

Abstract
Objectives
The role of vitamin D (VD) in innate and adaptive immune responses to tuberculosis is still unclear. Our research was aimed to uncover the effect of VD on Th17 cells and elucidate potential molecular mechanism.
Materials and methods
VDR‐deficient and wild‐type mice were used to obtain CD4 T cells. Th17 cells were induced and activated by Bacillus Calmette Guerin. Flow cytometry was used to analyse the apoptosis rate and degree of differentiation of Th17 cells in the treatment of 1,25(OH)2D3. The interaction between P65 and Rorc was determined by immunofluorescence assay, luciferase reporter assay, EMSA‐Super‐shelf assay and ChIP assay. Co‐IP assay was carried out to test the interaction between VDR and NF‐κB family proteins. qRT‐PCR and Western blot were also performed to detect the levels of P65, RORγt and IL‐17.
Results
The Th17 cells differentiation was suppressed by 1,25(OH)2D3 in vitro. We confirmed that Rorc was a downstream gene of the transcription factor P65. VDR interacts with P105/P50, P100/P52 and P65 NF‐κB family proteins. 1,25(OH)2D3 inhibited the expression of RORγt/IL‐17 by suppressing p65 transcription factor translocating to nucleus. In vivo experiments, the expression of IL‐17 and RANKL was suppressed by 1,25(OH)2D3 by VD receptor. Moreover, 1,25(OH)2D3 suppressed the inflammatory infiltrates and inhibited the expression of P65, RORγt and IL‐17 in the spleen tissues of model mice.
Conclusions
Together, 1,25(OH)2D3 suppressed the differentiation of Th17 cells via regulating the NF‐κB activity.
Keywords: 1,25(OH)2D3; IL‐17; NF‐κB; Th17 cells differentiation; VDR
1. INTRODUCTION
Tuberculosis (TB) is a disease spread by obligate intracellular organism Mycobacterium tuberculosis.1, 2 Around 1.5 million patients died of TB and 9 million people were infected every year. TB has become to be a major public health threat in the world, a new medicine to treat TB is desperately needed.3
Vitamin D was synthesized in human skin under the exposure of ultraviolet. Many researches showed that the VD was involved in bone metabolism and immunomodulatory.4, 5 The variation in the ability to synthesize vitamin D (VD), including polymorphisms in the VDR was a contributing factor to increase TB susceptibility.6 For many patients, the normal treatment regimens were ineffective for TB because of infection of new drug‐resistant strains.7, 8 Indeed, it was found that sun exposure was useful for TB treatment decades ago, but the mechanism was unclear. The phagocytosis ability of macrophages cells was increased by VD. While, the inflammatory cytokines level of T cells were showed to be decreased by VD.9 As we all know, 1,25(OH)2D3 could regulate the expression of different genes for immunoregulation by biding to VDR.
Th17 cell, a distinct population of the T helper cells that is controlled by Retinoid‐related orphan nuclear receptor γt (RORγt) which is encoded by gene Rorc. T‐bet, STAT4, GATA‐3 and STAT6 have been reported to regulate the Th1 and Th2 functions.10, 11 The cytokines IL‐22 produced by Th17 cells was early increased during TB infection and IL‐17 plays a protection role of TB infection. IL‐17 could also regulate the development of Th1 cells.12 In our findings, the differentiation of Th17 cells was inhibited by1,25(OH)2D3. However, underlying molecular mechanism was still needed to be revealed.
In the study, the activated VDR‐deficient and wild‐type (WT) Th17 were treated with 1,25(OH)2D3. 1,25(OH)2D3 changed the NF‐κB activity in VDR‐/‐ Th17 cells decreased and sequestered p65 in the cytoplasm. Furthermore, increased p65 translocated to the nucleus was found in VDR‐/‐ Th17 cells than WT Th17 cells both under the treatment of 1,25(OH)2D3, indicating that VDR played an important role of NF‐κB activity..
Rorc, a gene encodes RORγt,13 was identified to have a potential promoter‐binding sites biding to P65. We confirmed that Rorc was the direct target of transcription factor P65 in Th17 cells and the binding of P65 and Rorc promoter‐binding sites was inhibited by 1,25(OH)2D3. Then, 1,25(OH)2D3 suppressed the NF‐κB activity and level of RORγt/IL‐17 by inhibiting p65 transcription factor translocating to nucleus. In vivo, 1,25(OH)2D3 inhibited the inflammatory infiltrates and expression of P65, RORγt and IL‐17 in the spleen of model mice. These study results revealed the molecular mechanisms about the immune system regulation regulated by VD, and provided a foundation for targeting Th17 immunity by 1,25(OH)2D3 analogues.
2. MATERIALS AND METHODS
2.1. Animals
The VDR‐/‐ mice (B6/CBA genetic background) were backcrossed to C57BL/6J mice for 9 times. We maintained genetic background of the mice for 8 years.14 To induce the VD defective in C57BL/6 mice, mice were placed in ultraviolet light‐free cages and raised with a VD‐depleted die. Three months later, the blood was got and serum 1,25(OH)2D3 levels were measured. We chose the mice lacing 1,25(OH)2D3 as the experimental mice. The normal group mice were raised by VD‐abundant diet in the same ultraviolet light‐free environment. The mice were given tail intravenous injection with Bacillus Calmette Guerin (BCG) for 6 weeks. All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee.
2.2. Preparation of Th17 cells
The antigen‐presented cells were isolated from the spleen of VDR‐/‐ and WT mice as previous described.15 Briefly, the mononuclear lymphocytes got from spleen were incubated with CD4, CD8 antibody and complement to eliminate the T cells. Naive CD4 T cells were isolated from VDR‐/‐ and WT mice and cultured in RPMI‐1640 containing 10% FBS.
CD4 T cells isolated from mouse splenocytes by CD4 T‐cell isolation kit (Teterow, Germany ) following the manufacturer's protocol. The cell proportion of purified naive CD4+ T cells before or after isolation was tested by Flow cytometry. For Th17 cells, sorted TCR‐Tg CD4+ T cells and APC cells were mixed in a ratio(1:6) with BCG, TGF‐β and IL‐6 (2 ng/mL) for 3 days. Then, the cells were washed and IL‐23 (10 μg/mL) was added. Cells passage culture for 3 days a time and the third generation cells were mixed with APC cells (1:6) in culture medium contained BCG (1 μg/mL) for 5 hours for subsequent experiment. The fluorochrome‐conjugated antibodies were obtained from purchased from eBioscience (Thermo Fisher Scientific, Waltham, MA USA) and cytokines were purchased from R&D Systems Inc (Minneapolis, MN, USA).
2.3. Cell viability (MTT) assay
Briefly, the Th17 cells (2 × 103 cells/well) were incubated in triplicate in a 96‐well plate in the presence of various concentrations of 1,25(OH)2D3 1, 10 or 100 nmol/L or vehicle for 24, 48 or 72 hours. At different time points, 20 μL of 5 g/L MTT solution (Sigma, St Louis, MO, USA) was added and incubated for 4 hours. Finally, the optical absorbance (OD) was measured at 570 nm.
2.4. Enzyme‐linked immunosorbent assay
Enzyme‐linked immunosorbent assay (ELISA) Kits (Elabscience Biotechnology Co., Ltd, Wuhan, China) was used to measure the levels of VD, IL‐17 and RANKL in the serum were in strict accordance with the kit instructions. First, the ELISA Kits were placed at room temperature for 30 minutes. The standard sample and actual samples were diluted and prepared according to the instructions. Then, the samples were added and the liquid was mixed. The optical density values were determined under 450 nm. The content of VD, IL‐17 and RANKL in the serum can be detected with the graph.
2.5. Flow cytometry
Briefly, Th17 cells were washed, centrifuged after incubation for 48 hours with 1,25(OH)2D3. Then, the cell suspension was added 5 μL Annexin‐V‐FITC solution (BD Biosciences, Franklin Lakes, NJ, USA) and incubated for 20 minutes. The cell apoptosis was detected by the flow cytometer (BD Biosciences; FASCalibur).
2.6. Immunohistochemistry assay
Spleen sections were deparaffinized, hydrated, blocked for endogenous peroxidase using 3% H2O2/H2O and subsequently subjected to microwave epitope. The primary antibodies specific for P65, IL‐17 and RORγt (Abcam, Cambridge, UK) were used. The sections were incubated with the antibodies for half an hour at 37°C. Diluted normal human serum was used for negative control sections instead of primary antibodies. A universal biotinylated secondary antibody (DAKO) and peroxidase substrate 3, 3‐diaminobenzidine (Sigma‐Aldrich) were applied, counterstaining with Harris’ haematoxylin.
2.7. Immunofluorescence assay
The fixed VDR‐deficient (VDR−/−) and WT Th17 cells by 4% paraformaldehyde were stained with endogenous NF‐κB p65 and 4′,6‐diamidino‐2‐phenylindole for cell nucleus as previously described.16 P65 polyclonal antibody was purchased from Cell Signalling Technology (Danvers, MA). Secondary antibodies conjugated with Alexa Fluor® 647 and DAPI (1:1000; Sigma) were used. Cells were visualized by Olympus BX60 fluorescent microscope.
2.8. Western blot
Cells were washed and lysed in ice‐cold lysis buffer. BCA Protein Assay Kit (Pierce, Rockford, IL, USA) was used to detect the protein concentration. An amount of 50 μg of protein samples were separated on a 4%‐12% SDS‐PAGE gel and the protein was transferred onto PVDF membranes (Millipore, Billerica, MA, USA). TBST containing 2% BSA, antibodies against IKB (CST, 1:1000), P65 (CST, 1:1000), RORγt (CST, 1:1000), IL‐17 (CST, 1:1000), RANKL (CST, 1:1000), P105/P50 (CST, 1:1000), P100/P52 (CST, 1:1000), IκBα (CST, 1:1000) phosphorylated IκBα (CST, 1:1000) and GAPDH (CST, 1:2000) and goat anti‐rabbit‐HRP‐conjugate antibodies (Bio‐Rad; California, USA 1:2000) were used. Chemiluminescent kit (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to detect the protein bands.
2.9. RNA extraction and quantitative reverse transcription PCR (Q‐PCR)
RNA isolation and qRT‐PCR were performed as previously described.17 Trizol Reagent (Life Technologies, Grand Island, NY) was used for RNA extraction and SYBR green PCR Mix (iTAP; Bio‐Rad) was used for detection following the manufacturer's protocols.17 △△CT values were used to show the fold change and primers for quantitative reverse transcription PCR are listed in Table 1.
Table 1.
Primers used in this study
| Gene | Primer (5′‐3′) |
|---|---|
| GAPDH‐F | AGGTCGGTGTGAACGGATTTG |
| GAPDH‐R | CCTCTGAGGCCTCGCTGCG |
| P65‐F | CTTCTGGGCCTTATGTGGAG |
| P65‐R | TGTCTTGGTGGTATCTGTGCTT |
| RORγt‐F | GCAGCAACAGGAACAAGTGG |
| RORγt‐R | CCTCGGGTAGGTCAGGTGAG |
| IL17a‐F | TCAGACTACCTCAACCGTTCC |
| IL17a‐R | GGTGGTCCAGCTTTCCCT |
| RANKL‐F | ACCTGATGAAAGGAGGGAGC |
| RANKL‐R | AAGGGTTGGACACCTGAATG |
2.10. Transient transfection and small interfering RNA
For small interfering RNA (siRNA) assay, RiboBio (Guangzhou, China) synthesized the siRNA targeting human P65 and scrambled siRNA. The siRNA was transfected by Lipofectamine 2000 (Life Technologies) into targeting cells and cells were harvested at indicated time point for further assay.
2.11. Luciferase assays
Plasmid pcDNA‐p65, pGL3‐Rorc‐5′UTR‐WT and pGL3‐Rorc‐5′UTR‐MUT were constructed. Reporter was constructed by ligating Rorc putative promoter region and mutations to pGL3‐basic (pGL3‐Rorc‐5′UTR‐WT and pGL3‐Rorc‐5′UTR‐MUT reporter). Plasmid pCDNA3.1 and pGL3‐basic were used as control. Lipofectamine 2000 (Life Technologies) was used to transfect Th17 cells. The cells were transfected for 36 hours and luciferase activity was measured by Promega Turner Biosystems Modulus Multimode Reader luminometer.
2.12. Electrophoretic mobility shift assays
The electrophoretic mobility shift assays were performed in strict accordance with the protocol.18, 19 Nucleoprotein Extraction Kit (Novagen, Darmstadt, Germany) and 5′‐Cy5‐labelled cognate double‐stranded DNA elements containing motif sequence were used. Typhoon FLA‐7000 Phosphor Imager (FUJIFILM) scanned band.
2.13. Co‐immunoprecipitation (co‐IP)
Total cell extracts were prepared in a RIPA buffer (50 mmol/L Tris‐HCl, pH 7.5, 150 mmol/L sodium chloride, 1% Nonidet P‐40, and 0.5% sodium deoxycholate) for co‐immunoprecipitation. The experiments were carried out following the manufacturer's instructions (Pierce). Briefly, normal IgG or VDR protein‐specific Abs were incubated with protein G‐conjugated agarose beads at room temperature for 5 hours. The unbound antibodies were removed by washing with 1× PBS 3 times. The antibody‐bound agarose beads were then incubated with 300 μL of cell lysate at 4°C overnight. Immunocomplexes were resolved by SDS‐PAGE and Western blotting was performed by specific antibodies P105/P50, P100/P52 and P65. The antibodies were purchased from Cell Signalling Technology.
2.14. Chromatin immunoprecipitation
Cells were rinsed twice with cold PBS, harvested by gentle scraping, and pelleted by centrifugation at 2000 g for 4 minutes at 4°C, after which the pellets were washed once with 1× PBS. The cell pellets were resuspended in 300 μL of cell lysis buffer and incubated on ice for 10 minutes to release the nuclei. The chromatin fragmentation was done by sonication on ice for 4 cycles (30 seconds “ON”, 30 seconds “OFF” at 40% amplitude) to yield an average length of <600 bp. Chromatin immunoprecipitation (ChIP) assay was performed using Zymo‐Spin ChIP Kit (Irvine, Santa Ana, CA, USA) following the manufacturer's protocol. Briefly, the supernatants of the fragmented lysates were diluted 10‐fold with chromatin dilution buffer. Chromatin solutions were immunoprecipitated with P65 antibody (CST, USA) at 4°C overnight. ZymoMag Protein A beads were added to the lysate to isolate the antibody‐bound complexes. The elute was reverse cross‐linked by heating at 65°C for 30 minutes. Samples were then treated with proteinase K for 90 minutes at 65°C to digest the proteins that were immunoprecipitated. Purified DNA from each ChIP assay (0.5‐1 μl) was subjected to PCR. To identify the P65‐binding sequence, 2 sets of primers were used for PCR amplification of the Rorc promoter‐specific primers: primers for P65‐binding sites: Rorc‐CHIP‐F: 5′‐GTGAAGTTTTACGGAGGGTTAAC‐3′, Rorc‐CHIP‐R: 5′‐AATAAACAACAAAACAAAAACCGAC‐3′; and control primers: F: 5′‐TGTCCTCCCACACACAACTC‐3, R: 5′‐GAGGCAGGACTCTGGCTTTA‐3′.
2.15. Statistical analysis
All experiments are repeated at least 3 times and expressed as mean ± SEM, unless otherwise indicated. Two‐tailed Student's t test was used to compare and Graphpad prism software was used.
3. RESULTS
3.1. The Th17 cells differentiation was inhibited by 1,25(OH)2D3 in vitro
VDR−/− and WT Naive CD4+ T cells were isolated. The cell proportion of purified naive CD4+ T cells after isolation was tested and showed in Figure 1A. We first assessed the cell viability of Th17 cells under 1, 10 or 100 nmol/L 1,25(OH)2D3 or vehicle. 1,25(OH)2D3 stimulation affected the Th17 cell viability by a dose‐dependent manner; 10 nmol/L 1,25(OH)2D3 was chosen as the appropriate concentration in subsequent assay (Figure 1B). Then, we tested the apoptosis of Th17 cells under 1,25(OH)2D3 (10 nmol/L) or vehicle and found that 10 nmol/L 1,25(OH)2D3 did not influence the Th17 cell apoptosis (Figure 1C). In view of this, the 10 nmol/L was preferred as the suited concentration in subsequent assay. The effects of 10 nmol/L 1,25(OH)2D3 on Th17 in purified CD4+ T cells were assessed. 1,25(OH)2D3 was found to inhibit both WT CD4+/IL‐17+ T cells and VDR−/− CD4+/IL‐17+ T cells (Figure 1D).
Figure 1.
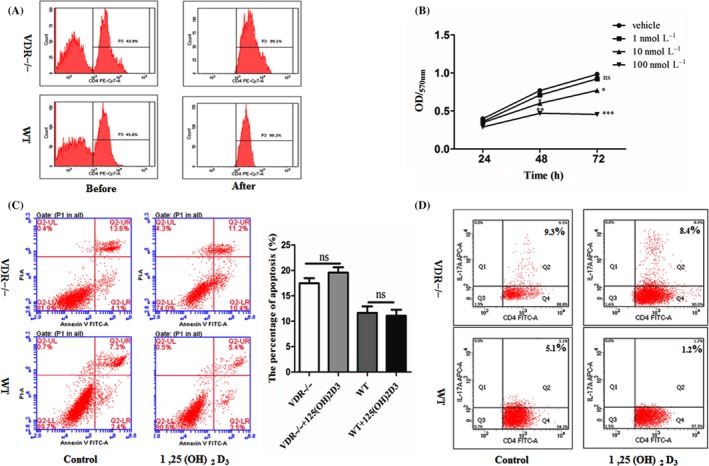
1,25(OH)2D3 inhibits the Th17 cells in vitro. A, The cell proportion of purified naive CD4+ T cells before or after isolation was tested by flow cytometry. B, Cell viability of Th17 cells in the presence of 1, 10, or 100 nmol/L 1,25(OH)2D3 or vehicle was tested by CCK8 assay. C, Apoptosis of Th17 cells under the presence of 10 nmol/L 1,25(OH)2D3 or vehicle for 3 d was analysed by flow cytometry. D, The purified CD4+ T cells from WT mice and VDR−/− mice were treated with 10 nmol/L 1,25(OH)2D3 and the proportion of CD4+/IL‐17+ Th17 cells was analysed by flow cytometry. NS P > .05, *P < .05, ***P < .001
3.2. 1,25(OH)2D3 inhibited the nuclear translocation of P65 and Rorc was a downstream gene of the transcription factor P65
In order to explore the relation between 1,25(OH)2D3 treatment and NF‐κB activity, both the activated WT and VDR−/− Th17 cells from mice were treated with 1,25(OH)2D3 and the p65 transcription factor was assessed. The enhanced fluorescence intensity of the p65 in the nucleus of Th17 cells stands for the robust NF‐κB activity. In the WT Th17 cells, 1,25(OH)2D3 treatment had less p65 translocation to the nucleus (Figure 2A). The NF‐κB activity in VDR−/− Th17 cells decreased and sequestered p65 in the cytoplasm under treatment of 1,25(OH)2D3 (Figure 2B). However, more p65 translocation to the nucleus was found in VDR−/− Th17 cells than WT Th17 cells both under the presence of 1,25(OH)2D3, indicating that VDR was involved in the inhibition of NF‐κB activity by 1,25(OH)2D3 (Figure 2A,B). The mammalian NF‐κB family consists of 5 members: NF‐κB 1 (p50 and p105), NF‐κB 2 (p52 and p100), RelA (p65), RelB and c‐Rel. These proteins share a Rel homology domain that mediates DNA binding, dimerization and interactions with specific inhibitory factors named IκBs, which retain NF‐κB dimers in the cytoplasm.20, 21 To further study the underlying mechanisms that 1,25(OH)2D3 inhibited the p65 translocation, we investigated whether VDR binds to the NF‐κB family proteins by Co‐IP assay. As shown in Figure 2F,H, P105/P50, P100/P52 and P65 can be clearly observed in the FLAG‐VDR but not the control immunoprecipitates. These data demonstrated that VDR interacts with P105/P50, P100/P52 and P65 NF‐κB family proteins. So, the VDR may form a ternary complex with NF‐κB and IκBα to inhibit the nuclear translocation of P65. We also studied the role of VDR in phosphorylation and/or ubiquitination of IkB and the level of NF‐κB 1 and NF‐κB 2. The results showed that the activation of VDR decreased the level of P105/P50, P100/P52 and phosphorylated IκBα in WT Th17 cells. The treatment of 1,25(OH)2D3 also decreased the level of P105/P50, P100/P52 and phosphorylated IκBα in WT Th17 cells (Figure S1A,B).
Figure 2.
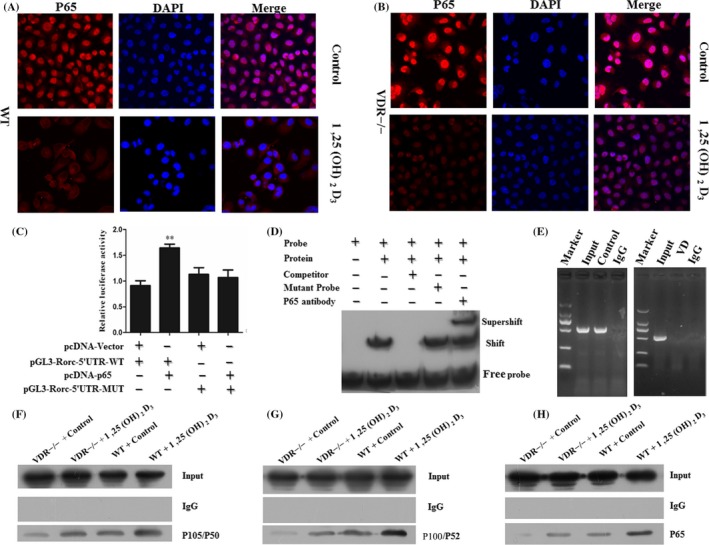
Rorc was a downstream gene of the transcription factor P65. (A, B) WT and VDR−/− Th17 cells were fixed and stained for DAPI (nucleus) and p65. Robust NF‐κB is represented by high fluorescence levels of the p65 transcription factor in the nucleus of VDR−/− Th17 cells with 1,25(OH)2D3 treatment. (C) Plasmid pcDNA‐Vector, pGL3‐Rorc‐5′UTR‐WT, pcDNA‐p65 and pGL3‐Rorc‐5′UTR‐MUT reporter were co‐transfected and the luciferase was measured. (D) EMSA‐Super‐shelf assay was performed to identify the P65‐binding sites in the Rorc promoter. Nuclear protein extracts are prepared from Th17 cells and were incubated with a biotin‐labelled DNA probe, followed by chemiluminescent EMSA. The unlabelled probe served as a cold competitor, and its mutant served as a negative control. The specificity of the P65‐binding motif was confirmed by the ability of P65‐specific antibody to block the DNA complex shift. An arrow indicates the P65 DNA‐binding complex. (E) ChIP DNA agarose gel results of DNA sequences immunoprecipitated by normal rabbit IgG or a rabbit anti‐P65 antibody amplified by Rorc promoter‐specific primers. Control represents the vehicle and VD represents the 1,25(OH)2D3. (F) Co‐IP experiments were carried out in WT and VDR−/− Th17 cells with or without 1,25(OH)2D3 treatment. Western blot analysis of whole cell lysate and co‐IP samples of IgG or anti‐P105/P50 antibody. (G) Western blot analysis of whole cell lysate and co‐IP samples of IgG or anti‐P100/P52. (H) Western blot analysis of whole cell lysate and co‐IP samples of IgG or anti‐P105/P50 antibody. Data represent 3 independent experiments (average and s.e.m of triplicate samples). **P < .01
RORγt, encoded by Rorc, is a key transcription factor for the development and function of Th17 cells. We have found that the p65 translocation from cytoplasmic to the nucleus was inhibited by 1,25(OH)2D3. We used Jaspar database to identify potential P65 and Rorc promoter‐binding sites. We did luciferase reporter assay and found luciferase activities were significantly enhanced in pcDNA‐p65 and pGL3‐Rorc‐5′UTR‐WT plasmid co‐transfected cells compared to the empty vector and pGL3‐Rorc‐5′UTR‐MUT plasmid co‐transfected cells. The luciferase activities were significantly reduced in pcDNA‐p65 and pGL3‐Rorc‐5′UTR‐WT plasmid co‐transfected cells compared to the pcDNA‐p65 and pGL3‐Rorc‐5′UTR‐MUT plasmid co‐transfected cells (Figure 2C). In addition, EMSA‐Super‐shelf assay was performed and demonstrated the validity of P65‐binding sites in the Rorc promoter (Figure 2D). To confirm P65 binding to Rorc promoter‐binding sites, ChIP assay was performed. The treatment of 1,25(OH)2D3 inhibited the binding of P65 and Rorc promoter‐binding site (Figure 2E). These results indicate that Rorc was the direct target of transcription factor P65 in Th17 cells and the binding of P65 and Rorc promoter‐binding sites was suppressed by 1,25(OH)2D3.
3.3. 1,25(OH)2D3 suppressed the p65 translocation to nucleus and expression of RORγt/IL‐17 by VDR
We further assessed the protein level of P65 in nuclear of Th17 cells and found 1,25(OH)2D3 treatment suppressed the p65 translocation to nucleus. Less P65 protein was translated to nucleus in WT Th17 cells compared with VDR−/− Th17 cells under presence of 1,25(OH)2D3 (Figure 3A). The P65 in VDR−/− and WT Th17 cells was knockdown, and treated with 1,25(OH)2D3 or vehicle. As shown in the Figure 3B, si‐P65 transfected decreased the mRNAs levels of P65, RORγt and IL‐17 in WT Th17 cells. The mRNAs levels of RORγt and IL‐17 in both si‐NC and si‐P65 groups were down‐regulated by 1,25(OH)2D3 compared with vehicle. Correspondingly, the similar results were found in protein level of P65, RORγt and IL‐17 both in si‐NC group (Figure 3C,D) and si‐P65 group (Figure 3E,F).
Figure 3.
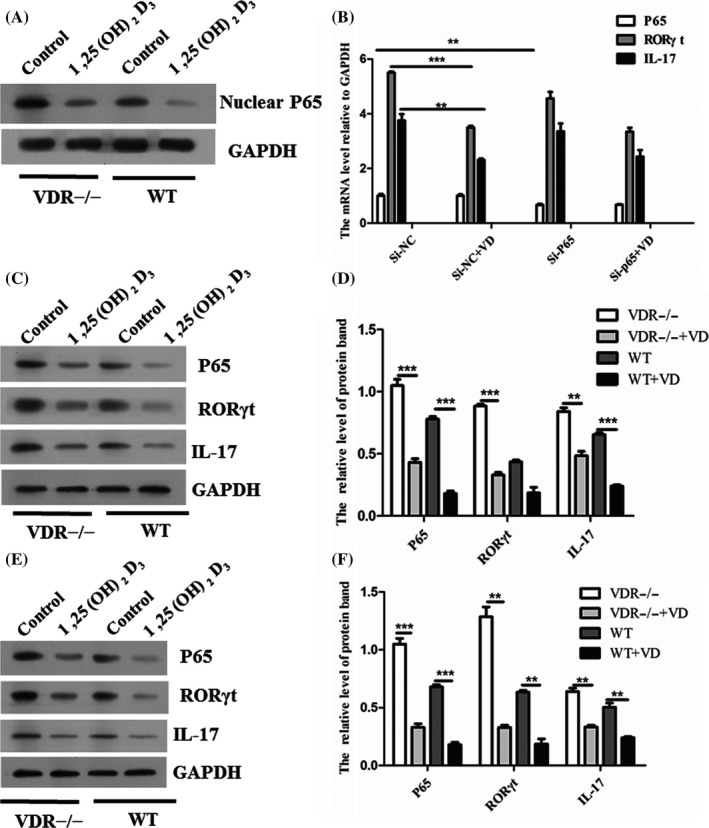
1,25(OH)2D3 inhibited the p65 translocation to nucleus and expression of RORγt/IL‐17 by VDR. A, The protein level of P65 in nucleus of VDR−/− and WT Th17 cells with the treatment of 1,25(OH)2D3 or vehicle. B, The siRNA targeting against P65 was used to knockdown the P65 in VDR−/− and WT Th17 cells. Then, the cells were treated with 1,25(OH)2D3 or vehicle and the mRNA level of P65, RORγt and IL‐17 was tested. C, The protein level of P65, RORγt and IL‐17 was tested in si‐NC group by Western blot. D, Densitometry plot of results from (C). The relative expression levels were normalized to GAPDH. Data represent the mean ± standard error (n = 3). E, The protein level of P65, RORγt and IL‐17 was tested in si‐P65 group by Western blot. F, Densitometry plot of results from (E). The relative expression levels were normalized to GAPDH (n = 3). Data represent 3 independent experiments (average and s.e.m of triplicate samples). **P < .01. ***P < .005
3.4. 1,25(OH)2D3 inhibited Th17 cells differentiation in vitamin D deficiency mouse model following vaccination with BCG
To reveal the role of VD in Th17 in vivo, we establish VD deficiency mouse model using yellow light and purified diet without VD. Measurement of the level of VD, RANKL and IL‐17 in the blood was performed. We found that the VD level was elevated with 1,25(OH)2D3 addition (Figure 4A). Intriguingly, 1,25(OH)2D3 treatment down‐regulated the level of RANKL and IL‐17 in the blood (Figure 4B,C). Then, we assessed the mRNA level of IL‐17, RORγt and RANKL in the spleen. We found that the addition of VD decreased the mRNA level of IL‐17 and RANKL both in VDR−/− and WT group (Figure 4D,E). However, 1,25(OH)2D3 decreased the expression of RORγt in WT group but not in VDR−/− group (Figure 4F). Then, we found the protein level of nuclear P65, IL‐17, RORγt and RANKL was significantly down‐regulated by 1,25(OH)2D3 both in VDR−/− and WT group (Figure 4G,H). In addition, the IKB and cytoplasm P65 protein level were significantly enhanced by 1,25(OH)2D3 both in VDR−/− and WT group (Figure 4G,H).
Figure 4.
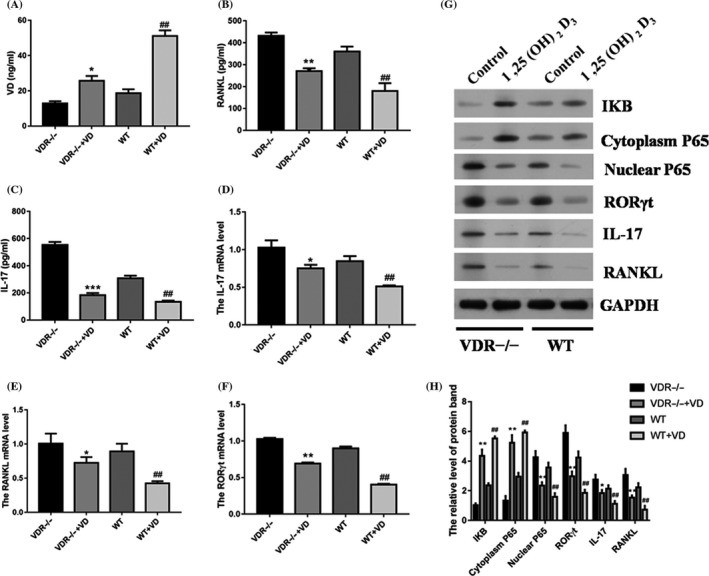
1,25(OH)2D3 inhibited Th17 cells in vitamin D deficiency mouse model. ELISA assay was performed to test the level of VD (A), RANKL (B) and IL‐17 (C) in the blood. Q‐PCR assessed the mRNA level of IL‐17, RORγt and RANKL in spleen (D‐F). (G) Western blot analysed the protein level of IKB, cytoplasm P65, nuclear P65, IL‐17, RORγt and RANKL in Th17 cells. (H) Densitometry plot of results from Figure 4G. The relative expression levels were normalized to GAPDH. Data represent the mean ± standard error (n = 3). *P < .05, **P < .01, ***P < .001 vs VDR−/− group. ## P < .01, vs WT group
3.5. 1,25(OH)2D3 inhibited the inflammatory infiltrates and expression of P65, RORγt and IL‐17 in the spleen of vitamin D deficiency mouse model
Haematein eosin and immunohistochemistry assay was performed in the spleen of mice. Histological analysis showed less intratumoural leukocyte populations in mice under 1,25(OH)2D3 treatment both in VDR−/− and WT group (Figure 5A). Immunohistochemical staining of P65 showed less P65‐positive cells in mice under 1,25(OH)2D3 treatment both in VDR−/− and WT group (Figure 5B). The treatment of 1,25(OH)2D3 also decreased the level of P105/P50, P100/P52 and phosphorylated IκBα in spleen of VD deficiency model mice (Figure S1C,D).
Figure 5.
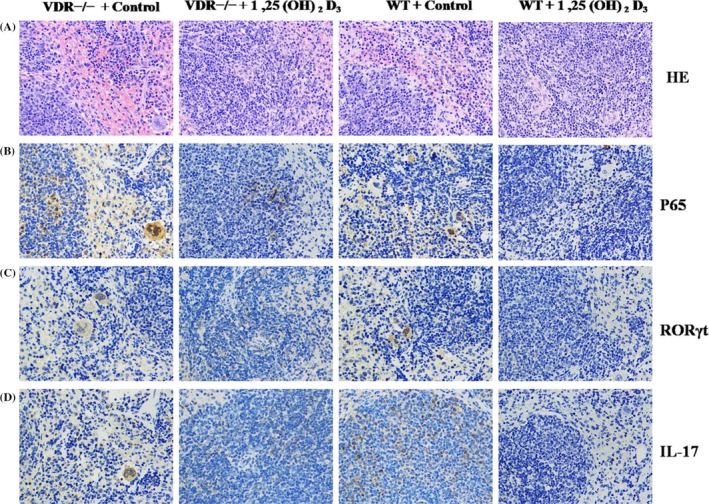
1,25(OH)2D3 inhibited the inflammatory infiltrates and expression of P65, RORγt and IL‐17 in the spleen of vitamin D deficiency mouse model. A, Representative images of haematoxylin and eosin staining in spleen of vitamin D deficiency model mice. B, Representative images of immunohistochemical (IHC) staining for P65. C, Representative images of IHC staining for RORγt. D, Representative images of IHC staining for IL‐17. Magnification, 400×
1,25(OH)2D3 decreased the expression of RORγt and IL‐17 in the spleen of VDR−/− and WT group (Figure 5C,D). In addition, VDR−/− mice showed the enhanced expression of P65, RORγt and IL‐17 in the spleen compared with WT mice under the presence of 1,25(OH)2D3 or control (Figure 5B‐D). Together, our findings showed that 1,25(OH)2D3 inhibited inflammatory damage by suppressing the differentiation of Th17 cells in VD deficiency mouse model.
4. DISCUSSION
Growing evidence indicated that VD deficiency was associated with susceptibility and TB reactivation.22, 23, 24 Patients with TB have a high rate of VD deficiency, both at diagnosis and during the course of treatment with anti‐TB drugs. Low VD level confers higher risk of progression from LTBI to active TB. The hypovitaminosis D can be regularly found in subjects with active TB.25 VD restricted M. tuberculosis growth by regulating the production of the antimicrobial peptide, cathelicidin in macrophages TLR activation of human macrophages up‐regulated expression of the VD receptor and the VD‐1‐hydroxylasegenes, leading to induction of the antimicrobial peptide cathelicidin and killing of intracellular M. tuberculosis.26 The antimicrobial effects of VD have been reported.The reduced VD status is known to be associated with the susceptibility to M. tuberculosis infection. 1,25(OH)2D3 has been reported to suppress the T‐cells proliferation.27 1,25(OH)2D3 was also been reported that selective inhibited the functions of Th1 over Th2 cells. 1,25(OH)2D3 was also found to suppress secretion of IFN‐gamma by Th1 clones with no obvious influence on secretion of IL‐4 by Th2.1 CD4+ CD25+ Treg expansion were induced by epidermal 1,25(OH)2D3. 28 In vitro assay, the frequency of murine CD4+ T cells was up‐regulated while the frequency of IFN‐γ‐producing cells was decreased by addition of 1,25(OH)2D3. 29 However, Staeva‐Vieira reported that IFN‐γ and IL‐4 production by murine CD4+ T cells was decreased by 1,25(OH)2D3. 30
With respect to Th17 cells, 1,25(OH)2D3 suppressed the differentiation of Th17 cells and the level of IL‐17 produced by human CD4+ T cells.31 In autoimmune encephalomyelitis, Th17 cell migration to the central nervous system was inhibited by 1,25(OH)2D3. 32 In vitro assay, we found 10 nmol/L 1,25(OH)2D3 suppressed the differentiation of Th17 cells. In addition, 1,25(OH)2D3 inhibited the NF‐κB activity and expression of RORγt/IL‐17 by inhibiting p65 transcription factor translocating to nucleus. Then, in VD deficiency mouse model, 1,25(OH)2D3 treatment decreased the mRNA level of IL‐17 and RANKL in vivo via VDR. In VD deficiency mouse model, the inflammatory infiltrates and level of P65, RORγt and IL‐17 in the spleen were suppressed by 1,25(OH)2D3. Our results were consistent with previous reports and we elucidate the underlying molecular mechanism. We found that Rorc was the direct target of transcription factor P65 in Th17 cells and 1,25(OH)2D3 inhibited the p65 translocation to nucleus by VDR. The binding of P65 and Rorc promoter‐binding sites was inhibited by 1,25(OH)2D3.
It has been reported that the production of IL‐17and IFN‐γ in VDR−/− CD8+ T cells was higher than WT CD8+ T cells following transfer to immunodeficient recipients. In vivo proliferation assay, the proliferation index of VDR−/− CD8+ T cells was higher than WT CD8+ T cells.33 In our study, the difference between VDR−/− mice and WT mice group was significant in Th17 cell differentiation, NF‐κB activity and relative gene expression.
Previous studies reported that experimental allergic encephalitis was exacerbated by activating Th1 and Th17 responses induced by NF‐kB activation (Cowan et al, 2010). NF‐kB signalling pathway was reported to directly promote the transcription of Foxp3 (Momparler, 2005). In our study, the activation of NF‐κB was inhibited by 1,25(OH)2D3. VDR interacts with P105/P50, P100/P52 and P65 NF‐κB family proteins. So, the VDR may form a ternary complex with NF‐κB and IκBα to inhibit the nuclear translocation of P65. Rorc was confirmed as a downstream gene of the transcription factor P65 in T cells. Collectively, 1,25(OH)2D3 suppressed the NF‐κB activity and expression of RORγt/IL‐17 by inhibiting p65 transcription factor translocating to nucleus. Our findings suggested that 1,25(OH)2D3 suppressed Th17 cells differentiation in TB via regulating the NF‐κB activity and expression of IL‐17.
CONFLICT OF INTEREST
The authors have no conflict of interest.
AUTHOR CONTRIBUTIONS
Dong Sun performed the experimental work. Fei Luo and Jun‐chao Xing participated in experimental design and prepare figures. Dong Sun, Fei Zhang, Jian‐zhong Xu and Ze‐hua Zhang contributed to interpretation of data and prepare papers. The article was written by Sun Dong. All authors reviewed the final manuscript.
Supporting information
ACKNOWLEDGEMENTS
This work was funded by grants from the National Science Foundation of China (Grant No.81401816).
Sun D, Luo F, Xing J‐c, Zhang F, Xu J‐z, Zhang Z‐h. 1,25(OH)2D3 inhibited Th17 cells differentiation via regulating the NF‐κB activity and expression of IL‐17. Cell Prolif. 2018;51:e12461 10.1111/cpr.12461
Correction added on 22 May 2018 after first online publication: The grant number was previously incorrect and has been corrected in this version of the article.
Contributor Information
Jian‐zhong Xu, Email: xjzslw@163.com.
Ze‐hua Zhang, Email: zhangzehuatmmu@163.com, Email: zhangzehua001612@126.com.
REFERENCES
- 1. Li R, Liang J, Ni S, et al. A mesenchymal‐to‐epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51‐63. [DOI] [PubMed] [Google Scholar]
- 2. Zeisberg M, Neilson EG. Biomarkers for epithelial‐mesenchymal transitions. J Clin Invest. 2009;119:1429‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The mouse snail gene encodes a key regulator of the epithelial‐mesenchymal transition. Mol Cell Biol. 2001;21:8184‐8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang R, Lan Y, Norton CR, Sundberg JP, Gridley T. The Slug gene is not essential for mesoderm or neural crest development in mice. Dev Biol. 1998;198:277‐285. [PubMed] [Google Scholar]
- 5. Waldmann J, Slater EP, Langer P, et al. Expression of the transcription factor snail and its target gene twist are associated with malignancy in pheochromocytomas. Ann Surg Oncol. 2009;16:1997‐2005. [DOI] [PubMed] [Google Scholar]
- 6. Bornman L, Campbell SJ, Fielding K, et al. Vitamin D receptor polymorphisms and susceptibility to tuberculosis in West Africa: a case‐control and family study. J Infect Dis. 2004;190:1631‐1641. [DOI] [PubMed] [Google Scholar]
- 7. Thakur S, Feng X, Qiao Shi Z, et al. ING1 and 5‐azacytidine act synergistically to block breast cancer cell growth. PLoS ONE. 2012;7:e43671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindner DJ, Wu Y, Haney R, et al. Thrombospondin‐1 expression in melanoma is blocked by methylation and targeted reversal by 5‐Aza‐deoxycytidine suppresses angiogenesis. Matrix Biol. 2013;32:123‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Samavarchi‐Tehrani P, Golipour A, David L, et al. Functional genomics reveals a BMP‐driven mesenchymal‐to‐epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64‐77. [DOI] [PubMed] [Google Scholar]
- 10. Mikyskova R, Indrova M, Vlkova V, et al. DNA demethylating agent 5‐azacytidine inhibits myeloid‐derived suppressor cells induced by tumor growth and cyclophosphamide treatment. J Leukoc Biol. 2014;95:743‐753. [DOI] [PubMed] [Google Scholar]
- 11. Park BV, Pan F. The role of nuclear receptors in regulation of Th17/Treg biology and its implications for diseases. Cell Mol Immunol. 2015;12:533‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Momparler RL, Ayoub J. Potential of 5‐aza‐2’‐deoxycytidine (Decitabine) a potent inhibitor of DNA methylation for therapy of advanced non‐small cell lung cancer. Lung Cancer. 2001;34(Suppl 4):S111‐S115. [DOI] [PubMed] [Google Scholar]
- 13. Eberl G. RORgammat, a multitask nuclear receptor at mucosal surfaces. Mucosal Immunol 2016;10:27‐34. [DOI] [PubMed] [Google Scholar]
- 14. Liu S, Song L, Yao H, et al. MiR‐375 is epigenetically downregulated by HPV‐16 E6 mediated DNMT1 upregulation and modulates EMT of cervical cancer cells by suppressing lncRNA MALAT1. PLoS ONE. 2016;11:e0163460. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15. Kokkinos MI, Murthi P, Wafai R, Thompson EW, Newgreen DF. Cadherins in the human placenta–epithelial‐mesenchymal transition (EMT) and placental development. Placenta. 2010;31:747‐755. [DOI] [PubMed] [Google Scholar]
- 16. Chen Y, Wang K, Qian CN, Leach R. DNA methylation is associated with transcription of Snail and Slug genes. Biochem Biophys Res Commun. 2013;430:1083‐1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ren J, Nie Y, Lv M, et al. Estrogen upregulates MICA/B expression in human non‐small cell lung cancer through the regulation of ADAM17. Cell Mol Immunol. 2015;12:768‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2016;389:299‐311. [DOI] [PubMed] [Google Scholar]
- 19. Maresso KC, Tsai KY, Brown PH, Szabo E, Lippman S, Hawk ET. Molecular cancer prevention: current status and future directions. CA Cancer J Clin. 2015;65:345‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aggarwal BB. Nuclear factor‐kappaB: the enemy within. Cancer Cell. 2004;6:203‐208. [DOI] [PubMed] [Google Scholar]
- 21. Escarcega RO, Fuentes‐Alexandro S, Garcia‐Carrasco M, Gatica A, Zamora A. The transcription factor nuclear factor‐kappa B and cancer. Clin Oncol (R Coll Radiol). 2007;19:154‐161. [DOI] [PubMed] [Google Scholar]
- 22. Li H, Chiappinelli KB, Guzzetta AA, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5‐azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Slominski AT, Brozyna AA, Skobowiat C, et al. On the role of classical and novel forms of vitamin D in melanoma progression and management. J Steroid Biochem Mol Biol. 2017;177:159‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gois PHF, Ferreira D, Olenski S, Seguro AC. Vitamin D and infectious diseases: simple bystander or contributing factor? Nutrients. 2017;9:651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arnedo‐Pena A, Juan‐Cerdan JV, Romeu‐Garcia A, et al. Vitamin D status and incidence of tuberculosis among contacts of pulmonary tuberculosis patients. Int J Tuberc Lung Dis. 2015;19:65‐69. [DOI] [PubMed] [Google Scholar]
- 26. Liu PT, Stenger S, Li H, et al. Toll‐like receptor triggering of a vitamin D‐mediated human antimicrobial response. Science. 2006;311:1770‐1773. [DOI] [PubMed] [Google Scholar]
- 27. Chen LH, Liu DW, Chang JL, et al. Methylation status of insulin‐like growth factor‐binding protein 7 concurs with the malignance of oral tongue cancer. J Exp Clin Cancer Res. 2015;34:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Watanabe K, Emoto N, Hamano E, et al. Genome structure‐based screening identified epigenetically silenced microRNA associated with invasiveness in non‐small‐cell lung cancer. Int J Cancer. 2012;130:2580‐2590. [DOI] [PubMed] [Google Scholar]
- 29. Gao Y, Zeng F, Wu JY, et al. MiR‐335 inhibits migration of breast cancer cells through targeting oncoprotein c‐Met. Tumour Biol. 2015;36:2875‐2883. [DOI] [PubMed] [Google Scholar]
- 30. Staeva‐Vieira TP, Freedman LP. 1,25‐dihydroxyvitamin D3 inhibits IFN‐gamma and IL‐4 levels during in vitro polarization of primary murine CD4 + T cells. J Immunol. 2002;168:1181‐1189. [DOI] [PubMed] [Google Scholar]
- 31. Chao YL, Shepard CR, Wells A. Breast carcinoma cells re‐express E‐cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010;9:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal‐to‐epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor‐2. Cancer Res. 2006;66:11271‐11278. [DOI] [PubMed] [Google Scholar]
- 33. Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol. 2013;425:3723‐3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials