

Abstract
Objectives
Alzheimer's disease (AD) is one of the most prevalent brain diseases among the elderly, majority of which is caused by abnormal deposition of amyloid beta‐peptide (Aβ). Galantamine, currently the first‐line drug in treatment of AD, has been shown to diminish Aβ‐induced neurotoxicity and exert favourable neuroprotective effects, but the detail mechanisms remain unclear.
Materials and methods
Effects of galantamine on Aβ‐induced cytotoxicity were checked by MTT, clone formation and apoptosis assays. The protein variations and reactive oxygen species (ROS) production were measured by western blotting analysis and dichloro‐dihydro‐fluorescein diacetate assay, respectively.
Results
Galantamine reversed Aβ‐induced cell growth inhibition and apoptosis in neuron cells PC12. Aβ activated the entire autophagy flux and accumulation of autophagosomes, and the inhibition of autophagy decreased the protein level of cleaved‐caspase‐3 and Aβ‐induced cytotoxicity. Meanwhile, galantamine suppressed Aβ‐mediated autophagy flux and accumulation of autophagosomes. Moreover, Aβ upregulated ROS accumulation, while ROS scavengers N‐acetyl‐l‐cysteine impaired Aβ‐mediated autophagy. Further investigation showed that galantamine downregulated NOX4 expression to inhibit Aβ‐mediated ROS accumulation and autophagy.
Conclusions
Galantamine inhibits Aβ‐induced cytostatic autophagy through decreasing ROS accumulation, providing new insights into deep understanding of AD progression and molecular basis of galantamine in neuroprotection.
1. INTRODUCTION
Alzheimer's disease (AD) is one of the most common mental disorders among elder patients, approximately 60%‐80% of which develop dementia eventually and lower the quality of lives.1 Galantamine is currently the first‐line pharmacological agent to treat AD, which has been shown to effectively improve the cognition of AD patients in a short time course.2, 3 The underling mechanism is that galantamine inhibits the function of cholinesterase, leading to the accumulation of acetylcholine and improve the condition. Although remarkable progression in the treatment of AD has been made, AD can be only temporarily relieved but not reversed; therefore, novel ideas are still urgently needed for successful treatment of AD.
Autophagy is characterized as an essential cellular self‐renewal process, which promote the degradation of damage components and recycling of building blocks to maintain energy homeostasis and facilitate cell survival under stress.4 This evolutionally conserved pro‐survival machinery was thought to play important roles in some conditions such as cancer development and cancer drug resistance.5 However, it has been reported that excessive autophagy could result in autophagic cell death, suggesting that autophagy functions diversely in different conditions.6 Importantly, autophagy has been also purposed as a double‐edged sword in the pathogenesis of AD.7, 8, 9 On the one hand, the amyloid beta‐peptide (Aβ) aggregates were shown to be degraded by autophagy‐lysosome pathway as other ready‐to‐degrade cargo, and the secretion of Aβ can also be inhibited by the activation of autophagy.10, 11 In vivo study found that the accumulation of Aβ and the consequent AD phenotype were accompanied by the downregulation of autophagy‐related gene expression during the aging of Drosophila melanogaster.12 These data indicate a neuroprotective role of autophagy in AD prevention. On the other hand, emerging evidences have reported that Aβ could be generated from autophagic vacuoles, suggesting the autophagy machinery may serve as a new source where Aβ was produced.13, 14 Targeting autophagy to explore the novel anti‐AD therapeutic strategies have been extensively investigated, but the results were largely disappointing at least partially due to the its complicated roles in AD progression.
Functioning as a stress‐relieving mechanism, autophagy is activated upon oxidative stress to restore cellular redox balance. Importantly, the accumulation of reactive oxygen species (ROS) in neuron cells has been characterized as another hallmark of AD progression. Protein oxidation by ROS can lead to their polymerization thereby form protein aggregation, resembling the abnormal deposition of Aβ peptides. In fact, the Met35 residue of Aβ is capable of being oxidized to form methionine sulfoxide, which renders conformational alterations and is associated with its neuron toxicity.15, 16 In addition, biomolecules other than proteins such as DNA and RNA have also been shown to be oxidized in AD brains.17, 18 Surprisingly, clinical studies have shown that antioxidant‐based treatments failed to cure the AD patients,19 suggesting that underlying mechanisms need to be further understood before this problem solved. Galantamine is a scavenger of ROS, which may contribute to its neuroprotective effect.20 However, the molecular events of ROS elimination by galantamine remain elusive, and whether autophagy plays any roles in this process is also unknown.
Here, we reported a new mechanism of treatment of AD with galantamine. We found that galantamine significantly inhibited the autophagy and apoptosis of neuron cell line PC12 induced by Aβ. Mechanistically, Aβ upregulates NOX4 expression and ROS accumulation, thereby stimulating excessive autophagy and consequent cell death of neuron cells. Galantamine profoundly inhibits the NOX4 expression to lower the ROS level, thus prevents the excessive activation of autophagy and protects neuron cells. Our data provide new insights of galantamine‐based AD treatment methods, and help to achieve a deep understanding of the molecular mechanisms underlying AD progression and therapy.
2. MATERIALS AND METHODS
2.1. Reagents
Galantamine was purchased from Xi'an Huilin Biological Technology Co., Ltd, China, Aβ1‐42 was purchased from Dalian Meilun Biological Technology Co., Ltd, China, 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐di‐phenyl (MTT), Hoechst 33342, Giemsa, chloroquine (CQ), 3‐methyladenine (3‐MA), lipopolysaccharide and N‐acetyl‐l‐cysteine (NAC) were purchased from Sigma (St. Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM) was purchased from Gibco, USA. Antibodies used in this study were against β‐actin and Beclin‐1 and p62 and LC3 and caspase‐3 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), NOX4 and NOIX2 and sheep anti‐mouse IgG‐HRP and sheep anti‐rabbit IgG‐HRP were purchased from Zen BioScience Co. Ltd, China.
2.2. Cell culture
Differentiated rat pheochromocytoma cell line PC12 was purchased from the ATCC and maintained in DMEM supplemented with 10% foetal bovine serum (BI), 100 U/mL penicillin (Invitrogen, Carlsbad, CA, USA) and 100 μg/mL streptomycin (Invitrogen). Cell culture was incubated in a humidified incubator under 5% CO2 atmosphere at 37°C. Cells were grown to 70% to 80% confluency in dishes or cell culture plates and treated under various conditions as indicated.
2.3. Aβ peptides
To prepare aggregated Aβ1‐42 fibrils, Aβ1‐42 was diluted to 5 mmol/L with phosphate‐buffered saline (PBS) and incubated at 37°C for 7 days. When using, it was diluted to different concentrations with serum‐free DMEM.
2.4. Cell viability assay
Cells were plated in 96‐well cell culture plates (3000 cells/well) and pretreated with various concentrations of galantamine (10, 20, 40, 80, 160 μmol/L) for 24 hours followed exposure to 2 μmol/L Aβ1‐42 for another 24 hours. Thereafter, cells were incubated with 5 mg/mL MTT (15 mL/well) for 4 hours. The supernatant then was removed and 150 mL of dimethyl sulfoxide was added. Absorbance was measured at 570 nm on an enzyme‐linked immunosorbent assay reader. For relative quantification, the value of absorbance in each group was normalized to that in the control group.
2.5. Colony formation assay
Colony formation assay was performed as reported previously with minor modifications. Briefly, 300 cells were seeded in a 60‐mm dish. The cells were continuously cultured in the presence or absence of galantamine and Aβ for 2 weeks. Clones were stained with Giemsa and counted using a microscope. Only those cell clusters containing more than 50 cells were considered as clones.
2.6. TUNEL assay
The cells were plated in 24‐well plate and fixed by 4% paraformaldehyde followed by TUNEL staining using the DeadEnd Fluorometric TUNEL system (Promega, USA) according to instruction.
2.7. Nuclear staining with Hoechst 33342
Nuclear condensation was observed using the chromatin dye Hoechst 33342. Cells were fixed in 4% paraformaldehyde in PBS and stained with 5 mg/mL Hoechst 33342 for 5 minutes. Nuclear morphology was visualized under a fluorescent microscopy (IX‐71; Olympus, Tokyo, Japan). Apoptotic cells displayed nuclear condensations and reduced nuclear size.
2.8. ROS measurement
Intracellular production of ROS in PC12 cells following Aβ1‐42 and galantamine treatment were determined by measuring DCF‐derived fluorescence with the use of flow cytometry. After treatment, PC12 cells were incubated with 10 μmol/L dichloro‐dihydro‐fluorescein diacetate in serum‐free DMEM at 37°C in a humidified incubator with 5% CO2 for 1 hour. The treated cells were then washed and resuspended in PBS. The cells were analysed by flow cytometry measuring DCF fluorescence (λex = 540/λem = 570 nm).
2.9. Western blotting analyses
Drug‐treated cells were lysed by RIPA buffer supplemented with protease and phosphatase inhibitor (Invivogen, USA) for protein extraction. Proteins (30‐60 μg) were fractionated by SDS‐PAGE, and then transferred to PVDF membranes (ISEQ00010; Millipore, USA). After blocking, the membranes were incubated with primary antibodies: LC3 (1:1000), β‐actin (1:5000), p62 (1:1000), Beclin‐1 (1:1000), caspase‐3 (1:1000), NOX4 (1:1000). After washing with Tris‐buffered saline containing 0.1% Tween 20 (Beyotime) (TBST), the blots were probed with secondary antibodies for 1 hour at room temperature. Target proteins were examined using enhanced chemiluminescence reagents (Millipore). Band intensity was determined by densitometry using ImageJ software. The relative intensity of each band was standardized to the corresponding internal control. For relative quantification, the intensity of each band was standardized to the corresponding internal control and normalized to the intensity of the band of control group.
2.10. Small RNA interference
siRNAs targeting ATG5 and NOX4, and negative control (siNC) were synthesized by Genephama, China. The sequences of siRNAs were as follows: rat ATG5 siRNA, sense: 5′‐GACGCUGGUA ACUGACAAATT‐3′ and antisense: 5′‐UUUGUCAGUU ACCAGCGUCTT‐3′; rat NOX4 siRNA, sense: 5′‐GCCCUUCAUU CAAUCUAGATT‐3′ and antisense: 5′‐UCUAGAUUGA AUGAAGGGCTT‐3′. PC12 cells were transfected with siRNA or siNC using Lipofectamine 3000 reagent (L3000015; Invitrogen) according to the manufacturer's protocol.
2.11. Immunofluorescence
Cells were fixed with 4% paraformaldehyde, and then incubated with PBS containing 3% albumin from bovine serum (BS043C; Biosharp, China) for 30 minutes. The slides were then probed with primary antibody at 4°C overnight, and subsequently FITC‐conjugated secondary antibody at room temperature for 1 hour. Images were captured using a fluorescence microscope (IX‐71; Olympus).
2.12. Statistical analysis
All data shown were reported as mean ± SD. Data analysis was performed using Prism 6.0 The statistical significance of the difference between experimental groups in instances of single comparisons was determined using the unpaired Student's t‐test of the means. Statistical significance was defined as *P < .05; **P < .01; ***P < .001.
3. RESULTS
3.1. Galantamine reversed the Aβ‐induced cytotoxic effect in PC12 cells
The cytotoxic effect of Aβ on neuron cells has been well documented.21 To verify it, MTT assay was performed to assess the viability of differentiated rat pheochromocytoma cell line PC12 under Aβ treatment. Consistent with previous reports, treatment of Aβ for 24 hours suppressed the cell viability in a dose‐dependent manner, where IC50 was approximately 2 μmol/L (Figure 1A). To ascertain the protective effect of galantamine against Aβ‐induced cell death, PC12 cells were treated with 2 μmol/L Aβ combined with different concentrations of galantamine followed by cell viability detection. As shown in Figure 1B, galantamine restored the cell viability in a dose‐dependent manner although limited side effect was observed under very high concentration. This protective effect was further supported by clone formation assay, in which treatment of galantamine significantly restored the proliferation of Aβ‐treated PC12 cells (Figure 1C).
Figure 1.

Galantamine protected neuron cells from amyloid beta‐peptide (Aβ)‐induced cytotoxicity. (A) Cell viability of PC12 was measured by MTT assay under indicated concentration of Aβ treatment for 24 h, (B) the protective effect of galantamine on cells was determined through combine treatment with Aβ (2 μmol/L) and various concentration of galantamine for 24 h. The cells were treated with Aβ (2 μmol/L), galantamine (40 μmol/L) or Aβ plus galantamine for 24 h, following, (C) colony formation assay was performed, (D) cell apoptosis rate was measured by TUNEL assay. (E) Morphological of apoptosis was evaluated by fluorescence microscopy, and (F) the expressions of caspase‐3 and cleaved caspase‐3 were detected by western blotting to verity the anti‐apoptosis effect of galantamine, β‐actin was used as a loading control. The results of western blotting were analysed using ImageJ (mean ± SD of 3 independent experiments)
Given that apoptosis has been thought to be one of the most common cell death patterns, we next detected the apoptotic rate through TUNEL assay. As expected, Aβ induced significant apoptosis in PC12 cells, which can be partly reversed by the treatment of galantamine (Figure 1D). Furthermore, nuclei were stained with Hoechst 33342 to evaluate the apoptotic level. As shown in Figure 1E, the nuclei exhibited highly concentrated and fragmented morphology, and co‐treatment of 40 μmol/L galantamine reduced the number of condensed and apoptotic cells. Additionally, we also determined the expression of cleaved caspase‐3, and the results were consistent with above TUNEL assay and Hoechst staining (Figure 1F). Together, our results suggested that galantamine restored the cell viability and alleviate the apoptosis of PC12 cells under cytotoxic Aβ treatment.
3.2. Galantamine inhibited the cytostatic autophagy induced by Aβ in PC12 cells
Apart from its pro‐survival function, autophagy has been regarded as a kind of cell death mechanism if it excessively proceeding.21 Therefore, we investigated whether autophagy played a role in the Aβ‐induced apoptosis and neuron protection by galantamine. LC3 turnover, in which LC3‐I is modified by phosphatidyl ethanolamine group thus transform into LC3‐II, is an essential process in autophagy activation. Treatment of PC12 cells with Aβ was shown to upregulate the rate of LC3 turnover, which suggested that Aβ may promote autophagosomes accumulation (Figure 2A). In line with it, immunofluorescence also confirmed the autophagosomes accumulation, for the LC3 dots were increased upon Aβ treatment (Figure 2F). Given that the increased autophagosomes could be due to either autophagy initiation or blocking of autophagic degradation,22 we also detected the expressions of Beclin1 and p62, marker of autophagy initiation and degradation, respectively (Figure 2A). The results turned out to be that Aβ activated the entire autophagy flux, but not induced autophagy arrest, which contributed to the accumulation of autophagosomes. It was further evidenced by the treatment of Aβ combined with autophagy inhibitor 3‐MA or CQ, where LC3 turnover was further down or upregulated (Figure 2C). Together, these data suggested that Aβ activated autophagy flux in the PC12 cells.
Figure 2.
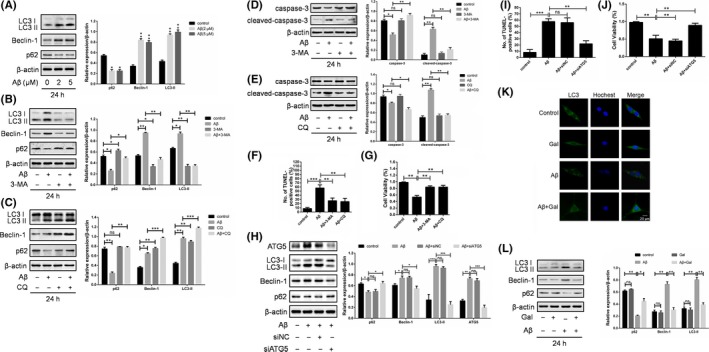
Galantamine inhibited the amyloid beta‐peptide (Aβ)‐induced cytostatic autophagy. (A) PC12 cells incubated with Aβ (2, 5 μmol/L) for 24 h were analysed by western blotting for LC3, BECLIN‐1, p62 and β‐actin. The effect of autophagy inhibitor 3‐MA (5 mmol/L) and CQ (10 μmol/L) on expressions of LC3, BECLIN‐1, p62 (B and C), caspase‐3 and cleaved‐caspase3 (D and E) were detected in PC12 cells treated with Aβ (2 μmol/L) for 24 h. (F) PC12 cells were treated with Aβ (2 μmol/L) for 24 h after pretreatment with 3‐MA (5 mmol/L) for 2 h or with CQ (10 μmol/L) for 6 h. Cell apoptosis rate was measured by TUNEL assay, and then, (G) cell proliferation was examined by the MTT assay. (H) PC12 cells were transfected with siNC or siATG5 for 24 h, and then treated with 2 μmol/L Aβ for another 24 h. The expression of ATG5, LC3, Beclin 1, p62 and β‐actin were examined by western blotting, (I) Cell apoptosis rate was measured by TUNEL assay and (J) cell viability was examined by the MTT assay. (K) The cells treated with 2 μmol/L Aβ or galantamine (40 μmol/L) for 24 h, then immunofluorescence assay was used to determine LC3 accumulation, and (L) western blotting was employed to assess the expression of LC3, BECLIN‐1, p62. β‐actin was used as a loading control. The results of western blotting were analysed using ImageJ (mean ± SD of 3 independent experiments)
Next, we determined whether autophagy was pro‐survival or pro‐death by Aβ treatment combined with or without autophagy inhibitors. As shown in Figure 2D‐G, the autophagy inhibitors 3‐MA and CQ partially reversed the apoptosis and growth inhibition induced by Aβ in PC12 cells. Furthermore, ATG5, an essential autophagy gene, was knock‐down using siRNA. The result showed that siATG5 blocked Aβ‐triggered autophagy, and reversed Aβ‐induced apoptosis and growth inhibition (Figure 2H‐J). Above data indicated that Aβ‐triggered cytotoxic autophagy contributed to the pro‐death role. Importantly, administration of galantamine significantly inhibited the LC3 turnover rate and autophagosomes accumulation (Figure 2K,L), suggesting that the neuroprotective effect of galantamine was due to the inhibition of cytotoxic autophagy.
3.3. Galantamine suppressed the ROS accumulation to inhibit Aβ‐induced cytostatic autophagy
Autophagy is activated by a variety of stimuli, including pathogen infection, protein aggregation, lack of nutrient and oxidative stress.23, 24, 25, 26 Moreover, it has been reported that ROS played crucial roles in the Aβ‐induced neuron toxicity and pathogenesis of AD.27 To determine the mechanism underlying galantamine‐induced autophagy inhibition, we evaluated the cellular ROS level upon Aβ or galantamine combined treatments. As shown in Figure 3A, Aβ dramatically increased the ROS accumulation in PC12 cells, which was mostly reversed by the treatment of galantamine. Then, we employed NAC and LPS as ROS antagonist and inducer, respectively, and the results ascertained that Aβ‐induced ROS elevation and galantamine‐induced ROS elimination were specific (Figure 3B,C).
Figure 3.
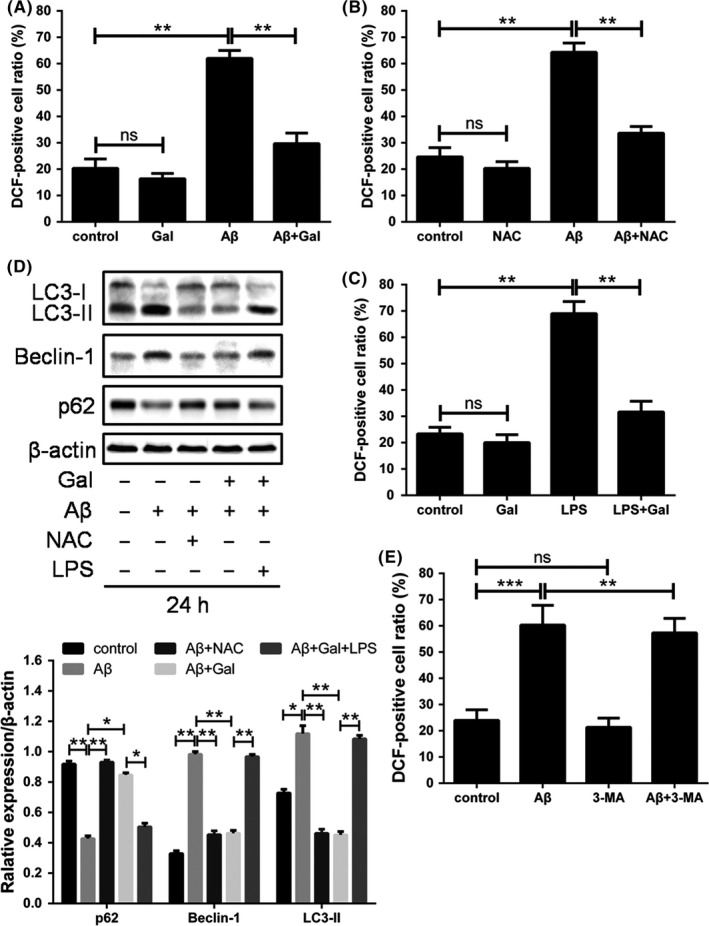
Galantamine eliminated reactive oxygen species (ROS) to suppress cytostatic autophagy in PC12 cells. Cells were treated with galantamine (40 μmol/L), amyloid beta‐peptide (Aβ) (2 μmol/L) and LPS (500 ng/mL) as indicated for 24 h and treated with Aβ (2 μmol/L) and LPS (500 ng/mL) for 24 h after a 2 h pre‐incubation with 5 mmol/L NAC, (A‐C) followed by ROS detection, and (D) western blotting for LC3, BECLIN‐1, p62 and β‐actin, the results of western blotting were analysed using ImageJ (mean ± SD of 3 independent experiments). (E) Cells were treated with Aβ (2 μmol/L) for 24 h after pretreatment with 3‐MA (5 mmol/L) for 2 h, followed by ROS detection
To determine whether the autophagy inhibition by galantamine was dependent on the regulation of ROS, autophagy activation was measured following ROS manipulation. As expected, NAC reduced the autophagy activated by Aβ; LPS abolished the galantamine‐induced autophagy suppression, while 3‐MA did not significantly change Aβ‐induced ROS generation. These results indicated that galantamine restored redox balance to prevent cytostatic autophagy in neuron cells (Figure 3D,E).
3.4. Galantamine downregulated the NOX4 expression to reduce Aβ‐induced ROS accumulation
Cellular ROS is generated from multiple resources such as mitochondria or metabolic enzymes, especially NADPH oxidase 4 (NOX4).28, 29 To further investigate the mechanism underlying the reversing effect of galantamine on Aβ‐induced oxidative stress, the expression of NOX4 in PC12 cells was measured upon Aβ and galantamine treatment. Our data showed that the expression of NOX4 was upregulated by Aβ, which can be mostly reversed by the galantamine treatment, indicating that the regulation of NOX4 expression was an important mechanism underlying galantamine‐induced ROS elimination (Figure 4A). Moreover, the LPS‐increased NOX4 expression was markedly impaired by galantamine (Figure 4B). Interestingly, the level of its isoform NOX2 did not show obvious alteration upon Aβ or galantamine treatments (Figure 4A,B), suggesting the regulation of ROS by NOX4 in this content was relatively specific. Furthermore, the knock‐down of NOX4 using siRNA inhibited the ROS accumulation and autophagy (Figure 4C,D). Together, galantamine inhibited the expression of NOX4 to eliminate excessive ROS induced by Aβ, thereby preventing the cytostatic autophagy and apoptosis of neuron cells.
Figure 4.
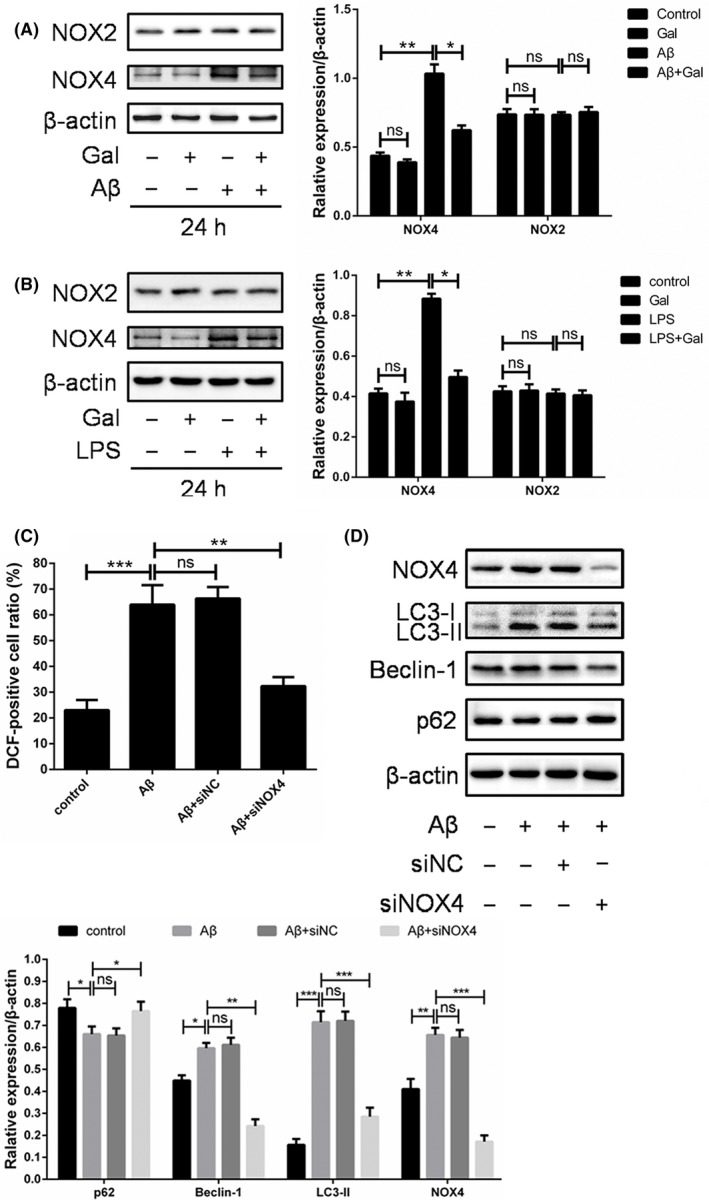
Galantamine reduced the expression of NOX4. (A) The expression of NOX4 and NOX2 were detected by western blotting following PC12 cells treated with amyloid beta‐peptide (Aβ) (2 μmol/L) and galantamine (40 μmol/L) for 24 h. (B) LPS (500 ng/mL) was used as positive control to ascertain that Aβ‐induced ROS accumulation, samples were analysed by western blotting for NOX4, NOX2 and β‐actin. PC12 cells were transfected with siNC or siNOX4 for 24 h, and then treated with 2 μmol/L Aβ for another 24 h, (C) followed by ROS detection, (D) western blotting for NOX4, LC3, Beclin 1, p62 and β‐actin. The results of western blotting were analysed using ImageJ (mean ± SD of 3 independent experiments)
4. DISCUSSION
Galantamine has long been used as the first‐line drug to delay the progression of AD, exhibiting favourable effect in numerous patients suffered from neurodegeneration. It was well documented that galantamine inhibited the cholinesterase to upregulate the level of acetylcholine within neuron cells.2, 3 However, the mechanism underlying galantamine‐mediated neuron protection remains incompletely understood during Aβ‐induced AD progression. Here, we found that Aβ prominently induced cytotoxic autophagy in PC12 cells, which can be partly reversed by galantamine, providing a novel mechanism by which AD progression and galantamine‐based therapeutic strategy.
Autophagy plays dual roles in cellular physiological processes. Generally, basal level of autophagy promotes the self‐renewal of organism, which helps to materials recycling and facilitates the cell survival under adverse environment.4, 30 On the other side, excessive autophagy causes structural damage of cellular components, leading to cell senescence or death.31 To determine the exact role of autophagy in a particular condition is of great value, especially to disease treatment. It has been reported that block of protective autophagy by autophagy inhibitors such as CQ can partly overcome cancer drug resistance, but on the contrary, some agents exhibited anticancer activity through inducing cytostatic autophagy.32 In this paper, we identified that Aβ induced cytotoxic, but not pro‐survival autophagy to lead to neuron cell death in rat pheochromocytoma cells, suggesting that inhibition of the autophagy may contributes to survival of neuron cells. However, some researchers have showed opposite results indicating that autophagy prevented neuron cells from Aβ‐induced cytotoxicity in human neuroblastoma cells, and the activation of autophagosome‐lysosome degradation pathway facilitated the clearance of Aβ aggregates.33 In addition, autophagy‐deficient mice (loss of Atg7) have been reported to develop neurodegeneration, suggesting autophagy impairment can be a primary cause of AD occurrence.34 These findings are opposite but not contradictory. We purpose that autophagy functions as a protein quality‐control process that facilitates Aβ clearance in the healthy body, which prevents the initiation of AD (in the early stage). However, when Aβ is accumulated extensively in the progressed AD brain, autophagy machinery is not adequate for the elimination of Aβ. Worse more, overactive autophagy renders damage to crucial cellular components and subsequent cell death before Aβ clearance, as is the case in this paper (in the late stage). Our hypothesis is supported by the finding that showed autophagy was upregulated in the brain of AD patients, but downregulated in that of normal aging cohorts, indicating that autophagy protects brain cell from aging in the physiologic process, but acts as a death mechanism when overactivated in the pathologic event.9, 35 The complexity of autophagy regulation not only gives remarkable challenges for the intervention of autophagy‐targeted AD therapeutic strategies, but also provides new opportunities to achieve personalized medicine without interfering normal physiologic process of brain. In addition, we found that galantamine reverses Aβ‐induced neuron cell death through attenuating cytostatic autophagy, which suggests a novel mechanism underlying its neuroprotective effects.
Among the various stimuli that activate autophagy, ROS is particularly notable due to its effectiveness. It has been recognized that ROS is more than a set of destructive species, but functions as second messengers to conduct countless cellular physiological processes, including the activation of autophagy.36, 37 ROS has been shown to regulate autophagy through several signalling pathways such as PI3K‐Akt, AMPK. These findings indicate that the regulation of autophagy by ROS is ubiquitous and comprehensive; however, the connection between ROS and autophagy in AD progression remains elusive. Some studies have indicated that oxidative stress is one of the most important factors contributing to AD progression, and administration of antioxidants such as ascorbic acid may be helpful to relieve conditions.38 We here reported that Aβ‐induced autophagy activation is dependent on the ROS accumulation, but detail mechanisms by which ROS activates autophagy need further exploration.
Moreover, we found that galantamine reduced the expression of NOX4, but not NOX2 thereby counteract the oxidative stress induced by Aβ, leading to the consequent autophagy inhibition and neuron cell survival. The NOX family members have been characterized as predominant sources of ROS, and each of them differs in expression, translocation and function.39 Particularly, NOX4 regulates autophagy in energy‐deprived cells, purposing a hypothesis of NOX4‐derived ROS stimulates excessive autophagy in AD brains. Increasing evidences implicate that NOX4 plays crucial roles in the oxidative damage and induced autophagy activation dependently of ROS accumulation during neuron diseases progression.40 In this study, galantamine was used as a ROS scavenger working through downregulating the expression of NOX4, suggesting that NOX4 can be an attractive therapeutic target in the treatment of AD.
Taken together, our research revealed that galantamine inhibited Aβ‐induced neuronal damage through alleviating oxidative stress and cytostatic autophagy in PC12 cells, providing new insights into deep understanding of AD progression and molecular basis of galantamine in neuroprotection.
ACKNOWLEDGEMENTS
This work was supported by National Natural Science Foundation of China (grant no. 81301919 and 31701212), Application and Basic Project of Science and Technology Department of Sichuan Province (grant no. 2017JY0166 and 2016JY0184), Scientific Research Foundation of Education Department of Sichuan Province (grant no. 16ZA0285), Scientific Research Foundation of Health and Family Planning commission of Sichuan Province (grant no. 17PJ584), Scientific Research Foundation of Collaborative Innovation Center of Elderly Care and Health (grant no. YLZBZ1504), and all support is gratefully acknowledged.
COMPETING INTERESTS
The authors have no competing interests.
Jiang S, Zhao Y, Zhang T, et al. Galantamine inhibits β‐amyloid‐induced cytostatic autophagy in PC12 cells through decreasing ROS production. Cell Prolif. 2018;51:e12427 10.1111/cpr.12427
Contributor Information
Kejian Pan, Email: pankejian2005@163.com.
Kun Zhang, Email: zhangkunyyo@163.com.
REFERENCES
- 1. Seltzer B. Galantamine‐ER for the treatment of mild‐to‐moderate Alzheimer's disease. Clin Interv Aging. 2010;5:1‐6. [PMC free article] [PubMed] [Google Scholar]
- 2. Corey‐Bloom J. Galantamine: a review of its use in Alzheimer's disease and vascular dementia. Int J Clin Pract. 2003;57:219‐223. [PubMed] [Google Scholar]
- 3. Burns A, Bernabei R, Bullock R, et al. Safety and efficacy of galantamine (Reminyl) in severe Alzheimer's disease (the SERAD study): a randomised, placebo‐controlled, double‐blind trial. Lancet Neurol. 2009;8:39‐47. [DOI] [PubMed] [Google Scholar]
- 4. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guillon‐Munos A, van Bemmelen MX, Clarke PG. Autophagy can be a killer even in apoptosis‐competent cells. Autophagy. 2006;2:140‐142. [DOI] [PubMed] [Google Scholar]
- 7. Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345‐357. [DOI] [PubMed] [Google Scholar]
- 8. Shafei MA, Harris M, Conway ME. Divergent metabolic regulation of autophagy and mTORC1‐early events in Alzheimer's disease? Front Aging Neurosci. 2017;9:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nilsson P, Saido TC. Dual roles for autophagy: degradation and secretion of Alzheimer's disease Abeta peptide. BioEssays. 2014;36:570‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tung YT, Wang BJ, Hu MK, et al. Autophagy: a double‐edged sword in Alzheimer's disease. J Biosci. 2012;37:157‐165. [DOI] [PubMed] [Google Scholar]
- 11. Zhao H, Wang ZC, Wang KF, Chen XY. Abeta peptide secretion is reduced by Radix Polygalae‐induced autophagy via activation of the AMPK/mTOR pathway. Mol Med Rep. 2015;12:2771‐2776. [DOI] [PubMed] [Google Scholar]
- 12. Omata Y, Lim YM, Akao Y, Tsuda L. Age‐induced reduction of autophagy‐related gene expression is associated with onset of Alzheimer's disease. Am J Neurodegen Dis. 2014;3:134‐142. [PMC free article] [PubMed] [Google Scholar]
- 13. Mizushima N. A(beta) generation in autophagic vacuoles. J Cell Biol. 2005;171:15‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno‐electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113‐122. [DOI] [PubMed] [Google Scholar]
- 15. Butterfield DA, Boyd‐Kimball D. The critical role of methionine 35 in Alzheimer's amyloid beta‐peptide (1‐42)‐induced oxidative stress and neurotoxicity. Biochim Biophys Acta. 2005;1703:149‐156. [DOI] [PubMed] [Google Scholar]
- 16. Kanski J, Aksenova M, Schoneich C, Butterfield DA. Substitution of isoleucine‐31 by helical‐breaking proline abolishes oxidative stress and neurotoxic properties of Alzheimer's amyloid beta‐peptide. Free Radic Biol Med. 2002;32:1205‐1211. [DOI] [PubMed] [Google Scholar]
- 17. Nunomura A, Perry G, Pappolla MA, et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959‐1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994;36:747‐751. [DOI] [PubMed] [Google Scholar]
- 19. Persson T, Popescu BO, Cedazo‐Minguez A. Oxidative stress in Alzheimer's disease: why did antioxidant therapy fail? Oxid Med Cell Longev. 2014;2014:427318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsvetkova D, Obreshkova D, Zheleva‐Dimitrova D, Saso L. Antioxidant activity of galantamine and some of its derivatives. Curr Med Chem. 2013;20:4595‐4608. [DOI] [PubMed] [Google Scholar]
- 21. Jana A, Pahan K. Fibrillar amyloid‐beta‐activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer's disease. J Neurosci. 2010;30:12676‐12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zois CE, Giatromanolaki A, Sivridis E, Papaiakovou M, Kainulainen H, Koukourakis MI. “Autophagic flux” in normal mouse tissues: focus on endogenous LC3A processing. Autophagy. 2011;7:1371‐1378. [DOI] [PubMed] [Google Scholar]
- 23. Chandra P, Kumar D. Selective autophagy gets more selective: uncoupling of autophagy flux and xenophagy flux in Mycobacterium tuberculosis‐infected macrophages. Autophagy. 2016;12:608‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim Y, Park H, Nah J, et al. Essential role of POLDIP2 in Tau aggregation and neurotoxicity via autophagy/proteasome inhibition. Biochem Biophys Res Commun. 2015;462:112‐118. [DOI] [PubMed] [Google Scholar]
- 25. Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress‐induced autophagy. Autophagy. 2013;9:1983‐1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pettersen K, Monsen VT, Hakvag Pettersen CH, et al. DHA‐induced stress response in human colon cancer cells ‐ Focus on oxidative stress and autophagy. Free Radic Biol Med. 2016;90:158‐172. [DOI] [PubMed] [Google Scholar]
- 27. Luo Y, Yue W, Quan X, Wang Y, Zhao B, Lu Z. Asymmetric dimethylarginine exacerbates Abeta‐induced toxicity and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic Biol Med. 2015;79:117‐126. [DOI] [PubMed] [Google Scholar]
- 28. Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer. 2014;14:709‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discovery. 2011;10:453‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song W, Ma Z, Zhang Y, Yang C. Autophagy plays a dual role during intracellular siRNA delivery by lipoplex and polyplex nanoparticles. Acta Biomater. 2017;58:196‐204. [DOI] [PubMed] [Google Scholar]
- 31. Ba MC, Long H, Cui SZ, et al. Mild hyperthermia enhances sensitivity of gastric cancer cells to chemotherapy through reactive oxygen species‐induced autophagic death. Tumour Biol. 2017;39:1010428317711952. [DOI] [PubMed] [Google Scholar]
- 32. Maes H, Kuchnio A, Peric A, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. 2014;26:190‐206. [DOI] [PubMed] [Google Scholar]
- 33. Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Abeta‐induced cytotoxicity. Autophagy. 2009;5:502‐510. [DOI] [PubMed] [Google Scholar]
- 34. Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880‐884. [DOI] [PubMed] [Google Scholar]
- 35. Lipinski MM, Zheng B, Lu T, et al. Genome‐wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci USA. 2010;107:14164‐14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. 2015;35:615‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao H, Zhang M, Zhou F, et al. Cinnamaldehyde ameliorates LPS‐induced cardiac dysfunction via TLR4‐NOX4 pathway: the regulation of autophagy and ROS production. J Mol Cell Cardiol. 2016;101:11‐24. [DOI] [PubMed] [Google Scholar]
- 38. Huang J, May JM. Ascorbic acid protects SH‐SY5Y neuroblastoma cells from apoptosis and death induced by beta‐amyloid. Brain Res. 2006;1097:52‐58. [DOI] [PubMed] [Google Scholar]
- 39. Miller FJ Jr. Nox4 NADPH oxidase: emerging from the veil of darkness. Eur Heart J. 2015;36:3457‐3459. [DOI] [PubMed] [Google Scholar]
- 40. Sciarretta S, Volpe M, Sadoshima J. NOX4 regulates autophagy during energy deprivation. Autophagy. 2014;10:699‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]