

Abstract
Autophagy is an evolutionarily conserved intracellular degradative function that is important for liver homeostasis. Accumulating evidence suggests that autophagy is deregulated during the progression and development of alcoholic and non-alcoholic liver diseases. Impaired autophagy prevents the clearance of excessive lipid droplets (LDs), damaged mitochondria, and toxic protein aggregates, which can be generated during the progression of various liver diseases, thus contributing to the development of steatosis, injury, steatohepatitis, fibrosis, and tumors. In this review, we look at the status of hepatic autophagy during the pathogenesis of alcoholic and non-alcoholic liver diseases. We also examine the mechanisms of defects in autophagy, and the hepato-protective roles of autophagy in non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD), focusing mainly on steatosis and liver injury. Finally, we discuss the therapeutic potential of autophagy modulating agents for the treatment of these two common liver diseases.
Keywords: Autophagy, Non-alcoholic fatty liver disease (NAFLD), Alcoholic liver disease (ALD), Liver injury, Steatosis
1. Introduction
Basal autophagy occurs in the liver to allow constitutive turn-over of cytosolic components.1 As an adaptive process, a number of stressors, including starvation, growth factor depletion, oxidative injury, and chemicals can stimulate autophagy. Among these stressors, starvation or fasting is the best-understood stimulus for autophagy. How cells sense the need to boost in autophagic activity has not yet well-defined. It probably depends on the nature of the stress. For example, amino acid starvation is perceived through the lysosomal nutrient sensing system to inactivate mammalian target of rapamycin complex 1 (mTORC1), so autophagy is de-repressed.2 Once autophagy signal is induced, autophagy machinery kicks in, leading to the formation of the autophagosome membrane, its elongation, cargo engulfment and trafficking to the lysosome, and ultimately the breakdown of the components inside the lysosome (Fig. 1).3
Fig. 1. Overview of cellular autophagy.
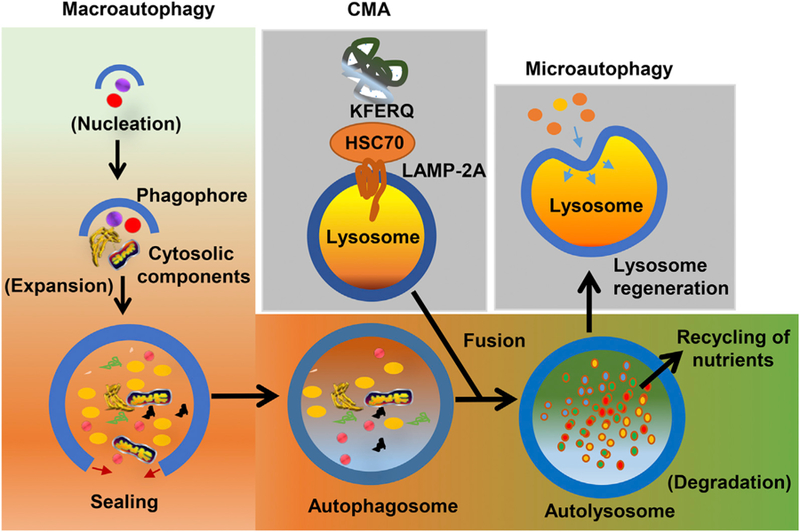
In macroautophagy, phagophore formation and elongation sequester the autophagic cargoes and forms autophagosomes. The autophagosome then fuses with the lysosome to form autolysosomes. In microautophagy, substrates are directly engulfed by the lysosomal membrane. In CMA, proteins with the pentapeptide motif KFERQ are selectively recognized by the HSC70 chaperone. Substrate-bound HSC70 then interacts with LAMP-2A in lysosome membrane and subsequently translocates into the lysosomal lumen. In all three processes, the cargos are ultimately degraded by lysosomal hydrolases, to release smaller biomolecules for reutilization. Abbreviations: CMA, chaperonin mediated autophagy; HSC70, heat shock cognate 70; LAMP-2A, lysosome-associated membrane glycoprotein 2A.
2. Concepts and signaling regulators of autophagy
2.1. General concepts of autophagy
The induction of autophagy requires a set of conserved factors called “autophagy-related (ATG)” proteins that function as complexes to induce autophagy.4 The ATG factors are grouped into functional units that includes unc-51 like autophagy activating kinase 1 (ULK1) tetrameric kinase complex (ULK1, RB1-inducible coiled-coil protein 1 (FIP200/RB1CC1), ATG13L, and ATG101), the class III phosphatidylinositol 3-phosphate kinase complex (PI3KC3) (VPS34, beclin-1, p115, and ATG14 or UV radiation resistance-associated gene protein (UVRAG)), WD-repeat protein interacting with phosphoinositides (WIPI) family, the ATG9A cycling system, the two conjugation systems (for ATG12/ATG5/ATG16 and the ATG8 family members), and the activating E1-like or E2-like enzymes (ATG7, ATG10, ATG3 required for two conjugation processes of ATG16-ATG5/12 and LC3-phosphatidylethanolamine (PE)/LC3-II).
In mammalian cells, autophagy begins with the formation of an expanding double-membranous, cup-shaped structure, called phagophore, the edges of which extend and sequester cytosolic components to form autophagosome vesicles. The phagophore membrane may be derived from the endoplasmic reticulum (ER), the mitochondria, the Golgi apparatus, or the plasma membrane.5 Formation of autophagosomes is the morphological signature of the induction of autophagy. Autophagosomes then fuse with lysosomes to deliver and break down its contents by lysosomal proteases.6
The initiation and formation of phagophore begin with the activation of the ULK1 kinase complex and the formation of ULK1 puncta at a discrete location on the ER. The activated ULK protein complex then recruits the VPS kinase complex to the phagophore, and phosphorylates the class III PI3K together with the ULK1 complex, generating a local pool of phosphatidylinositol 3-phosphate. This alteration in lipid composition changes the membrane curvature, further expands the phagophore, and recruits the oligomers of ATG12-conjugated ATG5 in complex with ATG16L. The formation of the ATG12-ATG5-ATG16 complex is mediated by ATG7 and ATG10. The ATG12-ATG5-ATG16 complex is then recruited to the autophagosome membrane. This complex facilitates the lipidation of ATG8/LC3 via conjugation to PE on the autophagosome membrane. The latter event causes further expansion and enclosure of the autophagosome membrane.
Autophagosomes are generally formed around the peripheral regions of cells, and hence, they are transported along microtubules towards the perinuclear regions, where lysosomes reside for the final degradation.7 The lysosome contains a repertoire of degradative enzymes, such as proteases, nucleases and glycosidase. The degradation products, including amino acids, sugars and lipids/free fatty acids (FFAs), are transported out of the autolysosome via the lysosomal permeases for reutilization of these simple biomolecules. During termination step of autophagy, lysosome is regenerated through autophagic lysosome reformation (ALR). During ALR, tubules extrude from autolysosomes, and small proto-lysosomal vesicles are regenerated. Eventually, proto-lysosome matures into functional lysosomes.8,9
Autophagy can also perform selective recognition and sequestration of cargo through a process, called selective autophagy. Depending on the nature of the sequestration of intracellular substrates, autophagy is termed mitophagy (damaged or dysfunctional mitochondria), proteophagy (protein aggregates), pexophagy (peroxisomes), lipophagy (excessive lipid droplets (LDs)), ferritinophagy (ferritin), or xenophagy (intracellular microorganisms).10,11 Selective autophagy relies on a plethora of selective autophagy receptors (SARs), such as p62/SQSTM1, neighbor of BRCA1 gene 1 (NBR1), NDP52 (nuclear domain 10 protein 52 kDa), and optineurin (OPTN). SAR generally binds to the cargo (often ubiquitinated) and key components of the autophagy machinery, especially the LC3 protein.10,12
In the context of intracellular trafficking pathways, conceptually, autophagy appears to function as a parallel pathway that carries intracellular cargo into the lysosome, in a similar fashion to the hepatocellular endocytic pathway, which internalizes and transports extracellular cargo (e.g. low density lipoprotein (LDL)/LDL receptor) into the lysosome. There could be a potential cross-talk between autophagy and the endocytic pathway, because (i) specific compartments are shared between them, (ii) both contribute to cellular proteolysis, (iii) in both cases the degradative executioner components is retained by lysosomal compartment and (iv) both pathways require regulated trafficking of vesicles that interact in a dynamic way, undergoing fission and fusion as part of their maturation process. Notably, whether autophagy or endocytic pathway related vesicular trafficking really impacts the lysosomal function, number, or structure is unknown.
2.2. Autophagy signaling regulators: mTORC1, AMP-activated protein kinase (AMPK), and transcription factor EB (TFEB)
Given the important roles of autophagy in the survival mechanism of mammalian cells, it is not surprising that alterations of autophagy occur in response to extracellular or intracellular stress, such as starvation, growth factor deprivation, ER stress, and pathogen infection.13 How different external stressors activate the same autophagy executioner complex is an interesting issue. We will discuss autophagy signaling in reference to the cellular nutrient status.
In liver, autophagy gets activated not only during starvation, but this process gets activated daily between meals to provide essential nutrient components and guarantee periodical cellular clearance. One of the best-characterized regulators of autophagy signaling is the mTORC1. In general, mTORC1 activation suppresses autophagy, whereas the suppression of mTORC1 activates autophagy.2,14 mTORC1 phosphorylates and actively sequesters the ULK1 in a complex with ATG13 and FIP200 in an inactive state. Nutrient starvation increases the cellular AMP/ATP ratio, activating cellular energy sensor AMPK, which inhibits mTORC1 through three molecular mechanisms: (i) direct phosphorylation of mTORC1, (ii) phosphorylation of tuberous sclerosis 2 (TSC2) and Raptor (mTORC1 component) to inhibit mTORC1, and (iii) phosphorylation of ULK1 at different residue to activate ULK1 kinase complex. All of these three different phosphorylation events inhibit mTORC1 activity and hence reduce ULK1 phosphorylation and promote its release from mTORC1. ULK1 kinase complex is then activated to recruit other autophagy proteins and induce autophagy.2,14 With the complete execution of autophagy process, lysosomal degradation of cellular components release the nutrients, including amino acids, which can reactivate mTORC1 and thereby attenuate the ULK1 dependent autophagy.14 This inhibitory feedback prevents excessive autophagy which may lead to apoptosis.
In addition to mTORC1 and AMPK mediated regulation of autophagy at the signaling level, the longer-term transcriptional regulation of autophagy is carried out by TFEB. TFEB is a basic helix-loop-helix (bHLH) leucine zipper transcription factor of the NITF family.15 It is a master transcriptional regulator of lysosomal biogenesis and autophagy. Under nutrient-rich conditions, mTORC1 phosphorylates TFEB at Ser211 and retains TFEB in the cytosol via the interaction with the members of the YWHA (14–3-3) family of proteins.16–18 However, nutrient deprivation leads to mTORC1 inhibition, dephosphorylation of S211 and its rapid translocation to the nucleus to initiate the downstream transcription of autophagy genes.19 There, TFEB induces its own expression and also binds directly to the promoters of a multitude of autophagy-related genes and drives rapid expression of autophagy genes such as ATG4, ATG9, LC3, p62, UVRAG and Wili1, which are involved in cargo sequestration, autophagosome membrane closure, and fusion with the lysosome. Besides, TFEB induces lysosome biogenesis, thus favoring autophagy degradation.17 Approximately 96 lysosomal genes termed the coordinated lysosomal expression and regulation (CLEAR) genes are regulated for lysosomal biogenesis and function. Interestingly, lysosomal Ca2+ release through mucolipin 1 (MCOLN1), which activates cytosolic calcineurin, is required for the dephosphorylation of TFEB mediated autophagy induction and lysosomal biogenesis.20 Nuclear localization and activity of TFEB are also regulated by phosphorylation (Ser142) by the extracellular signal-regulated kinase-2 (Erk2), the activity of which is tuned by the levels of extracellular nutrients.17 In support of these observations, overexpression of TFEB is sufficient to induce autophagy and liver-directed TFEB expression corrects hepatic disease by increased SERPINA1 polymer degradation via enhanced autophagy flux.21 Similar protective effect of TFEB overexpression has been noted in various genetic and dietary models of obesity and alcoholic liver disease (ALD) models.22,23
Next, we will detail the roles of autophagy in the pathogenesis of alcoholic and non-alcoholic fatty liver diseases. Basically, in the following section, we will take account of the autophagy-related basic questions, for example, what the status of autophagy is and what the molecular mechanism of alteration in autophagy in alcoholic and non-alcoholic liver diseases is. We will also discuss the role of autophagy in disease pathogenesis, particularly focusing on liver injury and steatosis. Finally, we will describe the current therapeutic potential of autophagy process for these two common liver diseases.
3. Autophagy in non-alcoholic fatty liver disease (NAFLD)
In NAFLD, hepatocyte has an excessive accumulation of triglycerides (TGs) and cholesterols in LDs. Autophagy can regulate the hepatocellular lipid accumulation by selective degradation.24 Based on this rationale, autophagy has been implicated to play a protective role during NAFLD. Hepatic autophagy can be inhibited or decreased in genetic and dietary rodent models of NAFLD. Similar observations have been made in human liver samples that have been diagnosed with non-alcoholic steatohepatitis (NASH), which is pathologically more advanced than NAFLD. Increased accumulation of LC3-II and p62 were observed in patients diagnosed with NASH.25 Interestingly, the accumulation of LC3-II and p62 correlates positively with the severity of the disease.25,26 Autophagy dysfunction is also associated with cathepsins expression and hence lysosomal function in livers from patients with NAFLD.27
3.1. Autophagy status in NAFLD
The current consensus is that autophagy is decreased in the fatty hepatocyte. Based on the in vitro studies in hepatic cell lines and the in vivo high fat diet (HFD) feeding model, autophagosomes accumulate in hepatic cells. The increase in autophagosomes is due to the blocking of the fusion between autophagosomes and lysosomes.28 Consistent with this observation, in NAFLD, there is a prominent accumulation of insoluble protein inclusions containing p62 and ubiquitinated protein aggregates, which are substrates of autophagy.29 Thus, blockade of autophagy flux (impaired lipophagy) probably further increases the intracellular fat accumulation. This vicious cycle of fat accumulation (due to excessive intake) and impairments in autophagic flux results in the development of hepatic steatosis. Whether blockade of autophagic flux is a major contributing factor to hepatic steatosis or merely a bystander effect of intracellular lipid accumulation is less clear. Moreover, the impact of autophagic dysregulation in the development and progression from steatosis to the steatohepatitis (NASH) is also undetermined.
3.2. Mechanism of autophagy deregulation in NAFLD
There are potentially four major mechanisms that may account for the impairment of autophagy in NAFLD. They are discussed below.
3.2.1. Downregulation of autophagy proteins
A decrease in the expression of key autophagy molecules, such as ATG7, can reduce autophagy process.30 Abnormal activation of proteases, such as calpain-2, can also reduce autophagy proteins levels, such as ATG3, ATG5, beclin-1 and ATG7, and hence reduce the autophagy process.30,31
3.2.2. Decreased lysosomal acidification
Decreased lysosomal acidification and lysosomal proteolytic activity due to reduction of hepatic cathepsin B and L levels impair the autophagic substrate degradation.27,32 Increased expression of asparagine synthetase (ASNS) due to ER stress and subsequent lysosomal calcium retention has also been reported to affect the lysosomal acidification and impair autophagy.33
3.2.3. Defective autophagosome-lysosome fusion
Increases in intracellular lipids alter the intracellular membrane lipid composition of both autophagosomes and lysosomes.34 This reduces the ability of autophagosomes to fuse with lysosomes and leads to a decrease in autophagic flux. Saturated fatty acids (FAs), such as palmitic acid (PA), post transcriptionally upregulate Rubicon (a beclin-1-interacting negative regulator for autophagosome-lysosome fusion), which then suppress the late state of autophagy fusion with lysosomes.28 The accumulation of saturated FA has also been recently proposed to inhibit hepatic ATP2A2/SERCA2, an ER calcium pump that is responsible for the influx of cytosolic calcium into the ER lumen.35 Inhibition of ATP2A2/SERCA2 causes chronic increases in cytosolic calcium levels, which then arrests autophagy process by blocking the autophagic flux.35 Interestingly, a certain lipid, phosphatidic acid, restores autophagic function and hence significantly reduces hepatic lipid accumulation in HFD-induced NAFLD.36
3.2.4. Other mechanisms
Elevated levels of metabolites, such as S-adenosylmethionine (SAMe) and methionine, during HFD can activate protein phosphatase 2A (PP2A) by methylation and hence block autophagic flux.37 Similarly, over activation of mTORC1 due to inhibition of AMPK1 via sirtuin (SIRT) 3 activation also suppresses autophagy.38
4. Autophagy in ALD
Liver is the main organ responsible for alcohol metabolism. Oxidative metabolism of alcohol by alcohol dehydrogenase (ADH) (cytosol), cytochrome P450 (Cyp)2e1/Cyp1A2/Cyp3A4 (ER, micro-somes) and catalase enzyme (peroxisomes) results in the generation of acetaldehyde and acetate.39 Oxidative metabolism of ethanol also produces reactive oxygen species (ROS), including hydroxyethyl, superoxide anion, and hydroxyl radicals all of which increase the risk of tissue damage.40 Alcohol oxidation generates a highly reduced cytosolic environment in liver cells (more nicotinamide adenine dinucleotide (reduced) (NADH)). Moreover, the NADH that is generated is oxidized by a series of chemical reactions in the mitochondrial electron transport chain, which further increases the free radicals and makes hepatocytes vulnerable to these toxic intermediates. The formation of protein adducts (by acetaldehyde, an alcohol metabolism intermediate) in hepatocytes impairs protein secretion, which has been proposed to play a role in the enlargement of the liver (i.e. hepatomegaly).40 Ethanol also causes the accumulation of LDs and damages mitochondria, both of which can be regulated by autophagy.
4.1. Autophagy status in ALD
The effect of ethanol on autophagy is complex. Acute and chronic alcohol exposure may differentially regulate hepatic autophagy.41 Hepatic autophagy is activated in acute ethanol treatment but can be suppressed in chronic or high dose treatment of alcohol.23,42 An analysis of autophagy in primary cultured hepatocytes and a hepatic cell line treated with ethanol (20–80 mM for 6–24 h) showed increased autophagosome formation and autophagy flux, suggesting activation of cellular autophagy.43 Similar observations were made in the livers of mice that were fed with the acute binge ethanol feeding model (4–6 g ethanol/kg body weight).41,43 Acute ethanol feeding enhances the content of autophagic vacuoles, autophagosome-lysosomal fusion, and autophagic flux. Interestingly, the enhanced autophagy in acute ethanol fed mice was associated with a higher hepatic nuclear content of TFEB, which is considered to be a master regulator of transcription of genes in lysosome biogenesis and autophagy.17,41
Conflicting results have been reported about the autophagy status in the chronic ethanol feeding conditions. Chronic ethanol feeding (Lieber-DeCarli model) for 4 weeks or 10 weeks increases autophagosome numbers in rodent liver, suggesting the induction of autophagy.44,45 However, in another similar model, a chronic ethanol treatment, in which mice were fed ethanol for 10 days and ethanol concentration (as percent of total calories) was gradually increased from 3.6% on day 1to 7.2% on day 4, 10.2% on day 6, 21.6% on day 8, 29.2% (5.2% ethanol by volume) on day 10, the contents of hepatic autophagic vesicles were enhanced, but autophagic flux was lowered.41 This suggests that the chronic ethanol impairs autophagic flux and hence increases autophagic vesicles in the mouse liver. Impairments in autophagic flux have been similarly observed in a chronic plus binge ethanol feeding model (Gao-binge model: ethanol liquid diet for 10-day followed by a single binge of ethanol 5 g ethanol/kg body weight).23 In this study, the authors proposed that chronic ethanol induce insufficient autophagy, a novel autophagic flux scenario in which cells have a decreased number of lysosomes resulting in the accumulation of early autophagosomes. Mechanistically, chronic ethanol feeding was found to decrease the hepatic TFEB and lysosomal biogenesis in mouse livers. Earlier studies have also indicated that chronic ethanol exposure disrupts lysosome function and biogenesis.41,46 Overall, the various chronic ethanol feeding models implicate that even ethanol induces autophagy, due to the impairment of lysosomal function (TFEB suppression), and that autophagy activity is ultimately impaired during chronic alcohol consumption.
4.2. Mechanism of autophagy deregulation in ALD
In contrary to the autophagy inhibition during chronic alcohol treatment, the induction of autophagy in murine primary hepatocytes exposed to ethanol-induced autophagy process dependent on its metabolism, ROS production, and mTORC1 inhibition. Furthermore, autophagy protects hepatocytes against the detrimental effects of ethanol likely by removing damaged mitochondria and accumulated LDs. Acute ethanol-induced autophagy by the following mechanisms.
4.2.1. Increased ROS production
The metabolism of ethanol in hepatocytes by ADH and Cyp2e1 generates ROS that promotes autophagy probably via inactivation of ATG4B.47
4.2.2. mTORC1 suppression
Acute ethanol treatment suppresses the mTORC1 complex.43 mTORC1 inhibition leads to activation of the downstream ULK1 complex to trigger autophagy, as described in Section 2.2.
Alcohol consumption also affects the TFEB, a transcriptional regulator of autophagy and lysosomal function. The differential effect of alcohol in autophagy process correlates with the cellular localization of TFEB.23,41 Acute ethanol treatment increases the nuclear protein levels of TFEB in mouse livers, correlating with the important role of TFEB in the induction of autophagy. In contrast, chronic alcohol treatment decreases nuclear TFEB proteins levels in mouse livers.41 A similar observation was made with the chronic feeding plus binge alcohol model, which mimicked consumption patterns of human alcoholics.23 Decreased TFEB was also observed in human ALD samples compared with healthy human donor liver samples.42 Mechanistically, ethanol was found to activate mTORC1 and decrease TFEB proteins and the TFEB-mediated lysosomal biogenesis, resulting in insufficient autophagy in mouse livers.23 How TFEB protein level is downregulated is unknown.
Besides the TFEB transcription factor, alcohol also affects the transcription factor forkhead box O3a (FoxO3a) to modulate hepatic autophagy process.48 FoxO3a belongs to the O subfamily of the Fork head box protein family.49 The activity of FoxO3a is largely regulated by its multiple post-transcriptional modifications, including phosphorylation, acetylation, ubiquitination, and methylation.49 Acute ethanol treatment induce FoxO3a dephosphorylation (via Akt inhibition) and acetylation (due to an increase in the NADH/nicotinamide adenine dinucleotide (oxidized) (NAD+) ratio and a decrease in hepatic SIRT 1) in mouse liver and increase nuclear translocation of FoxO3a. Nuclear FoxO3a then elevates the transcription of many autophagy-related genes, such as ULK1, ATG5, beclin-1 and ATG7, and thus induces autophagy in mouse liver.48 Interestingly, how FoxO3a coordinates with other autophagy-related transcription factors, such as TFEB and farnesoid X receptor (FXR), to regulate the expression of autophagy-related genes is unknown. However, the critical role of FoxO3 in ethanol-induced hepatic autophagy is well supported by the observation of blunted expression of autophagy-related genes, enhanced steatosis and liver injury in FoxO3a knock out mice.48
Chronic ethanol exposure was also reported to inhibit hepatocytes lipophagy through the inactivation of the small guanosine triphosphate Rab7 and reduced dynamin 2 activity, thereby causing depletion of lysosomes for LD breakdown.50,51 The inhibition of AMPK activity by chronic ethanol treatment may also be responsible for the suppression of autophagy.52 Mechanistically, in both acute and chronic alcohol treatment, there is an active involvement of ROS, mTOCR1, and TFEB in the regulation of autophagy process and lysosomal degradation function. The key role of ROS (nicotinamide adenine dinucleotide phosphate (reduced) (NADPH) oxidase-derived) in alcohol-induced liver disease is well established.53 Similarly, the counteracting relationship between the mTORC1 and TFEB is also well known (as described above in Section 2.2). However, how ROS generation in acute or chronic alcoholic condition systematically affects the mTORC1 activation or TFEB translocation is unclear. How ROS that are generated in acute alcohol treatment inhibits mTORC1, whereas chronic ethanol reactivates mTORC1 is also unknown. It is possible that the extent of ROS generation may modulate the activation status of mTORC1 and, hence, the autophagy process. Interventional studies using antioxidants, such as polyphenols and N-acetyl cysteine, which neutralize the ROS generation under alcohol liver conditions, may be helpful in answering these questions.
5. Role of autophagy in steatosis and liver injury associated with NAFLD and ALD
Recent studies suggest that deregulated autophagy could play a role in the pathogenesis of alcoholic and non-alcoholic liver diseases. However, whether the deregulated autophagy is a cause or a consequence of the disease pathogenesis is less clear. Hepatic steatosis is an early histological observation in both alcoholic and non-alcoholic liver disease. There are varying degrees of hepatocyte injury that could be associated with inflammation and fibrosis. Various lipid intermediates, such as FFAs (especially saturated), ceramides, cholesterol, diacyl-glycerol, phospholipid and lysophosphatidylcholine (LPC), which are significantly increased in liver tissue of NASH, have been observed to cause lipid toxicity.54 In chronic ethanol, hepatocyte injury is likely mediated by acetaldehyde-protein adducts and the hydroxyl free radical that are generated from ethanol metabolism.39 In general, hepatic lipid overload, mitochondrial dysfunction, ER stress, and the accumulation of protein aggregates have been implicated as important stimuli of hepatocyte cell death and thus liver injury. However, the extent of liver injury that is generally observed in vivo animal study or human clinical observation is mild. Recently, the concept of sublethal lipotoxic hepatocyte injury has been proposed.55 Lipid accumulation cause hepatocyte stress and dysfunction of an insufficient magnitude to cause cell death but enough to induce pro-apoptotic signaling that results in the release of various aberrant pro-inflammatory signaling molecules via extracellular vehicles (EVs).55
The primary role of autophagy is to degrade intracellular organelles and protein aggregates that are too large to be broken down by the proteasome. Thus, the defect in autophagy either due to the high-fat diet or the chronic alcohol can contribute to disease (initiation or progression) by two general mechanisms: (i) the accumulation of potentially dangerous autophagic substrates and (ii) the lack of potentially beneficial autophagic products or functions. Followings are three possible major routes through which autophagy could ameliorate the pathogenesis of alcohol or non-alcoholic liver disease (Fig. 2).
Fig. 2. Role of autophagy in steatosis and liver injury associated with NAFLD and ALD.
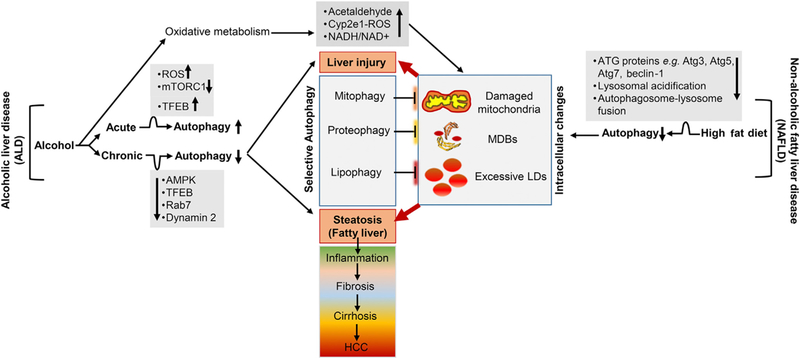
NAFLD and ALD are marked by hepatic steatosis, liver injury, inflammation, fibrosis, cirrhosis, and HCC. Alcohol consumption or a high-fat diet impairs autophagy, resulting in intracellular accumulation of damaged mitochondria, MDBs, and excessive LDs. Autophagy relieves hepatocellular steatosis and liver injury by degrading these intracellular toxic components. The up and down black arrows indicate increased and decreased in individual level or activity. Abbreviations: NAFLD, non-alcoholic fatty liver disease; ALD, alcoholic liver disease; HCC, hepatocellular carcinoma; MDBs, Mallory-Denk bodies; LDs, lipid droplets.
5.1. Autophagy lowers hepatic lipid load by lipophagy
A seminal study by Czaja and colleagues24 in 2009 demonstrated that hepatocytes consume intracellular lipid when they are starved. Intracellular lipids (triglyceride and cholesterol) are stored as LDs. Under conditions of stress, autophagy promotes lipid hydrolysis and generates FFAs by releasing the content of LDs to lysosomes for degradation. Based on this concept, inhibition of autophagy can potentially lead to the accumulation of fat, which is potentially toxic to cells. Alternatively, the activation of autophagy pharmacologically or genetically could alleviate the intrahepatic lipid load and hence decrease the cellular stress.44,56
5.2. Autophagy clears dysfunctional or damaged mitochondria by mitophagy
Mitochondria is actively involved in the hepatic metabolism of ethanol or lipid (from high-fat diet). There is a considerable evidence from both human and animal studies that, during the oxidative metabolism of ethanol or lipid, mitochondria are intimately involved in the generation of ROS. Mitochondria are themselves also targets of oxidative stress that can deregulate the cellular energy balance. The vicious cycle of ROS generation through oxidative metabolism and mitochondrial damage leads to the promotion of hepatocyte’s apoptotic or necrotic death and hence cause liver injury. Alternatively, activation of autophagy/ mitophagy can help to eliminate these dysfunctional or damaged mitochondria and hence reduce the ROS level and potentially alleviate the disease. The PTEN-induced kinase 1 (PINK1)-Parkin mediated mitophagy is well-studied pathway particularly in the context of ALD. Ethanol causes a marked increase in the number of mitophagic vacuoles in the hepatocytes following ethanol treatment, suggesting selective induction of hepatic mitophagy.43,57 Mitophagy is initiated by accumulation of PINK1 at the outer membrane of damaged mitochondria, resulting in the recruitment of cytoplasmic Parkin (an E3 ubiquitin ligase) to those mitochondria.58 The PINK1-Parkin protein interaction in damaged mitochondria initiates ubiquitination of mitochondrial outer membrane proteins and subsequent mitochondrial degradation by LC3-mediated autophagosomes. The important protective role of PINK1-Parkin mediated mitophagy in ALD is supported by the observation made with the ethanol-treated Parkin knock out mice. Parkin knock out mice had increased liver injury, oxidative stress, and steatosis after alcohol treatment compared with wild-type mice.59 At a cellular level, Parkin knock out hepatocytes had severely swollen and damaged mitochondria that lacked cristae, decreased mitophagy, β-oxidation, mitochondria respiration, and cytochrome c oxidase activity after acute ethanol treatment.59
5.3. Clearance of Mallory-Denk bodies (MDBs) by proteophagy
MDBs are cytoplasmic hyaline inclusion in the hepatocytes, generally considered as an indicator of histological severity in ALD and NAFLD.60,61 The major components of MDBs include keratin 8, keratin 18, ubiquitin, and p62.61 ROS released from the oxidative metabolism of ethanol or through excessive fat metabolism can potentially affect many intracellular proteins resulting in the formation of MDBs which could be potentially damaging to hepatic structure and function. Even though the mechanisms of MDBs formation is less clear, there is emerging evidence that autophagy apparently participates in the elimination of components of MDB.62,63 Pharmacological activation of autophagy by rapamycin prevents MDB formation and also causes MDB resorption.63
6. Pharmacological modulation of autophagy
Alterations in autophagy or autophagy related processes occur in fatty liver situation, which support the possibility that pharmacological activators of autophagy could be beneficial for NAFLD and ALD patients. The potential benefit of autophagy-enhancing agents to reduce fatty liver conditions and liver injury has been reported in murine models of NAFLD and ALD.44 Several pharmacological and nutritional supplements are available to modulate autophagy. However, these agents modulate autophagy though multipronged or hitherto uncharacterized molecular mechanisms. Moreover, recent studies indicate that autophagy may play a different role in hepatocytes and hepatic non-parenchymal cells, which results in opposite effects on liver disease.64,65 For example, in liver fibrosis, the activation of autophagy may be pro-fibrotic (due to stellate cell activation) as well as anti-fibrotic (Kupffer cells and hepatocytes). Thus, targeting autophagy in a hepatic cell-specific manner is one of the main challenges for future therapeutic approaches for liver diseases. Further characterization of dysfunctional autophagy in different stages of liver diseases will offer new avenues for future drug development.
The first preclinical study to show the potential utility of stimulating autophagy to improve liver pathologies in NAFLD and ALD was performed by Lin et al.44 using two well established autophagy inducers: carbamazepine and rapamycin. Carbamazepine and rapamycin induce autophagy in hepatocytes in vitro and in vivo. When mice that have been fed with HFD or chronic ethanol, are injected with carbamazepine or rapamycin, hepatic steatosis, liver injury, and other metabolic parameters, such as serum triglycerides and insulin resistance, are alleviated. In contrast, chloroquine, which blocks autophagy, exacerbates steatosis and injury in these animals.44 Besides, rapamycin, various rapalogs, such as Torin 1, PP242, AZD3147, Z1001, rottlerin, XL388, AP23573, and RAD001 have also been identified as autophagy inducers.66 However, the impact of these autophagy inducers in the prevention and treatment of NAFLD and ALD are yet to be tested in established preclinical models.
Several nutritional supplements, such as caffeine, resveratrol, Vitamin D and retinoic acid, improve hepatic steatosis by inducing autophagy.67–69 Vitamin D protects against HFD-induced liver steatosis by inducing autophagy by upregulating ATG16L1 in mice.69 Glycycoumarin (coumarin isolated from natural product licorice) inhibits hepatocyte lipoapoptosis through reactivation of impaired autophagy.70 Acutely activating autophagy by caloric restriction or physical exercise also improves hepatic steatosis and other various metabolic parameters (insulin sensitivity, body weight).71 In addition to pharmacological or nutritional activation of autophagy, genetic reconstitution of autophagy also attenuates steatosis.30,56
7. Conclusions
Autophagy plays a crucial role in liver homeostasis, and hence, the deregulation of autophagy could play a crucial role in the pathogenesis of alcoholic and non-alcoholic liver disease. However, it is still unclear how autophagy is deregulated under these conditions. Better understanding of the mechanisms of deregulation of autophagy during acute or chronic alcoholic conditions and NAFLD will further assist in designing and developing autophagy specific therapeutics in a near future.
Acknowledgements
This work was supported in part by the USA National Institutes of Health (NIH) grants R01AA021751 and R21AA021450 (to X.-M. Yin).
Footnotes
Edited by Yuxia Jiang, Peiling Zhu and Genshu Wang.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology 2008;47: 1773–1785. [DOI] [PubMed] [Google Scholar]
- 2.Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature 2015;517:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb cell 2016;3:588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009;10: 458–467. [DOI] [PubMed] [Google Scholar]
- 5.Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 2013;14:759–774. [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- 7.Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct 2008;33: 109–122. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Yu L. Autophagic lysosome reformation. Exp Cell Res 2013;319: 142–146. [DOI] [PubMed] [Google Scholar]
- 9.Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010;465:942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 2014;16:495–501. [DOI] [PubMed] [Google Scholar]
- 11.Okamoto K Organellophagy: eliminating cellular building blocks via selective autophagy. J Cell Biol 2014;205:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 2014;53:167–178. [DOI] [PubMed] [Google Scholar]
- 13.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015;125:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol 2016;32:255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy 2011;7:1379–1381. [DOI] [PubMed] [Google Scholar]
- 17.Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013;14:283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012;8:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015;17:288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pastore N, Blomenkamp K, Annunziata F, et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol Med 2013;5: 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid meta-bolism through a starvation-induced autoregulatory loop. Nat Cell Biol 2013;15:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chao X, Wang S, Zhao K, et al. Impaired TFEB-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology 2018. pii: S0016–S5085:34560–34568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Rodriguez A, Mayoral R, Agra N, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis 2014;5:e1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willy JA, Young SK, Mosley AL, et al. Function of inhibitor of Bruton’s tyrosine kinase isoform alpha (IBTKalpha) in nonalcoholic steatohepatitis links autophagy and the unfolded protein response. J Biol Chem 2017;292:14050–14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukuo Y, Yamashina S, Sonoue H, et al. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol Res 2014;44:1026–1036. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka S, Hikita H, Tatsumi T, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016;64:1994–2014. [DOI] [PubMed] [Google Scholar]
- 29.Cho CS, Park HW, Ho A, et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology 2017. 10.1002/hep.29742 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 30.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metabol 2010;11: 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Q, Guo Z, Deng W, et al. Calpain 2-mediated autophagy defect increases susceptibility of fatty livers to ischemia-reperfusion injury. Cell Death Dis 2016;7:e2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inami Y, Yamashina S, Izumi K, et al. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Biophys Res Commun 2011;412:618–625. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Zhang X, Chu ESH, et al. Defective lysosomal clearance of autophagosomes and its clinical implications in nonalcoholic steatohepatitis. FASEB J 2018;32:37–51. [DOI] [PubMed] [Google Scholar]
- 34.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J 2010;24:3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park HW, Park H, Semple IA, et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat Commun 2014;5:4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hur JH, Park SY, Dall’Armi C, et al. Phospholipase D1 deficiency in mice causes nonalcoholic fatty liver disease via an autophagy defect. Sci Rep 2016;6:39170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zubiete-Franco I, Garcia-Rodriguez JL, Martinez-Una M, et al. Methionine and S-adenosylmethionine levels are critical regulators of PP2A activity modulating lipophagy during steatosis. J Hepatol 2016;64:409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S, Dou X, Ning H, et al. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 2017;66: 936–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zakhari S Overview: how is alcohol metabolized by the body? Alcohol Res Health 2006;29:245–254. [PMC free article] [PubMed] [Google Scholar]
- 40.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology 2006;43: S63–S74. [DOI] [PubMed] [Google Scholar]
- 41.Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM Jr. Acute and chronic ethanol administration differentially modulate hepatic autophagy and transcription factor EB. Alcohol Clin Exp Res 2015;39:2354–2363. [DOI] [PubMed] [Google Scholar]
- 42.Lu Y, Cederbaum AI. Autophagy protects against CYP2E1/chronic ethanol-induced hepatotoxicity. Biomolecules 2015;5:2659–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding WX, Li M, Chen X, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010;139:1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin CW, Zhang H, Li M, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 2013;58:993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eid N, Ito Y, Maemura K, Otsuki Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. J Mol Histol 2013;44:311–326. [DOI] [PubMed] [Google Scholar]
- 46.Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM Jr. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochim Biophys Acta 1995;1245:421–429. [DOI] [PubMed] [Google Scholar]
- 47.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007;26:1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni HM, Du K, You M, Ding WX. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol 2013;183:1815–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci 2007;120: 2479–2487. [DOI] [PubMed] [Google Scholar]
- 50.Schulze RJ, Rasineni K, Weller SG, et al. Ethanol exposure inhibits hepatocyte lipophagy by inactivating the small guanosine triphosphatase Rab7. Hepatol Commun 2017;1:140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rasineni K, Donohue TM Jr, Thomes PG, et al. Ethanol-induced steatosis involves impairment of lipophagy, associated with reduced Dynamin2 activity. Hepatol Commun 2017;1:501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004;127:1798–1808. [DOI] [PubMed] [Google Scholar]
- 53.Kono H, Rusyn I, Yin M, et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest 2000;106:867–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirsova P, Ibrahim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res 2016;57: 1758–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018;67: 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiong X, Tao R, DePinho RA, Dong XC. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem 2012;287:39107–39114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eid N, Ito Y, Otsuki Y. Triggering of Parkin mitochondrial translocation in mitophagy: implications for liver diseases. Front Pharmacol 2016;7:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams JA, Ni HM, Ding Y, Ding WX. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol 2015;309:G324–G340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Basaranoglu M, Turhan N, Sonsuz A, Basaranoglu G. Mallory-Denk bodies in chronic hepatitis. World J Gastroenterol 2011;17:2172–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zatloukal K, French SW, Stumptner C, et al. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res 2007;313:2033–2049. [DOI] [PubMed] [Google Scholar]
- 62.Harada M Autophagy is involved in the elimination of intracellular inclusions, Mallory-Denk bodies, in hepatocytes. Med Mol Morphol 2010;43: 13–18. [DOI] [PubMed] [Google Scholar]
- 63.Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology 2008;47:2026–2035. [DOI] [PubMed] [Google Scholar]
- 64.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012;142:938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu K, Zhao E, Ilyas G, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015;11:271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov 2016;15: 249–274. [DOI] [PubMed] [Google Scholar]
- 67.Sinha RA, Farah BL, Singh BK, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 2014;59: 1366–1380. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y, Chen ML, Zhou Y, et al. Resveratrol improves hepatic steatosis by inducing autophagy through the cAMP signaling pathway. Mol Nutr Food Res 2015;59:1443–1457. [DOI] [PubMed] [Google Scholar]
- 69.Li R, Guo E, Yang J, et al. 1,25(OH)2 D3 attenuates hepatic steatosis by inducing autophagy in mice. Obesity (Silver Spring) 2017;25:561–571. [DOI] [PubMed] [Google Scholar]
- 70.Zhang E, Yin S, Song X, Fan L, Hu H. Glycycoumarin inhibits hepatocyte lipoapoptosis through activation of autophagy and inhibition of ER stress/GSK-3-mediated mitochondrial pathway. Sci Rep 2016;6:38138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov 2017;16:487–511. [DOI] [PMC free article] [PubMed] [Google Scholar]