the mean value is reported.
LLE: ligand-lipophilicity efficiency (calculated as pIC50 − c log P).
Class II Analogues.
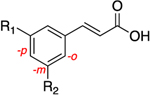
The central A-ring meta-substituted derivatives 19a, 26a, and 26h showed enhanced AKR1C3 inhibitory potency. The meta-ester derivative (19a) provided the best combination of potent inhibition (IC50 = 0.088 μM) and selectivity in all of the derivatives discussed thus far, with 261-fold selectivity for AKR1C3 (Table 3). The meta-amide derivative (26a) is the most potent AKR1C3 inhibitor identified among all of the baccharin derivatives synthesized to date (IC50 = 0.066 μM), equipotent with the most active reported AKR1C3 enzyme inhibitors.26 Selectivity is diminished over the parent scaffold, but the compound still retains a 109-fold selectivity for AKR1C3. Compounds that have midnanomolar potency for AKR1C3 and >100-fold selectivity are leads for further development. Moreover, 26a displayed an increase in LLE by 2-fold as compared to 1 and by 1.3-fold in comparison to its isomer 14a. Introduction of rigidity in the side chain amide chain via formation of a trans-alkene (26h) decreased the inhibitory potency for AKR1C3 (IC50 = 0.098 μM) as compared to 26a but resulted in 50% increased selectivity. In comparison with the parent scaffold (1), 26h retained equipotency but suffered a 3-fold loss of selectivity. This data suggests that free rotation on this side chain is essential for high selectivity. Replacement of the prenyl side chain with a 3-hydroxymethylphenyl group (28) decreased inhibitory potency (5-fold reduction over that of 1) and selectivity (34-fold loss over that of 1). Removal of the terminal methyl groups on the prenyl chain, to form a terminal alkene 19b, also diminished potency and selectivity, suggesting that the methyl groups extend into an open subpocket (SP) in the active site of AKR1C3 to confer potency and selectivity. Removal of the prenyl and ester side chains (17a) completely abrogated activity. Consistent with class I analogues, the amide derivatives (26a, 26h, and 28) exhibit more promising physicochemical traits as evidenced by their LLE values when compared to the ester derivatives (17a, 19a, and 19b) (Table 3).
Table 3.
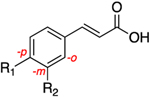 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 AKR1C3 (μM) |
IC50 AKR1C2 (μM) |
Selectivity 1C2:1C3 |
LLEb |
| 17a | -Br | -OH | 38 | 85.3 | 2 | 2.26 |
| 19a47 |  |
 |
0.088 | 23 | 261 | 1.36 |
| 19b | 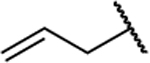 |
 |
0.273 | 44 | 161 | 1.80 |
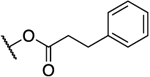
| 26a47 | 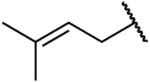 |
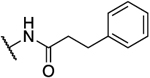 |
0.066 | 7.2 | 109 | 2.69 |
| 26h | 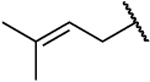 |
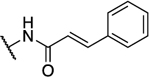 |
0.098 | 14 | 143 | 2.11 |
| 28 | 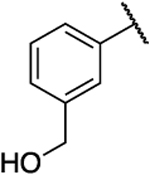 |
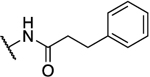 |
0.540 | 8.2 | 15 | 3.09 |
IC50 values were measured in quadruplicate, and technical replicates gave mean + SE. The SE is less than 10%, and the mean value is reported.
LLE: ligand-lipophilicity efficiency (calculated as pIC50 − c log P).
Based on the enhanced potency of 26a, we next sought to investigate the effect of substituents on the terminal B ring of the parent scaffold. Such derivatives exhibited a range of activities from compounds that were slightly more potent than 1 (26b–g) to a reduction of approximately 1.5-fold in AKR1C3 inhibition potency (26i–k). Introduction of fluorine displayed fairly similar potencies, comparable to 26a, with meta substitution (26c) showing a slight advantage over the ortho and para positions. However, the selectivity decreased by approximately 1.5-fold (26b–d). Interestingly, methoxy substitution at the para position (26g) increased isoform selectivity as compared to 26a and all substitution patterns conserved inhibitory potency (26e–g). The inhibition profiles of tosyl (26i and 26j) and hydroxy (26k and 26l) substituted analogues were comparable to those of 1 with only a marginal decrease in potency; however, selectivity decreased by 5–10fold. The LLE value remained similar to that of 26a among all of the derivatives, except hydroxy substituted compounds (26k and 26l), which displayed an increased LLE to more than 3 (a 2.4-fold increase over that of 1) (Table 4).
Table 4.
Inhibitory Properties of Class II Analogues on AKR1C3 and AKR1C2a
 | |||||
|---|---|---|---|---|---|
| compound | R | IC50 AKR1C3 (μM) |
IC50 AKR1C2 (μM) |
selectivity 1C2:1C3 |
LLEb |
| 26b | 2-F | 0.098 | 5.8 | 59 | 2.37 |
| 26c | 3-F | 0.076 | 6 | 79 | 2.49 |
| 26d | 4-F | 0.091 | 7.8 | 86 | 2.41 |
| 26e | 2-OMe | 0.081 | 7 | 86 | 2.68 |
| 26f | 3-OMe | 0.089 | 10 | 112 | 2.64 |
| 26g | 4-OMe | 0.094 | 16 | 170 | 2.62 |
| 26i | 3-OTs | 0.180 | 7.6 | 42 | 0.81 |
| 26j | 4-OTs | 0.190 | 16 | 84 | 0.78 |
| 26k | 3-OH | 0.140 | 23 | 164 | 3.03 |
| 26l | 4-OH | 0.110 | 14 | 127 | 3.13 |
IC50 values were measured in quadruplicate, and technical replicates gave mean + SE. The SE is less than 10%, and the mean value is reported.
LLE: ligand-lipophilicity efficiency (calculated as pIC50 − c log P).
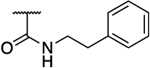
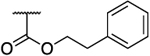
Rearrangement of the substituents on the central A ring to adopt a 1,3,5 substitution pattern greatly enhanced the selectivity for AKR1C3 inhibition over the highly related AKR1C1, 1C2, and 1C4 isoforms, as compared to 1. With the exception of 39b (Table 5), all compounds possessing this substitution pattern showed inhibitory potency in the low submicromolar range, with the majority exhibiting comparable or greater potency than 1 (39a, 49a–h) (Tables 5 and 6). The meta-amide 39a displayed inhibitory potency similar to that of 1 with a 1.5-fold increase in selectivity over AKR1C2, and the selectivity over other AKR isoforms also increased. Replacement of the prenyl side chain with a 3-hydroxymethylphenyl group (39b) diminished potency and selectivity over 1C2 by 100-fold and 21-fold, respectively. When the amide was retroinverted (49a), we observed isoform selectivity >2857-fold over AKR1C2, an increase of 5.6-fold as compared to 1. Selectivity over the other isozymes, AKR1C1 and AKR1C4, increased by 1.5 and 7-fold, respectively, when compared to 1. Moreover, 49a demonstrated an IC50 of 0.07 μM for AKR1C3, which is a 1.5-fold increase from that of 1. Additionally, 49a also exhibited a 1.6-fold increase in LLE as compared to the meta-amide 39a. Replacement of the prenyl side chain with an allyl group (50) resulted in a 7-fold reduction in isoform selectivity and 4-fold reduction in potency in agreement with prior data (19b). Although compound 50 did exhibit similar selectivity over AKR1C2 with a 2-fold reduction in potency as compared to our hit scaffold (1). Replacement of the prenyl chain with a 3-hydroxymethylphenyl moiety (51) reduced AKR1C3 inhibitory potency (3-fold from that of 1) as well as selectivity (1.7-fold from that of 1), although to a lesser extent than 39b and showed an improvement in LLE value (2-fold from that of 1). The meta-ester analogue (52) decreased AKR1C3 inhibitory potency by 1.7-fold and selectivity over AKR1C2 by 5-fold compared to its amide counterpart (49a), while maintaining comparable selectivity over AKR1C2 similar to 1 and equipotent inhibition for 1C3, although LLE reduction was dramatic (3.4-fold as compared to 49a). The ether analogues (59a and 59b) suffered a significant loss (18–46-fold) in inhibition selectivity over AKR1C2, a decrease in inhibition potency (2–4-fold), and a slight reduction of LLE (1.01–1.1-fold) when compared to 49a (Table 5).
Table 5.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 AKR isoform (μM) |
Fold Selectivity | LLE* | |||||
| 1C1 | 1C2 | 1C3 | 1C4 | 1C1: 1C3 |
1C2: 1C3 |
1C4: 1C3 |
||||
| 1 | 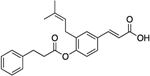 |
187 | 51 | 0.10 | 21 | 1870 | 510 | 210 | 1.29 | |
| 39a |  |
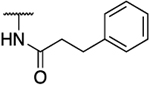 |
35.5% inhibition at 200 μM |
94 | 0.13 | 206 | >1538 | 723 | 1584 | 1.53 |
| 39b | 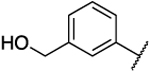 |
 |
N.D. | 41 | 1.20 | N.D. | N.D. | 34 | N.D. | 1.71 |
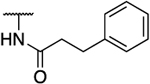
| 49a | 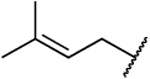 |
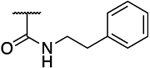 |
No inhibition at 200 μM |
39% inhibition at 200 μM |
0.07 | 99 | >2857 | >2857 | 1414 | 2.09 |
| 50 | 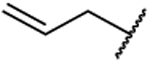 |
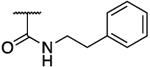 |
N.D. | 88 | 0.26 | N.D. | N.D. | 400 | N.D. | 2.45 |
| 51 | 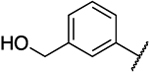 |
 |
N.D. | 40% inhibition at 100 μM |
0.33 | N.D. | N.D. | >300 | N.D. | 2.57 |
| 52 | 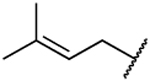 |
 |
47.0% inhibition at 200 μM |
62 | 0.12 | 15 | >1666 | 517 | 125 | 0.61 |
| 59a | 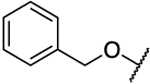 |
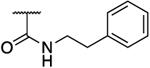 |
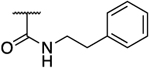
N.D. | 26 | 0.16 | N.D. | N.D. | 163 | N.D. | 2.05 |
| 59b | 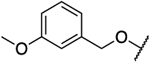 |
 |
N.D. | 18 | 0.29 | N.D. | N.D. | 62 | N.D. | 1.87 |
N.D.: Not determined.
AKR1C3 IC50 and fold selectivity over AKR1C2 highlighted in red.
IC50 values were measured in quadruplicate, and technical replicates gave mean + SE. The SE is less than 10%, and the mean value is reported.
LLE: ligand-lipophilicity efficiency (calculated as pIC50 − c log P).
Table 6.
Inhibitory Properties of Class II Analogues on AKR1C1 and AKR1C4a
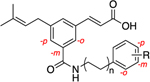 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | n | R | IC50 AKR isoform (μM) |
Selectivity | LLEb | |||||
| 1C1 | 1C2 | 1C3 | 1C4 | 1C1:1C3 | 1C2:1C3 | 1C4:1C3 | ||||
| 49b | 2 | 4-F | No inhibition at 200 μm |
148 | 0.10 | 140 | >2000 | 1480 | 1400 | 1.80 |
| 49c | 2 | 4-OMe | No inhibition at 200 μM |
136 | 0.11 | 72 | >1802 | 1236 | 649 | 0.98 |
| 49d | 1 | 4-OMe | No inhibition at 100 μM |
87 | 0.07 | 60 | >1315 | 1144 | 789 | 2.27 |
| 49e | 1 | 4-OCF3 | No inhibition at 200 μM |
165 | 0.11 | 151 | >1802 | 1500 | 1360 | 0.99 |
| 49f | 1 | 3-OCF3 | No inhibition at 200 μM |
129 66.5% |
0.13 | 133 | >1538 | 992 | 1023 | 0.93 |
| 49g | 1 | 4-Me | No inhibition at 200 μM |
No inhibition at 200 μM |
0.07 | 145 | >2817 | >2817 | 2042 | 1.72 |
| 49h | 1 | 4-NMe2 | No inhibition at 100 μM |
80 | 0.06 | 59 | >1493 | 1194 | 880 | 1.27 |
IC50 values were measured in quadruplicate, and technical replicates gave mean + SE. The SE is less than 10%, and the mean value is reported.
LLE: ligand-lipophilicity efficiency (calculated as pIC50 − c log P).
When substitutions on the B ring were coupled with homologation of the amide-containing chain, all of the substitutions were generally well tolerated, increasing the selectivity for AKR1C3 inhibition by 3–7-fold in comparison to 1 and maintaining the inhibition potency equivalent to or greater than that of 1 (49b–f) (Table 6). The 4-fluoro (49b) and 4-methoxy (49c) substitutions on the phenethyl side chain exhibited similar inhibitory potency to 1, increasing the selectivity over 1C2 by 3-fold and 2.4-fold, respectively, without any significant change in LLE over 1. Reduction of the carbon spacer length, along with substitution with N,N-dimethyl (49h), methoxy (49d), or methyl (49g) groups at the 4-position on the benzyl side chain further increased the inhibitory potency as compared to 1. Compounds 49h and 49d were 3-fold more selective than 1, whereas 49g exhibited the best combination of potency (IC50 = 0.07 μM) and selectivity (>2800, a 7-fold increase from that of 1). The 4-trifluoromethoxy benzyl containing analogue (49e) exhibited an inhibition and selectivity profile similar to that of 49b. However, movement of the –OCF3 group to the meta position (49f) diminished inhibitory potency by 1.2-fold and selectivity over AKR1C2 by 1.5-fold compared to 49e. With the exception of 49d, none of the derivatives displayed an increase in the LLE value as compared to 49a (Table 6).
Class III Analogues.
Boronic acid bioisosteres of carboxylic acids are expected to demonstrate increased binding affinity to their molecular targets since they would provide a strong counterion in the oxyanion (OX) hole as compared to ionic interactions with their carboxylic acid counterparts, as well as their ability to form covalent bonds with amino acid residues.60,61 Bioisosteric replacement of the carboxylic acid of our parent scaffold with a pinacol boronate (7) or boronic acid (7a) decreased AKR1C3 inhibitory potency by 66- and 23-fold, respectively, compared to 1, with a concurrent 500-fold decrease in selectivity, in addition to exhibiting a poor LLE profile. Removal of the prenyl moiety (24) abrogated bioactivity in agreement with our previous findings.44 Surprisingly, when the amide chain was placed ortho relative to the carboxylic acid group (33a), a 1.4-fold increase in potency was observed compared to 14a with similar selectivity and an LLE similar to that of 1. Replacement of the prenyl moiety in 33a with a 3-hydroxymethylphenyl moiety (33b) diminished potency and selectivity by 5-fold and 12-fold, respectively, in comparison to 33a without any appreciable impact on LLE (Table 7).
Table 7.
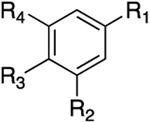
Inhibitory Properties of Class III Analogues on AKR1C3–AKR1C2a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50 AKR1C3 (μM) |
IC50 AKR1C2 (μM) |
Selectivity 1C2:1C3 |
LLEb |
| 7 | 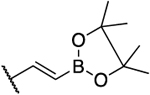 |
H | 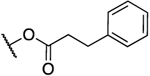 |
 |
6.6 | 4.7 | 0.71 | −2.89 |
| 7a | 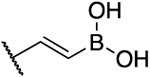 |
H | 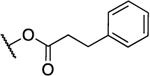 |
 |
2.3 | 3.8 | 1.7 | 0.26 |
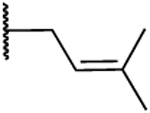
| 24 | 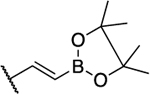 |
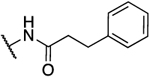 |
-Br | H | 16.0 | 109 | 6.8 | −0.93 |
| 33a | 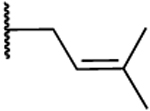 |
 |
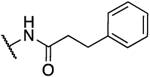 |
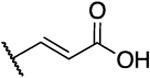
H | 0.2 | 39 | 163 | 1.26 |
| 33b | 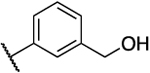 |
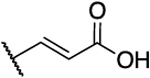 |
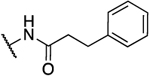 |
H | 1.0 | 14 | 14 | 1.79 |
IC50 values were measured in quadruplicate, and technical replicates gave mean + SE. The SE is less than 10%, and the mean value is reported.
LLE: ligand-lipophilicity efficiency (calculated as pIC50 − c log P).
In this study, the inhibitory potency and selectivity of several analogues of 1 against AKR1C3 were investigated. Compound 26a showed the most potent inhibition (IC50: 66 nM), whereas compound 49a displayed the highest selectivity for AKR1C3 over AKR1C2 (IC50 ratio: >2857). Analysis of crystal structures of AKR1C3-nicotinamide adenine dinucleotide phosphate (NADP+)–inhibitor complexes have revealed that the ligand binding site can be dissected into five subsites: the oxyanion site (consisting of catalytic residues Tyr55, His117, and cofactor NADP+), the steroid channel for the binding of steroid ligands, and three subpockets (SPs), namely, SP1, SP2, and SP3, to accommodate other ligands.23,26 In the AKR1C3-NADP+ 1 complex model (Figure 3A),43 the carboxylate group on the cinnamic acid was predicted to occupy the SP1 pocket and could form hydrogen bonds with the side chain of Ser118, which contributes to strong binding affinity. In addition, several polar amino acids (e.g., Ser308 and Tyr319) also reside in the SP1 pocket of AKR1C3, which provide the possibility for ligand–protein interactions. These polar interactions would be expected to increase the selectivity of 1 for AKR1C3 over the other AKR1C isoforms where the amino acids in the corresponding positions of the SP1 pocket are Phe118, Leu308, and Phe319.23,26 The docking model predicted that the benzyl moiety of the 4-dihydrocinnamoyloxy group was located in the SP3 pocket and could form hydrophobic interactions with the side chain of Gln222 or Phe306 to provide high binding affinity.43 In addition, recent studies have shown that polar substitutions (e.g., hydroxyl and carboxylate groups) on the phenyl ring of the cinnamic acid, or the phenyl ring of the 4-dihydroxycinnamoyloxy group, decreased inhibition for AKR1C3.43,45,62 For example, the displacement of the 4-dihydrocinnamoyloxy group by a hydroxyl group to form drupanin (1a) resulted in a loss of inhibitory potency (IC50: 15 μM) and only a 7-fold selectivity for AKR1C3 versus AKR1C2. An increase or decrease of the carbon spacer along with replacement of the B ring in class I analogues exhibited low or submicromolar inhibition activities (8h–n) but still exhibited >50-fold isoform selectivity. To impart hydrolytic stability to our derivatives, replacement of the ester functional group with an amide was performed. The corresponding amide bioisosteres displayed diminished potency and selectivity (14a–e), suggesting that the increased rigidity of the side chain amide is not tolerated at the para position of the A ring. The B-ring substitutions on the dihydrocinnamoyloxy moiety were very well tolerated, and only a marginal reduction of potency and selectivity was observed (8b–g).
Figure 3.
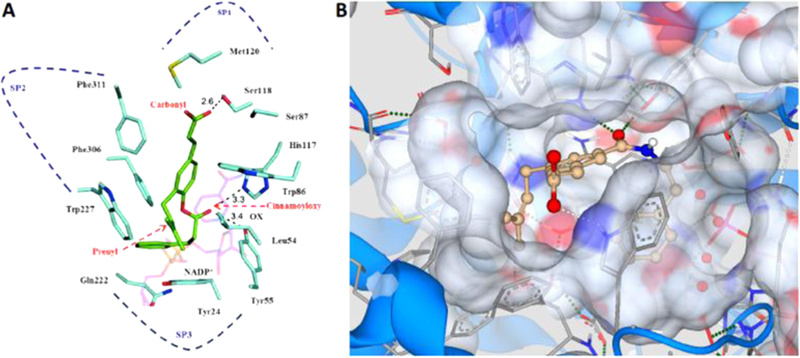
Modeling of the parent compound (1) and the most selective analogue 49a. (A) Model of the AKR1C3·NADP+·1 complex. AKR1C3 residues (light blue), NADP+ (pink), and baccharin (green). Dotted line: possible hydrogen bond, OX: oxyanion site, SP: subpocket. The docking model of AKR1C3·NADP+·1 complex was constructed by calculations using the program Glide 5.0. The generated docking structure showing the highest score was selected for further structural analysis. The crystal structure of the AKR1C3·NADP+ complex was chosen from the RCSB protein data bank (PDB code: 1S2C). Reproduced with permission from ref 43. (B) Quantitative structure–activity relationship prediction of analogue 49a (gold) with amino acid residues of the AKR1C3 binding site (gray). Green dotted line: strong predicted hydrogen bond, white dotted line: weak predicted hydrogen bond. The model was constructed with SeeSAR 8.1 (BioSolveIT GmbH) using AKR1C3 protein from the RCSB protein data bank (PDB code: 3UG8).
When the substitution pattern of the dihydrocinnamoyloxy side chain was changed to the meta position (analogues 19a and 26a), the inhibitory potency increased in comparison to that of 1. The presence of either a carbonyl or carboxylate group is required to anchor many ligands to the oxyanion site because it can form a strong hydrogen bond with Tyr55 and His117 and bring the ligands into close proximity of the nicotinamide head group of the cofactor.26 Based on the model of baccharin docked into the AKR1C3–NADP+ complex structure (Figure 3A),43 the carbonyl of the 4-dihydrocinnamoyloxy group of 1 was found to be close to the oxyanion site and could form a hydrogen-bond interaction with Tyr55 and His117, which could contribute to the high inhibitory potency. Thus, it seems plausible that in the meta-substituted analogues, the change in the position of the carbonyl group results in close proximity to the oxyanion site, imparting enhanced potency due to a shorter hydrogen-bond distance. The presence of a side chain amide (26a) increased potency even further, along with exhibiting good tolerability of B-ring substituents (26b–1). Further modification of the side chain substitution pattern did not increase potency or selectivity (33a and 33b). Contrary to expectation, the boronic acid analogue (7a) lost inhibition potency by more than 10-fold and abrogated isoform selectivity.
Modeling experiments performed on compound 49a (Figure 3B) predict that the designed derivatives with a retroinverted amide group (49a–h, 51, and 52) form a stronger hydrogenbond interaction in comparison to their noninverted counterparts (39a and 39b) within the oxyanion site, due to the switch in the position of the carbonyl group, resulting in formation of a shorter hydrogen bond with amino acid residues Tyr55 and His117, leading in turn to superior inhibitory potency and selectivity. Substituting the side chains in a 1,3,5 arrangement at the central A ring places the prenyl chain in close proximity to the hydrophobic amino acid residues of the SP2 subpocket, which imparts strong hydrophobic interactions and increases selectivity over the closely related isoforms AKR1C1 and AKR1C2 that do not possess this open pocket. This is further supported by the observation that removal of the terminal methyl groups to form the allyl derivative (50) results in reduced potency and selectivity as this would be expected to result in retraction of the chain from the open pocket, weakening the hydrophobic interactions. This model also predicts the formation of a hydrogen bond between the carboxylic acid of compound 49a and the Trp227 residue, rather than Ser118 residue as in the parent compound 1. Homologation of the carbon linker between the two phenyl rings to reduce the length results in little observable effect (compare 49a and 49g) as the area occupied by the terminal phenyl ring in analogues with either one or two methylene groups is rich in π systems that can maintain the predicted π–π stacking interactions.
Evaluation of AKR1C3 Inhibitors in Leukemic Cell Models.
The hit compound 1, m-amide analogue (26a), and 1,3,5 trisubstituted analogues 49a and 49g were selected for further studies in AML cell models as they represent a range of the most potent (AKR1C3 IC50 = 0.105, 0.066, 0.07, and 0.07 μM, respectively) and selective (AKR1C3 fold selectivity of 420, 109, 2857, and 2817, respectively) AKR1C3 inhibitors. Consistent with our previous findings,47 the inhibitors did not induce cytotoxicity in a panel of AML cell lines up to concentrations as high as 100 μM, with the exception of 26a, which did not reduce AML cell viability up to 50 μM (Figures S1–S3, Supporting Information (SI)). Low inhibitor concentrations of 0.1, 1, and 10 μM were chosen to evaluate the adjuvant effect of AKR1C3 inhibitor in combination with clinically approved chemotherapeutic agents. The AML cell lines HL-60 (M3 subtype),47 KG1a (M0 subtype), and THP-1 (M5 subtype), expressing varying levels of AKR1C3 along with AKR1C3 overexpressing (Figure S4) primary patient-derived T-ALL cells (Children’s Oncology Group (COG)-LL-317h and COG-LL-329h), were chosen to evaluate the effects of AKR1C3 inhibitors.
Synergistic Effect of Combination of AKR1C3 Inhibitor and Daunorubicin on AML Cell Lines.
Daunorubicin, an anthracycline used as a front-line treatment for AML63,64 and as induction chemotherapy for T-ALL,11 is most susceptible to AKR1C3-mediated metabolism among all of the anthracycline chemotherapeutic agents, rendering the agent inactive.65 To evaluate whether a combination of an AKR1C3 inhibitor with daunorubicin provides a synergistic effect, a dose–response curve of daunorubicin in AML cells was first obtained (Figure S5, SI) and AKR1C3 inhibitors were then dosed with daunorubicin in a fixed ratio of 100:1 (inhibitor/daunorubicin). Combination co-treatments of AKR1C3 inhibitors 1, 26a, 49a, or 49g with daunorubicin did not reduce cell viability beyond what was observed with daunorubicin treatment alone. The combination, when analyzed by the Chou–Talalay method,66 showed a slight antagonistic to additive effect, in HL-60 cells (M3) with all of the inhibitors tested (combination index (CI) = 1.2–2.3), with the exception of 49a, which was weakly synergistic (CI = 0.8). In KG1a cells (M0), which are daunorubicin resistant,67,68 none of the co-treatments of AKR1C3 inhibitors with daunorubicin reduced cell viability by more than 50%. Only concentrations of 0.1–10 μM AKR1C3 inhibitors with 0.01 μM daunorubicin were able to reduce cell viability to >50% in THP-1 cells (M5) (Figure S6, SI).
Pretreatment of AKR1C3 inhibitors for 24 h followed by daunorubicin exposure for a further 72 h enhanced the cytotoxicity of the anthracycline in HL-60 cells. A near-complete reduction in cell viability was observed following pretreatment with an AKR1C3 inhibitor followed by 0.1 μM daunorubicin. When the effect was quantified, a strong to moderate drug synergism was noted (CI = 0.2–0.7) (Figure 4A and the table inset). The daunorubicin-resistant KG1a cells with high AKR1C3 expression displayed a complete abrogation of cell viability when pretreated with AKR1C3 inhibitors at 0.1 μM concentration followed by 0.001 μM daunorubicin. Avery strong synergistic drug effect with all inhibitors was seen (CI = >0.01), providing a chemotherapeutic dose reduction of >100- fold (dose reduction index (DRI) > 100) and reducing the combination IC50 to <0.01 μM, indicating a complete reversal of daunorubicin resistance (Figure 4B and the table inset). A complete reduction in cell viability reduction was also observed in THP-1 cells following pretreatment with all AKR1C3 inhibitors followed by 0.1 and 1 μM daunorubicin treatment, and a 50% reduction in cell viability was seen with 0.01 μM daunorubicin when this concentration of daunorubicin alone had no effect on cell viability. A strong synergism was established in THP-1 cells (CI = 0.06–0.1) where the reduction in IC50 value of daunorubicin was greater than 7-fold. The more selective compounds 49a and 49g provided a greater reduction (DRI = 16) in chemotherapeutic dosing as compared to 1 and 26a (DRI = 6.6–8.3) (Figure 4C and the table inset). Furthermore, a direct correlation is observed between AKR1C3 expression in AML cell lines (KG1a ≫ THP-1 > HL-60) (Figure S4, SI) and the synergistic effect and dose reduction index. Overall, such high degree of drug synergism can be attributed to the inhibitory potency of compounds 1, 26a, 49a, and 49g for AKR1C3 24 h prior to daunorubicin exposure in pretreatment experiments as compared to the co-treatments where only an additive to moderate synergism was observed.
Figure 4.
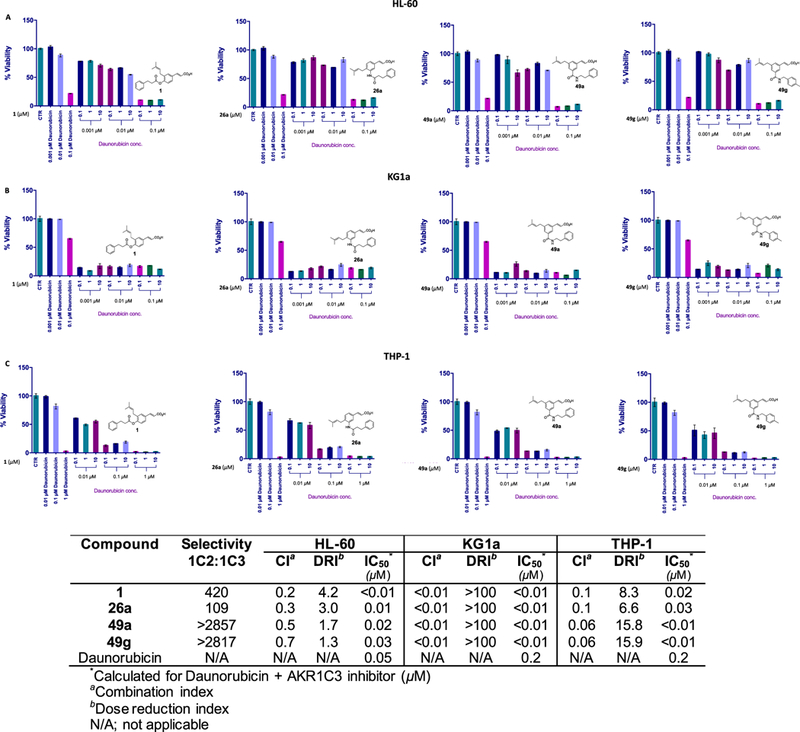
Combination treatment of AKR1C3 inhibitors with daunorubicin in AML cells. The percentage cell viability after 24 h pretreatment with 1, 26a, 49a, and 49g followed by a 72 h exposure of daunorubicin in (A) HL-60, (B) KG1a, and (C) THP-1 cells at indicated concentrations. The table inset shows the quantification of drug interactions.
Synergistic Effect of Combination of AKR1C3 Inhibitor and Cytarabine on AML Cell Lines.
The first-line AML chemotherapeutic cytarabine (AraC) was combined with AKR1C3 inhibitors under a similar dosing regimen as described above. To the best of our knowledge, no relationship has been established between the enzymatic activity of AKR1C3 and the sensitivity of leukemic cells to AraC. Upon co-treatment of AKR1C3 inhibitors with AraC in HL-60 cells, the change in cell viability reduction closely matched that of AraC alone (Figure S5, SI). Effects ranged from moderate synergism (compounds 1 and 7) (CI = 0.5) to slight antagonism (4) (CI = 3.4) to addition (6) (CI = 1). Similarly, KG1a and THP-1 cell lines displayed nearly identical dose–response curves for AraC in the presence and absence of all AKR1C3 inhibitors. Only an additive drug effect was observed for the combination that was consistent among all tested inhibitors (CI = 0.9–1.4) (Figure S7 and the table inset, SI).
The 24 h pretreatment experiments with AKR1C3 inhibitors followed by AraC exposure provided strong synergistic effects in HL-60 cells. Combination index values ranged from 0.02 to 0.09, which provided a dose reduction in AraC ranging from 10- to 33-fold (Figure 5A and the table inset). Similar to the observations made with pretreatment with AKR1C3 inhibitors followed by daunorubicin in the high-AKR1C3-expressing KG1a cells, AKR1C3 inhibitor pretreatments followed by AraC exposure also abrogated the cell viability at all compound concentrations, displaying a very strong degree of synergistic drug effect with all inhibitors (CI = <0.01). A chemotherapeutic DRI of >100-fold was achieved, which reduced the combination IC50 to <0.01 μM (Figure 5B and the table inset). Similarly, a strong synergism was observed in THP-1 cells (CI = 0.05–0.07), where the reduction in the IC50 value of AraC was greater than 13-fold (Figure 5C and the table inset).
Figure 5.
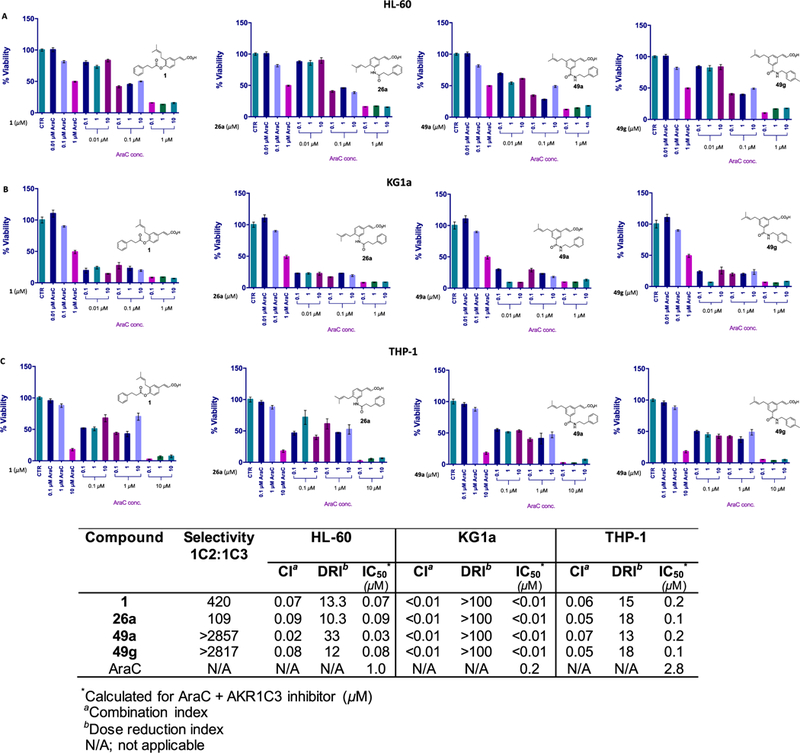
Combination treatment of AKR1C3 inhibitors with AraC in AML cells. The percentage cell viability after 24 h pretreatment with 1, 26a, 49a, and 49g followed by a 72 h exposure of AraC in (A) HL-60, (B) KG1a, and (C) THP-1 cells at indicated concentrations. The table inset shows the quantification of drug interactions.
Synergistic Effect of AKR1C3 Inhibition on Primary Patient-Derived T-ALL Cells.
For further evaluation of the adjuvant effects of AKR1C3 inhibitors, primary patient-derived T-ALL cells were employed as an in vitro model. The Children’s Oncology Group (COG)-317h cell line represents a cell line derived from samples of pediatric patients who relapsed after chemotherapy, whereas COG-329h cells were derived from samples of pediatric patients prior to chemotherapy treatment. After individual dose–response curves of 49a, daunorubicin, and AraC were obtained (Figure S8, SI), compound 49a, possessing the best combination of AKR1C3 inhibitory potency (IC50 = 0.07 μM) and isoform selectivity (>2857), demonstrated a very strong synergistic drug effect on both COG-317h and COG-329h cells, when co-treated with daunorubicin (DRI = 19.8 and 13.0, respectively) (Figure 6A and the table inset).
Figure 6.
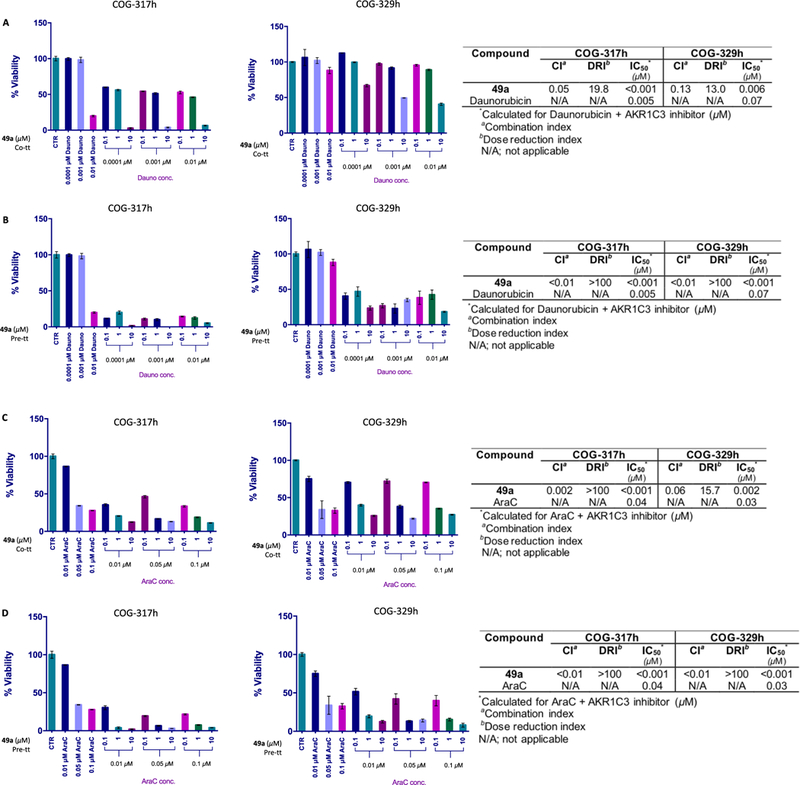
Combination treatment of AKR1C3 inhibitors with daunorubicin and AraC in patient-derived primary T-ALL cells. The percentage cell viability of indicated cell lines after treatment with the indicated concentrations of daunorubicin and AKR1C3 inhibitor 49a as (A) co-treatments and (B) 24 h pretreatments. The percentage cell viability of indicated cell lines after treatment with the indicated concentrations of AraC and 49a as (C) co-treatments and (D) 24 h pretreatments. The table inset shows the quantification of drug interactions.
The degree of drug synergism increased even further among pretreatment experiments (DRI > 100), and a near-complete abrogation of cell viability was noticed in COG-317h cells (Figure 6B and the table inset). Both co-treatment and pretreatments of 49a with AraC showed a very strong synergism in COG-317h cells (DRI > 100). In COG-329h cells, the effect increased from a DRI of 15.7, among co-treatments, to >100 in pretreatment experiments (Figure 6C,D and the table inset). Thus, the combination of our developed AKR1C3 inhibitors with either daunorubicin or AraC in patient-derived T-ALL cells reduces the IC50 of both clinically approved chemotherapeutics to less than one nanomolar.
Effect of AKR1C3 Inhibition on Primary Bone Marrow Mononuclear Cells (BMMNC).
To evaluate the selective chemotherapeutic potentiation of AKR1C3 inhibitor 49a toward leukemic cells, 24 h pretreatment experiments with 49a followed by incubation with AraC or daunorubicin were performed in primary bone marrow mononuclear cells. No chemotherapeutic potentiation was noted with either AraC or daunorubicin combination treatments (Figure S9, SI).
CONCLUSIONS
This study revealed a detailed SAR map for inhibition of AKR1C3 around the chemotype of 1. The 1,3,5-meta-substituted retroinverted amides are disclosed as the most selective inhibitors of AKR1C3 across all known inhibitor classes reported thus far, a first of their kind to exhibit exquisite selectivity (>2857-fold) for AKR1C3 inhibition with inhibitory potency in the nanomolar range. These agents carry the potential to be further developed into advanced lead compounds and span applicability across a diverse variety of malignancies that are AKR1C3-dependent.
Inhibitors of AKR1C3 derived from the modification of a natural product possess enhanced biological stability and exhibit extremely high selectivity and potency. A proof of concept that AKR1C3 inhibitors derived from 1 have a synergistic effect to sensitize AML and primary T-ALL cells to the chemotherapeutic effects of daunorubicin and AraC is demonstrated. These findings are in agreement with those of prior reports by us, and others, that establish an adjuvant effect of AKR1C3 inhibitors in combination with various chemotherapeutic agents.46,69 Treatment of the nontoxic AKR1C3 inhibitors in in vitro models of AML representing various French–American–British subtypes of AML (M0, M3, and M5), along with co-administration of a range of clinical chemotherapeutics, results in a synergistic drug action, potentiating cytotoxicity and reducing the IC50 value. This is contrary to recent reports detailing that selective AKR1C3 inhibitors do not perform an adjuvant role as compared to pan- AKR1C isoform inhibitors. This observation is further extended to the primary patient-derived T-ALL cells where a very strong potentiation of the effects of daunorubicin and AraC was established.
Further development of the inhibitors described herein represents a significant drug discovery opportunity for the identification of potent adjuvants to enhance the therapeutic index of chemotherapeutics in the hope of availing this treatment regime to pediatric and geriatric leukemia patients.
EXPERIMENTAL SECTION
Chemistry.
All reactions were carried out in oven- or flame-dried glassware under positive nitrogen pressure unless otherwise noted. Reaction progress was monitored by thin-layer chromatography carried out on silica gel plates (2.5 cm × 7.5 cm, 200 μm thick, 60 F254) and visualized using UV (254 nm) or by potassium permanganate and/or phosphomolybdic acid solution as an indicator. Flash column chromatography was performed with silica gel (40–63 μm, 60 Å) using the mobile phase indicated or on a Teledyne Isco (CombiFlash Rf 200 UV/vis). Solvents and reagents were purchased from Fisher Scientific (Houston, TX), Sigma-Aldrich (Milwaukee, WI) or for prenylboronic acid pinacol ester, Santa Cruz Biotechnology (Dallas, TX) and were used without further purification, except as indicated. Anhydrous solvents were purchased from Across Organics and stored under an atmosphere of dry nitrogen over molecular sieves.
1H, 13C, correlated spectroscopy, heteronuclear multiple quantum coherence, and distortionless enhancement by polarization transfer NMR spectra were recorded in the indicated solvent on a Bruker 400 MHz Avance III HD spectrometer at 400 and 100 MHz for 1H and 13C, respectively, with tetramethylsilane as an internal standard. Multiplicities are indicated by s (single), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are reported in hertz. High-resolution mass spectrometry (HRMS) was performed on an LC/MS IT-TOF (Shimadzu) using an electrospray ionization (ESI) source located at the University of Texas at Arlington, Shimadzu Center for Advanced Analytical Chemistry. High-pressure liquid chromatography (HPLC) was performed on a Gilson HPLC system with 321 pumps and a 155 UV/vis detector using Trilution software v2.1 with a Phenomenex Luna (C18 100 Å, 250 × 4.6 mm2) column. All final compounds were assessed to be of >96% purity and were consistent with their HRMS data.
General Procedure A. Synthesis of Ester Intermediates.
To a solution of 4-iodo-2-(3-methylbut-2-en-1-yl)phenol (3) (1 equiv) in DCM (5 mL), DMAP (0.1 equiv) was added followed by addition of substituted acid chloride (1.5 equiv) and NEt3 (1.5 equiv). The mixture was stirred overnight at RT. Saturated NaHCO3 was added and the layers were separated. The organic layer was washed with H2O, dried (Na2SO4), filtered, and concentrated. The crude product was purified by column chromatography using hexane/EtOAc gradient (10:1, 4:1, 2:1) and a solvent was evaporated in vacuo to provide pure esterified compounds.
General Procedure B. tert-Butyl Ester Hydrolysis.
To a solution of substituted tert-butyl intermediates in toluene, chromatography-grade silica gel was added and the mixture was refluxed with vigorous agitation overnight.52 Upon cooling, the reaction mixture was diluted with 10% methanol in DCM and filtered over a celite pad using 10% methanol in DCM as the solvent. The crude product was purified by column chromatography using DCM/MeOH gradient (20:1, 10:1) and a solvent was evaporated in vacuo to provide the final compounds.
(E)-3-(3-(3-Methylbut-2-en-1-yl)-4-((3-phenylpropanoyl)-oxy)phenyl)acrylic Acid (1).47
Compound 6a (280 mg, 0.6 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo and purified by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) to provide the title compound as a white solid (140 mg, 0.4 mmol, 66%). Rf 0.1 (hexane/EtOAc, 4:1) 1H NMR (400 MHz, CDCl3): δ 1.69 (3H, s, CH3), 1.77 (3H, s, CH3), 2.94 (2H, t, J = 7.7 Hz, CH2), 3.11 (2H, t, J = 7.6 Hz, CH2), 3.17 (2H, d, J = 7.4 Hz, CH2), 5.20 (1H, t, J = 7.3 Hz, CH), 6.40 (1H, d, J =16 Hz, CH), 7.00 (1H, d, J = 8.3 Hz, ArCH) 7.24–7.42 (7H, m, ArCH), 7.75 (1H, d, J =16 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 17.8, 25.7, 28.4, 30.8, 35.8, 116.7, 120.8, 122.8, 126.5, 126.8, 128.6, 130.1, 131.9, 133.9, 134.3, 139.9, 146.3, 150.6, 170.6, 171.0. ESI-HRMS (m/z): [M – H]– calcd for C23H24O4, 363.1602; found, 363.1620.
(E)-2-(3-Methylbut-2-en-1-yl)-4-(2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)vinyl)phenyl 3-Phenylpropanoate (7).
To a solution of 5a (350 mg, 0.8 mmol) in dry toluene (8 mL), PPh3 (27 mg, 0.1 mmol), Pd(OAc)2 (12 mg, 0.05 mmol), and NEt3 (0.3 mL, 2 mmol) were added. The mixture was stirred for 10 min, and 4,4,5,5-tetramethyl-2-vinyl-1,3,2-dioxaborolane (0.2 mL, 1 mmol) was added and the reaction mixture was refluxed overnight. The reaction mixture was allowed to cool and washed with saturated aqueous NH4Cl and water, extracted in DCM, dried (Na2SO4), filtered, and concentrated. Purification by column chromatography (hexane/EtOAc = 10:1, 4:1) provided the title compound as a yellow oil (70 mg, 0.15 mmol, 20%). Rf: 0.74 (hexane/EtOAc, 4:1) 1H NMR (400 MHz; CDCl3): δ 1.34 (12H, s, CH3), 1.68 (3H, s, CH3), 1.76 (3H, s, CH3), 2.92 (2H, t, J = 7.1 Hz, CH2), 3.11 (2H, t, J = 7.8 Hz, CH2), 3.14 (2H, d, J = 7.8 Hz, CH2), 5.20 (1H, t, J = 7.8 Hz, CH), 6.12 (1H, d, J = 18.4 Hz, CH), 6.93 (1H, d, J = 8.6 Hz, ArCH), 7.23–7.39 (8H, m, ArCH and CH). 13C NMR (100 MHz; CDCl3): δ 17.8, 24.8, 25.7, 28.4, 30.9, 35.8, 83.3, 121.1, 122.2, 125.5, 128.3, 128.3, 128.5, 128.6, 128.7, 133.5, 133.5, 135.4, 140.0, 148.7, 149.2, 171.1. ESI-HRMS (m/z): [M + Na]+ calcd for C28H35BO4, 469.2526; found, 469.2522.
(E)-(3-(3-Methylbut-2-en-1-yl)-4-((3-phenylpropanoyl)oxy)-styryl)boronic Acid (7a).
The solution of 7 (20 mg, 0.05 mmol) in 1:1 MeOH/H2O was stirred at 80 °C overnight. The reaction mixture was allowed to cool and washed with saturated aqueous NH4Cl and brine, extracted with DCM, dried (Na2SO4), filtered, and concentrated. Purification by column chromatography (hexane/EtOAc = 12:1, 9:1, 4:1, 2:1, 1:1) provided the title compound as a white solid (5 mg, 0.01 mmol, 20%). Rf: 0.1 (hexane/EtOAc, 4:1) 1H NMR (400 MHz; CDCl3): 5 1.68 (3H, s, CH3), 1.73 (3H, s, CH3), 2.94 (2H, t, J = 7.2 Hz, CH2), 3.06 (2H, t, J = 7.5 Hz, CH2), 3.11 (2H, d, J = 7.4 Hz, CH2), 5.16 (1H, t, J = 7.2 Hz, CH), 6.31 (1H, d, J = 18.0 Hz, CH), 6.90 (1H, d, J = 8.2 Hz, ArCH), 7.20–7.34 (7H, m, ArCH and CH), 7.39 (1H, dd, J1 = 2.1 Hz, J2 = 8.3 Hz, ArCH). 13C NMR (100 MHz; CDCl3): δ 16.5, 24.4, 28.2, 30.4, 35.2, 121.4, 122.2, 124.9, 126.0, 128.0, 128.1, 128.3, 132.6, 133.6, 135.8, 140.2, 147.1, 149.2, 171.5.
(E)-3-(4-((3-(2-Fluorophenyl)propanoyl)oxy)-3-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (8b).
Compound 6b (250 mg, 0.6 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (78 mg, 0.2 mmol, 35%). Rf: 0.16 (hexane/EtOAc, 4:1) 1H NMR (400 MHz, MeOD) δ 1.67 (3H, s, CH3), 1.72 (3H, s, CH3), 2.93 (2H, t, J = 7.3 Hz, CH2), 3.13 (2H, t, J = 7.1 Hz, CH2), 3.12 (2H, d, J = 7.1 Hz, CH2), 5.14 (1H, t, J = 7.1 Hz, CH), 6.41 (1H, d, J = 15.9 Hz, CH), 6.98 (1H, d, J = 8.2 Hz, ArCH), 7.05–7.13 (2H, m, ArCH), 7.23–7.28 (1H, m, ArCH), 7.33 (1H, t, J = 7.6 Hz, ArCH), 7.41–7.45 (2H, m, ArCH), 7.62 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, MeOD): δ 16.5, 23.9, 24.4, 28.2, 33.6, 114.7, 114.9, 117.9, 121.1, 122.7, 124.0, 126.3, 128.1, 129.5, 130.5, 132.2, 133.0, 134.2, 144.0, 150.4, 168.8, 171.0. ESI-HRMS (m/z): [M – H]– calcd for C23H23O4F, 381.1508; found, 381.1515.
(E)-3-(4-((3-(3-Fluorophenyl)propanoyl)oxy)-3-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (8c).
Compound 6c (260 mg, 0.6 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (103 mg, 0.3 mmol, 45%). Rf. 0.18 (hexane/EtOAc, 4:1) 1H NMR (400 MHz, MeOD): δ 1.67 (3H, s, CH3), 1.73 (3H, s, CH3), 2.98 (2H, t, J = 7.0 Hz, CH2), 3.07 (2H, t, J = 7.7 Hz, CH2), 3.12 (2H, d, J = 7.1 Hz, CH2), 5.15 (1H, t, J = 7.2 Hz, CH), 6.42 (1H, d, J = 15.9 Hz, CH), 6.93–6.99 (2H, m, ArCH), 7.06 (1H, d, J = 8.4 Hz, ArCH), 7.12 (1H, d, J = 7.6 Hz, ArCH), 7.32 (1H, q, J1 = 6.1 Hz, J2 = 7.9 Hz, ArCH), 7.43 (1H, s, ArCH), 7.46 (1H, dd, J1 = 2.1 Hz, J2 = 8.3 Hz, ArCH), 7.63 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, MeOD): δ 16.5, 24.4, 28.1, 29.9, 30.0, 34.7, 112.8, 114.9, 117.9, 121.0, 122.6, 123.9, 126.3, 129.5, 129.8, 132.3, 133.0, 134.2, 143.0, 144.0, 150.4, 161.7, 168.8, 171.1. ESI-HRMS (m/z): [M – H]– calcd for C23H23O4F, 381.1508; found, 381.1493.
(E)-3-(4-((3-(4-Fluorophenyl)propanoyl)oxy)-3-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (8d).
Compound 6d (160 mg, 0.36 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (72 mg, 0.2 mmol, 52%). Rf 0.16 (hexane/EtOAc, 4:1) 1H NMR (400 MHz, MeOD): δ 1.65 (3H, s, CH3), 1.72 (3H, s, CH3), 2.92 (2H, t, J = 7.2 Hz, CH2), 3.08 (2H, t, J = 7.0 Hz, CH2), 3.17 (2H, d, J = 7.4 Hz, CH2), 5.13 (1H, t, J = 7.0 Hz, CH), 6.40 (1H, d, J = 15.9 Hz, CH), 7.02 (2H, t, J = 8.6 Hz, ArCH), 7.29 (2H, t, J = 8.1 Hz, ArCH), 7.40–7.44 (2H, m, ArCH), 7.62 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, MeOD): δ 16.5, 24.4, 28.1, 29.5, 35.1, 114.6, 117.9, 121.0, 122.6, 129.7, 132.3, 133.0, 134.2, 136.1, 144.0, 150.4, 160.4, 162.8, 168.8, 171.1. ESI-HRMS (m/z): [M – H]– calcd for C23H23O4F, 381.1508; found, 381.1490.
(E)-3-(4-((3-(2-Methoxyphenyl)propanoyl)oxy)-3-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (8e).
Compound 6e (150 mg, 0.3 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (73 mg, 0.2 mmol, 56%). Rf 0.1 (hexane/EtOAc, 4:1) 1H NMR (400 MHz, MeOD): δ 1.71 (3H, s, CH3), 1.78 (3H, s, CH3), 2.93 (2H, t, J = 7.1 Hz, CH2), 3.10 (2H, t, J = 7.5 Hz, CH2), 3.20 (2H, d, J = 7.2 Hz, CH2), 3.88 (3H, s, CH3), 5.23 (1H, t, J = 7.2 Hz, CH), 6.41 (1H, d, J = 15.9 Hz, CH), 6.92 (2H, q, J = 6.4 Hz ArCH), 7.03 (1H, d, J = 8.9 Hz, ArCH), 7.23–7.28 (2H, m, ArCH), 7.42 (2H, s, ArCH), 7.678 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, MeOD): δ 17.8, 25.7, 26.2, 28.5, 34.1, 55.2, 110.2, 117.0, 120.5, 120.9, 122.9, 126.9, 127.9, 128.2, 130.1, 130.2, 131.8, 133.9, 134.3, 146.4, 150.8, 157.5, 171.5, 172.3. ESI-HRMS (m/z): [M – H]– calcd for C24H26O5, 393.1707; found, 393.1714.
(E)-3-(4-((3-(3-Methoxyphenyl)propanoyl)oxy)-3-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (8f).
Compound 6f (220 mg, 0.5 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (60 mg, 0.15 mmol, 32%). Rf 0.09 (hexane/EtOAc, 4:1) 1H NMR (400 MHz, MeOD): δ 1.65 (3H, s, CH3), 1.72 (3H, s, CH3), 2.91 (2H, t, J = 7.1 Hz, CH2), 3.00 (2H, t, J = 7.2 Hz, CH2), 3.09 (2H, d, J = 7.1 Hz, CH2), 3.76 (3H, s, CH3), 5.14 (1H, t, J = 7.2 Hz, CH), 6.40 (1H, d, J = 15.9 Hz, CH), 6.76–6.78 (1H, m, ArCH), 6.84 (2H, s, ArCH), 6.95 (1H, d, J = 8.2 Hz, ArCH), 7.21 (1H, t, J = 8.2 Hz, ArCH), 7.39–7.43 (2H, m, ArCH), 7.62 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, MeOD): δ 16.6, 24.5, 28.1, 30.4, 35.1, 54.1, 111.4, 113.8, 117.9, 120.3, 121.1, 122.7, 126.3, 129.1, 129.5, 132.3, 133.0, 134.2, 141.7, 144.0, 150.5, 159.9, 168.8, 171.3. ESI- HRMS (m/z): [M – H]– calcd for C24H26O5, 393.1707; found, 393.1717.
(E)-3-(4-((3-(4-Methoxyphenyl)propanoyl)oxy)-3-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (8g).
Compound 6g (200 mg, 0.4 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (73 mg, 0.2 mmol, 42%). Rf 0.07 (hexane/EtOAc, 4:1) 1H NMR (400 MHz, MeOD): δ 1.66 (3H, s, CH3), 1.74 (3H, s, CH3), 2.90 (2H, t, J = 7.1 Hz, CH2), 2.98 (2H, t, J = 7.0 Hz, CH2), 3.07 (2H, d, J = 7.2 Hz, CH2), 3.77 (3H, s, CH3), 5.14 (1H, t, J = 7.2 Hz, CH), 6.41 (1H, d, J = 15.9 Hz, CH), 6.86 (2H, d, J = 8.7 Hz, ArCH), 6.96 (1H, d, J = 8.2 Hz, ArCH), 7.20 (2H, d, J = 8.7 Hz, ArCH), 7.41 (1H, s, ArCH), 7.45 (1H, dd, J1 = 2.1 Hz, J2 = 8.3 Hz, ArCH), 7.62 (1H, d, J = 15.9 Hz, Ch). 13C NMR (100 MHz, MeOD): δ 16.5, 24.4, 28.0, 29.6, 35.4, 54.2, 133.5, 117.9, 121.0, 122.7, 126.2, 129.0, 129.4, 132.0, 132.3, 133.0, 134.2, 144.0, 150.5, 158.3, 168.8, 171.4. ESI-HRMS (m/z): [M – H]– calcd for C24H26O5, 393.1707; found, 393.1696.
(E)-3-(4-Acetoxy-3-(3-methylbut-2-en-1-yl)phenyl)acrylic Acid (8h).44
Compound 6h (100 mg, 0.3 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo and purified by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) to provide the title compound as a white solid (86 mg, 0.3 mmol, 99%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz, CDCl3): δ 1.73 (3H, s, CH3), 1.78 (3H, s, CH3), 2.34 (3H, s, CO2CH3), 3.27 (2H, d, J = 7.7 Hz, CH2), 5.24 (1H, t, J = 7.1 Hz, CH), 6.41 (1H, d, J = 15.9 Hz, CH), 7.09 (1H, d, J = 8.4 Hz, ArCH), 7.43 (2H, s, ArCH), 7.75 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 17.8, 20.8, 25.7, 28.6, 116.8, 120.8, 122.9, 126.9, 130.2, 132.0, 134.3, 146.1, 150.7, 169.0, 170.1, 206.9. ESI-HRMS (m/z): [M – H]– 273.0 (100%).
(2E)-3-[3-(3-Methylbut-2-en-1-yl)-4-[(4-phenylbutanoyl)-oxy]phenyl]prop-2-enoic Acid (8i).
Compound 6i (230 mg, 0.5 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo and purified by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) to provide the title compound as a white solid (12.2 mg, 0.03 mmol, 6%). Rf: 0.10 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; CDCl3): δ 1.68 (3H, s, CH3), 1.76 (3H, s, CH3), 2.12 (2H, q, J = 7.5 Hz, CH2), 2.62 (2H, t, J = 7.5 Hz, CH2), 2.78 (2H, t, J = 7.5 Hz, CH2), 3.25 (2H, d, J = 7.1 Hz, CH2), 5.23 (1H, t, J = 7.2 Hz, CH), 6.40 (1H, d, J = 15.0, CH), 7.06 (1H, d, J = 8.5 Hz, ArCH), 7.23–7.26 (3H, m, ArCH), 7.31–7.35 (2H, m, ArCH), 7.42–7.44 (2H, m, ArCH), 7.76 (1H, d, J = 15.95 Hz, ArCH). 13C NMR (100 MHz; CDCl3): δ 17.8, 25.7, 26.2, 26.4, 28.6, 33.4, 35.1, 117.0, 120.8, 122.9, 126.1, 126.9, 128.5, 130.2, 131.9, 134.1, 134.3, 141.0, 146.3, 150.7, 171.6, 171.9. ESI-HRMS (m/z): [M – H]– calcd for C24H26O4, 377.1758; found, 377.1761.
(2E)-3-{4-[(2-Cyclohexylacetyl)oxyH3-methylbut-2-en-1-yl)-phenyl}prop-2-enoic Acid (8j).
Compound 6j (370 mg, 0.85 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 4:1, 1:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (130 mg, 0.3 mmol, 40%). Rf: 0.14 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; CDCl3): δ 1.05–1.39 (7H, m, cyc-H), 1.72 (3H, s, CH3), 1.79 (3H, s, CH3), 1.84–1.96 (3H, m, cyc-H), 2.47 (2H, d, J = 7 Hz, CH), 3.24 (2H, d, J = 7.1 Hz, CH2), 5.23 (1H, t, J = 7.1 Hz, CH), 6.39 (1H, d J = 15.9, CH), 7.04 (1H, d, J = 8.8 Hz, ArCH), 7.39–7.36 (2H, m, ArCH), 7.75 (1H, d, J = 15.9, CH). 13C NMR (100 MHz; CDCl3): δ 17.9, 25.7, 26.0, 28.5, 33.0, 34.9, 41.9, 117.0, 120.8, 122.9, 126.8, 128.3, 128.5, 128.9, 130.1, 131.8, 134.0, 134.3, 146.4, 150.8, 171.1, 172.3. ESI-HRMS (m/z): [M – H]– calcd for C22H28O4, 355.1915; found, 355.1921.
(2E)-3-[3-(3-Methylbut-2-en-1-yl)-4-{[2-(naphthalen-1-yl)-acetyl]oxy}phenyl]prop-2-enoic Acid (8k).
Compound 6k (66.1 mg, 0.15 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (34 mg, 0.1 mmol, 60%). Rf: 0.20 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; CDCl3): δ 1.57 (3H, s, CH3), 1.70 (3H, s, CH3), 3.00 (2H, d, J = 7.3 Hz, CH2), 4.36 (2H, s, CH2), 5.03 (1H, t, J = 7.2 Hz, CH), 6.37 (1H, d, J = 15.9 Hz, CH), 7.03 (1H, d, J = 8.2 Hz, ArCH), 7.34–7.38 (2H, m, ArCH), 7.48–7.62 (4H, m, ArCH) 7.72 (1H, d, J = 15.9 Hz, CH), 7.87 (1H, d, J = 8.1 Hz, ArCH), 7.89 (1H, dd, J1 = 8.7 Hz, J2 = 21.7 Hz, ArCH), 8.14 (1H, d, J = 8.5 Hz, ArCH). 13C NMR (100 MHz; CDCl3): δ 17.7, 25.6, 28.2, 29.7, 39.3, 117.0, 120.5, 122.7, 123.6, 125.5, 125.9, 126.6, 126.8, 128.3, 128.4, 128.8, 129.7, 130.0, 131.9, 132.0, 133.8, 133.9, 134.3, 146.3, 150.6, 169.6, ESI-HRMS (m/z): [M + Na]+ calcd for C26H24O4, 423.1567; found, 423.1558.
(E)-3-(4-(Benzoyloxy)-3-(3-methylbut-2-en-1-yl)phenyl)-acrylic Acid (8l).
Compound 6l (72 mg, 0.2 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (32 mg, 0.09 mmol, 50%). 1H NMR (400 MHz; MeOD): δ 1.61 (3H, s, CH3), 1.72 (3H, s, CH3), 3.35 (2H, d, J = 7.0 Hz, CH2), 5.26 (1H, t, J = 7.5 Hz, CH), 6.45 (1H, d, J = 16.0 Hz, CH), 7.23 (1H, d, J = 8.2 Hz, ArCH), 7.49–7.57 (4H, m, ArCH), 7.68 (1H, t, J = 7.0 Hz, ArCH), 7.81 (1H, d, J = 15.9 Hz, CH), 8.23 (2H, d, J = 7.3 Hz, ArCH). 13C NMR (100 MHz; MeOD): δ 17.8, 25.6, 28.8, 117.0, 120.8, 123.0, 127.0, 128.6, 130.2, 130.3, 132.0, 133.7, 134.0, 134.6, 146.3, 151.0, 164.7, 171.1, 171.5. ESI-HRMS (m/z): [M – H]– calcd for C21H20O4, 335.1289; found, 335.1300.
(2E )-3-[3-(3-Methylbut-2-en-1-yl)-4-(pyridine-4-carbonyloxy)phenyl]prop-2-enoic Acid (8m).
Compound 6m (350 mg, 0.9 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a yellow solid (150 mg, 0.4 mmol, 50%). Rf: 0.2 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.55 (3H, s, CH3), 1.62 (3H, s, CH3), 3.36 (2H, d, J = 7.0 Hz, CH2), 5.19 (1H, t, J = 7.0 Hz, CH), 6.50 (1H, d, J = 16.0 Hz, CH), 7.26 (1H, d, J = 8.0 Hz, ArCH), 7.57 (1H, s ArCH), 7.59 (1H, d, J = 2.1 Hz, ArCH), 7.69 (1H, d, J = 15.9 Hz, CH), 8.12 (2H, d, J = 6.1 Hz, ArCH), 8.86 (2H, d, J = 5.1 Hz, ArCH). 13C NMR (100 MHz; MeOD): δ 16.5, 24.3, 28.9, 60.1, 118.4, 121.2, 122.8, 123.3, 126.6, 129.9, 133.0, 134.2, 143.7, 150.1, 150.3, 163.1, 168.8. ESI-HRMS (m/z): [M + H]+ calcd for C20H19NO4, 338.1387; found, 338.1389.
(E)-3-(4-(Hexanoyloxy)-3-(3-methylbut-2-en-1-yl)phenyl)-acrylic Acid (8n).
Following the general procedure B, the title compound was obtained as a white solid (50 mg, 0.15 mmol, 37%). 1H NMR (400 MHz; MeOD): δ 1.36–1.43 (7H, m, CH3 and CH2), 1.50–1.62 (2H, m, CH2), 1.69 (3H, s, CH3), 1.76 (3H, s, CH3), 2.57 (2H, t, J = 7.4 Hz, CH2), 3.18 (2H, d, J = 7.0 Hz, CH2), 5.19 (1H, t, J = 7.2 Hz, CH), 6.45 (1H, d, J = 15.7 Hz, CH), 7.01 (1H, d, J = 8.3 Hz, ArCH), 7.52–7.54 (2H, m, ArCH), 7.46 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz; MeOD): δ 12.8, 16.5, 22.0, 24.2, 24.4, 28.3, 31.0, 33.5, 121.2, 122.6, 126.1, 129.3, 132.8, 132.9, 134.1, 142.3, 15
0.2, 172.2. ESI-HRMS (m/z): [M + Na]+ calcd for C20H26O4, 353.1723; found, 353.1718.
(2E )-3-[3-(3-Methylbut-2-en-1-yl)-4-(3-phenylpropanamido)phenyl]prop-2-enoic Acid (14a).47
Compound 13a (20 mg, 0.04 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (9.3 mg, 0.02 mmol, 50%). Rf 0.01 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.70 (3H, s, CH3), 1.77 (3H, s, CH3), 2.72 (2H, t, J = 7.0 Hz, CH2), 3.03 (2H, t, J = 7.3 Hz, CH2), 3.21 (2H, d, J = 6.9 Hz, CH2), 5.19 (1H, t, J = 7.0 Hz, CH), 6.42 (1H, d, J = 15.8 Hz, CH), 7.14–7.30 (5H, m, ArCH), 7.34–7.46 (3H, m, ArCH), 7.61 (1H, d, J = 16.5 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.6, 24.4, 29.4, 31.3, 37.8, 117.7, 121.3, 125.6, 125.9, 126.1, 127.8, 128.1, 128.1, 128.8, 132.3, 133.2, 136.5, 137.0, 140.6, 144.2, 169.0, 172.6. ESI-HRMS (m/z): [M – H]- calcd for C23H25NO3, 386.1727; found, 386.1729.
(2E)-3-[4-Acetamido-3-(3-methylbut-2-en-1-yl)phenyl]-prop-2-enoic Acid (14b).
Compound 13b (51.0 mg, 0.5 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (20.3 mg, 0.07 mmol, 50%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.75 (3H, s, CH3), 1.78 (3H, s, CH3), 2.16 (3H, s, CH3), 3.37 (2H, d, J = 6.9 Hz, CH2), 5.25 (1H, t, J = 6.4 Hz, CH), 6.43 (1H, d, J = 16.0 Hz, CH), 7.43–7.58 (3H, m, ArCH), 7.63 (1H, d, J = 15.7 Hz, CH). 13C NMR (100 MHz; MeOD): δ 21.7, 24.4, 29.3, 29.6, 31.6, 117.7, 121.3, 125.7, 126.1, 132.3, 133.2, 136.4, 137.2, 144.2, 170.7. ESI-HRMS (m/z): [M – H]– calcd for C16H19NO3, 272.1292; found, 272.1292.
(2E)-3-[3-(3-Methylbut-2-en-1-yl)-4-(4-phenylbutanamido)-phenyl]prop-2-enoic Acid (14c).
Compound 13c (17 mg, 0.04 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (7.3 mg, 0.02 mmol, 50%). Rf: 0.01 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; CDCl3): δ 1.74 (3H, s, CH3), 1.75 (3H, s, CH3), 2.07 (2H, d, J = 7.5 Hz, CH2), 2.33 (2H, d, J = 7.0 Hz, CH2), 2.73 (2H, d, J = 7.1 Hz, CH2), 3.33 (2H, d, J = 6.5 Hz, CH2), 5.19 (1H, t, J = 6.3 Hz, CH), 6.39 (1H, d, J = 16.9 Hz, CH), 7.20–7.46 (7H, m, ArCH), 7.73 (1H, d, J = 16.3 Hz, CH), 8.17 (1H, d, J = 8.6 Hz, ArCH). 13C NMR (100 MHz; MeOD): δ 13.0, 16.6, 19.4, 24.4, 27.3, 29.8, 34.9, 35.4, 60.1, 119.3, 121.5, 125.6, 126.1, 127.8, 128.1, 128.8, 132.6, 133.2, 136.5, 141.5, 143.0, 173.3. ESI-HRMS (m/z): [M–H]– calcd for C24H27NO3, 376.1918; found, 376.1924.
(2E)-3-[4-(2-Cyclohexylacetamido)-3-(3-methylbut-2-en-1-yl)phenyl]prop-2-enoic Acid (14d).
Compound 13d (20 mg, 0.05 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (13.2 mg, 0.03 mmol, 93%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.03–1.38 (7H, m, CH2), 1.73 (3H, s, CH3), 1.78 (3H, s, CH3), 1.78–1.90 (4H, m, CH2), 2.29 (2H, d, J = 6.9 Hz, CH2), 3.37 (2H, d, J = 6.9 Hz, CH2), 5.27 (1H, t, J = 7.7 Hz, CH), 6.43 (1H, d, J = 15.9 Hz, CH), 7.43–7.49 (3H, m, ArCH), 7.63 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.6, 24.4, 25.8, 25.9, 29.3, 29.6, 32.8, 35.5, 44.0, 117.9, 121.4, 125.7, 126.2, 129.0, 132.3, 133.3, 136.5, 137.1, 144.1, 173.0. ESI-HRMS (m/z): [M–H]– calcd for C22H29NO3, 354.2075; found, 354.2089.
(2E)-3-[3-(3-Methylbut-2-en-1-yl)-4-[2-(naphthalen-1-yl)-acetamido]phenyl]prop-2-enoic Acid (14e).
Compound 13e (50 mg, 0.1 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (20.7 mg, 0.05 mmol, 52%). Rf: 0.01 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.80 (6H, s, CH3), 3.36 (2H, s, CH2), 3.45 (2H, d, J = 5.8 Hz, CH2), 5.23 (1H, t, J = 7.7 Hz, CH), 6.43 (1H, d, J = 16.4 Hz, CH), 7.43–7.64 (7H, m, ArCH and CH), 7.88–7.95 (3H, m, ArCH), 8.37 (1H, d, J = 7.9 Hz, ArCH). 13C NMR (100 MHz; MeOD): δ 24.5, 30.5, 72.7, 120.7, 122.7, 124.0, 124.8, 125.4, 125.7, 125.8, 126.3, 128.3, 128.6, 129.2, 131.3, 131.3, 133.5, 134.2, 134.8, 136.0, 137.1, 144.0, 172.4. ESI-HRMS (m/z): [M–H]– 397.9 (20%).
(E)-3-(4-Bromo-3-hydroxyphenyl)acrylic Acid (17a).
To a solution of 17 (330 mg, 1.3 mmol) in water (8 mL) and tetrahydrofuran (2 mL) was added NaOH (100 mg, 2.5 mmol) and the reaction refluxed overnight. The solution was allowed to cool and was acidified with AcOH, washed with water and extracted in DCM, dried (Na2SO4), filtered and concentrated. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1, 0:1) provided the title compound as a white solid (150 mg, 0.6 mmol, 45%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 6.39 (1H, d, J = 15.8 Hz, CH), 6.78 (1H, dd, J1 = 2.9 Hz, J2 = 8.7 Hz, ArCH), 7.15 (1H, d, J = 2.9 Hz, ArCH), 7.44 (1H, d, J = 8.7 Hz, ArCH), 7.97 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz; MeOD): δ 113.6, 113.7, 118.9, 120.5, 133.6, 134.6, 142.9, 157.7, 168.3.
(E)-3-(4-(3-Methylbut-2-en-1-yl)-3-((3-phenylpropanoyl)-oxy)phenyl)acrylic Acid (19a).47
To a solution of (18) (430 mg, mmol) in dry DMF (8 mL), CsCO3 (555 mg, 1.7 mmol) and Pd(dppf)Cl2 (50 mg, 0.06 mmol) were added. Prenylboronic acid pinacol ester (330 μL, 1.5 mmol) was added, and the flask was heated at 90 °C overnight. The reaction mixture was allowed to cool and filtered through a celite pad with EtOAc; the solvent was evaporated, and the residue was re-dissolved in DCM. The residual DMF was removed by washing with copious amounts of water in DCM, dried (Na2SO4), filtered, and concentrated. Purification by column chromatography (hexane/EtOAC = 9:1, 4:1, 2:1, 1:1, 0:1) provided the title compound as a yellow oil (120 mg, 0.3 mmol, 27%). Rf: 0.10 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.72 (3H, s, CH3), 1.77 (3H, s, CH3), 2.90 (2H, t, J = 7.2 Hz, CH2), 3.04 (2H, t, J = 7.2 Hz, CH2), 3.43 (2H, d, J = 6.7 Hz, CH2), 5.15 (1H, t, J = 5.9 Hz, CH), 6.29 (2H, d, J = 15.7 Hz, CH), 6.94 (1H, d, J = 7.2 Hz, ArCH), 7.16–7.33 (7H, m, ArCH), 7.93 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.6, 24.4, 30.5, 31.3, 35.3, 119.0, 119.9, 122.3, 122.9, 126.0, 128.14, 128.18, 130.4, 132.3, 134.0, 138.6, 140.2, 141.5, 149.3, 168.5, 171.7. ESI-HRMS (m/z): [M – H]– calcd for C23H24O4, 363.1602; found, 363.1609.
(E)-3-(4-Allyl-3-((3-phenylpropanoyl)oxy)phenyl)acrylic Acid (19b).
To a solution of (18) (110 mg, 0.3 mmol) in dry DMF (4 mL), CsCO3 (75 mg, 0.4 mmol) and Pd(dppf)Cl2 (13 mg, 0.01 mmol) were added. Allyl boronic acid pinacol ester (70 μL, 0.4 mmol) was added, and the flask was heated at 90 °C overnight. The reaction mixture was allowed to cool and filtered through a celite pad with EtOAc; the solvent was evaporated, and the residue was re-dissolved in DCM. The residual DMF was removed by washing with copious amounts of water in DCM, dried (Na2SO4), filtered, and concentrated. Purification by column chromatography (hexane/ EtOAC = 9:1, 4:1, 2:1, 1:1, 0:1) provided the title compound as a yellow oil (60 mg, 0.2 mmol, 60%). Rf: 0.10 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 2.93 (2H, t, J = 7.5 Hz, CH2), 3.11 (2H, t, J = 7.5 Hz, CH2), 3.53 (2H, d, J = 6.1 Hz, CH2), 5.01 (1H, dd, J1 = 1.5 Hz, J2 = 17.0 Hz, CH2), 5.12 (1H, dd, J1 = 1.5 Hz, J2 = 10.1 Hz, CH2), 5.90–6.00 (1H, m, CH), 6.33 (2H, d, J = 15.7 Hz, CH), 7.02 (1H, dd, J1 = 2.4 Hz, J2 = 8.3 Hz, ArCH), 7.20–7.38 (7H, m, ArCH), 8.03 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz; MeOD): δ 30.9, 35.9, 36.9, 116.7, 119.4, 119.5, 123.6, 126.5, 128.4, 128.6, 131.3, 134.1, 136.1, 137.0, 139.9, 143.4, 149.3 171.3, 171.5. ESI-HRMS (m/z): [M – H]– calcd for C21H20O4, 335.1289; found, 335.1292.
(E)-N-(2-Bromo-5-(2-(4,4,5,5-tetramethyl-1,3,2-dioxaboro-lan-2-yl)vinyl)phenyl)-3-phenylpropanamide (24).
To a solution of 21b (64 mg, 0.1 mmol) in dry DCM (5 mL), DMAP (2 mg, 0.01 mmol), a solution of 3-phenylpropanoyl chloride (50 mg, 0.3 mmol) in DCM (2 mL), and NEt3 (0.1 mL, 0.5 mmol) were added. The reaction mixture was heated to 70 °C and stirred overnight. The reaction mixture was allowed to cool and washed with saturated aqueous NaHCO3 and water, extracted in DCM, dried (Na2SO4), filtered, and concentrated. Purification by column chromatography (hexane/EtOAc = 12:1, 9:1, 4:1, 2:1) provided the title compound as a yellow oil (40 mg, 0.08 mmol, 87%). Rf: 0.3 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; CDCl3): δ 1.33 (12H, s, CH3), 2.67 (2H, t, J = 7.4 Hz, CH2), 3.05 (2H, t, J = 7.6 Hz, CH2), 6.10 (1H, d, J = 18.2 Hz, CH), 7.22–7.34 (6H, m, ArCH), 7.47 (1H, d, J = 8.6 Hz, ArCH), 7.59 (1H, d, J = 2.2 Hz, ArCH), 7.65 (1H, d, J = 18.0 Hz, CH). 13C NMR (100 MHz; CDCl3): δ 24.8, 31.4, 39.4, 83.5, 118.2, 118.7, 121.6, 126.4, 128.3, 128.6, 133.3, 137.2, 137.8, 140.4, 147.0, 170.3. ESI-HRMS (m/z): [M + Na]+ calcd for C23H27NO3BBr, 478.1164; found, 478.1164.
(E)-3-(4-(3-Methylbut-2-en-1-yl)-3-(3-phenylpropanamido)-phenyl)acrylic Acid (26a).46
Compound 25a (80 mg, 0.2 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (19 mg, 0.05 mmol, 27%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz, MeOD): δ 1.72 (3H, s, CH3), 1.79 (3H, s, CH3), 2.66 (2H, t, J = 7.4 Hz, CH2), 3.01 (2H, t, J = 7.4 Hz, CH2), 3.43 (2H, d, J = 6.6 Hz, CH2), 5.16 (1H, t, J = 5.9 Hz, CH), 6.33 (1H, d, J = 15.7 Hz, CH), 7.16–7.30 (6H, m, ArCH), 7.46 (1H, d, J = 8.0 Hz, ArCH), 7.80 (1H, s, ArCH), 7.96 (1H, d, J = 15.7 Hz, CH). 13C NMR (100 MHz, MeOD): δ 16.6, 24.4, 31.3, 31.3, 38.4, 117.5, 119.2, 121.7, 122.6, 125.8, 128.1, 129.9, 132.0, 133.2, 136.9, 137.0, 140.7, 142.3, 168.8, 172.2. ESI-HRMS (m/z): [M – H]– calcd for C23H25NO3, 362.1762; found, 362.1761.
(E)-3-(3-(3-(2-Fluorophenyl)propanamido)-4-(3-methylbut-2-en-1-yl)phenyl)acrylic Acid (26b).
Compound 25b (200 mg, 0.4 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (130 mg, 0.34 mmol, 85%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.72 (3H, s, CH3), 1.78 (3H, s, CH3), 2.68 (2H, t, J = 7.4 Hz, CH2), 3.05 (2H, t, J = 7.6 Hz, CH2), 3.41 (2H, d, J = 6.9 Hz, CH2), 5.14 (1H, t, J = 7.0 Hz, CH), 6.32 (1H, d, J = 15.7 Hz, CH), 7.03–7.11 (2H, m, ArCH), 7.16 (1H, d, J = 8.3 Hz, ArCH), 7.20–7.25 (1H, m, ArCH), 7.30 (1H, t, J = 7.6 Hz, ArCH), 7.45 (1H, dd, J1 = 1.8 Hz, J2 = 8.2 Hz, ArCH), 7.80 (1H, d, J = 2.1, ArCH), 7.96 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.6, 24.4, 24.6, 31.3, 36.7, 114.6, 114.8, 117.6, 119.2, 121.8, 122.6, 123.9, 127.9, 129.9, 130.5, 132.0, 133.2, 137.0, 142.3, 159.8, 1652.3, 168.9, 171.9. ESI-HRMS (m/z): [M – H]– calcd for C23H24NO3F, 380.1667; found, 380.1676.
(E)-3-(3-(3-(3-Fluorophenyl)propanamido)-4-(3-methylbut-2-en-1-yl)phenyl)acrylic Acid (26c).
Compound 25c (150 mg, 0.4 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (120 mg, 0.32 mmol, 80%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.72 (3H, s, CH3), 1.78 (3H, s, CH3), 2.68 (2H, t, J = 7.3 Hz, CH2), 3.02 (2H, t, J = 7.4 Hz, CH2), 3.41 (2H, d, J = 6.9 Hz, CH2), 5.15 (1H, t, J = 7.0 Hz, CH), 6.33 (1H, d, J = 15.7 Hz, CH), 6.92 (1H, t, J = 8.2 Hz, ArCH), 7.02 (1H, d, J = 9.8 Hz, ArCH), 7.08 (1H, d, J = 7.6 Hz, ArCH), 7.17 (1H, d, J = 8.3 Hz, ArCH), 7.29 (1H, q, J = 7.9 Hz, ArCH), 7.46 (1H, dd, J1 = 2.1 Hz, J2 = 8.2 Hz, ArCH), 7.79 (1H, d, J = 2.1, ArCH), 7.95 (1H, d, J = 15.7 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.6, 24.4, 30.9, 31.3, 37.9, 112.4, 112.6, 114.6, 114.8, 117.5, 121.7, 122.6, 123.9, 129.7, 129.8, 129.9, 132.0, 133.3, 136.8, 137.0, 161.7, 164.1, 171.9. ESI- HRMS (m/z): [M – H]– calcd for C23H24NO3F, 380.1667; found, 380.1678.
(E)-3-(3-(3-(4-Fluorophenyl)propanamido)-4-(3-methylbut-2-en-1-yl)phenyl)acrylic Acid (26d).
Compound 25d (66 mg, 0.15 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (50 mg, 0.13 mmol, 87%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.73 (3H, s, CH3), 1.79 (3H, s, CH3), 2.66 (2H, t, J = 7.4 Hz, CH2), 3.00 (2H, t, J = 7.4 Hz, CH2), 3.4 (2H, d, J = 7.4 Hz, CH2), 5.16 (1H, t, J = 7.8 Hz, CH), 6.33 (1H, d, J = 17.5 Hz, CH), 7.00 (2H, t, J = 8.6 Hz, ArCH), 7.18 (1H, d, J = 8.3 Hz, ArCH), 7.27 (2H, dd, J1 = 3.2 Hz, J2 = 5.5 Hz, ArCH), 7.46 (1H, dd, J1 = 2.1 Hz, J2 = 8.2 Hz, ArCH), 7.81 (1H, d, J = 2.0, ArCH), 7.96 (1H, d, J = 17.3 Hz, CH). 13C NMR (100 MHz; MeOD): δ 24.4, 29.2, 30.4, 31.3, 38.4, 114.5, 114.7, 117.5, 121.6, 122.6, 129.6, 129.7, 129.9, 132.2, 133.3, 136.6, 136.9, 136.9, 142.1, 162.7, 172.0. ESI-HRMS (m/z): [M – H]– calcd for C23H24NO3F, 380.1667; found, 380.1664.
(E)-3-(3-(3-(2-Methoxyphenyl)propanamido)-4-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (26e).
Compound 25e (290 mg, 0.6 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (110 mg, 0.3 mmol, 50%). Rf: 0.2 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.73 (3H, s, CH3), 1.79 (3H, s, CH3), 2.64 (2H, t, J = 7.5 Hz, CH2), 3.00 (2H, t, J = 7.6 Hz, CH2), 3.43 (2H, d, J = 6.9 Hz, CH2), 3.84 (3H, s, CH3), 5.15 (1H, t, J = 7.0 Hz, CH), 6.33 (1H, d, J = 15.8 Hz, Ch), 6.86 (1H, t, J = 7.5 Hz, ArCH), 6.93 (1H, d, J = 7.9 Hz, ArCH), 7.16–7.21 (3H, m, ArCH), 7.46 (1H, dd, J1 = 2.2 Hz, J2 = 8.3 Hz, ArCH), 7.81 (1H, d, J = 2.1 Hz, ArCH), 7.97 (1H, d, J = 15.7 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.6, 24.4, 26.3, 31.3, 36.8, 54.2, 109.9, 117.7, 119.1, 120.0, 121.8, 122.6, 127.3, 128.6, 129.5, 129.9, 132.0, 133.1, 136.9, 137.0, 142.4, 157.4, 168.7, 172.7. ESI-HRMS (m/z): [M – H]– calcd for C24H27NO4, 392.1867; found, 392.1863.
(E)-3-(3-(3-(3-Methoxyphenyl)propanamido)-4-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (26f).
Compound 25f (240 mg, 0.5 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (62 mg, 0.16 mmol, 31%). Rf: 0.2 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.73 (3H, s, CH3), 1.79 (3H, s, CH3), 2.67 (2H, t, J = 7.4 Hz, CH2), 2.99 (2H, t, J = 7.6 Hz, CH2), 3.43 (2H, d, J = 6.7 Hz, CH2), 3.76 (3H, s, CH3), 5.16 (1H, t, J = 7.0 Hz, CH), 6.33 (1H, d, J = 15.7 Hz, CH), 6.76 (1H, dd, J1 = 2.5 Hz, J2 = 7.9 Hz, ArCH), 6.83–6.85 (2H, m, ArCH), 7.16–7.22 (2H, m, ArCH), 7.46 (1H, dd, J1 = 2.2 Hz, J2 = 8.2 Hz, ArCH), 7.80 (1H, d, J = 2.1 Hz, ArCH), 7.97 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz; MeOD): S 16.6, 24.4, 31.3, 31.4, 38.3, 5.1, 111.3, 113.6, 117.6, 129.3, 120.3, 121.8, 122.6, 129.0, 129.9, 132.0, 133.2, 136.9, 137.0, 142.2, 142.3, 159.8, 168.8, 172.3. ESI-HRMS (m/z): [M – H]– calcd for C24H27NO4, 392.1867; found, 392.1869.
(E)-3-(3-(3-(4-Methoxyphenyl)propanamido)-4-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (26g).
Compound 25g (50 mg, 0.1 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (25 mg, 0.06 mmol, 53%). Rf: 0.25 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.73 (3H, s, CH3), 1.79 (3H, s, CH3), 2.64 (2H, t, J = 7.4 Hz, CH2), 2.95 (2H, t, J = 7.6 Hz, CH2), 3.44 (2H, d, J = 7.2 Hz, CH2), 3.76 (3H, s, CH3), 5.15 (1H, t, J = 8.4 Hz, CH), 6.33 (1H, d, J = 15.7 Hz, CH), 6.85 (2H, d, J = 8.6 Hz, ArCH), 7.18 (3H, d, J = 8.4 Hz, ArCH), 7.46 (1H, d, J1 = 2.3 Hz, J2 = 8.2 Hz, ArCH), 7.79 (1H, d, J = 2.1 Hz, ArCH), 7.96 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.5, 24.4, 30.5, 31.3, 38.7, 54.2, 113.4, 117.6, 119.4, 121.7, 122.6, 128.9, 129.9, 132.0, 132.6, 133.2, 136.9, 136.9, 142.2, 157.8, 158.2, 172.4. ESI-HRMS (m/z): [M – H]– calcd for C23H24NO3F, 380.1667; found, 380.1664. ESI-HRMS (m/z): [M + H]+ calcd for C24H27NO4, 394.2013; found, 394.2003.
(E)-3-(3-Cinnamamido-4-(3-methylbut-2-en-1-yl)phenyl)-acrylic Acid (26h).
Compound 25h (80 mg, 0.2 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1, 2:1, 1:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (20 mg, 0.05 mmol, 30%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz, MeOD): δ 1.74 (3H, s, CH3), 1.80 (3H, s, CH3), 3.47 (2H, d, J = 7.0 Hz, CH2), 5.19 (1H, t, J = 7.1 Hz, CH), 6.39 (1H, d, J = 15.8 Hz, CH), 6.81 (1H, d, J = 15.6 Hz, CH), 7.23 (1H, d, J = 8.3 Hz, ArCH), 7.39–7.46 (3H, m, ArCH), 7.61–7.64 (3H, m, ArCH), 7.69 (1H, d, J = 15.6 Hz, CH), 8.0 (1H, d, J = 15.7 Hz, CH), 8.0 (1H, d, J = 2.1 Hz, ArCH). 13C NMR (100 MHz, MeOD): δ 16.6, 24.5, 31.4, 99.9, 117.4, 119.5, 120.7, 121.6, 122.6, 127.5, 128.6, 129.6, 130.0, 132.1, 133.4, 134.8, 137.1, 137.1, 141.5, 142.2, 165.2. ESI-HRMS (m/z): [M + Na]+ calcd for C23H24NO3, 384.1570; found, 384.1564.
(E)-3-(4-(3-Methylbut-2-en-1-yl)-3-(3-(3-(tosyloxy)phenyl)-propanamido)phenyl)acrylic Acid (26i).
Compound 25i (290 mg, 0.5 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (110 mg, 0.2 mmol, 41%). Rf: 0.2 (EtOAC). 1H NMR (400 MHz; MeOD): δ 1.70 (3H, s, CH3), 1.76 (3H, s, CH3), 2.39 (3H, s, CH3), 2.61 (2H, t, J = 7.5 Hz, CH2), 2.94 (2H, t, J = 7.5 Hz, CH2), 3.40 (2H, d, J = 6.9 Hz, CH2), 5.13 (1H, t, J = 7.0 Hz, CH), 6.32 (1H, d, J = 15.7 Hz, CH), 6.75 (1H, d, J = 7.3 Hz, ArCH), 6.95 (1H, s, ArCH), 7.14–7.22 (3H, m, ArCH), 7.30 (2H, d, J = 8.0 Hz, ArCH), 7.47 (1H, dd, J1 = 8.2 Hz, J2 = 2.1 Hz, ArCH), 7.61 (2H, d, J = 8.3 Hz, ArCH), 7.81 (1H, d, J = 2.1 Hz, ArCH), 7.95 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.6, 20.2, 24.5, 30.7, 31.3, 37.7, 117.6, 119.4, 119.6, 121.7, 121.9, 122.6, 127.0, 128.2, 129.2, 129.5, 129.9, 132.0, 132.1, 133.2, 136.9, 137.0, 142.2, 143.0, 145.6, 149.7, 168.8, 171.6. ESI-HRMS (m/z): [M + H]+ calcd for C30H31NO6S, 534.1945; found, 534.1947.
(E)-3-(4-(3-Methylbut-2-en-1-yl)-3-(3-(4-(tosyloxy)phenyl)-propanamido)phenyl)acrylic Acid (26j).
Compound 25j (400 mg, 0.7 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (104 mg, 0.2 mmol, 28%). Rf: 0.4 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.72 (3H, s, CH3), 1.79 (3H, s, CH3), 2.39 (3H, s, CH3), 2.65 (2H, t, J = 7.5 Hz, CH2), 2.99 (2H, t, J = 7.5 Hz, CH2), 3.45 (2H, d, J = 6.8 Hz, CH2), 5.16 (1H, t, J = 7.0 Hz, CH), 6.34 (1H, d, J = 15.7 Hz, CH), 6.88 (2H, d, J = 8.5 Hz, ArCH), 7.18–7.31 (5H, m, ArCH), 7.44 (1H, d, J = 8.3 Hz, ArCH), 7.59 (2H, d, J = 8.2 Hz, ArCH), 7.82 (1H, d, J =1.8 Hz, ArCH), 7.97 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.5, 20.1, 24.4, 30.5, 31.3 38.0, 117.4, 121.6, 122.0, 122.6, 128.2, 129.3, 129.4, 129.9, 132.0, 132.1, 133.3, 136.9, 137.0, 140.0, 142.0, 145.6, 148.1. ESI-HRMS (m/z): [M + Na]+ calcd for C30H31NO6S, 556.1764; found, 556.1761.
(E)-3-(3-(3-(3-Hydroxyphenyl)propanamido)-4-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (26k).
Compound 26i (15 mg, 0.02 mmol) was dissolved in MeOH (2 mL) and refluxed with 1 M NaOH (2 mL) overnight. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1) provided the title compound as a white solid (8 mg, 0.02 mmol, 90%). Rf: 0.2 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.73 (3H, s, CH3), 1.79 (3H, s, CH3), 2.65 (2H, t, J = 7.5 Hz, CH2), 2.94 (2H, t, J = 7.5 Hz, CH2), 3.44 (2H, d, J = 7.0 Hz, CH2), 5.17 (1H, t, J = 7.0 Hz, CH), 6.35 (1H, d, J = 15.6 Hz, CH), 6.63 (1H, d, J1 = 2.1 Hz, J2 = 8.2 Hz, ArCH), 6.71 – 6.74 (2H, m, ArCH), 7.10 (1H, t, J = 7.4 Hz, ArCH), 7.17 (1H, d, J = 8.3 Hz, ArCH), 7.47 (1H, d, J1 = 2.1 Hz, J2 = 8.4 Hz, ArCH), 7.81 (1H, d, J = 2.4 Hz, ArCH), 7.95 (1H, d, J = 15.7 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.5, 24.4, 31.3, 31.3, 99.9, 122.7, 114.8, 117.6, 119.1, 121.7, 122.6, 129.0, 129.8, 132.0, 133.3, 136.9, 142.2, 157.1, 172.3. ESI-HRMS (m/z): [M + Na]+ calcd for C23H25NO4 402.1676; found, 402.1666.
(E)-3-(3-(3-(4-Hydroxyphenyl)propanamido)-4-(3-methyl-but-2-en-1-yl)phenyl)acrylic Acid (26l).
Compound 26j (80 mg, 0.15 mmol) was dissolved in MeOH (4 mL) and refluxed with 1 M NaOH (2 mL) overnight. Purification by column chromatography (hexane/EtOAc = 9:1, 4:1) provided the title compound as a white solid (50 mg, 0.12 mmol, 84%). Rf: 0.1 (hexane/EtOAc, 4:1). 1H NMR (400 MHz; MeOD): δ 1.73 (3H, s, CH3), 1.79 (3H, s, CH3), 2.62 (2H, t, J = 7.5 Hz, CH2), 2.92 (2H, t, J = 7.5 Hz, CH2), 3.44 (2H, d, J = 7.4 Hz, CH2), 5.16 (1H, t, J = 7.0 Hz, CH), 6.33 (1H, d, J = 15.7 Hz, CH), 6.71 (2H, d, J = 8.5 Hz, ArCH), 7.08 (2H, d, J = 8.5 Hz, ArCH), 7.17 (1H, d, J = 8.3 Hz, ArCH), 7.46 (1H, d, J1 = 2.1 Hz, J2 = 8.2 Hz, ArCH), 7.81 (1H, d, J = 2.1 Hz, ArCH), 7.97 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz; MeOD): δ 16.5, 24.4, 30.6, 31.3, 38.8, 114.8, 117.6, 121.7, 122.6, 128.9, 129.8, 131.4, 132.2, 133.2, 136.9, 142.2, 155.3, 168.9, 172.5. ESI-HRMS (m/z): [M – h]– calcd for C23H25NO4 378.1711; found, 378.1700.
(E)-3-(3′-(Hydroxymethyl)-2-(3-phenylpropanamido)-[1,1′-biphenyl]-4-yl)acrylic Acid (28).
Compound 27 (70 mg, 0.15 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (27 mg, 0.06 mmol, 45%). Rf: 0.3 (DCM/MeOH, 10:1). 1H NMR (400 MHz, MeOD): δ 2.72 (2H, t, J = 7.6 Hz, CH2), 3.04 (2H, t, J = 7.6 Hz, CH2), 4.68 (2H, s, CH2), 6.41 (1H, d, J = 15.8 Hz, CH), 7.18–7.23 (2H, m, ArCH), 7.28–7.32 (6H, m, ArCH), 7.39–7.45 (2H, m, ArCH), 7.62–7.66 (2H, m, ArCH and CH), 7.99 (1H, d, J = 2.0 Hz, ArCH). 13C NMR (100 MHz, MeOD): δ 31.3, 38.5, 63.6, 117.3, 119.5, 121.3, 125.6, 125.8, 127.9, 128.0, 128.1, 128.5, 130.5, 132.6, 138.1, 138.6, 139.7, 140.7, 141.6, 143.2, 172.4. ESI-HRMS (m/z): [M + Na]+ calcd for C25H23NO4, 424.1519; found, 424.1510.
(E)-3-(5-((E)-3-Methylbut-1-en-1-yl)-2-(3-phenylpropanamido)phenyl)acrylic Acid (33a).
Compound 32a (30 mg, 0.07 mmol) was hydrolyzed following the general procedure B. Purification by column chromatography (hexane/EtOAc = 10:1, 4:1, 2:1, 1:1) followed by solvent evaporation in vacuo provided the title compound as a white solid (18 mg, 0.05 mmol, 71%). Rf: 0.4 (DCM/MeOH, 10:1). 1H NMR (400 MHz, MeOD): δ 1.75 (3H, s, CH3), 1.74 (3H, s, CH3), 2.74 (2H, t, J = 7.6 Hz, CH2), 3.04 (2H, t, J = 7.4 Hz, CH2), 3.37 (2H, d, J = 8.7 Hz, CH2), 5.33 (1H, t, J = 8.8 Hz, CH), 6.42 (1H, d, J = 15.7 Hz, CH), 7.15–7.23 (3H, m, ArCH), 7.29–7.33 (4H, m, ArCH), 7.50 (1H, s, ArCH), 7.70 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, MeOD): δ 24.4, 31.5, 33.2, 37.7, 122.5, 125.8, 126.8, 128.0, 128.1, 129.9, 130.1, 132.6, 132.8, 133.6, 133.7, 139.1, 140.4, 140.6, 173.2. ESI-HRMS (m/z): [M + Na]+ calcd for C23H324NO3, 386.1727; found, 386.1724.
(E)-3-(3′-(Hydroxymethyl)-4-(3-phenylpropanamido)-[1,1′-biphenyl]-3-yl)acrylic Acid (33b).
Compound 32b (20 mg, 0.05 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (13 mg, 0.03 mmol, 73%). Rf: 0.5 (hexane/EtOAc, 1:1). 1H NMR (400 MHz, MeOD): δ 2.79 (2H, t, J = 7.3 Hz, CH2), 3.07 (2H, t, J = 7.8 Hz, CH2), 4.70 (2H, s, CH2), 6.57 (1H, d, J = 15.8 Hz, CH), 7.21 (1H, q, J = 4.4 Hz, ArCH), 7.31–7.47 (7H, m, ArCH), 7.57 (1H, d, J = 7.6 Hz, ArCH), 7.65–7.67 (2H, m, ArCH), 7.79 (1H, d, J = 15.8 Hz, CH), 7.94 (1H, s, ArCH). 13C NMR (100 MHz, MeOD): δ 31.4, 37.7, 63.7, 124.7, 125.0, 125.4, 125.8, 125.8, 126.9, 128.0, 128.1, 128.4, 128.6, 130.2, 135.1, 139.2, 139.4, 139.9, 140.6, 142.1 173.2. ESI-HRMS (m/z): [M + Na]+ calcd for C25H23NO4 424.1519; found, 424.1523.
(E)-3-(3-(3-Methylbut-2-en-1-yl)-5-(3-phenylpropanamido)-phenyl)acrylic Acid (39a).
Compound 38a (100 mg, 0.2 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (55 mg, 0.15 mmol, 75%). Rf: 0.2 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.75 (3H, s, CH3), 1.78 (3H, s, CH3), 2.68 (2H, t, J = 7.6 Hz, CH2), 3.01 (2H, d, J = 7.6 Hz, CH2), 3.35 (2H, d, J = 7.4 Hz, CH2), 5.33 (1H, t, J = 7.3 Hz, CH), 6.44 (1H, d, J = 15.9 Hz, CH), 7.13 (1H, s, ArCH), 7.17–7.21 (1H, m, ArCH), 7.29–7.32 (2H, m, ArCH), 7.38 (1H, s, ArCH), 7.55–7.70 (3H, m, ArCH). 13C NMR (100 MHz; MeOD): 27.2, 31.3, 33.5, 38.4, 110.3, 116.4, 118.6, 121.5, 122.3, 123.5, 125.8, 128.0, 128.0, 132.7, 139.0, 140.7, 143.2, 144.4, 172.3. ESI-HRMS (m/z): [M + Na]+ calcd for C23H25NO4 386.1727; found, 386.1739.
(E)-3-(3′-(Hydroxymethyl)-5-(3-phenylpropanamido)-[1,1′-biphenyl]-3-yl)acrylic Acid (39b).
Compound 38b (100 mg, 0.2 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (50 mg, 0.1 mmol, 60%). Rf: 0.2 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 2.72 (2H, t, J = 7.3 Hz, CH2), 3.02 (2H, t, J = 7.7 Hz, CH2), 6.54 (1H, d, J = 15.8 Hz, Ch), 7.17–7.22 (1H, m, ArCH), 7.28–7.29 (4H, m, ArCH), 7.37–7.39 (1H, m, ArCH), 7.44 (1H, t, J = 7.5 Hz, ArCH), 7.53–7.56 (2H, m, ArCH), 7.64 (1H, s, ArCH), 7.70 (1H, d, J = 16.0 Hz CH) 7.78 (1H, s, ArCH), 7.85 (1H, s, ArCH). 13C NMR (100 MHz; MeOD): 31.3, 38.5, 63.7, 117.5, 120.1, 122.2, 125.2, 125.5, 125.8, 126.0, 128.0, 128.1, 128.6, 135.5, 139.5, 140.0, 140.7, 142.1, 142.4, 144.5, 172.5. ESI-HRMS (m/z): [M + H]+ calcd for C25H23NO4 402.1700; found, 402.1701.
(E)-3-(3-(3-Methylbut-2-en-1-yl)-5-(phenethylcarbamoyl)-phenyl)acrylic Acid (49a).
Compound (45a) (50 mg, 0.12 mmol) was hydrolyzed following the general procedure B. The solvent was evaporated in vacuo to provide the title compound as a white solid (21.7 mg, 0.06 mmol, 50%). Rf: 0.27 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.77 (3H, s, CH3), 1.78 (3H, s, CH3), 2.94 (2H, t, J = 7.3 Hz, CH2), 3.43 (2H, d, J = 7.4 Hz, CH2), 3.59–3.64 (2H, m, CH2), 5.35 (1H, t, J = 7.4 Hz), 6.53 (1H, d, J = 16.0 Hz, CH), 7.20–7.32 (5H, m, ArCH), 7.54 (1H, s, ArCH), 7.63 (1H, s, ArCH), 7.68 (1H, d, J = 16.0 Hz, CH), 7.80 (1H, s, ArCH). 13C NMR (100 MHz; MeOD): δ 16.5, 24.5, 33.4, 35.0, 41.2, 119.1, 122.0, 123.7, 125.9, 128.1, 128.5, 128.6, 130.6, 133.0, 134.8, 135.2, 139.1, 143.1, 143.9, 168.3, 168.6. ESI-HRMS (m/z): [M + Na]+ calcd for C23H25NO3, 386.1727; found, 386.1736.
(E)-3-(3-((4-Fluorophenethyl)carbamoyl)-5-(3-methylbut-2-en-1-yl)phenyl)acrylic Acid (49b).
Following the general procedure B, the title compound was obtained as a white solid (20 mg, 0.05 mmol, 41%). Rf: 0.22 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.77 (3H, s, CH3), 1.79 (3H, s, CH3), 2.92 (2H, t, J = 7.3 Hz, CH2), 3.44 (2H, d, J = 7.2 Hz, CH2), 3.58–3.62 (2H, m, CH2), 5.35 (1H, t, J = 7.2 Hz, CH), 6.54 (1H, d, J = 16.0 Hz, CH), 7.03 (2H, t, J = 8.9 Hz, ArCH). 7.28 (2H, q, J1 = 5.4 Hz, J2 = 3.1 Hz, ArCH), 7.56 (1H, s, ArCH), 7.63 (1H, s, ArCH), 7.68 (1H, d, J = 15.9 Hz, CH), 7.81 (1H, s, ArCH). 13C NMR (100 MHz; MeOD): δ 16.5, 24.5, 33.4, 34.2, 41.2, 110.7, 114.5, 114.7, 122.0, 123.7, 128.5, 130.1, 130.1, 130.6, 133.1, 134.8, 135.1, 143.2, 147.0, 160.4, 168.3. ESI-HRMS (m/z): [M + H]+ calcd for C23H24NO3F, 382.1813; found, 382.1804.
(E)-3-(3-((4-Methoxyphenethyl)carbamoyl)-5-(3-methylbut-2-en-1-yl)phenyl)acrylic Acid (49c).
Following the general procedure B, the title compound was obtained as a white solid (26 mg, 0.06 mmol, 30%). Rf: 0.25 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.77 (3H, s, CH3), 1.79 (3H, s, CH3), 2.87 (2H, t, J = 7.4 Hz, CH2), 3.44 (2H, d, J = 7.2 Hz, CH2), 3.57 (2H, q, J1 = 7.3 Hz, J2 = 6.3 Hz, CH2), 3.77 (3H, s, CH3), 5.35 (1H, t, J = 7.3 Hz, CH), 6.54 (1H, d, J = 15.9 Hz, CH), 6.87 (2H, d, J = 8.6 Hz, ArCH), 7.18 (2H, d, J = 8.6 Hz, ArCH), 7.55 (1H, s, ArCH), 7.63 (1H, s, ArCH), 7.67 (1H, d, J = 16.0 Hz, Ch), 7.80 (1H, s, ArCH), 8.61 (1H, t, J = 5.5 Hz, –NH). 13C NMR (100 MHz; MeOD): δ 16.5, 24.5, 33.4, 34.2, 41.4, 54.2, 110.7, 113.4, 122.0, 123.6, 128.5, 129.4, 130.6, 131.0, 133.0, 134.8, 135.2, 135.3, 143.1, 149.9, 158.3, 168.3.
(E)-3-(3-((4-Methoxybenzyl)carbamoyl)-5-(3-methylbut-2-en-1-yl)phenyl)acrylic Acid (49d).
Following the general procedure B, the title compound was obtained as a white solid (12 mg, 0.03 mmol, 45%). Rf: 0.25 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.76 (3H, s, CH3), 1.78 (3H, s, CH3), 3.44 (2H, d, J = 7.3 Hz, CH2), 3.78 (3H, s, CH3), 4.52 (2H, d, J = 4.1 Hz, CH2), 5.35 (1H, t, J = 7.3 Hz, CH), 6.55 (1H, d, J = 16.0 Hz, CH), 6.90 (2H, d, J = 8.7 Hz, ArCH), 7.29 (2H, d, J = 8.7 Hz, ArCH), 7.55 (1H, s, ArCH), 7.68 (1H, d, J = 16.0 Hz, CH), 7.71 (1H, s, ArCH), 7.89 (1H, s, ArCH), 9.00 (1H, t, J = 5.6 Hz, –NH). 13C NMR (100 MHz; MeOD): δ 16.5, 24.4, 33.4, 42.6, 54.2, 110.7, 113.5, 119.4, 122.0, 123.8, 128.5, 130.6, 130.7, 133.0, 134.9, 135.1, 143.2, 143.7, 159.0, 168.0. ESI-HRMS (m/z): [M + Na]+ calcd for C23H25NO4, 402.1676; found, 402.1669.
(E)-3-(3-(3-Methylbut-2-en-1-yl)-5-((4-(trifluoromethoxy)-benzyl)carbamoyl)phenyl)acrylic Acid (49e).
Following the general procedure B, the title compound was obtained as a white solid (39 mg, 0.09 mmol, 75%). Rf: 0.25 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.77 (3H, s, CH3), 1.78 (3H, s, CH3), 3.45 (2H, d, J = 7.2 Hz, CH2), 4.61 (2H, s, CH2), 5.36 (1H, t, J = 7.3 Hz, CH), 6.56 (1H, d, J = 15.9 Hz, CH), 7.26 (2H, d, J = 7.9 Hz, ArCH), 7.47 (2H, d, J = 8.7 Hz, ArCH), 7.58 (1H, s, ArCH), 7.69 (1H, d, J =16.0 Hz, CH), 7.73 (1H, s, ArCH), 7.92 (1H, s, ArCH). NMR (100 MHz; MeOD): δ 16.5, 24.4, 33.4, 42.4, 119.3, 119.3, 120.7, 122.0, 123.8, 128.7, 128.8, 130.7, 133.1, 134.8, 134.9, 138.1, 143.3, 143.8, 148.1, 168.2. ESI-HRMS (m/z): [M + Na]+ calcd for C23H22NO4F3, 456.1393; found, 456.1403.
(E)-3-(3-(3-Methylbut-2-en-1-yl)-5-((3-(trifluoromethoxy)-benzyl)carbamoyl)phenyl)acrylic Acid (49f).
Following the general procedure B, the title compound was obtained as a white solid (18 mg, 0.04 mmol, 33%). Rf: 0.21 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.77 (3H, s, CH3), 1.78 (3H, s, CH3), 2.87 (2H, t, J = 7.4 Hz, CH2), 3.45 (2H, d, J = 7.5 Hz, CH2), 4.63 (2H, d, J1 = 4.0 Hz, CH2), 5.36 (1H, t, J = 7.3 Hz, CH), 6.56 (1H, d, J = 16.0 Hz, CH), 7.18 (2H, d, J = 8.1 Hz, ArCH), 7.29 (1H, s, ArCH), 7.37–7.47 (2H, m, ArCH), 7.58 (1H, s, ArCH), 7.73 (1H, s, ArCH), 7.70 (1H, d, J = 16.0 Hz, CH), 7.92 (1H, s, ArCH). NMR (100 MHz; MeOD): δ 16.5, 24.4, 33.4, 42.6, 119.2, 119.6, 122.0, 123.8, 125.8, 128.6, 129.8, 130.8, 133.1, 134.8, 134.9, 141.6, 143.3, 143.9, 149.3, 150.1, 168.2, 168.6. ESI-HRMS (m/z): [M + Na]+ calcd for C23H22NO4F3, 456.1393; found, 456.1387.
(E)-3-(3-((4-Methylbenzyl)carbamoyl)-5-(3-methylbut-2-en-1-yl)phenyl)acrylic Acid (49g).
Following the general procedure B, the title compound was obtained as a white solid (18 mg, 0.05 mmol, 30%). Rf: 0.20 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.76 (3H, s, CH3), 1.77 (3H, s, CH3), 2.31 (3H, s, CH3), 3.43 (2H, d, J = 7.3 Hz, CH2), 4.54 (2H, d, J = 5.4 Hz, CH2), 5.35 (1H, t, J = 7.3 Hz, CH), 6.54 (1H, d, J = 16.0 Hz, CH), 7.15 (2H, d, J = 7.8 Hz, ArCH), 7.24 (2H, d, J = 8.0 Hz, ArCH), 7.55 (1H, s, ArCH), 7.68 (1H, d, J = 16.0 Hz, CH), 7.71 (1H, s, ArCH), 7.90 (1H, s, ArCH), 9.03 (1H, t, J = 5.7 Hz, -NH). NMR (100 MHz; MeOD): δ 16.5, 19.7, 24.5, 33.4, 42.9, 119.1, 122.0, 123.8, 127.1, 128.7, 128.7, 130.6, 133.0, 134.8, 135.0, 135.6, 136.5, 143.2, 143.9, 168.0, 168.7. ESI-HRMS (m/z): [M + H]+ calcd for C23H25NO3, 364.1907; found, 364.1899.
(E)-3-(3-((4-(Dimethylamino)benzyl)carbamoyl)-5-(3-meth-ylbut-2-en-1-yl)phenyl)acrylic Acid (49h).
Following the general procedure B, the title compound was obtained as a white solid (15 mg, 0.04 mmol, 42%). Rf: 0.27 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 1.77 (3H, s, CH3), 1.78 (3H, s, CH3), 2.91 (6H, s, N(CH3)2), 3.44 (2H, d, J = 7.2 Hz, CH2), 4.48 (2H, d, J = 5.6 Hz, CH2), 5.36 (1H, t, J = 7.3 Hz, CH), 6.55 (1H, d, J = 16.0 Hz, CH), 6.78 (2H, d, J = 8.7 Hz, ArCH), 7.23 (2H, d, J = 8.7 Hz, ArCH), 7.55 (1H, s, ArCH), 7.67 (1H, d, J = 16.0 Hz, CH), 7.71 (1H, s, ArCH), 7.89 (1H, s, ArCH), 8.92 (1H, t, J = 5.9 Hz, –NH). NMR (100 MHz; MeOD): δ 16.5, 24.4, 33.4, 39.7, 42.8, 112.9, 119.4, 122.1, 123.7, 126.9, 128.2, 128.7, 130.5, 133.0, 134.9, 135.2, 143.1, 143.7, 150.2, 168.0, 168.9. ESI-HRMS (m/z): [M + H]+ calcd for C24H28N2O3, 393.2173; found, 393.2169.
(E)-3-(3-Allyl-5-(phenethylcarbamoyl)phenyl)acrylic Acid (50).
Following the general procedure B, the title compound, synthesized using 46, was obtained as a white solid (11 mg, 0.03 mmol, 27%). Rf: 0.26 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 2.94 (2H, t, J = 7.1 Hz, CH2), 3.48 (2H, d, J = 6.7 Hz, CH2), 3.60 (2H, t, J = 7.8 Hz, CH2), 5.11–5.17 (2H, m, CH2), 5.96–6.06 (1H, m, CH), 6.55 (1H, d, J = 16.0 Hz, CH), 7.20–7.32 (5H, m, ArCH), 7.59 (1H, s, ArCH), 7.66 (1H, s, ArCH), 7.70 (1H, s, ArCH), 7.82 (1H, d, J = 16.0 Hz, CH). 13C NMR (100 MHz; CDCl3): δ 35.0, 39.3, 41.2, 115.5, 124.0, 125.9, 127.6, 128.1, 128.5, 128.8, 129.4, 130.8, 134.9, 135.3, 136.5, 139.1, 141.5, 143.6, 168.1. ESI-HRMS (m/z): [M + H]+ calcd for C21H21NO3, 336.1594; found, 336.1595.
(E)-3-(3′-(Hydroxymethyl)-5-(phenethylcarbamoyl)-[1,1′-bi-phenyl]-3-yl)acrylic Acid (51).
Following the general procedure B, the title compound, synthesized using 47, was obtained as a white solid (14 mg, 0.03 mmol, 35%). Rf: 0.19 (DCM/MeOH, 20:1). 1H NMR (400 MHz; CDCl3): δ 2.97 (2H, t, J = 7.1 Hz, CH2), 3.65 (2H, t, J = 7.5 Hz, CH2), 4.72 (2H, s, CH2), 6.66 (1H, d, J = 1(5.0 Hz, CH), 7.22–7.50 (7H, m, ArCH), 7.62 (1H, d, J = 7.5 Hz, ArCH), 7.71 (1H, s, ArCH), 7.75 (1H, d, J = 15.9 Hz, CH), 7.99 (2H, d, J = 4.8 Hz, ArCH), 8.06 (1H, s, ArCH). 13C NMR (100 MHz; CDCl3): δ 35.0, 41.3, 63.6, 124.7, 124.8, 125.2, 125.6, 126.0, 126.3, 126.9, 128.1, 128.5, 128.7, 129.1, 129.6, 135.6, 135.7, 139.1, 139.5, 142.1, 142.2, 168.0. ESI-HRMS (m/z): [m + Na]+ calcd for C25H23NO4, 424.1519; found, 424.1513.
(E)-3-(3-(3-Methylbut-2-en-1-yl)-5-(phenethoxycarbonyl)-phenyl)acrylic Acid (52).
Following the general procedure B, the title compound, synthesized using 48, was obtained as a white solid (30 mg, 0.08 mmol, 51%). 1H NMR (400 MHz; CDCl3): δ 1.76 (3H, s, CH3), 1.80 (3H, s, CH3), 3.12 (2H, t, J = 7.0 Hz, CH2), 3.42 (2H, d, J = 7.2 Hz, CH2), 4.57 (2H, t, J = 6.9 Hz, CH2), 5.32 (1H, t, J = 7.3 Hz, CH), 6.51 (1H, d, J = 15.9 Hz, CH), 7.29–7.38 (5H, m, ArCH), 7.53 (1H, s, ArCH), 7.80 (1H, d, J = 15.9 Hz, CH), 7.87 (1H, s, ArCH), 8.02 (1H, s, ArCH). 13C NMR (100 MHz; CDCl3): δ 17.9, 25.7, 33.9, 35.2, 65.7, 118.1, 121.8, 126.7, 128.5, 128.9, 129.0, 131.1, 131.5, 132.5, 133.9, 134.3, 137.7, 143.1, 146.0, 166.0, 171.3. ESI-HRMS (m/z): [M + Na]+ calcd for C23H24O4, 387.1567; found, 387.1573.
(E)-3-(3-(Benzyloxy)-5-(phenethylcarbamoyl)phenyl)acrylic Acid (59a).
Following the general procedure B, the title compound was obtained as a white solid (8 mg, 0.02 mmol, 33%). Rf: 0.3 (DCM/MeOH, 20:1). 1H NMR (400 MHz; MeOD): δ 2.94 (2H, t, J = 7.0 Hz, CH2), 3.61 (2H, t, J = 6.0 Hz, CH2), 5.18 (2H, s, CH2), 6.55 (1H, d, J = 15.9 Hz, CH), 7.20–7.49 (12H, m, ArCH), 7.58 (1H, s, ArCH), 7.65 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz; MeOD): δ 35.0, 41.3, 69.9, 115.0, 116.8, 118.8, 125.9, 127.3, 127.6, 128.1, 128.1, 128.5, 136.2, 136.4, 136.7, 139.1, 143.4, 159.3, 167.8. ESI-HRMS (m/z): [M + Na]+ calcd for C25H23NO4, 424.1519; found, 424.1520.
(E)-3-(3-((3-Methoxybenzyl)oxy)-5-(phenethylcarbamoyl)-phenyl)acrylic Acid (59b).
Following the general procedure B, the title compound was obtained as a white solid (10 mg, 0.02 mmol, 38%). Rf: 0.2 (DCM/MeOH, 20:1). 1H NMR (400 MHz; CDCl3): δ 2.93 (2H, t, J = 7.0 Hz, CH2), 3.61 (2H, t, J = 6.0 Hz, CH2), 3.81 (3H, s, CH3), 5.15 (2H, s, CH2), 6.55 (1H, d, J = 15.9 Hz, CH), 6.89 (1H, dd, J1 = 2.1 Hz, J2 = 8.0 Hz, ArCH), 7.03–7.05 (2H, m, ArCH), 7.19–7.33 (6H, m, ArCH), 7.37 (1H, s, ArCH), 7.44 (1H, s, ArCH), 7.58 (1H, s, ArCH), 7.61 (1H, d, J = 16.0 Hz, CH). 13C NMR (100 MHz; CDCl3): δ 35.0, 41.3, 54.2, 69.7, 112.6, 113.1, 114.9, 116.7, 118.7, 119.3, 125.9, 128.1, 128.5, 129.2, 136.3, 136.4, 138.2, 139.1, 142.7, 159.2, 159.9, 167.8. ESI-HRMS (m/z): [M + Na]+ calcd for C26H25NO5, 454.1625; found, 454.1630.
Enzyme Inhibition Assay.
(S)-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol) was purchased from Sigma-Aldrich (St. Louis, MO). Nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+) were purchased from Roche Diagnostics (Indianapolis, IN). Homogeneous recombinant enzymes AKR1C1, AKR1C2, AKR1C3 and AKR1C4 were prepared and purified as previously described.70 The specific activities of AKR1C3 and AKR1C2 for the oxidation of S-tetralol are 2.0 and 1.5 μmol/(min mg), respectively.
Assay of Enzyme Activity.
The dehydrogenase activities of AKR1C3 and AKR1C2 were determined by measuring the UV absorption of NADH formation at 340 nm using a Beckman DU-640 spectrophotometer. A typical assay solution contained 100 mM potassium phosphate pH 7.0, 2.3 mM NAD+, 3.0 mM (S)-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol), and 4% acetonitrile (v/v). The mixtures were incubated at 37 °C for 3 min followed by adding a serial dilution of AKR1C1, AKR1C2, AKR1C3 or AKR1C4 solution to a final volume of 1 mL to initiate the reaction. After continuously monitoring for 5 min, we recorded the increase in UV absorption using different concentrations of an enzyme to calculate the initial velocity and determine the specific activity of the enzyme.
IC50 Value Determination.
The inhibitory potency for each compound was represented by IC50 values and measured as described before.24,34,71 The IC50 values of baccharin and baccharin analogues were determined by measuring their inhibition of the NADP+- dependent oxidation of S-tetralol catalyzed by AKR1C3 or AKR1C2. The concentrations of S-tetralol used in this assay for AKR1C3 and AKR1C2 were 165 μM and 15 μM, respectively, which were equal to the Km value for each enzyme isoform to make a direct comparison of IC50 values. The IC50 value of each compound was acquired from a single experiment with each inhibitor concentration run in quadruplicate and directly calculated by fitting the inhibition data to an equation [y = (range)/[1 + (I/IC50)S] + background] using Grafit 5.0 software. In this equation, “range” is the fitted uninhibited value minus the “background” and “S” is a slope factor. “I” is the concentration of the inhibitor. The equation assumes that y decreases with the increasing “I”.
Adjuvant Assay.
HL-60 (ATCC CCL240), KG1a (ATCC CCL246.1), and THP-1 (ATCC TIB-202) cells were procured from ATCC. Isocove’s modified Dulbecco’s medium supplemented with 20% fetal bovine serum (FBS) and penicillin/streptomycin (1%) was used to culture HL-60 and KG1a cells, whereas THP-1 cells were cultured using Roswell Park Memorial Institute-1640 medium, supplemented with 20% fetal bovine serum (FBS), 0.05 mM 2-mercaptoethanol, and penicillin/streptomycin (1%). Cells were maintained at a density of 0.1–1 × 106 cells/mL under 5% CO2 at 37 °C. To screen the test compounds, cells were seeded at a density of 0.1 × 106 cells/mL in 96-well plates containing 100 μL of cell suspension per well. Stock solutions of the test compounds, etoposide, daunorubicin, and cytarabine (AraC), were prepared in DMSO. Cells were treated at the indicated concentrations of test compounds with or without the chemotherapeutic agents, limiting the final DMSO concentration to less than 1%. After incubation at 37 °C and 5% CO2 for 24, 48, 72, or 96 h, 20 μL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl) −2-(4-sulfophenyl) −2H-tetrazolium reagent (CellTiter 96 AQueous One Solution Reagent) was added to each well and incubated at the above-mentioned conditions for 2–3 h. Plates were read at optical density 490 nm on a plate reader, and the viability of cells was plotted as a percentage of controls.
Quantification of the Degree of Synergism.
To quantify the degree of synergism, the results of the co-treatment and pretreatment experiments were analyzed by CompuSyn software (Paramus, NJ) based on the median-effect principle or “Chou–Talalay” method.72 The method is based on the median-effect equation that encompasses the Michaelis–Menten, Hill, Henderson–Hasselbach, and Scatchard equations to provide combination and dose reduction indices.66 The combination index (CI) and dose reduction index (DRI) values were calculated at a constant ratio of a chemotherapeutic to AKR1C3 inhibitor at 50% cytotoxic effect (Fa = 0.5). CompuSyn software was used to generate the CI and DRI values. CI < 1, synergism; CI > 1.1, antagonism; and CI = 1.1, additive.66
Western Blotting.
HL-60 and KG1a cells were treated with compound (4) over a period of 48 and 72 h at indicated concentrations after which they were harvested and pelleted. The whole cell lysates were prepared in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors (1 mM phenylmethylsulfonyl fluoride, 38 μg/mL aprotinin, 2.5 mM Na3VO4). Samples were incubated on ice for 30 min after which they were sonicated and centrifuged (16 000g), and the supernatant was collected. Protein concentration in each sample was estimated following the bicinchoninic acid (BCA) assay protocol using bovine serum albumin as standard (Pierce BCA protein kit). Protein samples (40 μg) containing loading dye (7 μL) were loaded onto 12% sodium dodecyl sulfate polyacrylamide gel and electrophoresed (80 V, 2 h) and then transferred onto a poly(vinylidene difluoride) membrane overnight (25 V, 4 °C). The membrane was blocked with 5% nonfat milk (1 h) and probed with human anti-AKR1C3 mouse monoclonal antibody (1:500, R&D Systems, MAB7678) and corresponding horseradish peroxidase conjugated anti-mouse secondary antibody followed by immunodetection using VersaDoc (MP 5000). The membrane was stripped and re-probed for β-actin (1:5000, Sigma-Aldrich, A5441). Quantity One software was used to analyze the band intensities, and fold change in AKR1C3 enzyme expression was determined based on β-actin controls.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported in part by the National Cancer Institute of the National Institutes of Health under Award Numbers R01 CA226436 (P.C.T.), P30ES013508 (T.M.P.) and by a research grant from Leukemia Texas (P.C.T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- AKR1C3
aldo–keto reductase 1C3
- ALL
acute lymphoblastic leukemia
- AR
androgen receptor
- AraC
cytarabine
- ATRA
all-trans retinoic acid
- COG
Children’s Oncology Group
- CRPC
castration-resistant prostate cancer
- CI
combination index
- DRI
dose reduction index
- LLE
ligand-lipophilicity efficiency
- MPA
medroxyprogesterone acetate
- OX
oxyanion site
- PG
prostaglandin
- SP
subpocket
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.9b00090.
PDB file for the docking model of 49a with AKR1C3 (Figure 3B) (PDB)
Molecular formula strings (CSV)
Cell viability curves of AKR1C3 inhibitors 1, 26a, 49a, and 49g, daunorubicin, and AraC single treatment in HL-60, KG1a, and THP-1 cells. AKR1C3 expression in leukemic cells; cell viability curves of leukemic cells upon co-treatment of AKR1C3 inhibitor and daunorubicin and AKR1C3 inhibitor and AraC; cell viability curves of COG T-ALL cells treated with inhibitor 49a, daunorubicin, and AraC; cell viability curves of BMMNC cells treated with combination AKR1C3 inhibitor 49a and either AraC or daunorubicin; synthetic details and characterization data for all intermediates, and 1H and 13C NMR spectra of final compounds (PDF)
The authors declare the following competing financial interest(s): A patent application has been filed describing the compounds in this paper. Highly selective AKR1C3 inhibitors and methods of use thereof. PCT Int. Appl. (2018) WO 2018148721 A1.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A Cancer statistics, 2018. CA—Cancer J. Clin. 2018, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Shipley JL; Butera JN Acute myelogenous leukemia. Exp. Hematol. 2009, 37, 649–658. [DOI] [PubMed] [Google Scholar]
- (3).Renneville A; Roumier C; Biggio V; Nibourel O; Boissel N; Fenaux P; Preudhomme C Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia 2008, 22, 915–931. [DOI] [PubMed] [Google Scholar]
- (4).Grimwade D; Enver T Acute promyelocytic leukemia: where does it stem from? Leukemia 2004, 18, 375–384. [DOI] [PubMed] [Google Scholar]
- (5).Brown G; Hughes P Retinoid differentiation therapy for common types of acute myeloid leukemia. Leuk. Res. Treat. 2012, 2012, No. 939021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Huang M; Ye Y.-c.; Chen S; Chai J-R; Lu J-X; Zhoa L; Gu L; Wang Z-Y Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [PubMed] [Google Scholar]
- (7).Deschler B; de Witte T; Mertelsmann R; Lubbert M Treatment decision-making for older patients with high-risk myelodysplastic syndrome or acute myeloid leukemia: problems and approaches. Haematologica 2006, 91, 1513–1522. [PubMed] [Google Scholar]
- (8).Kuendgen A; Germing U Emerging treatment strategies for acute myeloid leukemia (AML) in the elderly. Cancer Treat. Rev. 2009, 35, 97–120. [DOI] [PubMed] [Google Scholar]
- (9).Terwilliger T; Abdul-Hay M Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017, 7, No. e577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Van Vlierberghe P; Ferrando A The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Invest. 2012, 122, 3398–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Marks DI; Rowntree C Management of adults with T-cell lymphoblastic leukemia. Blood 2017, 129, 1134–1142. [DOI] [PubMed] [Google Scholar]
- (12).Desmond JC; Mountford JC; Drayson MT; Walker EA; Hewison M; Ride JP; Luong QT; Hayden RE; Vanin EF; Bunce CM The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003, 63, 505–512. [PubMed] [Google Scholar]
- (13).Manesh DM; El-Hoss J; Evans K; Richmond J; Toscan CE; Bracken LS; Hedrick A; Sutton R; Marshall GM; Wilson WR; et al. AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood 2015, 126, 1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sales KJ; Milne SA; Williams AR; Anderson RA; Jabbour HN Expression, localization, and signaling of prostaglandin F2α receptor in human endometrial adenocarcinoma: regulation of proliferation by activation of the epidermal growth factor receptor and mitogen-activated protein kinase signaling pathways. J. Clin. Endocrinol. Metab. 2004, 89, 986–993. [DOI] [PubMed] [Google Scholar]
- (15).Straus DS; Glass CK Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med. Res. Rev. 2001, 21, 185–210. [DOI] [PubMed] [Google Scholar]
- (16).Khanim FL; Hayden RE; Birtwistle J; Lodi A; Tiziani S; Davies NJ; Ride JP; Viant MR; Gunther UL; Mountford JC Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukaemia. PLoS One 2009, 4, No. e8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ji Q; Chang L; Stanczyk FZ; Ookhtens M; Sherrod A; Stolz A Impaired dihydrotestosterone catabolism in human prostate cancer: critical role of AKR1C2 as a pre-receptor regulator of androgen receptor signaling. Cancer Res. 2007, 67, 1361–1369. [DOI] [PubMed] [Google Scholar]
- (18).Penning TM; Bauman DR; Jin Y; Rizner TL Identification of the molecular switch that regulates access of 5a- DHT to the androgen receptor. Mol. Cell. Endocrinol. 2007, 265–266, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hofman J; Malcekova B; Skarka A; Novotna E; Wsol V Anthracycline resistance mediated by reductive metabolism in cancer cells: The role of aldo-keto reductase 1C3. Toxicol. Appl. Pharmacol. 2014, 278, 238–248. [DOI] [PubMed] [Google Scholar]
- (20).Heibein AD; Guo B; Sprowl JA; MacLean DA; Parissenti AM Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer 2012, 12, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Byrns MC; Penning TM Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase (AKR1C3): role in breast cancer and inhibition by non-steroidal anti-inflammatory drug analogs. Chem. Biol. Interact. 2009, 178, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Matsunaga T; Hojo A; Yamane Y; Endo S; El-Kabbani O; Hara A Pathophysiological roles of aldo-keto reductases (AKR1C1 and AKR1C3) in development of cisplatin resistance in human colon cancers. Chem. Biol. Interact. 2013, 202, 234–242. [DOI] [PubMed] [Google Scholar]
- (23).Adeniji AO; Chen M; Penning TM AKR1C3 as a target in castrate resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2013, 137, 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liedtke AJ; Adeniji AO; Chen M; Byrns MC; Jin Y; Christianson DW; Marnett LJ; Penning TM Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J. Med. Chem. 2013, 56, 2429–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rižner TL; Šmuc T; Rupreht R; Sinkovec J; Penning TM AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol. Cell. Endocrinol. 2006, 248, 126–135. [DOI] [PubMed] [Google Scholar]
- (26).Byrns MC; Jin Y; Penning TM Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J. Steroid Biochem. Mol. Biol. 2011, 125, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lan Q; Mumford JL; Shen M; DeMarini DM; Bonner MR; He X; Yeager M; Welch R; Chanock S; Tian L; et al. Oxidative damage-related genes AKR1C3 and OGG1 modulate risks for lung cancer due to exposure to PAH-rich coal combustion emissions. Carcinogenesis 2004, 25, 2177–2181. [DOI] [PubMed] [Google Scholar]
- (28).Skarydova L; Zivna L; Xiong G; Maser E; Wsól V AKR1C3 as a potential target for the inhibitory effect of dietary flavonoids. Chem. Biol. Interact. 2009, 178, 138–144. [DOI] [PubMed] [Google Scholar]
- (29).Davies NJ; Hayden RE; Simpson PJ; Birtwistle J; Mayer K; Ride JP; Bunce CM AKR1C isoforms represent a novel cellular target for jasmonates alongside their mitochondrial¬mediated effects. Cancer Res. 2009, 69, 4769–4775. [DOI] [PubMed] [Google Scholar]
- (30).Adeniji A; Uddin MJ; Zang T; Tamae D; Wangtrakuldee P; Marnett LJ; Penning TM Discovery of (R)-2-(6- methoxynaphthalen-2-yl) butanoic acid as a potent and selective aldo-keto reductase 1C3 inhibitor. J. Med. Chem. 2016, 59, 7431–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Liedtke AJ; Adeniji AO; Chen M; Byrns MC; Jin Y; Christianson DW; Marnett LJ; Penning TM Development of potent and selective analogues for the inhibition of AKR1C3 (Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J. Med. Chem. 2013, 56, 2429–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bauman DR; Rudnick SI; Szewczuk LM; Jin Y; Gopishetty S; Penning TM Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol. Pharmacol. 2005, 67, 60–68. [DOI] [PubMed] [Google Scholar]
- (33).Byrns MC; Steckelbroeck S; Penning TM An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β- HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem. Pharmacol. 2008, 75, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Adeniji AO; Twenter BM; Byrns MC; Jin Y; Chen M; Winkler JD; Penning TM Development of potent and selective inhibitors of aldo–keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure–activity relationships. J. Med. Chem. 2012, 55, 2311–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen M; Adeniji AO; Twenter BM; Winkler JD; Christianson DW; Penning TM Crystal structures of AKR1C3 containing an N-(aryl) amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg. Med. Chem. Lett. 2012, 22, 3492–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yin YD; Fu M; Brooke DG; Heinrich DM; Denny WA; Jamieson SM The activity of SN33638, an inhibitor of AKR1C3, on testosterone and 17^-estradiol production and function in castration-resistant prostate cancer and ER-positive breast cancer. Front. Oncol. 2014, 4, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Loriot Y; Fizazi K; Jones RJ; Van den Brande J; Molife RL; Omlin A; James ND; Baskin-Bey E; Heeringa M; Baron B; Holtkamp GM; Ouatas T; De Bono JS Safety, tolerability and anti-tumour activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest. New Drugs 2014, 32, 995–1004. [DOI] [PubMed] [Google Scholar]
- (38).Kikuchi A; Furutani T; Azami H; Watanabe K; Niimi T; Kamiyama Y; Kuromitsu S; Baskin-Bey E; Heeringa M; Ouatas T; Enjo K In vitro and in vivo characterisation of ASP9521: a novel, selective, orally bioavailable inhibitor of 17β-hydroxysteroid dehydro¬genase type 5 (17βHSD5; AKR1C3). Invest. New Drugs 2014, 32, 860–870. [DOI] [PubMed] [Google Scholar]
- (39).Yepuru M; Wu Z; Kulkarni A; Yin F; Barrett CM; Kim J; Steiner MS; Miller DD; Dalton JT; Narayanan R Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin. Cancer Res. 2013, 19, 5613–5625. [DOI] [PubMed] [Google Scholar]
- (40).Pippione AC; Carnovale IM; Bonanni D; Sini M; Goyal P; Marini E; Pors K; Adinolfi S; Zonari D; Festuccia C; et al. Potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the benzoisoxazole moiety: application of a bioisosteric scaffold hopping approach to flufenamic acid. Eur. J. Med. Chem. 2018, 150, 930–945. [DOI] [PubMed] [Google Scholar]
- (41).Mills K; Gilkes A; Sweeney M; Choudhry M; Woodgate L; Bunce C; Brown G; Burnett A Identification of a retinoic acid responsive aldoketoreductase expressed in HL60 leukaemic cells. FEBS Lett. 1998, 440, 158–162. [DOI] [PubMed] [Google Scholar]
- (42).Khanim F; Davies N; Velica P; Hayden R; Ride J; Pararasa C; Chong M; Gunther U; Veerapen N; Winn P; et al. Selective AKR1C3 inhibitors do not recapitulate the anti-leukaemic activities of the pan-AKR1C inhibitor medroxyprogesterone acetate. Br. J. Cancer 2014, 110, 1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Endo S; Matsunaga T; Kanamori A; Otsuji Y; Nagai H; Sundaram K; El-Kabbani O; Toyooka N; Ohta S; Hara A Selective inhibition of human type-5 17ß-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J. Nat. Prod. 2012, 75, 716–721. [DOI] [PubMed] [Google Scholar]
- (44).Zang T; Verma K; Chen M; Jin Y; Trippier PC; Penning TM Screening baccharin analogs as selective inhibitors against type 5 17β-hydroxysteroid dehydrogenase (AKR1C3). Chem. Biol. Interact. 2015, 234, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Endo S; Hu D; Matsunaga T; Otsuji Y; El-Kabbani O; Kandeel M; Ikari A; Hara A; Kitade Y; Toyooka N Synthesis of non-prenyl analogues of baccharin as selective and potent inhibitors for aldo-keto reductase 1C3. Bioorg. Med. Chem. 2014, 22, 5220–5233. [DOI] [PubMed] [Google Scholar]
- (46).Verma K; Gupta N; Zang T; Wangtrakuldee P; Srivastava SK; Penning TM; Trippier PC AKR1C3 inhibitor KV-37 exhibits antineoplastic effects and potentiates enzalutamide in combination therapy in prostate adenocarcinoma cells. Mol. Cancer Ther. 2018, 17, 1833–1845. [DOI] [PubMed] [Google Scholar]
- (47).Verma K; Zang T; Gupta N; Penning TM; Trippier PC Selective AKR1C3 inhibitors potentiate chemotherapeutic activity in multiple acute myeloid leukemia (AML) cell lines. ACS Med. Chem. Lett. 2016, 7, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tani H; Hasumi K; Tatefuji T; Hashimoto K; Koshino H; Takahashi S Inhibitory activity of Brazilian green propolis components and their derivatives on the release of cys-leukotrienes. Bioorg. Med. Chem. 2010, 18, 151–157. [DOI] [PubMed] [Google Scholar]
- (49).Bates RW; Gabel CJ; Ji J; Rama-Devi T Synthesis of phenolic natural products using palladium catalyzed coupling reactions. Tetrahedron 1995, 51, 8199–8212. [Google Scholar]
- (50).Heck RF Mechanism of arylation and carbomethoxylation of olefins with organopalladium compounds. J. Am. Chem. Soc. 1969, 91, 6707–6714. [Google Scholar]
- (51).Heck RF Palladium-catalyzed vinylation of organic halides. Org. React. 2004, 27, 345–390. [Google Scholar]
- (52).Jackson RW A mild and selective method for the cleavage of tert-butyl esters. Tetrahedron Lett. 2001, 42, 5163–5165. [Google Scholar]
- (53).Trippier PC; McGuigan C; Balzarini J Phenylboronic-acid- based carbohydrate binders as antiviral therapeutics: monophenylbor-onic acids. Antiviral Chem. Chemother. 2010, 20, 249–257. [DOI] [PubMed] [Google Scholar]
- (54).Trippier PC; Balzarini J; McGuigan C Phenylboronic-acid-based carbohydrate binders as antiviral therapeutics: bisphenylboronic acids. Antiviral Chem. Chemother. 2011, 21, 129–142. [DOI] [PubMed] [Google Scholar]
- (55).Morin J-F; Shirai Y; Tour JM En route to a motorized nanocar. Org. Lett. 2006, 8, 1713–1716. [DOI] [PubMed] [Google Scholar]
- (56).Fitton P; Rick EA The addition of aryl halides to tetrakis(triphenylphosphine)palladium (0). J. Organomet. Chem. 1971, 28, 287–291. [Google Scholar]
- (57).Miyaura N; Yamada K; Suzuki A A new stereospecific cross¬coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar]
- (58).Gerbino DC; Mandolesi SD; Schmalz HG; Podesta JC Introduction of allyl and prenyl side-chains into aromatic systems by Suzuki cross-coupling reactions. Eur. J. Org. Chem. 2009, 2009, 3964–3972. [Google Scholar]
- (59).Shetty RS; Lee Y; Liu B; Husain A; Joseph RW; Lu Y; Nelson D; Mihelcic J; Chao W; Moffett KK; et al. Synthesis and pharmacological evaluation of N-(3-(1H-indol-4-yl)-5-(2- methoxyisonicotinoyl)phenyl)methanesulfonamide (LP-261), a potent antimitotic agent. J. Med. Chem. 2011, 54, 179–200. [DOI] [PubMed] [Google Scholar]
- (60).Groll M; Berkers CR; Ploegh HL; Ovaa H Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [DOI] [PubMed] [Google Scholar]
- (61).Trippier PC; McGuigan C Boronic acids in medicinal chemistry: anticancer, antibacterial and antiviral applications. Med Chem Comm 2010, 1, 183–198. [Google Scholar]
- (62).Brožič P; Golob B; Gomboc N; Rižner TL; Gobec S Cinnamic acids as new inhibitors of 17β-hydroxysteroid dehydrogenase type 5 (AKR1C3). Mot. Cell. Endocrinol. 2006, 248, 233–235. [DOI] [PubMed] [Google Scholar]
- (63).Lowenberg B; Ossenkoppele JC; van Putten W; Schouten HC; Graux C; Ferrant A; Sonneveld P; et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [DOI] [PubMed] [Google Scholar]
- (64).Mandelli F; Vignetti M; Suciu S; Stasi R; Petti M-C; Meloni G; Muus P; Marmont F; Marie J-P; Labar B; et al. Daunorubicin versus mitoxantrone versus idarubicin as induction and consolidation chemotherapy for adults with acute myeloid leukemia: the EORTC and GIMEMA Groups Study AML-10. J. Clin. Oncol. 2009, 27, 5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Matsunaga T; Yamaguchi A; Morikawa Y; Kezuka C; Takazawa H; Endo S; El-Kabbani O; Tajima K; Ikari A; Hara A Induction of aldo-keto reductases (AKR1C1 and AKR1C3) abolishes the efficacy of daunorubicin chemotherapy for leukemic U937 cells. Anti-Cancer Drugs 2014, 25, 868–877. [DOI] [PubMed] [Google Scholar]
- (66).Chou T-C Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [DOI] [PubMed] [Google Scholar]
- (67).Laurent G; Jaffrezou J-P Signaling pathways activated by daunorubicin. Blood 2001, 98, 913–924. [DOI] [PubMed] [Google Scholar]
- (68).Bailly J; Skladanowski A; Bettaieb A; Mansat V; Larsen A; Laurent G Natural resistance of acute myeloid leukemia cell lines to mitoxantrone is associated with lack of apoptosis. Leukemia 1997, 11, 1523. [DOI] [PubMed] [Google Scholar]
- (69).Novotna E; Bukum N; Hofman J; Flaxova M; Kouklfkova E; Louvarova D; Wsol V Roscovitine and purvalanol A effectively reverse anthracycline resistance mediated by the activity of aldo-keto reductase 1C3 (AKR1C3): A promising therapeutic target for cancer treatment. Biochem. Pharmacol. 2018, 156, 22. [DOI] [PubMed] [Google Scholar]
- (70).Penning TM; Burczynski ME; Jez JM; Hung C-F; Lin H-K; Ma H; Moore M; Palackal N; Ratnam K Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Adeniji AO; Twenter BM; Byrns MC; Jin Y; Winkler JD; Penning TM Discovery of substituted 3-(phenylamino) benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3). Bioorg. Med. Chem. Lett. 2011, 21, 1464–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chou TC Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.