

Abstract
We have previously designed and synthesized small-molecule inhibitors that reduce Vibrio cholerae virulence in vitro by targeting the transcription factor ToxT. Here we report the synthesis and biological activity of derivatives of our previous bicyclic, fatty acid-like inhibitors. All of the synthesized derivatives show anti-virulence activity in vitro. For the most potent compounds, a concentration of 5 μM completely inhibited ToxT-mediated tcpA expression as measured in the β-galactosidase assay. One indole compound, 3-(1-butyl-1H-indol-7-yl)propanoic acid (8), was also effective at inhibiting intestinal colonization in the infant mouse. These modified compounds may serve as good candidates for further anti-cholera drug development.
INTRODUCTION
The cholera bacterium Vibrio cholerae is a gram-negative bacterium that naturally inhabits aquatic environments such as brackish water and estuaries and can contaminate drinking water reservoirs. In order to cause disease, V. cholerae must produce two primary virulence factors, the toxin co-regulated pilus (TCP) and cholera toxin (CT), which are responsible for intestinal colonization and diarrheal symptoms, respectively.1,2 These virulence factors are regulated at the transcriptional level by a coordinated network of proteins that respond to specific environmental signals.3 Among these proteins is ToxT, an AraC family regulator which binds the tcpA and ctx promoters and directly activates virulence gene expression.4–7 ToxT plays an essential role in V. cholerae virulence, and its expression and activity are highly regulated among epidemic V. cholerae strains.8 As a result, ToxT is an obvious target for potential cholera therapeutics.
The X-ray structure of ToxT revealed the presence of cis-palmitoleic acid at the interface between the N-terminal and C-terminal domains of ToxT.9 This unsaturated fatty acid (UFA) was more than just a crystallographic ligand, however, as it inhibited ToxT transcriptional activity in vitro.9 Regulation of ToxT by UFAs likely originates in the context of the host environment. By binding ToxT and downregulating virulence gene expression, bile and UFAs present in the intestinal lumen prevent virulence activation until V. cholerae has reached the intestinal crypts, the appropriate location for colonization and delivery of cholera toxin.10 Based on the ToxT-bound conformation of cis-palmitoleic acid, we recently designed and synthesized a number of compounds that significantly reduce V. cholerae infection in vitro.11 These compounds, which we will identify by a methyl group at the 8 position of 1,8-disubstituted naphthalene (Figure 1), reduce virulence gene expression by binding ToxT and inhibiting ToxT-DNA binding interactions. These ‘8-methyl’ inhibitors were comparatively more potent than virstatin (Figure 1), a small-molecule therapeutic identified via high throughput screening,12 demonstrating the benefits of rational structure-based drug design. Virstatin presumably inhibits ToxT dimerization, a required activity for activating virulence gene expression at the ctx promoter.13 The crystal structures of two ‘8-methyl’ compounds in complex with ToxT revealed additional opportunities for structural modifications of the small molecules in order to further improve their inhibitory potential.11 Thus, we now report the design, synthesis, and biological characterization of analogues of our previous ‘8-methyl’ inhibitors.
Figure 1.
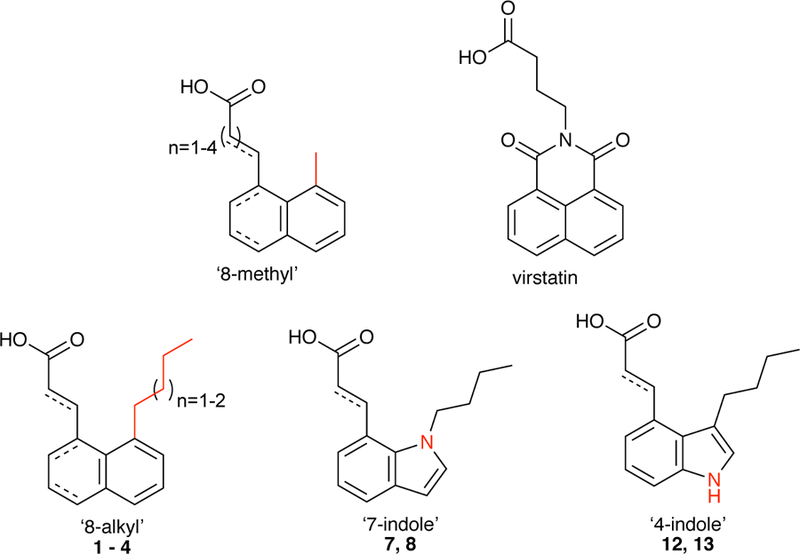
General structures of the ‘8-methyl’ compounds, virstatin, and newly synthesized derivatives.
The new disubstituted naphthalene and indole derivatives (Figure 1) are prepared using similar chemistry as we used for their 8-methyl-1-substituted naphthalene precursors, confirming that our synthetic design is amenable to additional manipulation. Furthermore, only a handful of reactions is necessary to achieve a variety of desired target molecules; this synthetic simplicity and robustness is an important criterion in drug design. More importantly, the second-generation derivatives are at least as effective as the previous ‘8-methyl’ compounds. Our newest lead compound 8 not only exhibits significantly improved activity in vitro, but protects against V. cholerae colonization in the infant mouse. Additionally, we explore the mechanism of ToxT inhibition by synthetically modifying the carboxylate moiety of one compound and by altering a critical lysine residue in ToxT, key factors involved in ToxT-inhibitor binding.
MATERIALS AND METHODS
Bacterial strains and media
Classical (O395) and El Tor (C6706) strains of V. cholerae are both virulent in humans. C6706 was used for the infant mouse colonization assay, as El Tor strains are more prevalent and more dominant in the current setting, and thus more relevant to in vivo colonization. Additionally, our methods were based on the C6706 inoculations reported by Hung et al. However, ToxT is more strongly expressed in O395 strains in vitro. Because we were monitoring a ToxT-mediated effect caused by the small-molecule inhibitors, O395 strains were used for the β-galactosidase assays.
All strains and plasmids used in this study are listed in Table 1. Strains were maintained at −80 °C in Luria-Bertani (LB) broth containing 30% (v/v) glycerol. All strains were grown in LB at 30 °C; cultures were aerated by rotating culture tubes on a roller drum. Antibiotics and additives were used in LB/agar plates at the following concentrations: kanamycin (Km), 45 μg/ml; gentamycin (Gm), 30 μg/ml; streptomycin (Sm), 100 μg/ml (for determining CFUs) or 1 mg/ml (for allelic exchange); 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), 40 μg/ml.
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Characteristic(s) | Reference |
|---|---|---|
| E. coli strains | ||
| S17–1 λpir | Tpr Smr recA, thi, pro, hsdR−M+ [RP4–2-Tc::Mu:Kmr Tn7] (λpir) | 14 |
| V. cholerae strains | ||
| AW40 | MBN142 toxT K230A | This work |
| AW46 | MBN017 toxT K230A | This work |
| C6706 str2 | El Tor Inaba, Smr | Laboratory collection |
| KSK1184 | C6706 str2 ΔtoxT | 15 |
| MBN017 | O395 Smr ΔtoxT | 16 |
| MBN135 | O395 Smr ΔlacZ tcpA-lacZ | 17 |
| MBN142 | MBN135 ΔtoxT | 17 |
| O395 Sm | Classical Ogawa, Smr | 1 |
| Plasmids | ||
| pAKW8 | pKAS154 tcpF-toxT K230A-tcpJ | This work |
| pKAS154 | pKAS32 derivative, Kmr | 18 |
| pMIN1 | pACYC184 Gm cassette, Gmr | 17 |
Strain and plasmid construction
The O395 tcpA-lacZ fusion strain carrying toxT mutant K230A was constructed using a gBlock Gene Fragment synthesized by IDT. The gBlock contained toxT K230A and 484 base pairs (bp) of the downstream gene tcpJ. The gBlock was amplified using primers ToxT F Sap and TcpJ R Not (Table S1). In addition, 450 bp of the upstream gene tcpF was amplified from O395 Sm using primers TcpF F Xba and ToxT R Sap (Table S1). The PCR fragments were digested with appropriate restriction enzymes and ligated into pKAS154,18 which had been pre-cut with XbaI and NotI enzymes. The resulting ligation, pAKW8, was electroporated into E. coli S17–1 λpir.14 Kmr colonies were screened for the insert via PCR. The mutant toxT gene was then introduced into V. cholerae strain MBN142 by allelic exchange,19 generating strain AW40. Strain MBN142 carries plasmid pMIN1, which confers Gmr as a counter-selection for conjugation.17 The ToxT K230A mutant was confirmed by DNA sequencing.
The O395 strain carrying toxT K230A was constructed for TcpA immunoblotting (Figure S1) via allelic exchange. Plasmid pAKW8 in E. coli S17–1 λpir was introduced into MBN017 (carrying plasmid pMIN1) to generate strain AW46. The ToxT K230A mutant was confirmed by DNA sequencing.
β-galactosidase assays
Strains were grown in LB pH 6.5 with aeration for 14 hours. Small-molecule inhibitors were added at the time cultures were inoculated. Cultures were diluted 1:10 in fresh LB 6.5 and β-galactosidase activity was determined by the method of Miller.20 Results are the average of three or more independent (biological) experiments, each performed in duplicate (2 technical replicates for each sample).
Infant mouse colonization assay
Mouse colonization assays were performed in accordance with Dartmouth College’s Institutional Animal Care and Use Committee (IACUC). Three-to four-day-old CD1 mice (Charles River) were orogastrically inoculated with ~105 CFU bacteria (50 μl of V. cholerae C6706 cultures that had been grown overnight then diluted 1:1,000 in fresh LB). Mice had been removed from their mothers and mixed with non-littermates 3 hours prior to inoculation. For mice receiving the wild-type C6706 strain, DMSO or the compound was added to the inoculum. Serial dilutions of the inocula were plated onto LB agar supplemented with 100 μg/ml Sm to determine input CFU. Infected mice were incubated at 30 °C for 16–18 h, then sacrificed and dissected. Small intestines were removed and homogenized with a Tissue Tearor in 4 ml sterile LB media containing 10% glycerol. Intestinal homogenates were serially diluted and plated onto LB agar/Sm as before to enumerate the CFUs recovered. The colonization index (CI) was reported as the ratio of output to input CFUs. Experiment was repeated.
In vitro cytotoxicity assay
Samples were prepared identically to mice inocula: compounds were added to cultures of V. cholerae C6706 that had been grown overnight and diluted. Cultures were then grown at 30 °C. Samples taken after one hour and overnight growth were serially diluted and plated. The colony forming units (CFUs) of cultures grown in the presence of compounds were compared to that of the wild-type culture. Compounds were tested at various concentrations until in vitro growth was not at all attenuated.
Western blot
V. cholerae strains O395 Sm, O395 Sm ΔtoxT (MBN017), and O395 Sm toxT K230A (AW46) were grown in LB pH 6.5 with aeration for 14 hours at 30 °C. Whole-cell extracts were assayed for total protein concentration using the Pierce BCA Protein Assay Kit (Thermo Scientific). Samples were subjected to SDS-PAGE on 16% Tris-Glycine gels (Invitrogen) and transferred to a nitrocellulose membrane using iBlot (Invitrogen). The membrane was blocked with 3% bovine serum albumin in TBST (1x Tris-buffered saline, 0.1% Tween), incubated with anti-TcpA antibody, and washed in TBST. After incubation with horseradish peroxidase-conjugated secondary antibody (Bio-Rad), the membrane was washed in TBS. Blots were visualized using the Pierce ECL detection system (Thermo Scientific) according to the manufacturer’s protocols.
RESULTS AND DISCUSSION
Synthesis and biological screening of derivatives
The crystal structures of the ‘8-methyl’ compounds in complex with ToxT revealed significant unoccupied space above the methyl group of the compounds (Figure 2).11 We hypothesized that the ToxT-effector binding pocket could easily accommodate an extended alkyl chain at the 8 position, and that increasing the aliphatic chain would exclude water from the primarily hydrophobic pocket. This is also consistent with the original crystal structure of ToxT, which accommodated a 16-carbon monounsaturated fatty acid and showed sufficient space for an 18-carbon UFA.9 We thus used the crystal structure to design the ‘8-alkyl’ derivatives.
Figure 2.
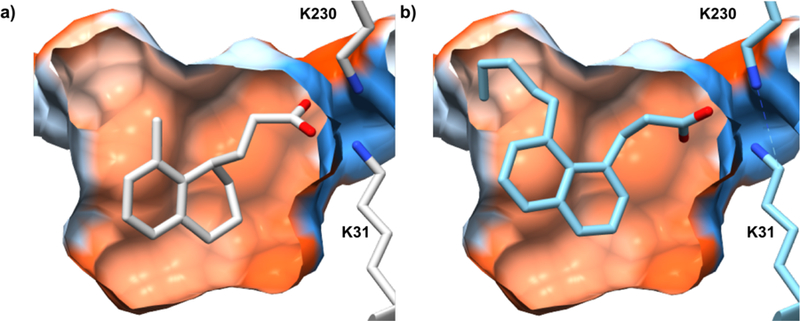
A cross-section of the ToxT inhibitor-binding pocket containing an ‘8-methyl’ compound (from the crystal structure, PDB code 5SUW) (a) and an ‘8-alkyl’ derivative (predicted by AutoDock21) (b). The surface of the pocket is colored by amino acid hydrophobicity: most hydrophilic (blue), neutral (white), most hydrophobic (orange red). Pocket calculated by CASTp.22
For 1,8-disubstituted naphthalenes 1 – 4, we employed the synthetic procedures used previously.11,23 Variation of the alkyl group at the 8 position of 8-methyl-1-substituted naphthalene was obtained by de novo synthesis from a suitable 2-alkylfuran; Diels-Alder cycloaddition using 2-n-butyl- and 2-n-pentylfurans in place of 2-methylfuran gave the corresponding epoxides. The reactions favored the formation of the syn-cycloadduct, the regioselectively major product, consistent with previous findings.24 Diimide reduction and subsequent acid-catalyzed dehydration quantitatively gave the bromonaphthalene ‘anchors’ for Heck coupling. Catalytic hydrogenation and ester hydrolysis gave the desired final ‘8-alkyl’ products.
The biological activity of compounds 1 – 4 was screened using the β-galactosidase assay.20 Cultures of O395 V. cholerae strains harboring chromosomal tcpA-lacZ fusions were grown in the presence of the synthesized compounds and assayed as described previously.11 Expression of tcpA was markedly decreased when treated with the ‘8-alkyl’ compounds (Figure 3). In fact, all four compounds inhibited tcpA to a greater extent than virstatin and all ten ‘8-methyl’ inhibitors.
Figure 3.
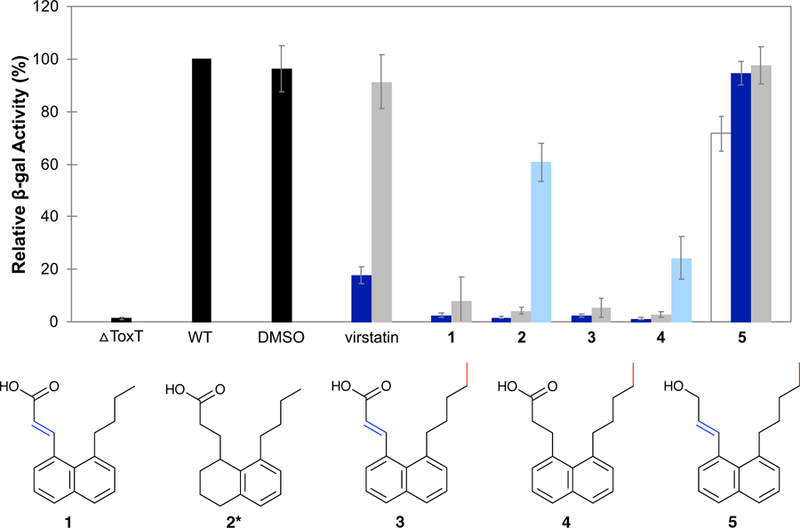
Effect of the ‘8-alkyl’ derivatives on tcpA-lacZ expression. Activity of the untreated wild-type strain (WT) is set to 100%.Relative β-gal activities of the negative control (△ToxT) and solvent control (DMSO) are calculated as a percentage of WT; samples containing inhibitor are calculated relative to DMSO. Final concentrations of inhibitor are 50 µM (white), 5 µM (dark blue), 0.5 µM (grey), and 0.05 µM (light blue). Error bars represent standard deviation where n is 3. *Compound 2 is a mixture of tetralins (see supplementary material).
As illustrated in Figure 2, the anionic carboxylate of the compound forms electrostatic interactions with the side chains of two lysine residues of ToxT. In order to support our hypothesis that the carboxylate moiety is required for the compounds’ inhibitory activity, we reduced the carboxylic acid of 3 to an alcohol using lithium aluminum hydride and tested the pure product as before. The resulting alcohol 5 is essentially inactive at 5 µM and shows minimal inhibitory activity even with a 10-fold increase in concentration (Figure 3). The reduced head group (of 5) must be responsible for this loss in activity, as the remainder of the molecule is identical to its acid predecessor 3. Although expected, this result nonetheless confirms our original assumption that the carboxylate is necessary for binding and inhibiting ToxT.
A second group of derivatives – the indole-based mimics – were designed with drug metabolism in mind. The indole moiety is ubiquitous in biology, and an indole-containing inhibitor may have advantages in vivo, such as reduced toxicity of its degradation products. Utilization of the indole moiety in place of the naphthalene may not necessarily improve the compounds’ ability to inhibit ToxT, but may nevertheless result in a more effective drug. Additionally, the free indole NH in the 4-indole derivatives serves as a potential handle for further substitution and provides a hydrogen-bond donor. Functionalization at this NH may provide opportunities to increase solubility.
Synthesis of the 7-indole derivatives is outlined in Scheme 1. Heck coupling of the commercially available 7-bromoindole and methyl acrylate, following the procedure employed previously,11,25 gave 6. Compound 7 was synthesized from 6 in two steps since alkylation of 626 was always accompanied by partial hydrolysis. While the acid and ester alkylation products were separable, we found it convenient to convert the ester to the desired acid instead. The crude mixture from step one was subjected to saponification (step two) to yield 7, which was reduced to 8.
Scheme 1.
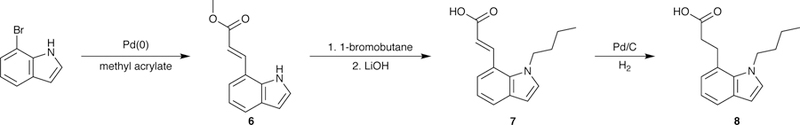
Synthesis of 7-indole derivatives.
Synthesis of the 4-indole derivatives was achieved starting from commercially available 4-bromoindole (Scheme 2). Acylation of the starting material26 followed by reduction of the ketone 9 provided 10. Heck coupling of 10 and methyl acrylate gave the disubstituted analogue 11, which was subjected to Pd-catalyzed hydrogenation and ester hydrolysis11 to yield final products 12 and 13. To facilitate a more facile purification, 13 was made from its corresponding ester (the reduced product of 11) rather than directly from 12.
Scheme 2.
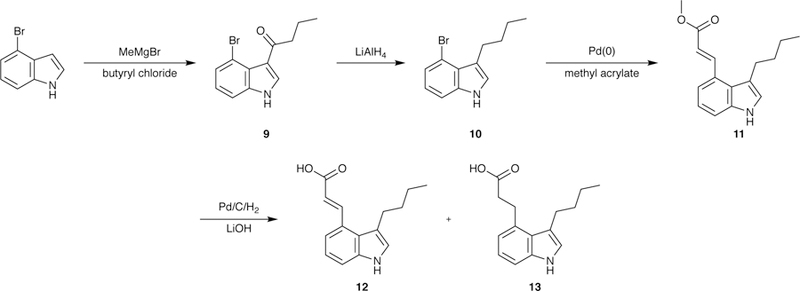
Synthesis of 4-indole derivatives.
The expression of tcpA was decreased significantly in the presence of 5 µM 7, 8, and 13 (Figure 4). Compound 12 did not inhibit tcpA transcription at the concentrations tested, which we believe were too low for activity. Given the effectiveness of compound 13 and its similarity to 12, we favor this explanation over the argument that 12 is unable to bind ToxT. In fact, compound 12 was moderately effective at higher concentrations (50 µM), reducing tcpA transcript levels to 60%.
Figure 4.
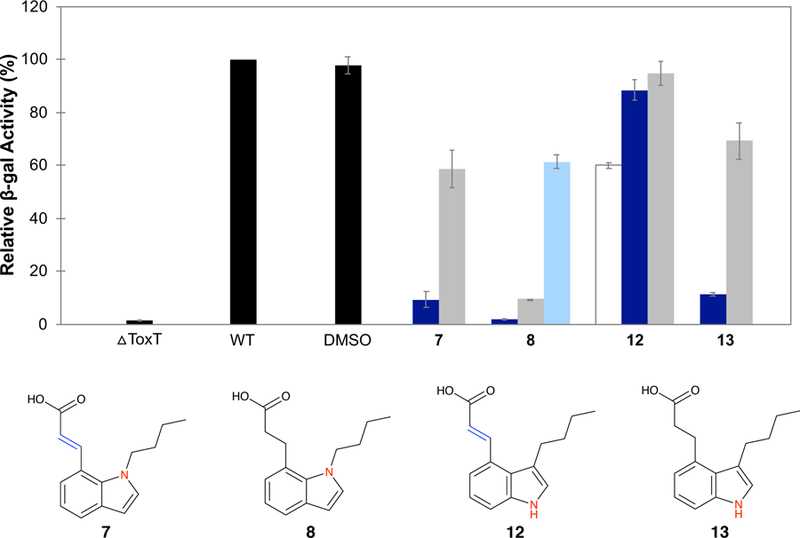
Effect of the indole derivatives on tcpA-lacZ expression. Relative β-gal activity is calculated as in Figure 3. Final concentrations of inhibitor are 50 µM (white), 5 µM (dark blue), 0.5 µM (grey), and 0.05 µM (light blue). Error bars represent standard deviation where n is 3–4.
As a whole, the introduction of the indole motif was less effective in improving the inhibitory activity of the molecules in vitro compared to the extension of the alkyl chain. If the improvement of the ‘8-alkyl’ derivatives over the ‘8-methyl’ precursors is due to the increased number of carbons, however, any benefit provided by the presence of the nitrogen in the indole may be counteracted by the smaller indole ring (C10 for naphthalene vs. C8 for indole). More detailed analyses are required for insights into the contributions of the presence of the N/NH, the position of the nitrogen (the 4-vs. the 7-postion), and the increase in the angle between the alkyl chain and the indole (which is more splayed than the alkyl chain-naphthalene angle).
In vivo activity of virstatin and compounds 4 and 8
We next tested the effectiveness of the new ToxT inhibitors to protect against V. cholerae infection in the suckling mouse. We used a prior study identifying virstatin as an effective ToxT inhibitor in vivo as a starting point for our experiment12 and chose compounds 4 and 8 to test alongside virstatin. In order to test our inhibitors at the high concentrations suggested in the previous study (upwards of 50 mM, three orders of magnitude higher than we have tested in the β-gal assays), we first measured cytotoxicity in vitro. Samples were prepared identically to those used for inoculating mice: untreated V. cholerae strains were grown overnight in non-inducing conditions, cultures were diluted to the appropriate CFU, compounds were introduced, and samples were cultured overnight. Not only was a high concentration of 8 lethal to V. cholerae, but virstatin reduced growth by 6 logs (Table 2). Higher concentrations of virstatin and compound 4 were also lethal to O395 cultures either immediately or after a few hours of growth (data not shown). Based on these results, it is unclear how additional 17.65 mM doses of virstatin (50 µg in 10 µl) administered post-inoculation could be tolerated by V. cholerae,12 when in vitro growth is severely attenuated at 1 mM (Table 2). Furthermore, the concentration of virstatin used previously was higher than the minimal bactericidal concentration determined for V. cholerae strain C6706.12 The data in Table 2 suggests that the decrease in in vivo colonization in the presence of virstatin measured previously may be an artificial effect caused by virstatin’s bactericidal effects on V. cholerae in the inoculum.
Table 2.
CFUs of in vitro cultures grown for 1 hour and overnight in the presence of inhibitors.
| 1 hour | overnight | |
|---|---|---|
| DMSO | 3.11 × 107 | 1.45 × 1010 |
| 1 mM virstatin | 3.48 × 107 | 2.00 × 104 |
| 0.1 mM virstatin | 2.83 × 107 | 2.37 × 1010 |
| 1 mM compound 4 | 4.43 × 107 | 1.21 × 109 |
| 0.1 mM compound 4 | 2.80 × 107 | 2.98 × 1010 |
| 10 mM compound 8 | 0 | 0 |
| 1 mM compound 8 | 5.98 × 107 | 1.77 × 1010 |
Because of the above considerations, virstatin, 4, and 8 were tested in vivo at concentrations that did not attenuate V. cholerae growth in vitro (0.1 mM for virstatin and 4; 1 mM for 8; see Table 2) (Figure 5). Mice were not given an additional “boost” of drug post-infection, as we were cautious to stay below lethal limits, and also wanted to avoid any deleterious effects caused by additional handling and successive oral gavages. As shown in Figure 5, mice treated with compound 8 exhibited a significant decrease in intestinal colonization by V. cholerae C6706, whereas no measurable effect was observed for compound 4. Colonization was slightly inhibited for virstatin-treated mice, but the difference was not statistically significant (p=0.1433 by Tukey-Kramer HSD means comparison). Notably, 8, an indole derivative designed to more closely resemble bioactive natural products and therefore possess greater therapeutic potential, was indeed less toxic to V. cholerae than 4 and virstatin. This allowed for administration of a higher dose, which was effective at inhibiting ToxT in vivo.
Figure 5.
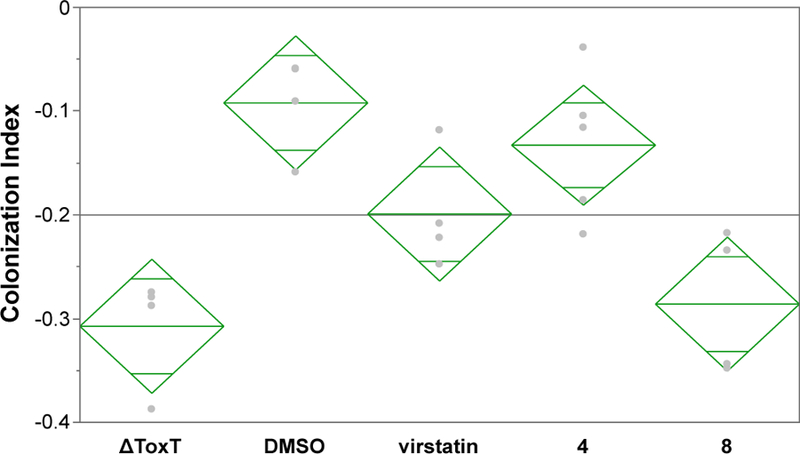
Effects of inhibitors on intestinal colonization. Mice were given the negative control strain (△ToxT), the wild-type strain containing a solvent control (DMSO), or the wild-type strain containing an inhibitor (0.1 mM virstatin, 0.1 mM compound 4, or 1 mM compound 8). A Box-Cox transformation of the colonization index raw data (output/input CFUs) was performed to normalize the data distribution (Figure S2). Pairwise means comparison using Tukey-Kramer HSD indicates statistically significant differences between 2 pairs: DMSO/△ToxT (p=0.0011) and DMSO/8 (p=0.0029). Note: A decrease in CI for compound 8 was also statistically significant when the untransformed (raw) data was analyzed using the nonparametric Kruskal-Wallis test (Figure S2).
Activity of ToxT K230A mutant
We further explored the interactions between the small-molecule inhibitors and ToxT by modifying ToxT. The most obvious residues for mutagenesis were Lys31 and Lys230, which interact with the carboxylate head of the inhibitor.9,11 Mutations were introduced into a tcpA-lacZ strain via allelic exchange.19 As shown in Figure 6, the K230A mutation caused a ~20% decrease in tcpA expression but had essentially no effect on the response of ToxT to its inhibitors. Surprisingly, virstatin and compound 8 inhibited tcpA expression in the K230A background, while the alcohol 5 had no effect, similar to the findings for the WT ToxT background. Oleic acid, a known ToxT inhibitor,9 also inhibited tcpA expression in the mutant background (Figure 6).
Figure 6.
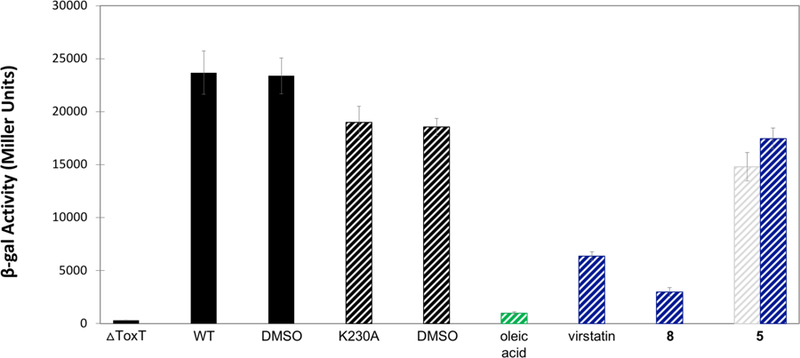
Influence of a toxT mutation on the expression of tcpA-lacZ in the presence or absence of inhibitors. β-gal activity was assayed in the wild-type toxT background (solid) or the toxT K230A background (striped). Samples were grown alone (WT and K230A), with a solvent control (DMSO), and in the presence of various inhibitors. Final concentrations of inhibitor are 708 µM (green), 50 µM (white/grey), and 5 µM (blue). Error bars represent standard deviation where n is 3. See Figure S1 for western blot.
We could not successfully obtain the K31A mutant or the K31A/K230A double mutant, however; V. cholerae would acquire the mutant alleles under antibiotic pressure, but strains containing toxT K31A were not recoverable under the conditions used for growth. It is possible that V. cholerae selects against toxT K31A and K31A/K230A by consistently excising the mutant copy along with the plasmid. An alternative explanation is that allelic exchange was successful, but cells containing the mutant toxT were non-viable and no colonies containing toxT K31A or K31A/K230A could be observed. While there is precedence for functional ToxT K31A, the previous construct was MBP-toxT K31A expressed from a plasmid.27
CONCLUSIONS
Structural modifications to our previously identified ToxT inhibitors were conducted in order to improve antivirulence efficacy and drug-like properties. Substituting an indole ring for naphthalene decreased cytotoxicity, and one indole derivative significantly reduced V. cholerae colonization in vivo. We further explored protein-ligand binding interactions by modifying the carboxylate head group of the compound and a key residue in the effector-binding pocket of ToxT. Compound 5, the alcohol derivative, showed no inhibitory activity in vitro, confirming the carboxylate moiety is essential for inhibiting ToxT. Therefore, it seems logical that amino acids interacting with the carboxylate – lysines 31 and 230 – are also critical for ToxT-inhibitor binding. Surprisingly, mutating lysine 230 had no effect; ToxT K230A behaved like wild-type in the presence of inhibitors. This suggests that, if K230 is not required for inhibition, K31 must be important. We were unable to confirm the importance of lysine 31, however, as we could not produce a V. cholerae strain carrying toxT K31A. One explanation for this result is that the K31A mutant renders ToxT insoluble and prone to aggregation, causing impaired cell growth and/or toxicity. Alternatively, V. cholerae may exert strong negative selection on toxT K31A if the mutant reduces fitness or is otherwise deleterious. Regardless of the explanation, it seems that lysine 31 must be conserved. Given our hypothesis that this residue is critical for inhibitor binding, it is therefore unlikely that V. cholerae will develop resistance to our compounds. Finally, the finding that K230 is not required for ToxT activity highlights the importance of future studies aimed at refining the structural model for ToxT activation/inhibition, which thus far has involved both lysine residues.9,11
Supplementary Material
Acknowledgements
We thank Karen Skorupski for providing laboratory resources for this study, as well as feedback on the manuscript. We gratefully acknowledge Carol S. Ringelberg of the Genomics and Molecular Biology Shared Resources (GMBSR), Geisel School of Medicine, for statistical analysis.
Funding
Research was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under award number F31AI124549 (to A.K.W.). This work was also supported by Donors of the Petroleum Research Fund administered by the American Chemical Society (to G.W.G.), and NIH grants AI072661 (to F.J.K.), AI025096 (to Karen Skorupski in the Department of Microbiology and Immunology, Geisel School of Medicine), and AI120068 (to Karen Skorupski and F.J.K.).
A patent based on molecules included in this manuscript has been awarded to AKW, EOO, FJK, and GWG.
Footnotes
ASSOCIATED CONTENT
Supporting Information
TcpA western blot (Figure S1); Oligonucleotides used in this study (Table S1); Colonization Index (CI) data (Table S2); Histograms and boxplot of the CI data (Figure S2); Experimental methods for synthesis of compounds; Experimental spectra of synthesized compounds (PDF)
References
- (1).Taylor RK, Miller VL, Furlong DB, and Mekalanos JJ (1987) Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proceedings of the National Academy of Sciences 84, 2833–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sánchez J, and Holmgren J (2008) Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cellular and Molecular Life Sciences 65, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Matson JS, Withey JH, and DiRita VJ (2007) Regulatory networks controlling Vibrio cholerae virulence gene expression. Infection and Immunity 75, 5542–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Higgins DE, Nazareno E, and DiRita VJ (1992) The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J. Bacteriol 174, 6974–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Champion GA, Neely MN, Brennan MA, and DiRita VJ (1997) A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Molecular Microbiology 23, 323–331. [DOI] [PubMed] [Google Scholar]
- (6).DiRita VJ, Parsot C, Jander G, and Mekalanos JJ (1991) Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A 88, 5403–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ogierman MA, and Manning PA (1992) Homology of TcpN, a putative regulatory protein of Vibrio cholerae, to the AraC family of transcriptional activators. Gene 116, 93–97. [DOI] [PubMed] [Google Scholar]
- (8).Childers BM, and Klose KE (2007) Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiology 2, 335–344. [DOI] [PubMed] [Google Scholar]
- (9).Lowden MJ, Skorupski K, Pellegrini M, Chiorazzo MG, Taylor RK, and Kull FJ (2010) Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proceedings of the National Academy of Sciences 107, 2860–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schuhmacher DA, and Klose KE (1999) Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J. Bacteriol 181, 1508–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Woodbrey AK, Onyango EO, Pellegrini M, Kovacikova G, Taylor RK, Gribble GW, and Kull FJ (2017) A new class of inhibitors of the AraC family virulence regulator Vibrio cholerae ToxT. Scientific Reports 7, 45011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hung DT, Shakhnovich EA, Pierson E, and Mekalanos JJ (2005) Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310, 670–674. [DOI] [PubMed] [Google Scholar]
- (13).Shakhnovich EA, Hung DT, Pierson E, Lee K, and Mekalanos JJ (2007) Virstatin inhibits dimerization of the transcriptional activator ToxT. Proceedings of the National Academy of Sciences 104, 2372–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).de Lorenzo V, and Timmis KN (1994) [31] Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5-and Tn10-derived minitransposons, in Methods in Enzymology, pp 386–405. Elsevier. [DOI] [PubMed] [Google Scholar]
- (15).Kovacikova G, Lin W, Taylor RK, and Skorupski K (2017) The fatty acid regulator FadR influences the expression of the virulence cascade in the El Tor biotype of Vibrio cholerae by modulating the levels of ToxT via two different mechanisms. Journal of Bacteriology (DiRita VJ, Ed.) 199, e00762–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Stonehouse EA (2000, October 15) Virulence and motility gene regulation in Vibrio cholerae PhD thesis, Dartmouth College. [Google Scholar]
- (17).Nye MB, Pfau JD, Skorupski K, and Taylor RK (2000) Vibrio cholerae H-NS Silences virulence gene expression at multiple steps in the ToxR regulatory cascade. Journal of Bacteriology 182, 4295–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kovacikova G, and Skorupski K (2002) Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter: Vibrio cholerae quorum sensing. Molecular Microbiology 46, 1135–1147. [DOI] [PubMed] [Google Scholar]
- (19).Skorupski K, and Taylor RK (1996) Positive selection vectors for allelic exchange. Gene 169, 47–52. [DOI] [PubMed] [Google Scholar]
- (20).Miller JH (1972) Experiments in Molecular Genetics Cold Spring Harbor Laboratory, Cold Spring Harbor, New York. [Google Scholar]
- (21).Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, and Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, and Liang J (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Research 34, W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Onyango EO, Kelley AR, Qian DC, and Gribble GW (2015) Syntheses of 1-Bromo-8-methylnaphthalene and 1-Bromo-5-methylnaphthalene. The Journal of Organic Chemistry 80, 5970–5972. [DOI] [PubMed] [Google Scholar]
- (24).Gribble GW, Keavy DJ, Branzt SE, Kelly WJ, and Pals MA (1988) Unexpected regioselective diels-alder cycloaddition reactions between 3-fluorobenzyne and 2-alkylfurans. Tetrahedron Letters 29, 6227–6230. [Google Scholar]
- (25).Tønder JE, and Tanner D (2003) Toward the enantioselective total synthesis of lyngbyatoxin A: on the stereocontrolled introduction of the quaternary stereogenic centre. Tetrahedron 59, 6937–6945. [Google Scholar]
- (26).Singh J, Zeller W, Zhou N, Hategan G, Mishra RK, Polozov A, Yu P, Onua E, Zhang J, Ramírez JL, Sigthorsson G, Thorsteinnsdottir M, Kiselyov AS, Zembower DE, Andrésson T, and Gurney ME (2010) Structure−Activity Relationship Studies Leading to the Identification of (2 E )-3-[l-[(2,4-Dichlorophenyl)methyl]-5-fluoro-3-methyl-l H -indol-7-yl]-N -[(4,5-dichloro-2-thienyl)sulfonyl]-2-propenamide (DG-041), a Potent and Selective Prostanoid EP3 Receptor Antagonist, as a Novel Antiplatelet Agent That Does Not Prolong Bleeding. Journal of Medicinal Chemistry 53, 18–36. [DOI] [PubMed] [Google Scholar]
- (27).Childers BM, Weber GG, Prouty MG, Castaneda MM, Peng F, and Klose KE (2007) Identification of residues critical for the function of the Vibrio cholerae virulence regulator ToxT by scanning alanine mutagenesis. Journal of Molecular Biology 367, 1413–1430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.