

Abstract
PURPOSE
Ductal prostate cancer (dPC) is a rare variant of prostatic adenocarcinoma associated with poor outcomes. Although its histopathologic features are well characterized, the underlying molecular hallmarks of this aggressive subtype are not well described. We sought to provide a comprehensive overview of the spectrum of mutations associated with dPC.
METHODS
Three case series across multiple institutions were assembled. All patients had a diagnosis of dPC, and histopathologic classification was confirmed by an expert genitourinary pathologist. Case series 1 included men who were prospectively enrolled in a tumor sequencing study at the University of Washington (n = 22). Case series 2 and 3 included archival samples from men treated at Johns Hopkins Hospital (n = 21) and University of Calgary (n = 8), respectively. Tumor tissue was sequenced on a targeted next-generation sequencing assay, UW-OncoPlex, according to previously published methods. The frequency of pathogenic/likely pathogenic mutations are reported.
RESULTS
Overall, 25 patients (49%) had at least one DNA damage repair gene alteration, including seven (14%) with a mismatch repair gene mutation and 16 (31%) with a homologous repair mutation. Germline autosomal dominant mutations were confirmed or suspected in 10 patients (20%). Activating mutations in the PI3K pathway (n = 19; 37%), WNT pathway (n = 16; 31%), and MAPK pathway (n = 8; 16%) were common.
CONCLUSION
This study strongly suggests that dPCs are enriched for actionable mutations, with approximately 50% of patients demonstrating DNA damage repair pathway alteration(s). Patients with dPC should be offered next-generation sequencing to guide standard-of-care treatment (eg, immune checkpoint inhibitors) or triaged toward an appropriate clinical trial (eg, poly [ADP-ribose] polymerase inhibitors).
INTRODUCTION
Ductal prostate cancer (dPC) is a rare prostate cancer variant characterized by large glands lined by tall, pseudostratified, columnar, neoplastic epithelial cells, typically arranged over fibrovascular cores or cribriform glands and associated with an aggressive clinical course.1-3 Outcomes for dPC generally mirror those of Gleason score 4 + 4 = 8 carcinomas, and tumors with at least 10% ductal morphology have been found to associate with a higher stage and suboptimal response to androgen deprivation.2,3 Overall, approximately 3% of all prostate cancers have some component of ductal histology.2,4
CONTEXT
Key Objective
The key objective was to provide an overview of the genomic alterations associated with ductal prostate cancers.
Knowledge Generated
Ductal prostate cancers are associated with a high incidence of mutations in DNA repair genes. Affected genes include those mediating homologous recombination and mismatch repair pathways.
Relevance
Because mutations in genes involved in DNA repair are highly actionable, sequencing all patients with ductal prostate cancer should be considered.
Although the histologic features of dPC are well described, there is relatively little information regarding the underlying molecular alterations associated with this prostate cancer subtype. Fluorescence in situ hybridization studies have found that TMPRSS2:ERG fusions are present in 10% to 50% of patients with dPC, and ERG protein expression (consistent with TMPRSS2:ERG fusions) is also present in this range.5-8 Limited gene expression profiling studies have found similarities between dPC and patients with acinar tumors, and there is molecular evidence that concurrent ductal and acinar tumors are clonally related.4,9,10 More recent immunohistochemical profiling studies have demonstrated that positive phospho–mammalian target of rapamycin staining correlated with risk of biochemical recurrence in patients with ductal carcinoma.11 In a separate study, it was found that loss of PTEN protein expression occurred more frequently in dPC compared with acinar adenocarcinoma, again, potentially implicating mammalian target of rapamycin signaling pathway in the pathobiology of dPC. However, these data remain controversial, because other studies have suggested a lower rate of PTEN protein loss in ductal carcinomas compared with Gleason score 8 acinar carcinomas.6 More recently, a study evaluating genomic and transcriptomic differences between foci of ductal and acinar prostatic carcinoma from the same individual found enrichment for mutations in CTNNB1 and PTEN within the ductal foci, with associated WNT- or PI3K-pathway activation.9
Given the rarity of dPC and the relative lack of information regarding the associated molecular features, we compiled a multi-institutional, international cohort of patients with dPC for targeted next-generation sequencing (NGS). We previously reported the NGS results from a small series characterizing patients with dPC at our institution (University of Washington [UW]).12 In that preliminary study, we observed a high rate of DNA damage repair (DDR) mutations, including loss-of-function mutations in mismatch repair (MMR) genes. Building from our initial case series, we now report sequencing results from an expanded multi-institutional collaborative cohort of 51 patients with dPC.
METHODS
Study Populations
We assembled three case series comprising 51 patients with dPC from institutions in the United States and Canada (Data Supplement). Histopathologic classification of all tumors was confirmed by an expert genitourinary pathologist at each institution. All tumor tissue was sequenced on the targeted NGS assay UW-OncoPlex according to previously published methods.12,13
Case series 1 consisted of prostate cancer specimens (radical prostatectomies and needle biopsies of prostate and metastatic tumors) from 22 men actively receiving treatment at the University of Washington/Seattle Cancer Care Alliance who were prospectively identified as having a diagnosis of dPC. Tissue used for sequencing was acquired between January 2015 and March 2017. Preliminary sequencing results from this series have been previously published.12 All men provided informed consent to have their tissue sequenced as part of this study.
Case series 2 included 21 radical prostatectomy samples. A subset was obtained from a tissue microarray composed of primary prostatectomy specimens from men with either dPC (n = 51) or Gleason pattern 4 acinar prostatic adenocarcinoma (n = 75) treated at Johns Hopkins Hospital (JHH) between 1984 and 2004. Details regarding this tissue microarray have been previously published.14 Additional patients with a ductal carcinoma in the dominant nodule were procured from consecutive radical prostatectomies performed at JHH. Case series 3 included archival tissue from eight men treated by transurethral resection of the prostate at the University of Calgary.
Blinded Morphologic Evaluation
To reevaluate the morphologic classification of all patients in a blinded manner, an expert genitourinary pathologist (J.I.E.) examined scanned digital images of a representative slide from each patient corresponding to the formalin-fixed paraffin-embedded (FFPE) block that was macrodissected for sequencing. Each patient was scored for percentage of the tumor that had ductal morphology overall, as well as the percentage of several described morphologic subtypes of ductal carcinoma: cribriform, papillary, gland-like, prostatic intraepithelial neoplasia–like and solid.
Macrodissection of Tumor Tissue
All tissue was previously FFPE. Hematoxylin and eosin–stained sections served as templates for either macrodissecting the dPC component from 10-micron sections or obtaining 5.0-×-0.6-mm punches from regions with the highest percentage of dPC.
Next-Generation Sequencing
For all patients, DNA was extracted from macrodissected FFPE samples and sequenced using the targeted NGS platform UW-OncoPlex, as that which interrogates approximately 1.8 Mb of DNA encompassing 262 genes. Briefly, genomic libraries were made from 500 ng of genomic DNA extracted from prostate tumor FFPE tissue and a custom Agilent (Santa Clara, CA) SureSelect XT capture set used for target enrichment and sequenced on an Illumina (San Diego, CA) NextSEquation 500 instrument with paired-end 101 bp reads. A custom bioinformatics pipeline detects single nucleotide variants, indels of all sizes, structural rearrangements, PMS2 pseudogene disambiguation, and copy number changes. Sequencing interpretation was performed by an expert molecular pathologist (C.C.P.). Reported alterations were limited to those deemed pathogenic or likely pathogenic (eg, loss of function mutations in tumor suppressors, activating mutations in genes involved in oncogenic signaling pathways).
All 262 genes in UW-OncoPlex were thoroughly reviewed for potential pathogenic germline mutations by an expert in clinical germline cancer predisposition testing (C.C.P.). All reported confirmed or suspected germline variants were carefully vetted by a team expert in variant classification comprising at least three individuals, per the usual clinical process. Of 18 suspected pathogenic germline mutations, 13 were confirmed in nontumor tissue. Of the five suspected germline mutations that did not have matched nontumor DNA, one additional patient was felt to represent a germline mutation after expert molecular pathologist review through cross-referencing against the ClinVar database and by multivariable analysis of the variant allele fraction in the context of tumor content, ploidy, and loss of heterozygosity status.15,16
RESULTS
Patient Characteristics
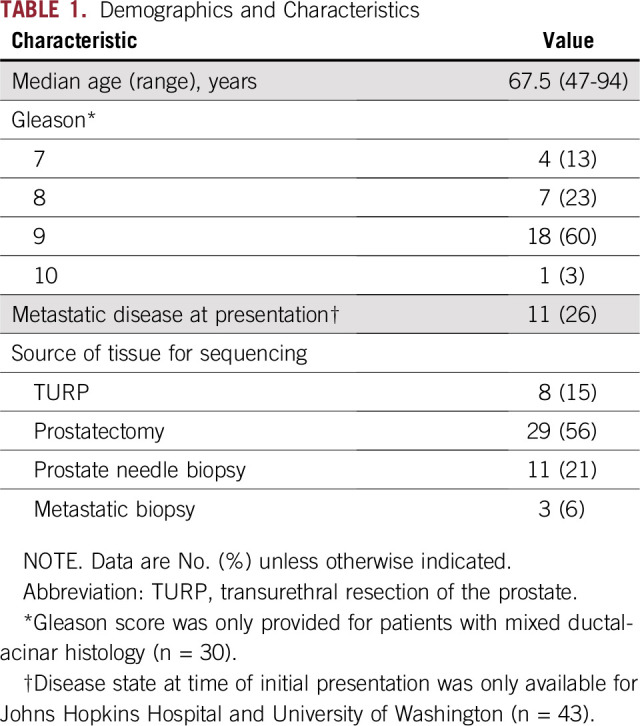
Across the three cohorts (Ntotal = 51), the median age at the time of diagnosis/tissue acquisition was 67.5 years. The majority of patients with data on tumor stage (23 of 40; 57.5%) had T3 or higher disease. For patients with clinical follow-up (UW and University of Calgary, n = 28), seven (25%) were deceased, and 12 (43%) had developed metastatic disease during long-term follow-up. Additional demographics details are listed in Table 1.
TABLE 1.
Demographics and Characteristics
dPC Genomics
Overall, our combined cohort of patients with dPC demonstrated a high number of recurrent genomic alterations (Fig 1; Data Supplement). These included alterations in genes involved in DDR repair (n = 24; 47%), PI3K pathway (n = 19; 37%), WNT-signaling pathway (n = 16; 31%), and MAPK signaling (n = 8; 16%). A large number of patients also had mutations in FOXA1 (n = 17; 33%), TP53 (n = 9; 18%), and SPOP (n = 6; 12%).
FIG 1.

Landscape of genomic alterations across 51 patients with ductal prostate cancer. Each column represents one patient. Pathogenic mutations were those predicted to either activate oncogenic signaling pathways (eg, WNT- or PI3K-signaling) or inactivate tumor suppressors (eg, DNA damage repair [DDR] genes, TP53). HR, homologous recombination; MMR, mismatch repair; VUS, variant of uncertain significance.
Recurrent DDR alterations.
Twenty-five (49%) of 51 patients had at least one alteration in a DDR pathway gene. Overall, seven of 51 patients (14%) had evidence of MMR alterations, six of whom had evidence of hypermutation (ie, ≥ 10 mutation per megabase), consistent with deficient MMR (one patient with monoallelic loss of MSH2 was not hypermutated). Three patients with MMR alterations also had concurrent secondary mutations in homologous recombination (HR) pathway genes. Sixteen patients (31%) had an HR mutation in the absence of a concurrent MMR mutation. An additional patient with a hotspot POLD1 mutation was ultramutated (ie, > 100 mutations per megabase).17,18 There were 10 patients (20%) with evidence of a pathogenic autosomal dominant germline alteration in a DDR gene, which is significantly higher than reported by The Cancer Genome Atlas for patients with primary prostate cancer (20% v 5%; P < .001) and numerically higher than unselected patients with metastatic prostate cancer (20% v 12%; P = .105).19,20 Of note, two individuals in our dPC cohort were carriers of recessive germline alterations in DDR pathway genes—one with an ERCC2 alteration and the other with an MUTYH alteration. Neither had evidence that the second allele was affected in tumor tissue. Compared with published genomic data from men with localized and castration-resistant prostate cancer (CRPC), our combined cohort of men with dPC was significantly enriched for mutations in DDR genes (both MMR and HR genes), as well as other genes of interest (Table 2).20,21
TABLE 2.
Recurrent Genomic Alterations in Patients With Ductal Prostate Cancer Compared With Men With Sporadic Localized and Castration-Resistant Prostate Cancer 22,23

Additional recurrent genomic alterations.
Similar to localized and metastatic CRPC, mutations in FOXA1 were frequently observed (n = 17; 33%).20,21 Interestingly, mutations in genes involved in WNT signaling were also frequent (n = 16; 31%). This includes activating/stabilizing mutations in CTNNB1 (n = 4), as well as inactivating mutations in APC (n = 12), a negative regulator of WNT pathway activation. Similar to a prior report, we found that PTEN alterations were generally mutually exclusive of mutations in genes associated with WNT-signaling activation (eg, APC and CTNNB1 mutations), although there was one patient with a pathogenic MSH2 mutation that had secondary alterations in APC, PTEN, and PIK3R1.9 There were three additional patients in whom WNT-pathway alterations co-occurred with PI3K-pathway alterations, including one patient with an APC and PIK3CA mutation, and two patients with CTNNB1 and PIK3CA mutations. Compared with patients with CRPC, there were fewer PTEN alterations and more PIK3CA mutations in our dPC cohort, with an overall similar incidence of PI3K-pathway alterations (Table 2). Alterations in AR were infrequent (n = 4; 8%); however, the majority of tissue samples sequenced were primary prostate tissue that had not been exposed to hormonal therapies. ETS fusions were significantly less common in our dPC cohort compared with patients with both localized and CRPC (Table 2).
Pathology correlates.
We have previously reported discordance between expert genitourinary pathologists in diagnosing dPC.24 As such, we performed a secondary pathologic review on available patients. Overall, 46 of the patients (94%) included in this analysis had slides available for central pathologic review (performed by J.I.E. at JHH). Of five ductal histologic subtypes (ie, cribriform, papillary, gland-like, prostatic intraepithelial neoplasia–like, and solid), cribriform (n = 22), and papillary (n = 23) were the most common, and 16 patients showed multiple histologic subtypes present. Although the quantity of dPC in each patient varied, there was no evidence that percentage of ductal involvement correlated with the underlying mutational profile, and a high frequency of DDR mutations was observed regardless of the overall quantity of dPC reported on secondary pathology review (Table 3).
TABLE 3.
Key Genomic Features Stratified by Ductal Involvement

DISCUSSION
To our knowledge, this is the largest cohort of dPC to be examined by NGS. We confirmed that dPCs are enriched for actionable mutations and, remarkably, found that almost half had at least one alteration in a DDR pathway gene. When compared with contemporary prostate cancer cohorts, we noted significant differences in the mutational profile of dPCs. For instance, in contrast to the Stand Up 2 Cancer–Prostate Cancer Foundation International Prostate Cancer Dream Team discovery set, which reported pathogenic DDR mutations in approximately 25% of unselected patients with metastatic CRPC, we found a significantly higher frequency of DDR alterations in dPCs, including loss-of-function mutations in MMR genes.21 In addition, because the vast majority of patients included in this analysis had primary tissue sequenced (48 of 51 patients), it is likely that DDR alterations are early, truncal events, which can be easily identified by sequencing archival tissue, negating the need to obtain fresh metastatic tissue in men with more advanced disease. Although we did not intentionally sequence the acinar carcinoma component in patients with mixed ductal-acinar tumors, the fact that concurrent ductal and acinar carcinomas share common ERG rearrangements and other alterations suggests that the DDR alterations are likely shared between these components as well.4,9 This study adds to the literature suggesting that aggressive histologic subtypes of localized prostate cancer (eg, primary Gleason pattern 5 acinar carcinomas, small cell carcinomas, and now dPCs) may be enriched for MMR defects.25
It is also notable that in addition to DDR alterations, there were a number of recurrently mutated genes, including those involved in WNT- and PI3K-signaling pathways. Interestingly, and consistent with prior work from our group, we found that PI3K-signaling alterations occurred more commonly in ductal carcinoma via PIK3CA mutations than by PTEN gene alterations, which is in contrast to unselected patients with metastatic CRPC.6,21 These findings are also consistent with a recent report examining genomic and transcriptomic differences between dPC and acinar prostate cancer foci from the same individual.9 However, in that report, it is worth noting that the authors did not observe enrichment for MMR alterations, and DDR alterations were relatively infrequent. Whether this is due to differences between the two series or the fact that their sample size was relatively small (10 patients) is not clear. We also confirmed that ETS gene rearrangements were significantly less common in patients with dPC compared with both primary (The Cancer Genome Atlas) and metastatic (Stand Up 2 Cancer–Prostate Cancer Foundation) patients with prostate cancer.4 Importantly, because the NGS panel used in this study (UW-OncoPlex) provides intronic gene coverage for rearrangement hotspot areas in TMPRSS2 and other recurrently rearranged genes, it has a higher degree of sensitivity for detecting ETS fusions and other complex genomic rearrangements that could be missed by panels that only sequence exonic regions.13,26 These findings indicate that alternative drivers may underlie dPC biology.
Another interesting observation was the apparent enrichment in patients with dPC for germline DDR gene alterations. A previous study had suggested that men with germline DDR mutations (particularly HR mutations) were more likely to harbor components of ductal or intraductal histology than those with pure acinar histology (48% v 12%; P < .01), although it is recognized that ductal and intraductal features are considered to be histologically distinct.27 In the current analysis, the prevalence of pathogenic or likely pathogenic germline DDR lesions was 20% (10 of 51), significantly higher than that observed in primary prostate cancers (20% v 5%; P < .001) or even in unselected sporadic metastatic prostate cancers (20% v 12%; P = .105).19,20 Taken together with the previous study, these findings suggest that patients with dPC should be preferentially offered germline genetic testing even in the absence of metastatic disease. Indeed, knowledge of germline status in the localized disease setting could afford opportunities for modifying management and improving outcomes before metastases develop.
There were important differences between the cohorts included in this analysis. The JHH and Calgary cohorts were assembled from archival tissue and were highly selected. In contrast, the UW cohort comprised prospectively identified men receiving care for prostate cancer in the clinic, and thus represented a real-world example of selecting patients for sequencing on the basis of histology. Another key difference is that the JHH cohort only included patients who underwent prostatectomy, and no long-term follow-up data were available. Given that all patients in the JHH cohort were considered to be prostatectomy candidates, this raises the possibility that these men had less aggressive disease, especially in light of the fact that 70% of men in the Calgary and UW cohorts either died or developed metastatic disease.
It is also notable that the UW cohort included a number of patients who were disputed in terms of whether dPC was present, and overall, 12 patients (26%) included in this series who underwent secondary pathology review were felt to not contain significant ductal features (n = 11 from UW and n = 1 from JHH). It is important to bear in mind, however, that all patients included in this analysis were believed to contain a component of ductal histology by at least one expert genitourinary pathologist. In addition, because slides from UW were not scanned before macrodissecting the ductal component, we cannot exclude the possibility that the slides sent for secondary review were not representative of the slides used for sequencing. In addition, the observed interpathologist variance is consistent with prior experience evaluating patients with dPC.28 Importantly, there were no clear differences in mutational profiles for patients felt to not possess a clear ductal component on secondary pathology review or between patients with pure versus mixed ductal histology (Table 3). Larger studies aimed at evaluating differences between patients with pure and mixed dPC and studies evaluating differences between ductal and acinar foci from the same patient are warranted but are beyond the scope of this study.
In conclusion, despite the heterogeneity of our cohort, these results indicate that patients with any fraction of dPC should be offered NGS, given that the presence of dPC histology can serve as a rapid means to select patients enriched for actionable mutations, particularly in MMR and HR genes. On the basis of this analysis, nearly half would qualify for treatment with investigational poly (ADP-ribose) polymerase inhibitors, platinum-based chemotherapy, and/or an immune checkpoint inhibitor.29-34
Footnotes
Supported by National Cancer Institute Grant No. P30 CA015704 and Pacific Northwest Prostate Cancer Grant No. SPORE CA097186. M.T.S. is supported by a Prostate Cancer Foundation Young Investigator Award and Department of Defense Award W81XWH-16-1-0484. C.C.P. is supported in part by the Prostate Cancer Foundation, a Department of Defense Prostate Cancer Research Program Award PC131820, and the Institute for Prostate Cancer Research. E.S.A. is partially supported by National Institutes of Health Grants No. R01 CA185297 and P30 CA006973 and Department of Defense Prostate Cancer Research Program Grant No. W81XWH-15-2-0050. A.C.H. is a V Foundation Scholar and is funded by a NextGen Grant for Transformative Cancer Research from the American Association for Cancer Research and a Burroughs Wellcome Fund Career Award for Medical Scientists.
AUTHOR CONTRIBUTIONS
Conception and design: Michael T. Schweizer, Emmanuel S. Antonarakis, Peter S. Nelson, Tamara L. Lotan, Colin C. Pritchard
Financial support: Michael T. Schweizer, Peter S. Nelson, Colin C. Pritchard
Administrative support: Michael T. Schweizer, Nola Klemfuss, Colin C. Pritchard
Provision of study materials or patients: Michael T. Schweizer, Emmanuel S. Antonarakis, Nola Klemfuss, Elahe A. Mostaghel, Peter S. Nelson, Evan Y. Yu, R. Bruce Montgomery, Tamara L. Lotan
Collection and assembly of data: Michael T. Schweizer, Tarek A. Bismar, Liana B. Guedes, Heather H. Cheng, Maria S. Tretiakova, Funda Vakar-Lopez, Nola Klemfuss, Eric Q. Konnick, Elahe A. Mostaghel, Andrew C. Hsieh, Evan Y. Yu, R. Bruce Montgomery, Lawrence D. True, Tamara L. Lotan, Colin C. Pritchard
Data analysis and interpretation: Michael T. Schweizer, Emmanuel S. Antonarakis, Liana B. Guedes, Heather H. Cheng, Eric Q. Konnick, Peter S. Nelson, Evan Y. Yu, R. Bruce Montgomery, Lawrence D. True, Jonathan I. Epstein, Tamara L. Lotan, Colin C. Pritchard
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Michael T. Schweizer
Consulting or Advisory Role: Janssen
Research Funding: Janssen (Inst), AstraZeneca (Inst), Roche (Inst), Pfizer (Inst), Zenith Epigenetics (Inst), Madison Vaccines (Inst)
Emmanuel S. Antonarakis
Honoraria: Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, Astellas Pharma, Merck, AstraZeneca, Clovis Oncology
Consulting or Advisory Role: Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, Astellas Pharma, Merck, AstraZeneca, Clovis Oncology
Research Funding: Janssen Biotech (Inst), Johnson & Johnson (Inst), Sanofi (Inst), Dendreon (Inst), Aragon Pharmaceuticals (Inst), Exelixis (Inst), Millennium (Inst), Genentech (Inst), Novartis (Inst), Astellas Pharma (Inst), Tokai Pharmaceuticals (Inst), Merck (Inst), AstraZeneca (Inst), Clovis Oncology (Inst), Constellation Pharmaceuticals (Inst)
Patents, Royalties, Other Intellectual Property: Co-inventor of a biomarker technology that has been licensed to Qiagen
Travel, Accommodations, Expenses: Sanofi, Dendreon, Medivation
Heather H. Cheng
Research Funding: Inovio Pharmaceuticals (Inst), Sanofi (Inst), Astellas Medivation (Inst), Janssen (Inst), Clovis Oncology (Inst), Color Foundation (Inst)
Eric Q. Konnick
Honoraria: Ventana Medical Systems
Travel, Accommodations, Expenses: Ventana Medical Systems
Elahe A. Mostaghel
Research Funding: Context Therapeutics (Inst)
Andrew C. Hsieh
Honoraria: Hotspot Therapeutics
Research Funding: eFFECTOR (Inst)
Patents, Royalties, Other Intellectual Property: MTOR modulators and uses thereof–patent number: 9629843, use of translational profiling to identify target molecules for therapeutic treatment–publication number: 20140288097
Peter S. Nelson
Consulting or Advisory Role: Janssen Oncology, Astellas Pharma, Genentech
Expert Testimony: Veneble-Fitzpatrick Law Firm
Travel, Accommodations, Expenses: Janssen Oncology
Evan Y. Yu
Consulting or Advisory Role: Janssen, Bayer, Merck, AstraZeneca, EMD Serono, Churchill Pharmaceuticals, Incyte, Amgen, Tolmar, QED, Dendreon, Seattle Genetics, Agensys (Inst), Astellas Pharma (Inst), Dendreon (Inst), Genentech (Inst), Bayer (Inst), Merck (Inst), Seattle Genetics (Inst)
R. Bruce Montgomery
Research Funding: ESSA, Medivation/Astellas, Janssen Oncology, Ferring, AstraZeneca
Lawrence D. True
Stock and Other Ownership Interests: Lightspeed Micro
Research Funding: Ventana Medical Systems
Patents, Royalties, Other Intellectual Property: Lens on an open-top lightsheet microscope
Jonathan I. Epstein
Consulting or Advisory Role: Dianon, PathAI, Knowerror
Tamara L. Lotan
Consulting or Advisory Role: Janssen Oncology
Research Funding: Ventana Medical Systems
No other potential conflicts of interest were reported.
REFERENCES
- 1. WHO: Classification of Tumours of the Urinary System and Male Genital Organs (ed 4). Geneva, Switzerland, World Health Organization, 2016. [Google Scholar]
- 2.Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012;60:59–74. doi: 10.1111/j.1365-2559.2011.04039.x. [DOI] [PubMed] [Google Scholar]
- 3.Brinker DA, Potter SR, Epstein JI. Ductal adenocarcinoma of the prostate diagnosed on needle biopsy: Correlation with clinical and radical prostatectomy findings and progression. Am J Surg Pathol. 1999;23:1471–1479. doi: 10.1097/00000478-199912000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Lotan TL, Toubaji A, Albadine R, et al. TMPRSS2-ERG gene fusions are infrequent in prostatic ductal adenocarcinomas. Mod Pathol. 2009;22:359–365. doi: 10.1038/modpathol.2008.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han B, Mehra R, Suleman K, et al. Characterization of ETS gene aberrations in select histologic variants of prostate carcinoma. Mod Pathol. 2009;22:1176–1185. doi: 10.1038/modpathol.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morais CL, Herawi M, Toubaji A, et al. PTEN loss and ERG protein expression are infrequent in prostatic ductal adenocarcinomas and concurrent acinar carcinomas. Prostate. 2015;75:1610–1619. doi: 10.1002/pros.23042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vinceneux A, Bruyere F, Haillot O, et al: Ductal adenocarcinoma of the prostate: Clinical and biological profiles. Profile 77:1242-1250, 2017. [DOI] [PubMed]
- 8.Chaux A, Albadine R, Toubaji A, et al. Immunohistochemistry for ERG expression as a surrogate for TMPRSS2-ERG fusion detection in prostatic adenocarcinomas. Am J Surg Pathol. 2011;35:1014–1020. doi: 10.1097/PAS.0b013e31821e8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gillard M, Lack J, Pontier A, et al: Integrative genomic analysis of coincident cancer foci implicates CTNNB1 and PTEN alterations in ductal prostate cancer. Eur Urology Focus 10.1016/j.euf.2017.12.003 [epub ahead of print on December 8, 2017] [DOI] [PMC free article] [PubMed]
- 10.Sanati S, Watson MA, Salavaggione AL, et al. Gene expression profiles of ductal versus acinar adenocarcinoma of the prostate. Mod Pathol. 2009;22:1273–1279. doi: 10.1038/modpathol.2009.103. [DOI] [PubMed] [Google Scholar]
- 11.Jeong SU, Kekatpure AK, Park JM, et al. Diverse immunoprofile of ductal adenocarcinoma of the prostate with an emphasis on the prognostic factors. J Pathol Transl Med. 2017;51:471–481. doi: 10.4132/jptm.2017.06.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schweizer MT, Cheng HH, Tretiakova MS, et al. Mismatch repair deficiency may be common in ductal adenocarcinoma of the prostate. Oncotarget. 2016;7:82504–82510. doi: 10.18632/oncotarget.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16:56–67. doi: 10.1016/j.jmoldx.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herawi M, Epstein JI. Immunohistochemical antibody cocktail staining (p63/HMWCK/AMACR) of ductal adenocarcinoma and Gleason pattern 4 cribriform and noncribriform acinar adenocarcinomas of the prostate. Am J Surg Pathol. 2007;31:889–894. doi: 10.1097/01.pas.0000213447.16526.7f. [DOI] [PubMed] [Google Scholar]
- 15.Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shirts BH, Casadei S, Jacobson AL, et al: Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet Med 18:974-981, 2016. [DOI] [PubMed]
- 17.Rayner E, van Gool IC, Palles C, et al. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 2016;16:71–81. doi: 10.1038/nrc.2015.12. [DOI] [PubMed] [Google Scholar]
- 18. Shlien A, Campbell BB, de Borja R, et al: Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nature Genet 47:257-262, 2015. [DOI] [PubMed]
- 19.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Cancer Genome Atlas Research Network The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson D, Van Allen EM, Wu YM. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. True L, Gulati R, Lange J, et al: Histological patterns of ductal adenocarcinoma of prostate correlates with mutations in DNA repair genes and may aid in selecting the type of systemic therapy for castration-resistant prostate carcinoma. Presented at the USCAP Annual Meeting. San Antonio, TX, March 4-10, 2017. [Google Scholar]
- 25.Guedes LB, Antonarakis ES, Schweizer MT, et al. MSH2 loss in primary prostate cancer. Clin Cancer Res. 2017;23:6863–6874. doi: 10.1158/1078-0432.CCR-17-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pritchard CC, Morrissey C, Kumar A, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun. 2014;5:4988. doi: 10.1038/ncomms5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Isaacsson Velho P, Silberstein JL, Markowski MC, et al: Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate 78:401-407, 2018. [DOI] [PMC free article] [PubMed]
- 28.Seipel AH, Delahunt B, Samaratunga H, et al. Diagnostic criteria for ductal adenocarcinoma of the prostate: Interobserver variability among 20 expert uropathologists. Histopathology. 2014;65:216–227. doi: 10.1111/his.12382. [DOI] [PubMed] [Google Scholar]
- 29.Cheng HH, Pritchard CC, Boyd T, et al. Biallelic inactivation of BRCA2 in platinum-sensitive metastatic castration-resistant prostate cancer. Eur Urol. 2016;69:992–995. doi: 10.1016/j.eururo.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20:764–775. doi: 10.1158/1078-0432.CCR-13-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. US Food and Drug Administration: FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication, https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm560040.htm.
- 33.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]