

SUMMARY
Analysis of cancer-derived extracellular vesicles (EVs) in biofluids potentially provides a source of disease biomarkers. At present there is no procedure to systematically identify which antigens should be targeted to differentiate cancer-derived from normal host cell-derived EVs. Here, we propose a computational framework that integrates information about membrane proteins in tumors and normal tissues from databases: UniProt, The Cancer Genome Atlas, the Genotype-Tissue Expression Project, and the Human Protein Atlas. We developed two methods to assess capture of EVs from specific cell types. (1) We used palmitoylated fluorescent protein (palmtdTomato) to label tumor-derived EVs. Beads displaying antibodies of interest were incubated with conditioned medium from palmtdTomato-expressing cells. Bound EVs were quantified using flow cytometry. (2) We also showed that membrane-bound Gaussia luciferase allows the detection of cancer-derived EVs in blood of tumor-bearing animals. Our analytical and validation platform should be applicable to identify antigens on EVs from any tumor type.
Graphical Abstract
In Brief
Cancer cell-derived extracellular vesicles (EVs) can be used in diagnostics, but their enrichment remain s challenging. Zaborowski et al. identify membrane proteins enriched on the surface of cancer cells compared with normal tissues using TCGA, the Human Protein Atlas, and GTEx and present methods to measure immunocapture of cancer EVs in vitro and in animal models.
INTRODUCTION
Extracellular vesicles (EVs) are nanosize, membrane-encased vesicles that transport DNA, RNA, lipids, proteins, and metabolites among cells (Abels and Breakefield, 2016; Skog et al., 2008; Tkach and Théry, 2016; Valadi et al., 2007; Zaborowski et al., 2015). The profile of cargo within EVs released into the extracellular space corresponds to the status of the cell of origin. EVs from tumor cells carry a distinctive RNA profile that is detectable in peripheral blood (Balaj et al., 2011; Noerholm et al., 2012; Skog et al., 2008). The detection of defined mutations, as well as changes in levels of extracellular RNA or DNA, has the potential to become a highly sensitive diagnostic tool (Quinn et al., 2015). For example, RT-PCR has been used in the detection of rare mutant copies of epidermal growth factor receptor variant III (EGFRvIII) mRNA in EVs in serum and cerebral spinal fluid from glioblastoma (GBM) patients (Figueroa et al., 2017; Skog et al., 2008).
The detection of EVs that originate specifically from tumor cells within the pool of highly abundant vesicles derived from normal host cells in biofluids remains challenging. It is anticipated that enrichment of cancer-derived EVs and downstream analysis of their contents will increase the sensitivity and specificity of diagnostic assays, irrespective of their type (mRNA, microRNA [miRNA], protein, lipids, metabolites, or DNA). The repertoire of antigens on the cellular plasma membrane is partially reflected in surface proteins on EVs (Raposo and Stoorvogel, 2013). Consequently, antigens expressed on the surface of cancer cells are typically present on the surface of their EVs, providing the ability to isolate them selectively (Im et al., 2014). Indeed, the expression of many antigens, including epithelial cell adhesion molecule (EpCAM), CD24, EGFR, claudin 3, platelet-derived growth factor receptor alpha (PDGFR-α), and podoplanin (PDPN), is highly consistent among ovarian cancers, as are some of the same and other antigens for GBM cells and the EVs released by them (Im et al., 2014; Peterson et al., 2013; Reátegui et al., 2018; Runz et al., 2007; Shao et al., 2012; Yang et al., 2017). The strategy of EV enrichment by antibody capture combined with analysis of mRNA in serum EVs proved informative in monitoring of the response of GBM patients to temozolomide (TMZ) chemotherapy (Shao et al., 2015).
Designing assays on the basis of immunocapture of cancer-derived EVs poses technical challenges: (1) it remains unclear what is the best way to select a set of antigens to target, and selection is typically based on assumptions from cell culture-based studies; (2) it is critical to have highly specific antibodies to tumor-enriched antigens on the surface of EVs; (3) few readout strategies are available to quantitatively and specifically detect the number of tumor-derived EVs captured, which makes optimization of methods difficult; (4) although numerous antigens can be tested on tumor cells grown in culture, it remains unknown what their levels are on corresponding EVs in biofluids; and (5) the observation that certain ‘‘free’’ antigens are elevated in biofluids, such as peripheral blood in cancer patients as opposed to healthy controls, though informative, is not conclusive and may vary among patients with the same type of cancer. Antigens and miRNAs detected in biofluids may also originate from host cells (e.g., leukocytes, platelets, red blood cells, endothelial cells) in response to tumor growth. In such situations, analysis of the collective contents of all EVs in the biofluid could be misleading, and diagnostic assays may give false-positive results, for example, if other disorders trigger similar host cell responses. For example, miR21 is elevated in plasma of patients with GBM (Akers et al., 2017), lymphoma (van Eijndhoven et al., 2016), cholangiocarcinoma (Correa-Gallego et al., 2016), and Crohn’s disease (Adams et al., 2014).
In this study we define a computational method on the basis of public databases that indicates which membrane proteins are highly expressed in a given tumor type while being present at low levels in other healthy tissues. We applied this algorithm to high-grade serous ovarian tumors. We also designed two independent validation methods to determine the efficiency of capture of cancer-derived EVs and to track the release of cancer EVs in the course of tumor growth in vivo. For this we used two reporter proteins, a palmitoylated fluorescent protein (palmtdTomato) (Lai et al., 2015) and membrane-bound Gaussia luciferase (mbGluc; Lai et al., 2014), which serve to label EVs derived only from the cells of interest. We demonstrate that EVs captured with various antibodies against antigens exposed on cancer cells can be selectively detected by fluorescent or bioluminescence signals, respectively, reflecting the efficiency of capture. The strength of these signals allowed enhanced detection of cancer cell-derived EVs in the serum in xenograft mouse tumor models, thereby defining a strategy for evaluating diagnostic and prognostic value of tumor-derived EVs in pre-clinical animal studies.
RESULTS
In Silico Identification of Tumor-Enriched Membrane Proteins
In order to define a set of surface proteins, we selected all human proteins localized to membranes on the basis of UniProt database description (The UniProt Consortium, 2017) (Figure 1A). mRNA expression of membrane proteins (on the basis of UniProt description) was assessed in ovarian cancer samples from a group of 489 patients on the basis of microarray data available through TCGA (Cancer Genome Atlas Research Network, 2011). EVs released from cancer cells into peripheral blood intermix with the numerous populations of EVs from other tissues. Therefore, we aimed to determine surface proteins highly expressed in tumors that at the same time have minimal expression in all other normal tissues. We computed a mean mRNA expression level across all normal tissues on the basis of data available through the Genotype-Tissue Expression Project (GTEx) (Lonsdale et al., 2013). To target cell surface proteins, we further sorted out proteins that were either experimentally confirmed or computationally predicted to contain an extracellular domain on the basis of the UniProt database (The UniProt Consortium, 2017), which narrowed down the final set from 4,255 to 1,451 genes. In this way we excluded membrane proteins that are anchored at the internal plasma membrane or reside within membranes of intracellular organelles. Although various proteins or other molecules can be presented on the surface of EVs that are not transmembrane proteins, use of antibodies against intracellular proteins could result in the capture of freely floating proteins coming from cells that have undergone apoptosis or necrosis. To avoid this confounding factor, we focused on those membrane proteins for which the structure predicts an extracellular domain. Among the genes representing membrane proteins highly expressed in normal tissues and not enriched in tumors, we identified CD63 and CD81 (Figure 1B), which are ubiquitous EV markers expressed by the majority of cells (Kowal et al., 2016). It should be noted that because of post-transcriptional and post-translational processing, the level of an mRNA does not necessarily correlate directly with its protein level. For this reason we evaluated protein levels in normal tissues and ovarian tumors as determined by immunohistochemistry available through the Human Protein Atlas (Uhlén et al., 2015). Our aim was to identify proteins with high staining in ovarian tumors and minimal staining in normal tissues (Figure 1C). Within a set of proteins with high differences between tumor and normal tissues, we found, among others, those that are well known to be overexpressed in ovarian cancer tumors, such as EpCAM, CLDN8, and the clinically used biomarker CA125 (MUC16). Finally, we defined the group of candidate surface proteins enriched in tumors compared with normal tissues that were overexpressed in terms of both mRNA and protein levels (Figure 1D). The final set of genes encoding extracellular antigenic epitopes with the highest difference between ovarian tumors and normal tissues consisted of membrane receptors (TFRC, OPRK1, EBAG9, HTR5A, GABRB1, and CD47), adhesion proteins (EpCAM, CLDN18, and CLDN1), and molecule transporters (KCNG1, KCNH2, and SLC22A11) (Figure 1E). The candidate sets we identified overlapped substantially with expression levels in these tumors on the basis of both microarray and RNA sequencing TCGA data (Figure S1).
Figure 1. Algorithm for Identifying Tumor-Enriched Membrane Proteins.
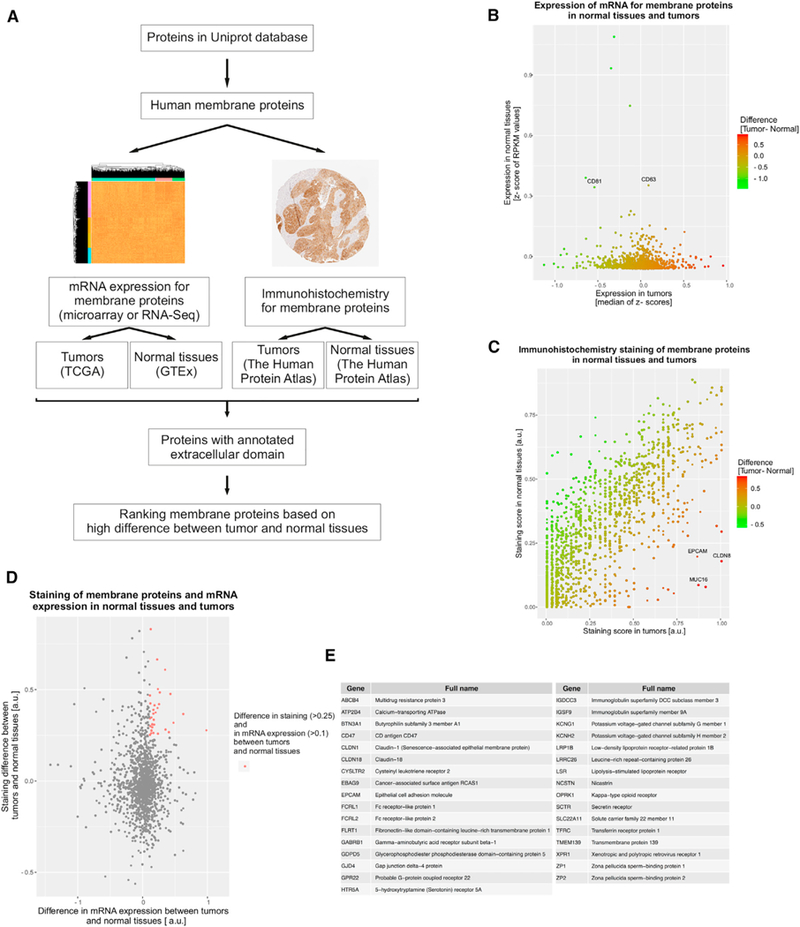
(A) Summary of computational steps.
(B) Z scores of membrane proteins expression averaged across patients with ovarian cancer from TCGA are plotted against Z score of RPKM (reads per kilobase of transcript per million mapped reads) values from RNA sequencing (RNA-seq) across all normal tissues from healthy volunteers included in the GTEx database. Plot shows the proteins with Z scores of RPKM values in normal tissues less than 1, because the analysis is focused on the proteins with low expression in normal tissues.
(C) Immunohistochemical staining of membrane proteins from normal tissues and ovarian cancer tumors on the basis of the Human Protein Atlas.
(D) Merge of transcriptome and immunohistochemistry data to identify membrane proteins with the highest expression in tumors and the lowest in all other healthy tissues.
(E) List of membrane proteins with high expression in tumors and low expression in all other healthy tissues on the basis of immunohistochemistry and mRNA data (red dots in plot D).
See also Figure S1.
Efficiency of EV Isolation Can Be Measured Using Palmitoylated tdTomato Labeling
We developed two validation methods on the basis of membrane-bound reporter proteins. Both can be used to assess expression of membrane proteins on the surface of EVs. One uses a palmitoylated fluorescent protein (palmtdTomato) that labels all cell membranes (Lai et al., 2015). Our previous work demonstrated that cells transduced with a lentiviral vector expressing palmtdTomato release labeled EVs (Figure 2A) (Lai et al., 2015). Indeed, different ovarian cancer cell lines transduced with an expression cassette for palmtdTomato showed strong membrane expression of this fluorescent protein (Figure 2B). We used a set of antibodies to capture antigens (CD24, MUC18, and EpCAM) shown to be expressed on ovarian cancer EVs (Im et al., 2014). Following 300 × g centrifugation to remove cells, we incubated conditioned medium containing EVs from palmtdTomato-positive ovarian tumor cells with streptavidin-coated polystyrene beads conjugated with biotinylated antibodies (Figure 2C). We visualized foci of increased palmtdTomato fluorescence on the beads covered with anti-CD24 antibodies corresponding to EVs. That signal was not observed with a control for unspecific binding with beads covered with IgG antibody (Figure 2D). To quantify the bound EVs, we counted fluorescence signal per bead by means of flow cytometry. Indeed, we observed that incubation with antibodies to antigens expressed on DF30 and Kuramochi ovarian cancer cells led to the capture of more fluorescently labeled beads in comparison with unspecific IgG binding (Figure 2E). We also observed that the percentage of beads considered as positive on the basis of the threshold set using background IgG signal, though relatively low, was highly reproducible across replicates (Figure 2F). This is consistent with single-vesicle analysis showing that EVs from the same cancer cells express only a subset of antigenic markers (Lee et al., 2018). Previous studies have shown that profiles of proteins on EVs reflect their expression in cells of origin (Im et al., 2014; Shao et al., 2012). We found that the percentage of positive beads for which the fluorescence signal exceeded threshold (on the basis of IgG background) overlapped well with the level of protein expression on the cell surface as measured by flow cytometry in a panel of five ovarian cancer cell lines (Figures 2G and 2H). Thanks to the quantitative nature of this assay, it should prove useful for testing candidate antigens for immunocapture of EVs and determining which panel of antigens performs optimally.
Figure 2. Validation Method Based on Palmitoylated tdTomato to Estimate Expression of Membrane Proteins on EVs.
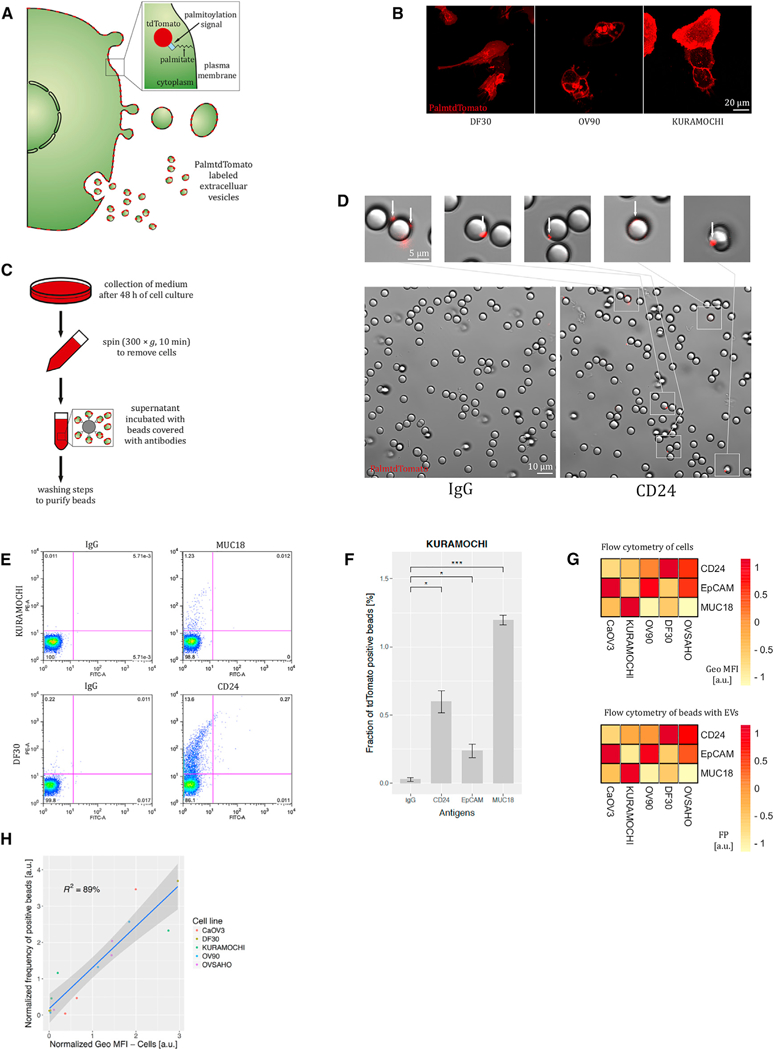
(A) Schematic diagram of cell membrane and EV labeling with palmtdTomato.
(B) Membrane expression of palmtdTomato in ovarian cancer cells. Confocal microscopy; scale bar, 20 μm.
(C) Experimental flowchart to capture EVs from conditioned medium by antibody-coated beads.
(D) Images of beads after incubation with the medium conditioned by palmtdTomato-positive cells. Scale bars, 10 and 5 μm in the big and small images, respectively.
(E) Flow cytometry quantification of the beads incubated with the medium conditioned with palmtdTomato-positive DF30 and Kuramochi cells (charts representative of three experiments).
(F) Fraction of palmtdTomato-positive beads depending on the surface antigen expression on EVs from Kuramochi cells (n = 3).
(G) Comparison of membrane protein expression on the ovarian cancer cells (flow cytometry) and on EVs as quantified by flow cytometry of beads (n = 3). Geo MFI, geometric mean fluorescence intensity; FP, fraction of palmtdTomato-positive beads out of all input beads. The ranges of geo MFI and FP of all antigens from one cell line (column-wise) were transformed to the range [−1, 1].
(H) Relationship between geometric mean fluorescence intensity (geo MFI) in flow cytometry of cells and fraction of palmtdTomato-positive beads (FP) (n = 3). Data presented in (B)–(H) come from cells transduced with lentivirus to stably express palmtdTomato.
Membrane-Bound Gaussia Luciferase Enables Assessment of EV Capture In Vitro
As an independent method to estimate the number of cancer cell-derived EVs, we used an mbGluc (also referred to as GlucB) reporter (Figure 3A) (Niers et al., 2012). Gaussia luciferase (Gluc) is a highly sensitive reporter protein whose expression is linear with respect to cancer cell proliferation in culture and in vivo (Badr and Tannous, 2011; Tannous, 2009). In the mbGluc construct, the transmembrane domain of the PDGFR is fused to Gluc, with Gluc exposed on the cell surface (Figure 3A) (Niers et al., 2012). As a result, all membrane-derived structures released from the cell, including EVs, are labeled with mbGluc (Figure 3B) (Lai et al., 2014). This construct also expresses GFP, and therefore cells transduced with a lentivirus vector bearing this mbGluc construct can be enriched using fluorescence-activated cell sorting (FACS) in order to achieve a more homogeneous population in terms of high levels of transgene expression. We stably transduced two ovarian cancer cell lines, OVCAR5 and A2780, with mbGluc. To confirm that expression of mbGluc was proportional to cell proliferation, we used a double reporter system with firefly luciferase (Fluc) and mbGluc (Figure 3B). We corroborated that structures pelleted upon 100,000 × g centrifugation (100K pellet) have the morphology of EVs when analyzed using transmission electron microscopy (TEM) and can be immunolabeled with antibodies to CD63, a marker of EVs, and antibodies to Gluc (Figure 3C; lower magnification images are presented in Figure S2). We confirmed by western blot analysis that mbGluc was present in structures pelleted at 2,000, 10,000, and 100,000 × g centrifugation, which correspond to EVs of various sizes (Figure 3D).
Figure 3. Membrane-Bound Gaussia Luciferase for Validation of EV Capture In Vitro.
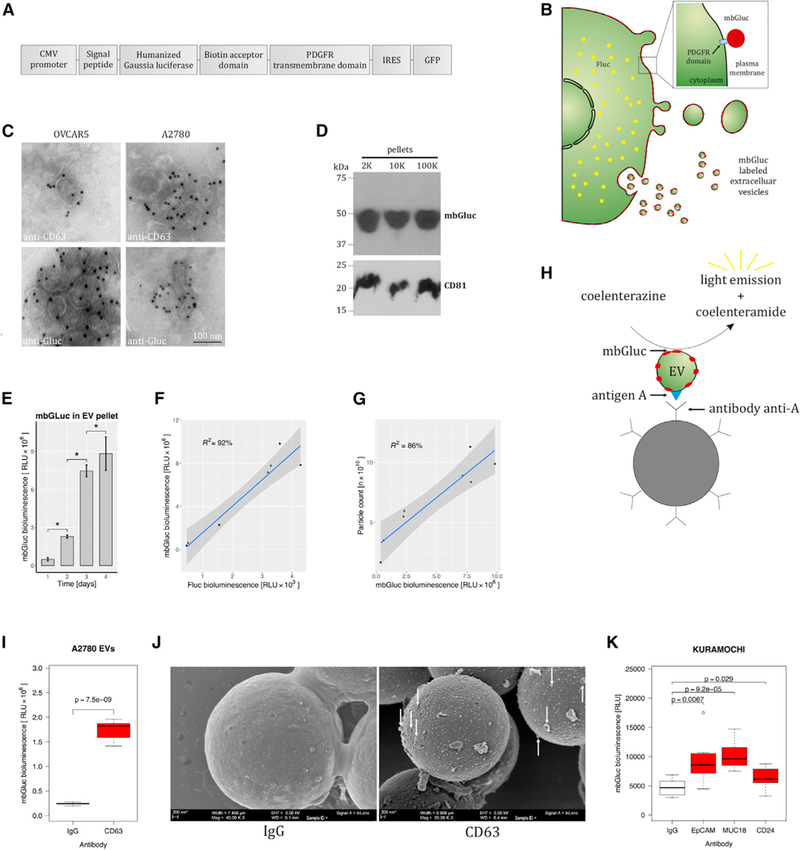
(A) Gaussia luciferase (Gluc) was fused to a transmembrane domain in order to incorporate it onto membrane-derived structures, including EVs.
(B) Schematic diagram of cell membrane and EV labeling with mbGluc.
(C) Transmission electron micrographs. EVs were isolated by ultracentrifugation (100,000 × g, 2 h) from OVCAR5 and A2780 cells and immunolabeled with anti-CD63 and anti-Gluc. See also Figure S2.
(D) Western blot analysis of proteins extracted from 2,000 × g (2K), 10,000 × g (10K), and 100,000 × g (100K) pellets isolated from conditioned medium of A2780 mbGluc-positive cells.
(E) mbGluc activity in the 100K pellets collected from A2780 cells after 1, 2, 3, or 4 days of culture (n = 2, each five technical replicates).
(F) mbGluc activity in the 100K pellets in relationship to Fluc activity that reflects A2780 cell number (n = 2, each three technical replicates).
(G) Relationship between mbGluc activity in the 100K pellets and EV number estimated by nanoparticle tracking analysis (NTA) from A2780 cells (n = 2, each five technical replicates).
(H) Schematic illustrating EV with antigen A on its surface binding to an antibody to antigen A that is attached to a bead. Reaction catalyzed by mbGluc generates bioluminescence, which in principle is proportional to number of captured EVs.
(I) mbGluc activity of EVs isolated by ultracentrifugation and captured by anti-CD63 tetraspanin, compared with control unspecific binding with IgG (n = 3).
(J) Scanning electron microscopy of beads with EVs captured with either IgG or anti-CD63 antibodies; scale bar, 200 nm.
(K) mbGluc activity from beads with EVs captured with antibodies against EpCAM, MUC18, or CD24 antigens compared with IgG from conditioned medium of Kuramochi cells (n = 3).
Data presented in (C)–(K) come from cells transduced with lentivirus to stably express mbGluc.
Subsequently, we verified that the mbGluc bioluminescent signal in the 100K pellet correlated with the number of EVs. We collected EVs from media conditioned by cells after 1, 2, 3, and 4 days in culture. The mbGluc signal in the 100K pellet derived from these cultures increased over time (Figure 3E) and was proportional to the Fluc signal from cells (Figure 3F). EV number monitored using nanoparticle tracking analysis (NTA) and mbGluc activity in the 100K pellet were also positively correlated (Figure 3G). Next, we hypothesized that if antibody capture via an antigen expressed on the extracellular surface is effective, it should result in a higher yield of EVs and thus emit more bioluminescence from mbGluc compared with non-specific binding (Figure 3H). Indeed, we observed elevated bioluminescence from the 100K pellet incubated with beads covered with antibodies to a marker of EVs, CD63, in comparison with the same sample captured non-specifically with IgG (Figure 3I). Scanning electron microscopy (SEM) of the same specimen confirmed more effective capture of EVs with CD63 antibodies than with IgG (Figure 3J). Measurement of bioluminescence from EVs captured directly from the conditioned medium of ovarian tumor cells (after removal of cells by 300 × g centrifugation for 10 min) with anti-EpCAM, anti-MUC18, and anti-CD24 antibodies also demonstrated enriched binding (Figure 3K). We conclude that measurement of mbGluc in EVs isolated by ultra-centrifugation reflects EVs number and, indirectly, cell number and is useful for measuring effectiveness of immunocapture in conditioned medium without additional sample concentration.
mbGluc Bioluminescence Reflects Efficacy of Cancer-Derived EV Isolation In Vivo
In order to test whether mbGluc can be used to monitor tumor growth in vivo, we established xenograft mouse models by intraperitoneal injection of OVCAR5 (mbGluc+, Fluc+) and A2780 (mbGluc+, Fluc+) cell lines, as well as a PBS control group. Tumor growth estimated by bioluminescent imaging of Fluc signal and activity of mbGluc in serum were measured weekly for 5 weeks (Figure 4A). Within 1 week after tumor implantation, the activity of mbGluc measured in 10 μL of serum had increased significantly above background level, indicating high sensitivity of our assay (Figure 4B). Weekly measurements of mbGluc activity in serum revealed a gradual increase over time in both xenograft models, with very low background in control animals (Figures 4C and 4D). The tumor growth as measured by in vivo Fluc bioluminescence imaging correlated with the mbGluc activity in serum (Figures 4E and 4F; r > 0.8). From these experiments we conclude that detection of mbGluc activity in peripheral blood can be used as a sensitive measure of tumor growth.
Figure 4. Monitoring of Tumor Growth and EV Release In Vivo by mbGluc.
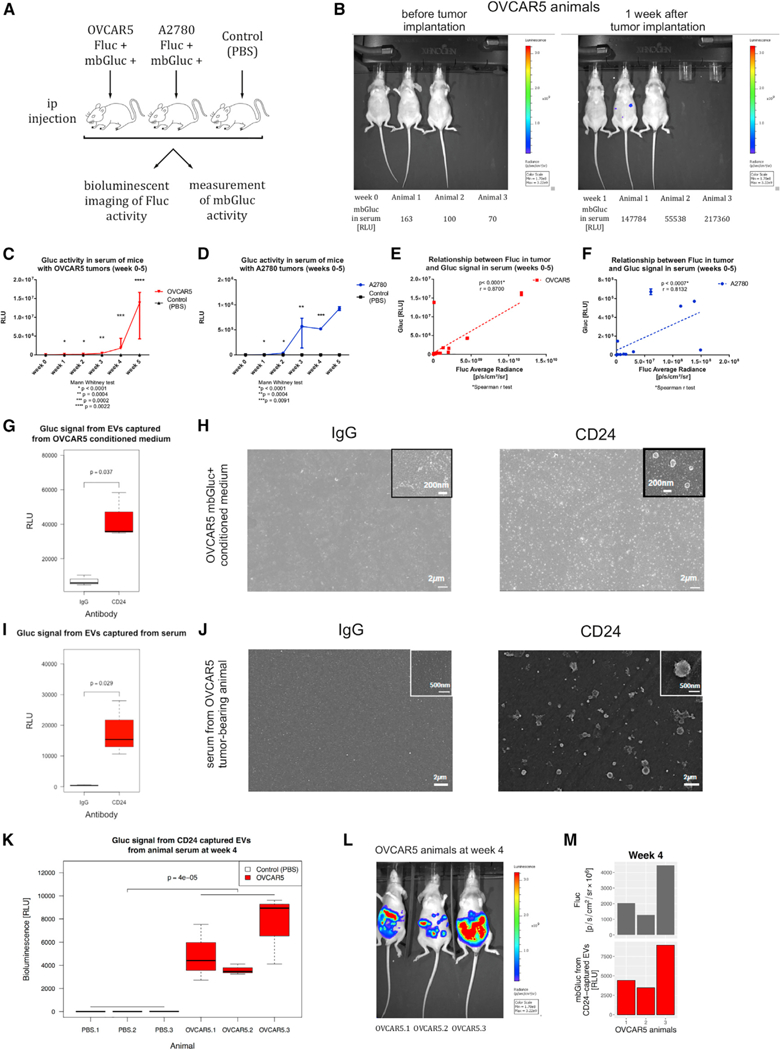
(A) OVCAR5 and A2780 ovarian cancer cell lines were injected intraperitoneally (i.p.) into nude mice (A2780, n = 3; OVCAR5, n = 3; PBS injection as a control, n = 3). Tumor growth was tracked once a week by Fluc in vivo bioluminescence imaging. In parallel, serum was collected to detect mbGluc signal (n = 3).
(B) Fluc in vivo bioluminescence imaging (top) and mbGluc activity measured in serum (without processing to isolate EVs) of individual OVCAR5 tumor-bearing animals (bottom) 1 week after implantation (n = 3).
(C and D) mbGluc signal in serum tracked during tumor progression in OVCAR5 (C) and A2780 (D) animals (n = 3 in each group).
(E and F) Correlation between tumor growth estimated by Fluc signal and mbGluc bioluminescence measured in serum of tumor-bearing animals with OVCAR5, (E) and A2780 (F) cells (n = 3 in each group).
(G) mbGluc bioluminescence signal from EVs captured by anti-CD24 antibody compared with unspecific binding (IgG) from OVCAR5 conditioned medium (n = 3).
(H) Scanning electron microscopy of structures captured by either IgG or anti-CD24 antibodies from OVCAR5 conditioned medium on a flat surface covered with neutravidin (magnification 8,000x; scale bar, 2 mm).
(I) mbGluc activity from EVs captured by anti-CD24 antibody compared with IgG in serum of OVCAR5 tumor-bearing animal (n = 4).
(J) Scanning electron microscopy of EVs isolated by either IgG or anti-CD24 from serum of OVCAR5 tumor-bearing animals on a flat surface covered with neutravidin (magnification 6,200x; scale bar, 2 mm).
(K) mbGluc activity of EVs isolated by anti-CD24 antibody from serum of control (PBS) (n = 3) and OVCAR5 (n = 3) animals at week 4 of tumor growth.
(L) Imaging of tumors by bioluminescence of Fluc in OVCAR5 animals (n = 3) at week 4 of tumor growth.
(M) Comparison of tumor size as measured by Fluc with mbGluc activity of EVs isolated by anti-CD24 antibody from serum of OVCAR5 animals (n = 3) at week 4 of tumor growth.
Cells potentially also release non-membrane-bound Gluc. Membrane-bound proteins with extracellular domain such as mbGluc may be subject to protease-induced cleavage that results in freely floating non-EV-bound proteins. For example, it has been demonstrated that EpCAM protein can be affected by such a cleavage (Rupp et al., 2011). In order to specifically measure the signal corresponding to EVs, we captured EVs with an antibody that detects a conventional membrane-associated antigen. To demonstrate that we could use a surface protein to capture EVs and measure bioluminescence, we chose CD24 antigen, which was reported in previous studies to be expressed in ovarian cancer cells (Im et al., 2014; Runz et al., 2007). We incubated conditioned medium from OVCAR5 mbGluc-positive cells with anti-CD24- or IgG-coated chips to control for non-specific binding. An enrichment of bioluminescent signal indicated that CD24 antigen was on the same EVs as mbGluc (Figure 4G). Captured structures (Figure 4H) visualized on SEM resembled EVs in size and morphology on the basis of previous studies (Raposo and Stoorvogel, 2013). SEM pictures also confirmed more numerous EVs on CD24 than on IgG chips, being barely detectable on the latter (Figure 4H). To test if this approach works for tumors in vivo, we incubated 7 μL of serum from OVCAR5 mbGluc-positive tumor-bearing animal with anti-CD24 antibody- and IgG-coated glass chips. We confirmed that capturing EVs with anti-CD24, compared with non-specific IgG, enriched both mbGluc bioluminescent signal (Figure 4I) and the number of EVs observed on SEM (Figure 4J). The bioluminescent signal from CD24-captured EVs was detected in all three tumor-bearing animals with very low background in control animals (Figure 4K). In addition, we observed that signal from EVs corresponded to tumor volume as measured by Fluc imaging (Figures 4L and 4M). Taken together, these data indicate that mbGluc signal reflects tumor volume and can be used as a measure of EV isolation efficiency in biofluids in xenograft models.
CD47, CD71, and EpCAM Are Expressed on Ovarian Cancer-Derived EVs
Next, we applied both palmtdTomato- and mbGluc-based assays to test selected surface antigens identified by our computational pipeline. We observed that both anti-CD47 and anti-CD71 (TFRC), but not anti-placental-like alkaline phosphatase (PALP) antibodies, were efficient in capture of EVs derived from ovarian cancer lines on the basis of the flow cytometry bead assay (Figure 5A). We demonstrated that both CD47 and CD71 were effective in EV capture from all five tested lines (Figure 5B). Bioluminescent assay using mbGluc confirmed good performance of CD47 and CD71 antibodies in this regard (Figure 5C). By means of flow cytometry we showed that a panel of antibodies consisting of anti-CD47, anti-CD71, and anti-EpCAM outperformed single antibodies (Figure 5D). Using the mbGluc-based method, we demonstrated that both anti-CD47 and anti-CD71 were effective in capturing tumor-derived EVs from peripheral blood serum (7 μL) in the OVCAR5 xenograft model (Figure 5E). To elucidate how these antibodies would perform in specific capture of cancer-derived EVs in human peripheral blood, we spiked in conditioned medium from Kuramochi cell line concentrated using a 100 kDa filter into plasma from a healthy donor (Figure S3A). On the basis of flow cytometry assay, we were able to isolate Kuramochi cancer cell-derived EVs from human plasma (Figure S3B). We isolated EVs from peritoneal fluid from patients affected with ovarian cancer and from individuals with benign conditions complicated with ascites, such as cirrhosis and hepatitis. After serial centrifugation and filtration steps, EVs were lysed and protein expression was analyzed using western blots. All samples contained both CD63 and beta(B)-actin (Figure 5F). We demonstrated, however, that surface protein EpCAM was detectable only in EVs isolated from patients with ovarian cancer, as opposed to the group with benign liver conditions.
Figure 5. CD47, CD71, and EpCAM Are Expressed on Ovarian Cancer-Derived EVs.
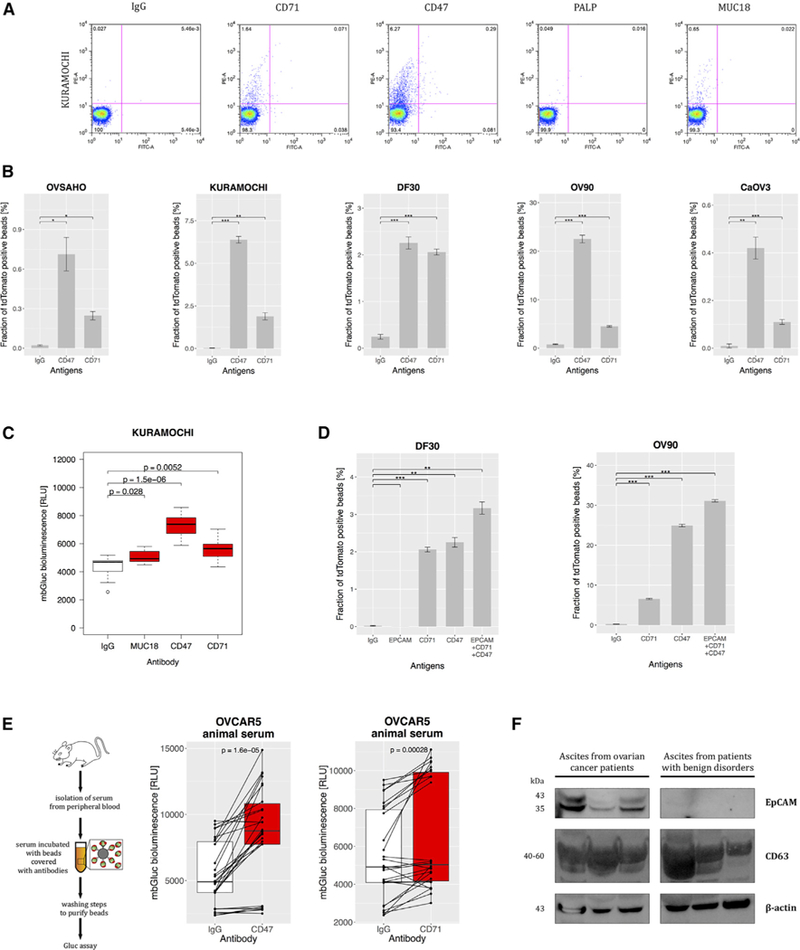
(A) Flow cytometry of the beads covered with IgG and antibodies against CD71, CD47, PALP, or MUC18 incubated with the medium conditioned by Kuramochi cells (palmtdTomato positive) (charts representative of three experiments).
(B) Fraction of palmtdTomato-positive beads depending on CD47 and CD71 expression on EVs from the panel of five cell lines (n = 3).
(C) mbGluc activity from beads with EVs captured by antibodies against MUC18, CD47, and CD71 from conditioned medium of Kuramochi cells (following removal of cells; n = 3).
(D) Fraction of palmtdTomato-positive beads depending on antigen expression on EVs from DF30 and OV90 cell lines (n = 3).
(E) Diagram of in vivo experiment to measure capture of EVs from serum of OVCAR5 tumor-bearing animal (left) and mbGluc activity from beads with EVs depending on the antigen expression (right) (n = 3 animals, two serum samples were processed from each animal, bioluminescence read in three to five technical replicates).
(F) EVs were isolated by differential centrifugation and filtration steps from ascites from patients with ovarian cancer (n = 3) and with benign conditions such as cirrhosis or hepatitis (n = 3). Proteins were resolved by SDS-PAGE and stained with antibodies to EpCAM, CD63, and B(beta)-actin and detected with chemiluminescence.
See also Figure S3.
DISCUSSION
Analysis of EVs which are derived from tissues of interest, especially from cancer cells, offers a promising diagnostic approach. Many EV isolation techniques have been developed using antibodies to EV surface antigens and different detection mechanisms (Im et al., 2014; Jeong et al., 2016; Yoshioka et al., 2014). The success of these methods depends on the choice and quality of antibodies. In many studies the panel of antigens is selected on the basis of an analysis of literature. In this work we presented a computational approach for identification of antigens tailored to the tumor type. Once an antigen is chosen, however, there is usually a variety of antibodies available that target different, but not necessarily extracellular, epitopes and may have poorly defined affinity. To address this point, we designed two validation strategies that can be applied both to in vitro and in vivo derived samples. Thanks to expression of proteins with palmitoylation signal or mbGluc, we were able to verify whether chosen antibodies effectively bound EVs derived from ovarian cancer cells.
Identification of membrane proteins enriched on cancer cells is potentially beneficial both for diagnostic and therapeutic applications. For example, detection of folate receptor alpha has been used for fluorescence-guided surgery in ovarian cancer (Tummers et al., 2016). Determination of new, more specific cancer-enriched membrane proteins can provide a step toward improvement of those approaches. In our study we screened for 1,451 proteins potentially highly expressed on ovarian cancer cells and having low or non-detectable expression in healthy tissues. We identified three major groups of proteins: membrane receptors, adhesion proteins, and molecule transporters. Interestingly, some of them included neuronal receptors, such as that for opioid receptor kappa 1 (OPRK1), 5-hydroxytryptamine (serotonin) 5A (HTR5A), and gamma-aminobutyric acid (GABA) subunit beta-1 (GABRB1). Importantly, some of these molecules have been implicated in aggressive cancer phenotypes. For instance, the expression of serotonin receptors has been associated with increased proliferation index and tumor size in hepatocellular carcinoma (Soll et al., 2012). Proteins from the group of adhesion proteins, claudins appear to increase invasiveness and survival of ovarian cancer cells (Agarwal et al., 2005). Another study corroborated that CLDN18 was expressed almost exclusively in pancreatic, esophageal, ovarian, and lung cancer tissues (Sahin et al., 2008). The group of solute transporters has been shown to be deregulated and associated with prognosis in patients with pancreatic cancer (Mohelnikova-Duchonova et al., 2013; Pedersen and Stock, 2013). For example, SLC28A1 transporter expression was related to poor overall patient survival (Mohelnikova-Duchonova et al., 2013; Pedersen and Stock, 2013). Taken together, our algorithm identified many proteins biologically relevant in cancer, suggesting that less studied proteins identified by this method should be good candidates for future research as surface proteins important in neoplastic cell expansion.
The identified antigens for EVs are not entirely unique to ovarian cancer tissue and are detected in some degree in other types of cancer, normal tissues, and biofluids. On the basis of our computational analysis, there were no normal proteins that were expressed only in cancer cells and completely absent in normal tissues, although some cancers express neoantigens unique to the tumor. On the basis of our results, for normal proteins it is not a question of the presence or absence of expression but rather of relative quantity. Our aim was to identify a set of proteins that would allow enrichment of tumor-derived EVs on the basis of their higher expression in cancer than in normal tissues. This type of approach has been successfully applied in the field of circulating tumor cells with respect to selected proteins (Alix-Panabières and Pantel, 2014). For instance, though EpCAM can be found in some normal epithelial cells, many research groups use it as one of antigens to enrich circulating tumor cells (Alix-Panabières and Pantel, 2016). Correspondingly, we observe that EpCAM is detectable in peritoneal fluid in patients with ovarian cancer and not in individuals with benign conditions (Figure 5F).
Studying proteins exposed on the surface of cells requires biochemical approaches that enrich for membrane proteins. Plasma membrane proteins on intact cells can be first biotinylated and then the isolated fraction subjected to mass spectrometry (Ghosh et al., 2017; Shin et al., 2003). Membranes and all their constituents can be also extracted from lysed cells by means of a Percoll/sucrose density gradient (Lund et al., 2009). Those methods, though very informative, require special preparation of a sample as well as time- and cost-consuming downstream processing. In our study, we proposed a systems approach on the basis of the search of membrane proteins using publicly available transcriptomic and immunohistochemical datasets. This method offers a number of advantages as a platform to enrich our knowledge about surface proteins expressed by cancer cells. First, we have performed a screen in samples from large groups of patients querying numerous proteins with an extracellular domain, which increases the chance of discovering less known surface proteins (Figures 1A–1E). Indeed, along with well-characterized species such as EpCAM and CA125, we have identified numerous other antigens, including neuronal receptors and solute transporters enriched on ovarian cancer cells. Second, it is important to consider that biofluids, such as peripheral blood, contain a mixture of antigens from many tissues. Our approach compared expression of surface proteins in tumors with those in all healthy tissues thanks to integration of TCGA and GTEx databases. This type of analysis would be highly challenging to perform using methods on the basis of mass spectrometry for so many samples in a unique standardized way. Third, ovarian cancers, similar to other tumor types, are highly heterogeneous, and their profiles vary from patient to patient. Thanks to analyzing a large patient population, we were able to identify subgroups of patients with overexpression of particular antigens. In this way our method enabled design of a panel of antibodies that cover different subgroups, reflecting the heterogeneity of tumors in this cancer patient population. The goal of this study was to identify antigens suitable for isolation of tumor-derived EVs in biofluids, but information obtained from our analysis can also indicate promising candidates for antibody-mediated targeted therapies as well as specific capture of circulating tumor cells.
Our approach is based on the premise that a repertoire of membrane proteins on EVs is a derivative of proteins expressed on the cell surface, as supported by many in vitro and clinical studies (Im et al., 2014; Jeong et al., 2016; Shao et al., 2012). It should be underscored, however, that there can be certain proteins that are favorably packaged in EVs, and the proportions between cellular and EV profiles might be disturbed. Our computational analysis ideally would be performed with the use of data on surface proteins identified directly in EVs. Initially, we considered using EV-specific databases such as ExoCarta (Mathivanan and Simpson, 2009) or Vesiclepedia (Kalra et al., 2012). Those resources have facilitated research and promoted standardization in the field of EVs. Although highly informative, they remain mainly qualitative in nature. It is their considerable advantage that they list studies in which a particular protein has been identified and provide information on the method of study. Unfortunately, in the current version there is no information about the level of a given cargo protein in EVs from specific cell types. Furthermore, given the variety of methods used for protein detection (western blot, ELISA, mass spectrometry, etc.) and EV isolation (ultracentrifugation, precipitation, antibody isolation, commercial kits, etc.), it is not possible to compare levels of proteins among studies. This information, though highly useful, does not form a quantitative and uniform dataset that could be used for the purpose of this study. Therefore, we focused our analysis on the profiles of membrane proteins in tissue databases that were generated in a standardized fashion, such as TCGA (The Cancer Genome Atlas), the GTEx, and the Human Protein Atlas. In these cases, although the data were generated in several research centers, standardized protocols were followed. Thus, levels of mRNA representative of membrane proteins are comparable across numerous patients affected with cancer and healthy donors. It should be kept in mind, however, that an actual level of protein in EVs might differ from expression of mRNA for that protein in the tissue. Because our analysis is based on expression in tissues and not directly in EVs, being aware of that limitation we used our in silico prediction as an indicator of potential candidate proteins that need to be further validated and we provided methods to test them.
Biomarker analysis restricted to comparison of RNA expression across tumors and tissues may have limitations, because post-transcriptional modifications and RNA processing may significantly affect the final protein level. For this reason, we included in our discovery platform the information about immunohistochemistry staining, which was available, albeit from a small number of patients. Small patient numbers contributing information about actual protein concentrations are a clear limitation that can affect final results of our analysis. A future version of this method would ideally include more proteomic data from both tumor and normal tissues. The analysis can become even more informative if it takes into consideration data on protein variants and post-translational modification, such as glycosylation patterns. Another potential limitation comes from lack of clarity about which cell types/tissues contribute the most and in what proportions to the pool of EVs in a given biofluid. When this is established, we will be able to improve the algorithm by assigning weights to tissue types. Being aware of those limitations in our in silico prediction, we developed validation techniques to enable future testing of selected antigens.
The knowledge of surface antigen repertoire is not sufficient to design a cancer detection assay. We have demonstrated validation techniques on the basis of the labeling methods developed in our group (Lai et al., 2014, 2015; Niers et al., 2012). Antibodies against membrane proteins may not necessarily target epitopes located on extracellular domains and as such may not be useful in an identification assay without a denaturation step, which is not desired if the contents of EVs are to be analyzed downstream. Therefore, we think that the presented algorithm should be used in combination with two complementary validation methods, as proposed in this study. Another argument in support of our validation methods is that the affinity of antibodies may not be sufficient to capture EVs efficiently. More important, it remains unclear how antibodies that are characterized by other means will perform in biofluids, where they may encounter many other potentially similar epitopes. Although other available assays have demonstrated the binding of EVs to antibodies, they were not designed to establish whether bound EVs are tumor derived or come from the tissue harboring the tumor or biofluids (i.e., some antigens could be released by normal cells in response to tumor but not by cancer cells) (Im et al., 2014). Our validation approach proved useful not only in in vitro experiments with EVs from conditioned medium but also in a xenograft model of ovarian cancer. This technique can be equally applicable in testing new biomarkers in pre-clinical studies with patient-derived xenograft models. Another future application of our validation assays would be the ability to track the changes in surface antigens in the course of tumor progression and response to therapy. Taken together, our validation methods are able to identify antibodies that can capture EVs derived specifically from cancer cells in biofluids.
Our aim was to design a computational approach and validation methods that would allow the evaluation of candidate antigens for capture of EVs in vitro and in pre-clinical (in vivo) models. To date researchers approaching design of EV biomarker assays have not demonstrated a way to systematically address these points. We are aware that developing a biomarker assay is a long process that is hard to encompass in a single study, especially when it includes a systems approach to identify surface epitopes. Our aim was not to design a final biomarker assay but to narrow down a search space by indicating promising candidates for future studies and providing tools to test them. We are aware that proper validation of a set of markers will require numerous groups of controls and patients.
In this study we have presented an in silico screen combined with validation techniques that together constitute a discovery platform for surface antigens enriched on cancer cells and EVs derived from them. Through this methodology we have both confirmed the utility of well-established antigens and identified other promising surface proteins for ovarian cancer. Our system uses data from a large group of patients with ovarian cancer and thus may systematically identify interesting antigen candidates that so far have been identified mainly on the basis of literature searches. We hope that the surface protein identification method described here will facilitate future studies in search of an optimal panel of antibodies to capture cancer-derived EVs from biofluids.
STAR✶METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mikołaj Piotr Zaborowski.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
Human ovarian cancer cell lines, CaOV3 (female), OV90 (female) and OVCAR5 (female) were obtained from American Type Culture Collection. A2780 (female) was acquired from European Collection of Cell Cultures. Kuramochi (female) and OVSAHO (female) cells were a generous gift from Dr. Kristi Egland (Sanford Research, South Dakota). Primary high grade ovarian cancer cells (DF30, female) were isolated as previously described (Davidowitz et al., 2014). CaOV3 were cultured in Dulbecco’s modified essential medium (Corning, catalog No. 10–013-CV). OV90, OVCAR5 and A2780 were grown in RPMI-1640 medium (Corning, catalog No. 10–040-CM). KURAMOCHI, OVSAHO, DF30 (Iwanicki et al., 2016) were cultured in 1:1 mixture of MCDB 105 (Cell Applications, catalog No. 117–500) medium and Medium 199 (GIBCO, catalog No. 11150059). All media were supplemented with fetal bovine serum (Gemini Bio-products, catalog No. 900–208) and penicillin-streptomycin solution (Corning, catalog No. 30–002-Cl) at the final concentrations 10% and 1%, respectively. Cells were kept in a humidified incubator with 5% CO2 at 37°C. All cells were tested for mycoplasma infection (MycoAlert Mycoplasma Detection Kit, Lonza, catalog No. LT07–218) and found to be negative. Cells were counted by means of Bright-Line Hemacytometer (Sigma, catalog No. Z359629).
Animal Protocol and Collection of Animal Samples
Female athymic nude mice aged 5–7 weeks of weight 25–30 g were housed in the MGH Animal Facility and handled under the policies of the MGH Review Board. Xenograft models were established by intraperitoneal injection of 3 × 106 cells thoroughly washed free of the culture medium with cold PBS by three centrifugation cycles at 1,000 rpm for 5 min and resuspension in 1 mL PBS. Control animals were injected with 1 mL PBS. All mice were subject to bioluminescence imaging for detection of Fluc signal (in autoexposition mode, IVIS Caliper LS system, Preseton Brook Runcorn, UK) once a week after intraperitoneal injection of D-luciferin (50 mg/kg). The Fluc signal was expressed as an average photon flux (photons/sec/cm2/surface area). Following submandibular vein incision with a lancet around 60–150 mL of peripheral blood was collected into BD Microtainer tube with no additive (Becton Dickinson, catalog No. 365957), as described (Golde et al., 2005). Blood was also collected by cardiac puncture at the time of animal sacrifice. Blood was centrifuged at 1,200 × g for 15 min at room temperature. The supernatant was centrifuged at 1,200 × g for 5 min at room temperature and the supernatant of that spin was referred to as a serum. Serum was stored in −80°C and analyzed collectively after completion of the study.
Clinical Samples
Patients were enrolled according to an Institutional Review Board approved protocol with informed consent. Ascites fluid samples were collected from female patients aged between 35 and 81 years in Massachusetts General Hospital Abdominal Imaging and Intervention suites. We analyzed 6 ascites samples, three from ovarian cancer patients (P1 - P3) and three from non-cancer patients (P4 - P6) with ascites induced by benign conditions such as cirrhosis or hepatitis. Cancer diagnoses were confirmed by histological examination and clinical imaging. Due to various substances in ascites, e.g., floating tissues, cell debris, and fat clogs, EVs were obtained through serial centrifugation steps combined with filtration: (1) 3,500 × g for 20 min at 4°C; (2) filtration of supernatant through 40 μm filter; (3) filtrate centrifuged at 14,000 × g for 20 min at 4°C; (4) filtration of supernatant through 40 mm filter; 0.45 mm filter and 0.22 mm filter; (5) filtrate centrifuged at 24,200 rpm for 70 min at 4°C; (6) pellet suspended in PBS centrifuged at 24,200 rpm for 70 min at 4°C. After ultracentrifugation, the EV pellet was resuspended in 300 μL PBS and stored in −80°C until analysis. The EV size and concentration were measured by NanoSight LM10 (Table below). Samples were further lysed by adding RIPA buffer (Thermo Scientific) supplemented with Halt protease inhibitor cocktail (Thermo Scientific). The total protein amount was assessed by Qubit protein assay (Invitrogen) (Table below). Plasma for the experiment presented in Figure S3A was received from an unidentified healthy donor from Blood Bank of Massachusetts General Hospital. 10 mL conditioned medium from Kuramochi cell line after cell removal (300 × g for 10 min at room temperature) was concentrated using a 100 kDa filter (Amicon Ultra-15 Centrifugal Fliters, catalog No. UFC910008) by centrifugation at 4,000 × g for 20 min at room temperature. The concentrate was spiked into 2 mL plasma mixed with 2 mL PBS. 300 μL plasma with spiked-in EVs per each replicate was incubated with beads covered with antibodies.
| Patient ascites | P1 | P2 | P3 | P4 | P5 | P6 |
|---|---|---|---|---|---|---|
| EV size (nm) | 138.5 | 199.4 | 232.8 | 272.4 | 285.8 | 278.7 |
| EV concentration (EV/mL) | 6.73E+09 | 8.94E+08 | 1.23E+09 | 4.54E+08 | 2.28E+07 | 4.14E+08 |
| Protein concentration (mg/mL) | 8.65 | 10.24 | 7.32 | 12.02 | 8.23 | 6.34 |
| EV numbers used in western blot | 1.56E+08 | 1.75E+07 | 3.36E+07 | 7.55E+06 | 5.54E+05 | 1.31E+07 |
METHOD DETAILS
Algorithm to Identify Tumor-enriched Membrane Proteins
Names of human membrane proteins were retrieved from Uniprot database (The UniProt Consortium, 2017) by using filtering criteria: << locations:(location’’:Cell membrane [SL-0039]’’) AND organism’’:Homo sapiens (Human) [9606]’’ >> . Isoform names were removed. Transcriptome (microarray and RNA sequencing) data were obtained from TCGA using http://www.cbioportal.org as CGDS object compatible with downstream analysis with R programming (Cerami et al., 2012). For analysis of microarray data from ovarian cancer tumors, values provided as ‘‘ov_tcga_pub_mrna_median_Zscores’’ in the study labeled as ‘‘ov_tcga_pub’’ were selected (The Cancer Genome Atlas Research Network, 2011). Patients with missing values or genes defined only in a subset of patients were excluded from the analysis. In order to analyze RNA expression in normal tissues, the ‘‘GTEx_Analysis_v6_RNA-seq_RNA-SeQCv1.1.8_gene_rpkm.gct’’ file was downloaded from GTEx Consortium website (Lonsdale et al., 2013). It included RPKM values from RNA sequencing of organs and tissues from healthy volunteers. Genes referring to membrane proteins (as defined by Uniprot filters above) were selected for further analysis. For each gene, a mean RPKM value was calculated across all tissues and organs and all volunteers enrolled in the GTEx project. Obtained values were scaled to Z-scores across all genes in the analysis to determine those with the lowest expression. Immunohistochemistry values for all normal tissues and ovarian tumors were downloaded from The Human Protein Atlas Consortium website (Uhlén et al., 2015) as files ‘‘normal_tissue_ProteinAtlas.csv’’ and ‘‘cancer.csv,’’ respectively. Staining described as ‘‘High,’’ ‘‘Medium,’’ ‘‘Low,’’ ‘‘Undetectable’’ was assigned a score 3, 2, 1 and 0, respectively. The mean immunohistochemistry staining score was calculated across all patients and scaled to range [0–1]. Proteins with an extracellular domain were further selected from all gene names for both transcriptome and immunohistochemical analyses based on the description of the category ‘‘Topological.domain’’ from Uniprot database. For comparison of RNA-Seq and microarray input, ovarian cancer tumor values provided as ‘‘ov_tcga_rna_seq_v2_mrna_median_Zscores’’ in the study labeled as ‘‘ov_tcga’’ were selected. All data processing was performed in R programming language (version 3.4.1) using RStudio (version 0.98.1060).
Bioluminescence Assay
Gluc activity of in vitro samples (Figures 3E–3G) was measured in 10 μL loaded in triplicates onto a white 96-well luminometer plate (Greiner Bio-One International, catalog No. 655075). After 17 s from automated injection at the rate 47 μL/s of 50 μL of coelenterazine (Nanolight, 303–10, stock solution dissolved in methanol at 5 mg/mL) dissolved in PBS at 1.6 μg/mL, bioluminescence was detected during 1 s integration time in FlexStation 3 Reader (Molecular Devices). Animal serum samples were loaded in replicates of 10 μL and measured using the same procedure with coelenterazine at 50 μg/mL. Although for most of the samples three replicates were included, for some time points only two or one measurement were performed due to insufficient biofluid volume. Gluc activity of the structures captured on the antibody-coated chip was read separately in individual wells of a 24-well plate, right after manual injection of 50 μL coelenterazine at 50 μg/mL. Samples isolated with antibody-coated beads were divided in three replicates of 10 μL and measured in the white 96-well luminometer plate. Measurement of bioluminescence was performed after automated injection of coelenterazine at the rate 250 μL/s at 50 μg/mL in Synergy HTX multi-mode reader (Biotek Instruments). Fluc assay of in vitro samples was performed in cells pelleted at 300 × g for 5 min, lysed with 5X Reporter Lysis buffer (Promega, E397A) that was diluted to 1X solution in water. Triplicates of 60 μL were loaded on the white 96-well luminometer plate and after addition of 50 μL D-luciferin solution [D-luciferin (250 μg/mL) and ATP (0.5 mM) diluted in PBS], the luminescence was detected over 500 ms integration times.
Preparation of Beads Covered with Antibodies
Forty μl of beads (Spherotech, Streptavidin polystyrene, SVP-50–5) per sample were washed four times with 1 mL 2% BSA (centrifugation at 3,000 × g for 5 min). 1.5 μg biotinylated antibodies were added to beads dissolved in 100 μL 2% BSA. Following antibodies were used in the assay: anti-EpCAM (Abcam, catalog No. ab79079, clone VU-1D9, biotinylated), anti-CD47 (Biolegend, catalog No. 323104, biotinylated), anti-CD71 (eBioscience, catalog No. 13–0719, biotinylated), IgG1 (Biolegend, catalog No. 400102, clone MOPC-21, unconjugated), anti-CD24 (eBioscience, catalog No. 14–0247-82, clone eBioSN3, SN3 A5–2H10, unconjugated), anti-MUC18 (R&D Systems, catalog No. MAB932, clone 128018, unconjugated), anti-CD63 (Ancell, catalog No. 215–030, clone AHN16.1/46–4-5, biotinylated) and rabbit anti-PALP (Abcam, catalog No. ab118856, unconjugated). Unconjugated antibodies were biotinylated according to the procedure described below. The biotinylated antibodies were mixed with the beads overnight in HulaMixer® Sample Mixer (Thermo Fisher Scientific) at 4°C. Beads with antibodies were washed four times with 1 mL 2% BSA (centrifugation at 3,000 × g for 5 min) leaving 100 μL solution after each washing step. Three hundred mL conditioned cell medium after cell removal (300 × g for 10 min at room temperature) or 300 μL plasma with spiked-in EVs (experiment in Figure S3A) or 7 μL animal serum per replicate were mixed with beads (in 100 μL 2% BSA) covered with antibodies overnight at 4°C (cold room) in HulaMixer® Sample Mixer. After incubation with a sample, beads were washed four times with 1 mL 2% BSA (centrifugation at 3,000 × g for 5 min). The beads were resuspended in PBS to reach the same volume in all tubes. Ten μL was subsequently resuspended in 400 μL PBS and subject to flow cytometry. Three volumes of 10 μL were used for bioluminescence assay in triplicates.
Biotinylation of Antibodies
The antibodies were biotinylated by Sulfo-NHS-biotin (10 mM, Pierce) solution in PBS, following manufacturer’s instructions. Briefly, the mixture of antibody and Sulfo-NHS-biotin was incubated overnight at 4°C. Unreacted sulfo-NHS-biotin was removed using a Zeba spin desalting column (7K MWCO, Thermo Scientific).
Preparation of Glass Substrate Covered with NeutrAvidin
The glass substrate was immersed in 4% (v/v) 3-mercaptopropyl trimethoxysilane (Sigma-Aldrich) in ethanol for 30 min, followed by 0.01 M N-γ-maleimidobutyryl-oxysuccinimide ester (Sigma-Aldrich) in ethanol for 20 min. After each step, the glass substrate was immersed in ethanol for 5 min. The glass substrate was then incubated with 200 μg/mL NeutrAvidin (Sigma-Aldrich) in 0.2% BSA (Sigma-Aldrich) overnight at 4°C and washed and immersed in 0.2% BSA buffer for 5 min. Then polydimethylsiloxane (PDMS) block with 3 mm diameter hole was attached on top of the glass substrate and used as a well for the sample.
Transduction
After 24 hr of culture, the medium was replaced with the fresh culture medium with hexadimethrine bromide (Polybrene, Sigma, catalog No. H9268) at final concentration 4 μg/mL to enhance transduction efficiency. Previously described, mbGluc (CSCW-GlucB-IRES-GFP; Niers et al., 2012) or palmtdTomato (Lai et al., 2015) lentivirus vectors generated by MGH Vector Core (Boston, MA USA) were added to the media (300 μL at a titer 9.6 × 107 and 300 mL at a titer 3.8 × 107, respectively). To promote virus infection, cells were centrifuged at 1,800 rpm (Thermo Scientific Sorvall Four-Place Swinging Bucket Rotor, radius 117 mm) for 90 min at 4°C and transferred to the 37°C incubator. Medium was changed the next day. Transduction efficiency was monitored by GFP or tdTomato signal using an inverted epifluorescence microscope (TE 200-U, Nikon, Melville, NY). After a few days of culture, depending on proliferation rate, cells were FACS sorted with regard to GFP or tdTomato expression to select population with a homogeneous level of strong transgene expression.
EV Isolation from Conditioned Media
For the purpose of EV isolation, fetal bovine serum (Gemini Bio-products, catalog No. 900–208) was EV-depleted by ultracentrifugation at 40,400 rpm in 70 Ti rotor (fixed angle, average radius: 65.7mm, k-Factor: 44, Beckman, catalog No. 337922) in Optima L-90K ultracentrifuge (Beckman Coulter, catalog No. 365670) for 17 hr at 4°C. To collect EVs, fresh medium with 5% EV-depleted FBS was added to cultures in four 15 cm plates and collected 48 hr later when cells were at approximately 90% confluency. This conditioned medium was subject to serial centrifugation steps combined with filtration: (1) 300 × g for 10 min at 4°C (Thermo Scientific Sorvall Four-Place Swinging Bucket Rotor); (2) 2,000 × g for 10 min at 4°C (Thermo Scientific Sorvall Four-Place Swinging Bucket Rotor); (3) filtration through 0.8 μm filter (Millipore®, catalog No. SLAA033SS); (4) filtrate centrifuged at 40,400 rpm in 70 Ti rotor for 2 hr at 4°C in polypropylene tubes (Beckman Coulter, catalog No. 342414). The pellet was resuspended in 150 μL cold PBS previously filtered twice through 0.22 μm (Millipore® catalog No. SLGP033RS), unless otherwise specified. The resulting suspension was referred to as the ‘‘100K pellet’’.
Transmission Electron Microscopy and Immunolabeling
The 100K pellet was centrifuged at 20,000 × g for 30 min at 4°C and, after gentle removal of supernatant, fixed for 2 hr in 500 μL of 4% paraformaldehyde (32% aqueous solution, EM grade, Electron Microscopy Sciences, 15714-S) diluted in PBS. Fixed pellets were cryosectioned and immunolabeled with anti-Gluc (mouse; Nanolight) or anti-CD63 (mouse; BD Biosciences) followed by rabbit anti-mouse (Cappel/MP Biomedicals, LLC) and 5 nm protein A-gold (University Medical Center, Utrecht, the Netherlands). Images were captured using Tecnai G2 Spirit Bio TWIN transmission electron microscope.
Flow Cytometry
After trypsinization and addition of culture medium, 1 × 106 cells were centrifuged at 500 × g for 5 min and resuspended in 100 μL PBS with 2% BSA. Primary mouse monoclonal antibodies, anti-CD24 (clone eBioSN3, SN3 A5–2H10, eBioscience, 14–0247-82), anti-EpCAM (clone VU-1D9, Abcam, ab79079), anti-MUC18 (clone 128018, R&D Systems, MAB932) and IgG1, κ isotype control (clone MOPC-21, Biolegend, 400102) were added to final concentrations of 5 μg/mL. Following 45 min incubation on ice, cells were washed twice with PBS and resuspended for 20 min in 100 μL of goat anti-mouse IgG secondary antibody conjugated to Alexa Fluor® 647 (Invitrogen, A-21235) diluted in PBS to 5 μg/mL. After washing, the cell pellet was dissolved in 500 μL PBS and analyzed in LSRII flow cytometer (Becton Dickinson) applying the same voltage for detection of Alexa 647 across all samples. FlowJo (version 8.7) software was used to calculate geometric mean of fluorescence. Relative fluorescence intensity was expressed as a ratio of the geometric mean of fluorescence of each antigen to the geometric mean of fluorescence of isotype control. The range of relative fluorescence intensities of all antigens from a cell line was transformed to the range [−1,1] and plotted in a heatmap (Figure 2G, upper panel). Fluorescence of beads was analyzed in Fortessa X-20 (Becton Dickinson) flow cytometer. For each replicate 20,000 beads were analyzed. Fraction of palmtdTomato-positive beads (out of all input beads) was treated as a measure of capture intensity. The range of that intensity of all antigens from one cell line was transformed to the range [−1,1] and plotted in a heatmap (Figure 2G, lower panel). Normalized geometric mean of fluorescence (Figure 2H) was expressed as a ratio of relative fluorescence intensity of an antigen to the mean relative fluorescence intensities of all antigens from a cell line. Normalized frequency of positive beads (Figure 2H) was expressed as a ratio of fraction of palmtdTomato-positive beads for an antigen to the mean of fractions of palmtdTomato-positive beads of all antigens from a cell line.
Confocal Microscopy
Samples were imaged with an LSM710 inverted confocal microscope and a 63x oil (Zeiss Plan-Apochromat SF25 DIC, 1.4NA) or 20x (Zeiss, Plan-Apochromat, DIC, 0.8 NA) objectives (Zeiss). Images were processed with ImageJ (1.48v).
Western Blot of Cell Culture Samples
Samples were lysed in M-PER Mammalian Protein Extraction Reagent (Thermo Scientific, catalog No. 78501) buffer containing protease inhibitors (complete, Mini, Roche Diagnostics, catalog No. 04693159001). Protein concentration was determined by Bradford protein assay (Bio-Rad). Thirty micrograms of total protein was boiled for 5 min in SDS sample buffer (Boston BioProducts, catalog No. BP-110R), resolved by the NuPAGE™ gradient 4%–12% Bis-Tris Gel (Invitrogen, catalog No. NP0321BOX) with molecular weight standards (Precision Plus Protein Standards, Bio-Rad, catalog No. 161–0374), and transferred onto nitrocellulose membranes (0.2 mm, Bio-Rad, catalog No. 162–0112). The membranes were blocked with 5% non-fat milk (LabScientific, catalog No. M08425) and incubated overnight with anti-Gluc (rabbit polyclonal, Nanolight, catalog No. 401P) antibody at the dilution 1:1000 and anti-CD81 (mouse monoclonal, Santa Cruz Biotechnology, catalog No. sc-166029) antibody at the dilution 1:200. This was followed by binding of secondary antibodies conjugated to horseradish peroxidase (HRP; donkey, anti-Rabbit IgG, GE Healthcare, NA934–1ML and sheep anti-Mouse IgG, GE Healthcare, NA931–1ML) at the dilution 1:7500 and signal detection with a chemiluminescent substrate (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific, catalog No. 34077).
Western Blot of Clinical Ascites Samples
Twenty micrograms of total proteins (Bradford assay) were denatured at 70°C for 10 min in NuPAGE® LDS sample buffer (Invitrogen, catalog No. NP0007) supplemented by β-mercaptoethanol (Sigma, catalog No. M6250), resolved by the NuPAGE™ gradient 4%–12% Bis-Tris Gel (Invitrogen, catalog No. NP0321BOX) with molecular weight standards (Precision Plus Protein Standards, Bio-Rad, catalog No. 161–0374), and transferred onto nitrocellulose membranes (iBlot 2 Transfer Stacks, Invitrogen, catalog No. IB23001). The membranes were blocked with 5% non-fat milk (LabScientific, catalog No. M08425) and incubated overnight with anti-CD63 antibody (mouse monoclonal, R&D, catalog No. MAB 5048) at the dilution 1:1000, biotinylated anti-EpCAM antibody (mouse monoclonal, Abcam, catalog No. ab-79079) at the dilution 1:1000, and anti-β-actin antibody (mouse monoclonal, Sigma, catalog No. A5441) at the dilution of 1:5000. This was followed by binding of secondary antibodies conjugated to HRP (donkey, anti-Mouse IgG, GE Healthcare, NA931–1ML and streptavidin-horseradish peroxidase, Pierce, 21130–1ML) at the dilution 1:2000 and signal detection with a chemiluminescent substrate (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific, catalog No. 34077).
Nanoparticle Tracking Analysis
The resuspended 100K pellet was analyzed by means of Nanosight LM10 instrument (Malvern, Framingham, MA) equipped with AVT MARLIN F-033B IRF camera (Allied Vision Technologies) and NTA 3.1 Build 3.1.46 software. Samples were diluted in 1 mL freshly prepared (double filtered through 0.22 μm filter) PBS kept at room temperature and were measured in the dilution range 1:50–1:100. Minimum track length and blur were set automatically. All measurements were performed with temperature set to 22°C. Each sample was imaged in at least 5 technical replicates. All movies were recorded for 60 s with a screen gain and camera level set to 9 and 8, respectively. Processing of images was performed with detection threshold 2 and screen gain 10. All movies had at least 2,000 valid tracks.
Scanning Electron Microscope Sample Preparation
The EV immobilized glass substrate and beads were fixed using Karnovsky’s fixative and dehydrated with ethanol. Dehydrated samples were dried via a critical point dryer (Autosamdri 931, Tousimis) and coated with platinum and palladium (20/80) using a sputter coater (EMS300T-D, EMS). The samples were then imaged with a scanning electron microscope (Ultra Plus FESEM, Carl Zeiss).
QUANTIFICATION AND STATISTICAL ANALYSIS
Normality of distribution was verified with Shapiro test. Groups with normal distribution were compared with t-Student test. Groups with distribution deviating from normal were compared using Mann-Whitney test. A paired Wilcoxon test (Wilcoxon signed rank test) was performed to analyze data in animal experiment presented in Figure 5E. The results were considered significant for p values < 0.05. p values were either specified in the figure or denoted as asterisks: * p < 0.05; ** p < 0.005; *** p < 0.0005. Analysis of data was performed in R programming language (version 3.4.1) using RStudio (version 0.98.1060). Data for Figures 4C–4F were analyzed and plotted in GraphPad Prism (version 7.03). Plot whiskers extend to the most extreme data point which is within 1.5 times the interquartile range from the box in the boxplots in Figures 3I, 3K, 4G, 4I, 4K, 5C, and 5E. Plot whiskers extend to standard deviation from the bar in the barplots in Figures 2F, 3E, 5B, and 5D. Numbers of replicates are stated in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-EpCAM | Abcam | Abcam Cat# ab79079; RRID:AB_1603294 |
| Mouse monoclonal anti-CD47 | BioLegend | BioLegend Cat# 323104; RRID:AB_756134 |
| Mouse monoclonal anti-CD71 | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# 13–0719-82; RRID:AB_466504 |
| Mouse monoclonal IgG1 | Biolegend | Cat# 400102, clone MOPC-21 |
| Mouse monoclonal anti-CD24 | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# 14–0247-82; RRID:AB_467173 |
| Mouse monoclonal anti-MUC18 | R and D Systems | R and D Systems Cat# MAB932; RRID:AB_2143503 |
| Mouse monoclonal anti-CD63 | Ancell | Ancell Cat# 215–030; RRID:AB_2665375 |
| Rabbit polyclonal anti-PALP | Abcam | Abcam Cat# ab118856; RRID:AB_10900125 |
| Goat anti-mouse IgG secondary antibody conjugated to Alexa Fluor® 647 | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# A-21235; RRID:AB_2535804 |
| Rabbit polyclonal anti-Gluc | Nanolight | Nanolight Cat# 401P; RRID:AB_2572411 |
| Mouse monoclonal anti-CD81 | Santa Cruz Biotechnology | Santa Cruz Biotechnology Cat# sc-166029; RRID:AB_2275892 |
| Donkey anti-rabbit IgG | GE Healthcare | GE Healthcare Cat# NA934; RRID:AB_772206 |
| Sheep anti-mouse IgG | GE Healthcare | GE Healthcare Cat# NA931; RRID:AB_772210 |
| Mouse monoclonal anti-CD63 | R and D Systems | R and D Systems Cat# MAB5048; RRID:AB_2275726 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Sigma-Aldrich Cat# A5441; RRID:AB_476744 |
| Mouse monoclonal anti-Gluc | Nanolight | Nanolight Cat# 401M; RRID:AB_2572413 |
| Bacterial and Virus Strains | ||
| CSCW-GlucB-IRES-GFP lentivirus | MGH vector Core (Boston, MA USA) Niers et al.,2012 | N/A |
| PalmtdTomato lentivirus | MGH Vector Core (Boston, MGA USA); Lai et al 2015 | N/A |
| Biological Samples | ||
| Human plasma from unidentified healthy donors | Blood Bank of Massachusetts General Hospital | N/A |
| Ascites fluid from female patients aged between 35 and 81 years | Massachusetts General Hospital Abdominal Imaging and Intervention suites | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Streptavidin polystyrene beads | Spherotech | Cat# SVP-50–5 |
| Coelenterazine | Nanolight | Cat# 303–10 |
| D-luciferin | Gold Biotechnology | Cat# LUCNA-1 |
| (3-mercaptopropyl)trimethoxysilane | Sigma-Aldrich | Cat# 175617 |
| N-γ-maleimidobutyryl-oxysuccinimide ester | Sigma-Aldrich | Cat# 63175 |
| M-PER™ Mammalian Protein Extraction Reagent | Thermo Scientific | Cat# 78501 |
| 5 nm protein A-gold | University Medical Center, Utrecht, the Netherlands | N/A |
| Polybrene | Sigma | Cat# H9268 |
| 4% paraformaldehyde (32% aqueous solution, EM grade) | Electron Microscopy Sciences | Cat# 15714-S |
| Critical Commercial Assays | ||
| Sulfo-NHS-biotin | Thermo ScientificPierce | Cat# 21217 |
| Zeba spin desalting column | Thermo Scientific | Cat# 89882 |
| NuPAGE™ gradient 4%- 12% Bis-Tris Gel | Invitrogen | Cat# NP0321BOX |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Scientific | Cat# 34077 |
| Experimental Models: Cell Lines | ||
| CaOV3 (Female | ATCC | ATCC Cat# HTB-75; RRID:CVCL_0201 |
| OV90 (Female) | ATCC | ATCC Cat# CRL-11732; RRID:CVCL_3768 |
| OVCAR5 (Female) | ATCC | NCI-DTP Cat# OVCAR-5; RRID:CVCL_1628 |
| A2780 (Female) | European Collection of Cell Cultures | NCI-DTP Cat# A2780; RRID:CVCL_0134 |
| Kuramochi (Female) | gift from Dr. Kristi Egland (Sanford Research, South Dakota) | JCRB Cat# JCRB0098; RRID:CVCL_1345 |
| OVSAHO (Female) | gift from Dr. Kristi Egland (Sanford Research, South Dakota) | JCRB Cat# JCRB1046; RRID:CVCL_3114 |
| DF30 (Female) | Davidowitz et al., 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| Female athymic nude mice (age 5–7 weeks, weight 25–30 g) | Charles River Lab | N/A |
| Software and Algorithms | ||
| FlowJo (version 8.7) | https://www.flowjo.com | N/A |
| ImageJ (1.48v) | https://imagej.nih.gov/ij/download.html | N/A |
| R (version 3.4.1) | https://cran.r-project.org | N/A |
| R studio (version 0.98.1060) | https://www.rstudio.com/products/rstudio/download/ | N/A |
| GraphPad Prism (version 7.03) | https://www.graphpad.com/scientific-software/prism/ | N/A |
| NTA 3.1 Build 3.1.46 software | Malvern, Framingham, MA | N/A |
| Other | ||
| BD Microtainer tube with no additive for collection of animal serum | Becton Dickinson | Cat# 365957 |
| 0.8 µm filter | Millipore® | Cat# SLAA033SS |
| polypropylene tubes for ultracentrifugation | Beckman Coulter | Cat# 342414 |
Highlights.
Membrane proteins on cancer cells can be identified in TCGA and Human Protein Atlas
Capture of EVs by targeted membrane proteins can be measured by palmtdTomato labeling
Isolation of cancer-derived EVs from animal serum can be evaluated by mbGluc signal
ACKNOWLEDGMENTS
This publication is part of the NIH Extracellular RNA Communication Consortium paper package and was supported by the NIH Common Fund’s exRNA Communication Program. We would like to thank Ms. Suzanne McDavitt for her supportive editorial assistance, Zofia Zaborowska for the graphical preparation of selected schematics, Amelia Burke for support in accessing facilities useful for this study, and Maria Ericsson for performing electron microscopic imaging. This work has been supported by NIH National Cancer Institute (NCI) grants P01CA069246, R35 CA232103, and U19 CA179563 through the Office of Strategic Coordination/Office of the NIH Director and the Richard Floor Biorepository Fund (M.P.Z., X.O.B., and C.P.L.). M.P.Z. received a scholarship from the Kosciuszko Foundation and is thankful for the support of the Poznan’ University of Medical Sciences. H.L. was supported in part by NIH grants R01CA229777 and U01CA233360 and the Massachusetts General Hospital (MGH) Scholar Fund. H.-Y.L. was supported by the MGH Fund for Medical Discovery Fellowship. M.J.B. was supported by U.S. Department of Defense (DOD) grant W81XWH-16–1-0593. We would like to thank K. Conway, M. Zinter, and E. Tabet for the production of lentiviruses (MGH Vector Core, Boston, MA, supported by NIH/National Institute of Neurological Disorders and Stroke [NINDS] grant P30NS045776 [B.A.T. and X.O.B.]). Part of the computational analysis was conducted on the Orchestra High Performance Compute Cluster at Harvard Medical School (National Center for Research Resources [NCRR] 1S10RR028832–01). The Genotype-Tissue Expression Project (GTEx) was supported by the Common Fund of the Office of the Director of the NIH, the NCI, the National Human Genome Research Institute (NHGRI), the National Heart, Lung, and Blood Institute (NHLBI), the National Institute on Drug Abuse (NIDA), the National Institute of Mental Health (NIMH), and NINDS. Cytometric findings reported here were performed in the MGH Department of Pathology Flow and Image Cytometry Research Core, which obtained support from the NIH Shared Instrumentation Program with grants 1S10OD012027–01A1, 1S10OD016372–01, 1S10RR020936–01, and 1S10RR023440–01A1. Confocal microscopy was performed in the MGH Cancer Center/Molecular Pathology Confocal Core.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.003.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abels ER, and Breakefield XO (2016). Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell. Mol. Neurobiol 36, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams AT, Kennedy NA, Hansen R, Ventham NT, OʼLeary KR, Drummond HE, Noble CL, El-Omar E, Russell RK, Wilson DC, et al. (2014). Two-stage genome-wide methylation profiling in childhood-onset Crohn’s Disease implicates epigenetic alterations at the VMP1/MIR21 and HLA loci. Inflamm. Bowel Dis 20, 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal R, D’Souza T, and Morin PJ (2005). Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res 65, 7378–7385. [DOI] [PubMed] [Google Scholar]
- Akers JC, Hua W, Li H, Ramakrishnan V, Yang Z, Quan K, Zhu W, Li J, Figueroa J, Hirshman BR, et al. (2017). A cerebrospinal fluid microRNA signature as biomarker for glioblastoma. Oncotarget 8, 68769–68779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alix-Panabières C, and Pantel K (2014). Challenges in circulating tumour cell research. Nat. Rev. Cancer 14, 623–631. [DOI] [PubMed] [Google Scholar]
- Alix-Panabières C, and Pantel K (2016). Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov 6, 479–491. [DOI] [PubMed] [Google Scholar]
- Badr CE, and Tannous BA (2011). Bioluminescence imaging: progress and applications. Trends Biotechnol 29, 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaj L, Lessard R, Dai L, Cho Y-J, Pomeroy SL, Breakefield XO, and Skog J (2011). Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun 2, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Gallego C, Maddalo D, Doussot A, Kemeny N, Kingham TP, Allen PJ, D’Angelica MI, DeMatteo RP, Betel D, Klimstra D, et al. (2016). Circulating plasma levels of microRNA-21 and microRNA-221 are potential diagnostic markers for primary intrahepatic cholangiocarcinoma. PLoS ONE 11, e0163699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidowitz RA, Selfors LM, Iwanicki MP, Elias KM, Karst A, Piao H, Ince TA, Drage MG, Dering J, Konecny GE, et al. (2014). Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Invest 124, 2611–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa JM, Skog J, Akers J, Li H, Komotar R, Jensen R, Ringel F, Yang I, Kalkanis S, Thompson R, et al. (2017). Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro-oncol 19, 1494–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Funk CC, Caballero J, Shah N, Rouleau K, Earls JC, Soroceanu L, Foltz G, Cobbs CS, Price ND, and Hood L (2017). A cell-surface membrane protein signature for glioblastoma. Cell Syst 4, 516–529.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde WT, Gollobin P, and Rodriguez LL (2005). A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim. (NY) 34, 39–43. [DOI] [PubMed] [Google Scholar]
- Im H, Shao H, Park YI, Peterson VM, Castro CM, Weissleder R, and Lee H (2014). Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat. Biotechnol 32, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanicki MP, Chen H-Y, Iavarone C, Zervantonakis IK, Muranen T, Novak M, Ince TA, Drapkin R, and Brugge JS (2016). Mutant p53 regulates ovarian cancer transformed phenotypes through autocrine matrix deposition. JCI Insight 1, e86829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S, Park J, Pathania D, Castro CM, Weissleder R, and Lee H (2016). Integrated magneto-electrochemical sensor for exosome analysis. ACS Nano 10, 1802–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, Bond VC, Borràs FE, Breakefield X, Budnik V, et al. (2012). Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol 10, e1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, and Théry C (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U S A 113, E968–E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CP, Mardini O, Ericsson M, Prabhakar S, Maguire C, Chen JW, Tannous BA, and Breakefield XO (2014). Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 8, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CP, Kim EY, Badr CE, Weissleder R, Mempel TR, Tannous BA, and Breakefield XO (2015). Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun 6, 7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Fraser K, Ghaddar B, Yang K, Kim E, Balaj L, Chiocca EA, Breakefield XO, Lee H, and Weissleder R (2018). Multiplexed profiling of single extracellular vesicles. ACS Nano 12, 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, et al. ; GTEx Consortium (2013). The Genotype-Tissue Expression (GTEx) Project. Nat. Genet 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund R, Leth-Larsen R, Jensen ON, and Ditzel HJ (2009). Efficient isolation and quantitative proteomic analysis of cancer cell plasma membrane proteins for identification of metastasis-associated cell surface markers. J. Proteome Res 8, 3078–3090. [DOI] [PubMed] [Google Scholar]
- Mathivanan S, and Simpson RJ (2009). ExoCarta: a compendium of exosomal proteins and RNA. Proteomics 9, 4997–5000. [DOI] [PubMed] [Google Scholar]
- Mohelnikova-Duchonova B, Brynychova V, Hlavac V, Kocik M, Oliverius M, Hlavsa J, Honsova E, Mazanec J, Kala Z, Melichar B, and Soucek P (2013). The association between the expression of solute carrier transporters and the prognosis of pancreatic cancer. Cancer Chemother. Pharmacol 72, 669–682. [DOI] [PubMed] [Google Scholar]
- Niers JM, Chen JW, Lewandrowski G, Kerami M, Garanger E, Wojtkiewicz G, Waterman P, Keliher E, Weissleder R, and Tannous BA (2012). Single reporter for targeted multimodal in vivo imaging. J. Am. Chem. Soc 134, 5149–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noerholm M, Balaj L, Limperg T, Salehi A, Zhu LD, Hochberg FH, Breakefield XO, Carter BS, and Skog J (2012). RNA expression patterns in serum microvesicles from patients with glioblastoma multiforme and controls. BMC Cancer 12, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SF, and Stock C (2013). Ion channels and transporters in cancer: pathophysiology, regulation, and clinical potential. Cancer Res 73, 1658–1661. [DOI] [PubMed] [Google Scholar]
- Peterson VM, Castro CM, Chung J, Miller NC, Ullal AV, Castano MD, Penson RT, Lee H, Birrer MJ, and Weissleder R (2013). Ascites analysis by a microfluidic chip allows tumor-cell profiling. Proc. Natl. Acad. Sci. U S A 110, E4978–E4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JF, Patel T, Wong D, Das S, Freedman JE, Laurent LC, Carter BS, Hochberg F, Van Keuren-Jensen K, Huentelman M, et al. (2015). Extracellular RNAs: development as biomarkers of human disease. J. Extracell. Vesicles 4, 27495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, and Stoorvogel W (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol 200, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reátegui E, van der Vos KE, Lai CP, Zeinali M, Atai NA, Aldikacti B, Floyd FPH Jr., H Khankhel A, Thapar V, Hochberg FH, et al. (2018). Engineered nanointerfaces for microfluidic isolation and molecular profiling of tumor-specific extracellular vesicles. Nat. Commun 9, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz S, Keller S, Rupp C, Stoeck A, Issa Y, Koensgen D, Mustea A, Sehouli J, Kristiansen G, and Altevogt P (2007). Malignant ascites-derived exosomes of ovarian carcinoma patients contain CD24 and EpCAM. Gynecol. Oncol 107, 563–571. [DOI] [PubMed] [Google Scholar]
- Rupp A-K, Rupp C, Keller S, Brase JC, Ehehalt R, Fogel M, Moldenhauer G, Marmé F, Sültmann H, and Altevogt P (2011). Loss of EpCAM expression in breast cancer derived serum exosomes: role of proteolytic cleavage. Gynecol. Oncol 122, 437–446. [DOI] [PubMed] [Google Scholar]
- Sahin U, Koslowski M, Dhaene K, Usener D, Brandenburg G, Seitz G, Huber C, and Türeci O (2008). Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin. Cancer Res 14, 7624–7634. [DOI] [PubMed] [Google Scholar]
- Shao H, Chung J, Balaj L, Charest A, Bigner DD, Carter BS, Hochberg FH, Breakefield XO, Weissleder R, and Lee H (2012). Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med 18, 1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Chung J, Lee K, Balaj L, Min C, Carter BS, Hochberg FH, Breakefield XO, Lee H, and Weissleder R (2015). Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun 6, 6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, Zhao R, Puravs E, Tra J, Michael CW, et al. (2003). Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem 278, 7607–7616. [DOI] [PubMed] [Google Scholar]
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr., Carter BS, Krichevsky AM, and Breakefield XO (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soll C, Riener M-O, Oberkofler CE, Hellerbrand C, Wild PJ, DeOliveira ML, and Clavien P-A (2012). Expression of serotonin receptors in human hepatocellular cancer. Clin. Cancer Res 18, 5902–5910. [DOI] [PubMed] [Google Scholar]
- Tannous BA (2009). Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat. Protoc 4, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res 45 (D1), D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, and Théry C (2016). Communication by extracellular vesicles: where we are and where we need to go. Cell 164, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Tummers QRJG, Hoogstins CES, Gaarenstroom KN, de Kroon CD, van Poelgeest MIE, Vuyk J, Bosse T, Smit VTHBM, van de Velde CJH, Cohen AF, et al. (2016). Intraoperative imaging of folate receptor alpha positive ovarian and breast cancer using the tumor specific agent EC17. Oncotarget 7, 32144–32155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, and Lötvall JO (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 9, 654–659. [DOI] [PubMed] [Google Scholar]
- van Eijndhoven MAJ, Zijlstra JM, Groenewegen NJ, Drees EEE, van Niele S, Baglio SR, Koppers-Lalic D, van der Voorn H., Libregts SFWM, Wauben MHM, et al. (2016). Plasma vesicle miRNAs for therapy response monitoring in Hodgkin lymphoma patients. JCI Insight 1, e89631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KS, Im H, Hong S, Pergolini I, Del Castillo AF, Wang R, Clardy S, Huang C-H, Pille C, Ferrone S, et al. (2017). Multiparametric plasma EV profiling facilitates diagnosis of pancreatic malignancy. Sci. Transl. Med 9, eaal3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka Y, Kosaka N, Konishi Y, Ohta H, Okamoto H, Sonoda H, Nonaka R, Yamamoto H, Ishii H, Mori M, et al. (2014). Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun 5, 3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborowski MP, Balaj L, Breakefield XO, and Lai CP (2015). Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience 65, 783–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.