

Abstract
Objectives
Prolyl hydroxylases (PHDs) play essential roles in oxygen‐sensing system, whereas the effects of PHDs on inflammation have not been totally uncovered. Our study aimed to investigate the role of PHDs in lipopolysaccharide (LPS)‐induced inflammation of human gingival fibroblasts (HGFs) and clarify the potential mechanisms.
Materials and methods
A pan hydroxylase inhibitor, dimethyloxallyl glycine (DMOG), and RNA interference were used to explore the role of PHDs in inflammation. Cytotoxic effect of DMOG was determined by cell‐counting kit‐8 and flow cytometry respectively. The secretion levels of IL‐6 and IL‐8 were assessed by ELISA. The mRNA levels of inflammatory cytokines, Toll‐like receptor (TLR) 4 and MyD88 were evaluated by quantitative real‐time PCR. The activation of NF‐κB, mitogen‐activated protein kinase (MAPK) and PI3K/AKT pathways were detected by western blot and the nuclear translocation of NF‐κB p65 was examined by immunofluorescence. Downregulation of PHD1 and PHD2 was performed with siRNA transfection.
Results
Dimethyloxallyl glycine inhibited LPS‐induced inflammatory cytokine, TLR4 and MyD88 expression in gene level and the elevated secretion of IL‐6 and IL‐8 was also downregulated. Additionally, LPS‐induced activation of NF‐κB, MAPK and AKT pathways was abolished by DMOG treatment. Importantly, LPS‐induced inflammatory cytokine expression was merely suppressed by PHD2 knockdown.
Conclusions
Prolyl hydroxylases acted as a positive regulator in LPS‐induced inflammation of HGFs via TLR4/MyD88‐mediated NF‐κB, MAPK and AKT signalling pathways and PHD2 among three isoforms was principally responsible for the effects.
1. INTRODUCTION
The pathogenesis of periodontitis is a complicated process implicated in the interactions among bacteria challenge, host immuno‐inflammatory response as well as environmental and genetic risk factors.1, 2 Gram‐negative anaerobic bacteria, especially “red complex” comprising Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia are closely related to the aetiology of periodontitis.3 Porphyromonas gingivalis, long considered as a main aetiological factor for periodontal diseases, has been confirmed to have strong association with pathogenesis of periodontitis.4, 5 Lipopolysaccharide (LPS) of P. gingivalis is the main virulence factor inducing inflammation and the destruction of periodontal tissues.6 As an important category of pattern recognition receptors, Toll‐like receptors (TLRs) could recognize LPS and transduce a series of signals through nuclear factor kappa B (NF‐κB) and mitogen‐activated protein kinase (MAPK) signalling pathway to upregulate proinflammatory cytokine expression.7
Human gingival fibroblasts (HGFs), the most abundant resident cells in periodontal connective tissues, play a key role in sustaining inflammation by triggering secretion of proinflammatory cytokines in response to LPS challenge.8 Porphyromonas gingivalis LPS‐induced interleukin (IL)‐1, IL‐6 and IL‐8 release in HGFs was owing to TLR4/myeloid differentiation primary response protein 88 (MyD88) mediated activation of NF‐κB and MAPK.9, 10, 11, 12 Furthermore, phosphoinositide‐3‐kinase (PI3K)/protein kinase B (PKB/AKT) pathway, an upstream regulator of NF‐κB, has been reported to be activated by LPS‐induced TLRs signals.13 Although upregulation of proinflammatory cytokines during inflammatory condition is a critical defensive mechanism against invading pathogens, the excessive inflammatory mediators result in periodontal tissue destruction.14 Therefore, downregulation of inflammatory mediator production by blocking the aforementioned intracellular signalling pathways may relieve the inflammatory responses13 and consequently restrict the progression of periodontitis.
Chronic inflammation is generally associated with hypoxia as a result of increased oxygen consumption by the metabolically active infiltrating immune cells and inflamed resident tissue cells.15 Prolyl hydroxylase domain containing proteins (PHDs), a family of 2‐oxoglutarate dependent dioxygenases, act as an important sensor of molecular oxygen and exist in three isoforms which are known as PHD1 (also called EGLN2), PHD2 (EGLN1) and PHD3 (EGLN3).16 Activity of PHDs depends entirely on the available oxygen. Under normal oxygen, PHDs hydroxylate hypoxia‐inducible factor (HIF)‐α subunits thereby mediating the inactivation of HIF which is an oxygen‐dependent transcription factor regulating gene expression to adapt hypoxia.17, 18 PHD/HIF axis regulates gene expression suited for hypoxia and a comprehensive crosstalk exists between oxygen‐sensing hydroxylases and inflammatory signalling pathways.15, 19 Currently, the role of PHDs in inflammation is still controversial because the effect of PHDs on inflammatory response is cell type‐specific, playing both pro‐ and anti‐inflammatory roles. It has been indicated that individual PHD isoforms knockdown or pharmacological inhibitor DMOG induced the activation of NF‐κB due to decreased PHD‐dependent hydroxylation of IκB kinase‐β (IKKβ) in HeLa cells.20 In HEK293T cells, PHD3 negatively regulated IKK‐NF‐κB signalling through inhibition of IKKγ ubiquitination.21 On the contrary, some studies indicated that inhibition of PHDs significantly diminished LPS‐induced inflammatory mediator production via blocking NF‐κB pathway in macrophages and promoted their skewing towards an anti‐inflammatory M2 phenotype.22, 23 Moreover, silencing of PHD2/3 attenuated the expression of matrix metalloproteinases and cyclooxygenase (COX)‐2 in TNF‐α‐induced nucleus pulposus cells, which was independent of HIF‐1α accumulation.24, 25 On the other hand, it has been reported that DMOG, through activation of HIF‐1α, played a protective role in the development of apical periodontitis via downregulation of NF‐κB, proinflammatory cytokine production, macrophages differentiation into M1 cells.26 An increasing number of studies have shown that DMOG was profoundly beneficial in alleviating different inflammatory diseases indicating its potential as a therapeutic target in inflammation.15, 27, 28, 29 However, the effects of PHD inhibition on HGFs and treatment of periodontal diseases are still not fully understood. Therefore, our study aimed to explore the role of PHDs in LPS‐induced inflammation of HGFs by employing DMOG or RNA interference and to clarify the underneath mechanisms thereby finding a novel therapeutic target for treatment of periodontal diseases.
2. MATERIALS AND METHODS
2.1. Tissue collection and cell culture
Healthy gingival tissues were harvested from three systemically healthy donors following the extraction of an impacted tooth. The signed information consent forms were obtained from all participators. The study protocol was approved by the Medical Ethical Committee of School of Stomatology, Shandong University (Protocol Number: GR201603). All the protocols were performed according to the guidelines of the Helsinki Declaration of 1975, as revised in 2013.
Before delivered to the labs, the isolated gingival tissues were stored in Dulbecco's modified Eagle's medium (DMEM; Hyclone, Logan, UT, USA) with 5% antibiotics (100 U/mL of penicillin, 100 mg/mL of streptomycin; Sigma Aldrich, St Louis, MO, USA) in ice bath. Immediately, the tissues were washed three times with phosphate‐buffered saline (PBS; Hyclone) and then were minced into fragments (1‐3 mm2) and digested with collagenase I (3 mg/mL; Sigma) and dispase II (4 mg/mL; Sigma) for 2 hours at 37°C. After termination of the digestion, the single‐cell suspension was seeded into 25 cm2 air‐permeable flasks and cultivated with DMEM supplemented with 20% foetal bovine serum (FBS; BioInd, Kibbutz, Israel) and 1% antibiotics at 37°C in a 5% CO2 incubator. The culture medium was changed once every 3 days and cells were passaged with 0.25% Trypsin‐EDTA (Solarbio, Beijing, China) solution till 80%‐90% confluent monolayer cells. The passaged HGFs were then cultured in 10% FBS DMEM without antibiotics. Cells at passages 4‐6 were used for the whole experiment.
2.2. Cell viability assay
The effect of DMOG on cell viability was assessed by cell‐counting kit‐8 (CCK8; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. HGFs were treated with 10, 50, 100, 250, 500 and 1000 μmol/L DMOG (Sigma) for 24 hours. Then, the culture medium of each well was replaced with 100 μL of 10% CCK‐8 reagent‐DMEM medium and the cells were incubated for another 2.5 hours at 37°C in CO2 incubator. The optical absorbance value at 450 nm wavelength was measured using a microplate reader (SPECTROstar Nano; BMG Labtech, Offenburg, Germany).
2.3. Flow cytometry
The cytotoxicity effect of DMOG was also detected by AnnexinV‐FITC apoptosis detection kit (MultiSciences, Hangzhou, China). HGFs were treated with 10, 50 and 100 μmol/L DMOG for 24 hours respectively. Then, the cells were harvested by trypsinization and washed with PBS twice. After centrifugation, the cells were resuspended in 500 μL of 1× binding buffer and 5 μL of Annexin V‐FITC and 1 μL of propidium iodide (PI) were added according to the manufacturer's protocols. After incubation at room temperature for 5 minutes in the dark, the cells were detected by flow cytometry (BD Biosciences, San Diego, CA, USA).
2.4. RNA isolation and quantitative real‐time polymerase chain reaction
Human gingival fibroblasts were pretreated with 10, 50 and 100 μmol/L DMOG for 24 hours and then were stimulated with LPS (5 μg/mL; InvivoGen, San Diego, CA, USA) in serum‐free DMEM for another 2, 6, 12 and 24 hours. The culture supernatants were aspirated for the follow‐up assay and total RNA of the cells was isolated by TRIzol reagent (Takara, Kusatsu, Japan) according to the manufacturer's protocols. The concentration and purity of RNA for each sample was evaluated using Nanodrop 2000 ultramicro spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Afterwards, RNA was reverse‐transcribed to complementary DNA with the PrimeScript® RT reagent kit with gDNA Eraser (Takara). Quantitative real‐time PCR assays were performed with LightCycler 96 Real‐Time PCR System (Roche, Basel, Switzerland) using SYBR® Premix Ex Taq™ II (Takara) to detect the gene level of IL‐6, IL‐8, TNF‐α, IL‐1β, TLR4 and MyD88. The primers used for amplification of target genes and the housekeeping gene glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) were listed in Table 1.
Table 1.
Primer sequences for qRT‐PCR
| Gene | Forward (5′‐3′) | Reverse (5′‐3′) |
|---|---|---|
| GAPDH | ACCACAGTCCATGCCATCAC | ACCACCCTGTTGCTGTA |
| IL‐6 | ATAACCACCCCTGACCCAAC | CCCATGCTACATTTGCCGAA |
| IL‐8 | TCAGAGACAGCAGAGCACAC | GGCAAAACTGCACCTTCACA |
| IL‐1β | CTTTGAAGCTGATGGCCCTAAA | AGTGGTGGTCGGAGATTCGT |
| TNF‐α | CCCAGGGACCTCTCTCTAATCA | GCTTGAGGGTTTGCTACAACAG |
| TLR4 | TGGTGTCCCAGCACTTCATC | CTGTCCTCCCACTCCAGGTA |
| MyD88 | ACTTGGAGATCCGGCAACTG | ATCCGGCGGCACCAATG |
| PHD1 | AACTGGGACGTTAAGGTGCAT | CATAGGCTGGCTTCACCTCG |
| PHD2 | AAGCCCAGTTTGCTGACATTG | CTCACACCTTTTTCACCTGTTAGA |
| PHD3 | AGGCAATGGTGGCTTGCTAT | ATCCCACCATGTAGCTTGGC |
2.5. Enzyme‐linked immunosorbent assay
The conditional medium collected from quantitative real‐time polymerase chain reaction (qRT‐PCR) assay was used for determining the protein levels of IL‐6 and IL‐8. After centrifugation to eliminate the bacterial and dead cell pellets, the supernatant was detected with enzyme‐linked immunosorbent assay (ELISA) kits (BioLegend, San Diego, CA, USA) according to the manufacturer's instructions. The optical absorbance values at 450 and 570 nm wavelength were measured using a microplate reader (SPECTROstar Nano; BMG Labtech). Moreover, the absorbance at 570 nm wavelength was subtracted from the absorbance at 450 nm when analysing the data.
2.6. Protein isolation and western blot analysis
Human gingival fibroblasts were pretreated with 10, 50 and 100 μmol/L DMOG for 24 hours, and stimulated with 5 μg/mL LPS for 1 hour. The whole protein was extracted with RIPA lysis buffer (Solarbio, Beijing, China) supplemented with 1% phenylmethanesulfonyl fluoride (Solarbio) and 1% phosphatase inhibitor (Boster, Wuhan, China). The concentration of protein was determined by a BCA Protein Assay Kit (KeyGEN BioTECH, Nanjing, China). Equal loading quantity of protein (20 μg/lane) was separated by 10% SDS‐PAGE gels and subsequently transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). After blocking with 5% nonfat milk for 1 hour, membranes were probed with the primary antibodies overnight at 4°C and incubated with horseradish peroxidase‐conjugated secondary antibodies (1:10 000; Proteintech, Chicago, IN, USA) for 1 hour at room temperature. The protein bands were visualized with enhanced chemiluminescence reagents (Millipore) and scanned using an extra‐sensitive imager (Amersham Imager 600; GE Healthcare Life Sciences, Pittsburgh, PA, USA). Image J 1.44 software (NIH, USA) was used to quantify the protein expression. The primary antibodies and dilution ratio were as follows: rabbit anti‐NF‐κB p65 (1:1000; Cell Signaling Technology, Danvers, MA, USA), rabbit anti‐IκBα (1:1000; Cell Signaling Technology), rabbit anti‐phospho‐NF‐κB p65 (1:1000; Cell Signaling Technology), rabbit anti‐phospho‐IκBα (1:1000; Cell Signaling Technology), rabbit anti‐p38 (1:1000; Cell Signaling Technology), rabbit anti‐phospho‐p38 (1:1000; Cell Signaling Technology), rabbit anti‐JNK (1:1000; Cell Signaling Technology), rabbit anti‐phospho‐JNK (1:1000; Cell Signaling Technology), rabbit anti‐ERK1/2 (1:1000; Cell Signaling Technology), rabbit anti‐phospho‐ERK1/2 (1:1000; Cell Signaling Technology), rabbit anti‐AKT (1:1000; Cell Signaling Technology), rabbit anti‐phospho‐AKT (1:1000; Cell Signaling Technology), rabbit anti‐PI3K p85α (1:1000; Abcam, Cambridge, UK), rabbit anti‐phospho‐PI3K p85α (1:500; Affinity Biosciences, Cincinnati, OH, USA) and GAPDH (1:10 000; Proteintech).
2.7. Immunocytochemistry
HGFs were pretreated with 10, 50 and 100 μmol/L DMOG for 24 hours, and stimulated with 5 μg/mL LPS for 1 hour. Cells were fixed with 4% paraformaldehyde for 10 minutes and then permeabilized using 0.5% Triton X‐100 (Solarbio) for 10 minutes. After blocking with 10% normal goat serum, cells were incubated with an anti‐NFκB RelA/p65 primary antibody (1:400; Cell Signaling Technology) at 4°C overnight, washed with PBS three times, and incubated with Alexa Fluor 594‐conjugated goat anti‐rabbit IgG secondary antibody (1:500; Proteintech) in the dark for 1 hour at 37°C. Nuclei were stained with 2‐(4‐Amidinophenyl)‐6‐indolecarbamidine dihydrochloride (DAPI; Proteintech). Images were observed under a fluorescence microscope (OLYMPUS IX73, Tokyo, Japan) in the darkroom and captured by the camera and imaging software (OLYMPUS cellSens Standard 1.17).
2.8. siRNA transfection and effects of PHD1/2 knockdown on inflammatory cytokine expression
About 30%‐50% confluent HGFs were transfected with small interfering RNA with fluorescence (FAM‐siRNA; GenePharma, Shanghai, China) using Lipofectamine 2000 (Invitrogen) to determine the transfection efficiency as previously described.30 To obtain the optimal PHD1/2 gene knockdown effect, three different small‐interfering sequences were respectively designed for PHD1 (478, 1364, 1608) and PHD2 (3758, 4216, 4405). The siRNA sequences targeting PHD1 and PHD2 were listed in Table 2. After transfection for 6 hours, the reduced‐serum medium (Opti‐MEM; Gibco, Grand Island, NY, USA) was replaced with 10% FBS DMEM and cultured for additional 24 or 48 hours. The silencing efficiency of PHD1/2 at 24 and 48 hours after transfection was determined by qRT‐PCR. The sequences used for the following assays were siPHD1 1608 and siPHD2 4216 respectively.
Table 2.
Sequences for small‐interfering RNA
| RNA oligo | Sense (5′‐3′) | Antisense (5′‐3′) |
|---|---|---|
| Negative control | UUCUCCGAACGUGUCACGUTT | ACGUGACACGUUCGGAGAATT |
| siPHD1‐487 | CCUCAGUUACCAGGGUCUUTT | AAGACCCUGGUAACUGAGGTT |
| siPHD1‐1364 | GCAUCACCUGUAUCUAUUATT | UAAUAGAUACAGGUGAUGCTT |
| siPHD1‐1608 | GCUAGCAUCAGGACAGAAATT | UUUCUGUCCUGAUGCUAGCTT |
| siPHD2‐3758 | GCAUGAACAAGCACGGCAUTT | AUGCCGUGCUUGUUCAUGCTT |
| siPHD2‐4216 | GCUGACAUUGAACCCAAAUTT | AUUUGGGUUCAAUGUCAGCTT |
| siPHD2‐4405 | CCUUCAGAUUCGGUCGGUATT | UACCGACCGAAUCUGAAGGTT |
Human gingival fibroblasts were separately transfected with siRNA PHD1 and siRNA PHD2 (30 nmol/L) using Lipofectamine 2000 (6 μL/well). After transfection for 6 hours, cells were stimulated with 5 μg/mL P. gingivalis LPS for 2, 6, 12 and 24 hours. Cell supernatants were collected to determine the protein levels of IL‐6 and IL‐8 by ELISA. The expression of IL‐6, IL‐8, TNF‐α and IL‐1β in gene level was detected through qRT‐PCR.
2.9. Statistical analysis
Data were presented as mean ± standard deviation (SD) and differences between more than two experimental groups and negative control group were analysed by one‐way ANOVA followed by Tukey HSD comparison test and variance between two groups was compared by two‐way t‐test with GraphPad Prism software (version 6, by MacKiev Software, Boston, MA, USA). P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Low concentrations of DMOG showed no cytotoxic effect on HGFs
The cytotoxic effect of various concentrations of DMOG on HGFs was detected by CCK‐8 assay and FCM. The results showed that 10, 50 and 100 μmol/L DMOG had no effect on HGFs proliferation after treating for 24 and 48 hours (Figure 1A,B). However, higher concentrations of DMOG at 250, 500 and 1000 μmol/L showed significant inhibitory effects on cell proliferation (Figure 1A,B). Furthermore, FCM results indicated that 10, 50 and 100 μmol/L DMOG treatment for 24 hours did not affect cell viability through counting PI‐positive dead cells (Figure 1C,D). Therefore, 10, 50 and 100 μmol/L DMOG were used for the further studies.
Figure 1.
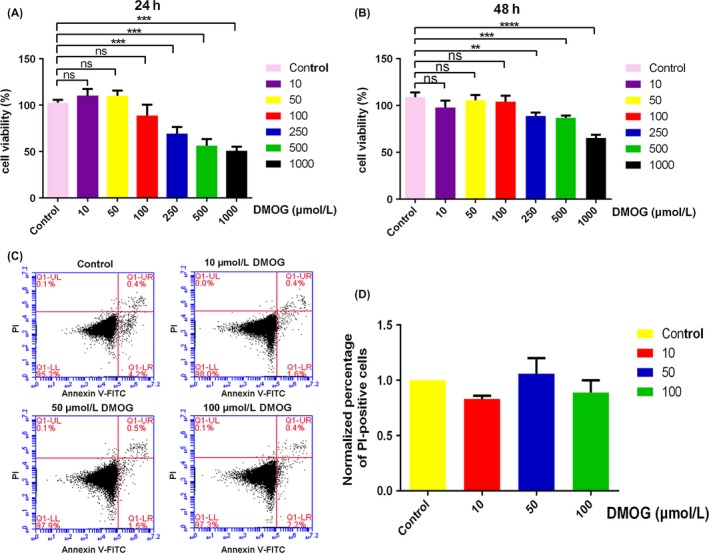
Effects of DOMG on HGFs viability. 10, 50 and 100 μmol/L DMOG exhibited no cytotoxic effect on HGFs. (A, B) The cytotoxic effect of 10, 50, 100, 250, 500 and 1000 μmol/L DMOG on HGFs was detected by CCK‐8 assay after treatment for 24 (A) and 48 h (B). The optical density was normalized to a relative value of 100% for untreated cells. (C) The cytotoxicity effect of DMOG was also detected by FCM. (D) PI‐positive dead cell percentage was normalized to the group without DMOG treatment. Data were represented as means ± standard deviation (SD) from three independent experiments. ****P < 0.0001, ***P < 0.001, and **P < 0.01 compared with control group
3.2. DMOG inhibited LPS‐induced inflammatory cytokines, TLR‐4 and MyD88 expression
RT‐PCR analysis was used to determine the effect of DMOG on LPS‐induced inflammatory response and LPS‐related receptor expression. The results showed that LPS significantly augmented the expression of IL‐6, IL‐8, TNF‐α and IL‐1β in gene level, while DMOG dose‐dependently inhibited the upregulated inflammatory cytokine expression for different timings of LPS challenge (Figure 2A,B,E,F). ELISA results revealed that LPS‐induced IL‐6 and IL‐8 production was suppressed by DMOG though there were no obvious increase of secreted IL‐6 with LPS for 2 hours and IL‐8 with LPS for 2, 6 and 12 hours (Figure 2C,D). Because the secretion level of TNF‐α and IL‐1β in the serum‐free culture supernatants was too low to be detected accurately, the data for the two cytokines was not shown. For the mRNA level of TLR4 and MyD88, qRT‐PCR analysis demonstrated that the dramatically elevated expression of TLR‐4 and MyD88 with LPS stimulation was also downregulated by DMOG pretreatment (Figure 2G,H).
Figure 2.
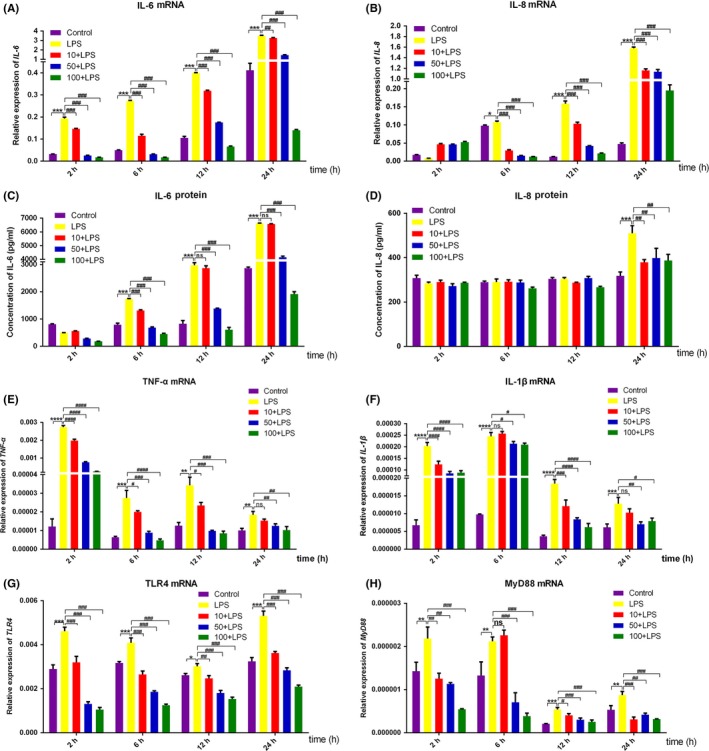
Effects of DMOG on LPS‐induced inflammatory cytokines, TLR‐4 and MyD88 expression. HGFs were pretreated with 10, 50 and 100 μmol/L DMOG for 24 h, followed by LPS stimulation for 2, 6, 12 and 24 h. (A, B, E‐H) The gene level of IL‐6 (A), IL‐8 (B), TNF‐α (E), IL‐1β (F), TLR4 (G) and MyD88 (H) were determined with qRT‐PCR. (C, D) Culture supernatants were collected to evaluate the protein level of IL‐6 (C) and IL‐8 (D) by ELISA. Histograms represented means ± SD of relative quantification from three independent experiments. ****P < 0.0001, ***P < 0.001, **P < 0.01 and *P < 0.05 compared with control group; ####P < 0.0001, ###P < 0.001, ##P < 0.01, and #P < 0.05 compared with LPS alone group
3.3. DMOG suppressed LPS‐induced activation of NF‐κB, MAPK and AKT signalling pathways
To clarify the mechanism of the inhibitory effect of DMOG, we detected the activation of NF‐κB, MAPK and PI3K/AKT signalling pathways which are responsible for LPS‐induced inflammatory cytokine production. The activation of NF‐κB was determined with the degradation and phosphorylation of IκBα, phosphorylation of RelA/p65 and subsequent translocation of NF‐κB p65 from the cytoplasm to the nucleus. Western blot indicated that LPS significantly promoted the degradation of IκBα and increased the expression of phospho‐IκBα (p‐IκBα) and phospho‐NF‐κB p65 (p‐p65) (Figure 3A). DMOG downregulated activation of NF‐κB compared with LPS group (Figure 3A). As shown in Figure 3B, nuclear translocation of NF‐κB p65 was examined employing immunofluorescence staining. NF‐κB p65 of untreated cells was predominantly localized in the cytoplasm and partly translocated to the nucleus when stimulated with LPS. However, when pretreated with DMOG, LPS‐induced nuclear translocation was blocked.
Figure 3.
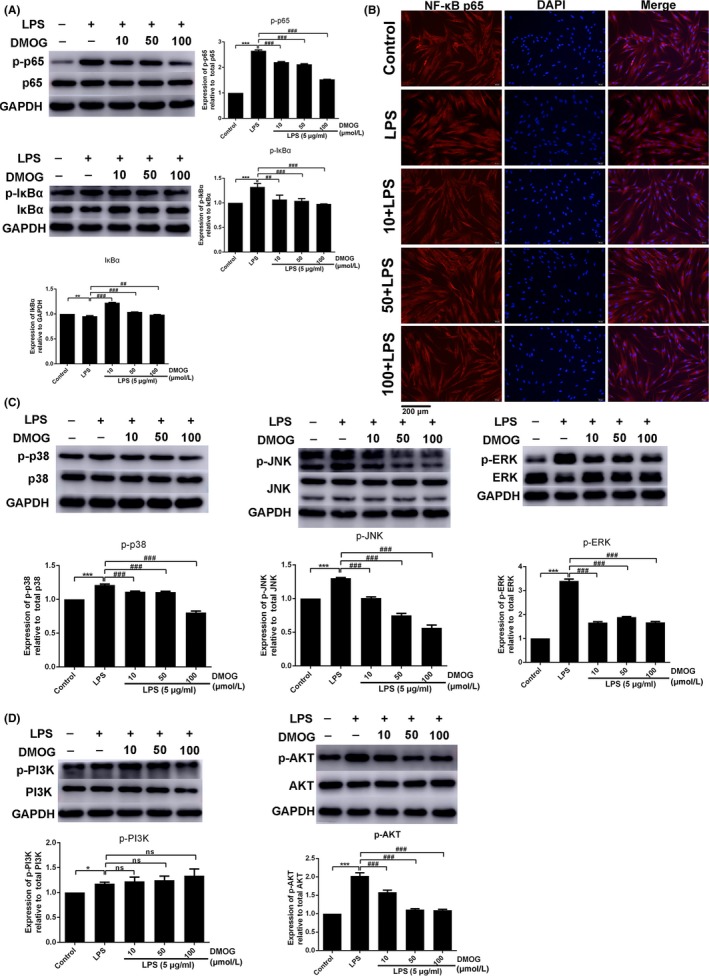
Effects of DMOG on LPS‐induced activation of NF‐κB, MAPK and PI3K/AKT pathways. HGFs were pretreated with 10, 50 and 100 μmol/L DMOG for 24 h, followed by LPS stimulation for 1 h. (A, C, D) The protein level of NF‐κB (A), MAPK (C) and PI3K/AKT (D) were determined by western blot. (B) The nuclear translocation of NF‐κB p65 was evaluated by immunofluorescence staining (NF‐κB p65, red fluorescent signals; DAPI, blue signals; magnification: ×200). Histograms showed means ± SD of relative quantification from three independent experiments. ***P < 0.001 and *P < 0.05 compared with control group; ###P < 0.001 and ##P < 0.01 compared with LPS alone group
To investigate whether MAPK pathway including p38, c‐Jun N‐terminal kinase (JNK), and extracellular signal regulated kinase (ERK), and PI3K/AKT pathway are also involved in the anti‐inflammatory effect of DMOG, phosphorylation of these kinases, crucial marker of their activation, were also evaluated by western blot. The phosphorylation levels of p38, JNK and ERK were elevated in LPS‐induced HGFs, whereas DMOG pretreatment markedly declined the phosphorylation level of these three kinases in the presence of LPS (Figure 3C). Similarly, LPS stimulation strongly activated PI3K and AKT, and DMOG significantly inhibited the activation of AKT (Figure 3D). Nevertheless, the phosphorylation of PI3K was not attenuated (Figure 3D). We concluded that DMOG suppressed LPS‐induced activation of NF‐κB, MAPK and AKT signalling pathways.
3.4. Effects of PHD1 and PHD2 knockdown
To confirm that anti‐inflammatory effect of DMOG is attributed to PHD inhibition, the gene expression of PHD was knocked down through siRNA transfection. PHD homologues (PHD1, PHD2 and PHD3) exhibited tissue‐specific expression patterns.31 Relative expression of three PHD isoforms was assessed by qRT‐PCR in HGFs. The results demonstrated that PHD2 was the most abundant isoform, followed by PHD1, whereas the expression level of PHD3 was pretty low (Figure 4A). Therefore, siRNA targeting PHD1 and PHD2 were used to transfect the cells. According to the result by FAM‐siRNA transfected assay, 30 nM was chosen as the optimal transfected concentration of siRNA for further experiment (Figure 4D‐F). To obtain the sequences possessing the optimal silencing efficiency for PHD1 and PHD2, qRT‐PCR was employed to evaluate the expression level at 24 and 48 hours after transfection. The results showed that sequences siPHD1‐1364 and siPHD2‐4405 displayed the highest silencing efficiency and reduced PHD1 and PHD2 mRNA expression by 78.4% and 91.9% compared to negative control respectively (Figure 4B,C). The two sequences were used for the following experiments.
Figure 4.
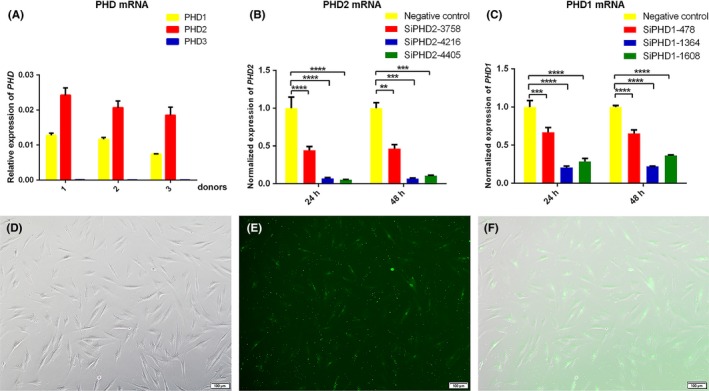
The relative expression of three PHD isoforms and effects of PHD1/2 knockdown. (A) The relative expression of three PHD isoforms was assessed with qRT‐PCR in HGFs from three donors. (D) HGFs transfected with 30 nmol/L FAM‐siRNA were observed under an inverted microscope. (E) Representative image under a fluorescence microscope after transfection. (F) Representative merge images of (D) and (E) (Magnification ×100, scale bar 100 μm). (B, C) HGFs were transfected with siRNA for 6 h and total RNA were extracted after 24 and 48 h. The relative expression of PHD1 (C) and PHD2 (B) were normalized to negative control group. Data represented means ± standard deviation (SD) from three independent experiments. ****P < 0.0001, ***P < 0.001, and **P < 0.01 compared with negative control group
3.5. Effect of PHD1 and PHD2 knockdown on LPS‐induced inflammatory cytokine expression
To investigate whether PHD knockdown attenuates LPS‐induced inflammatory response, PHD1‐ and PHD2‐depleted HGFs were treated with LPS for different timings. The results revealed that PHD2 knockdown strongly inhibited LPS‐induced IL‐6, IL‐8, TNF‐α and IL‐1β upregulation in gene level for different timings of LPS treatment (Figure 5A‐D). For the secretion of IL‐6 and IL‐8, PHD2 depletion slightly downregulated protein level of the two cytokines (Figure 6A,B). However, LPS‐induced gene upregulation of IL‐6, IL‐8, TNF‐α and IL‐1β (Figure 5E‐H) and the secretion of IL‐6 and IL‐8 (Figure 6C,D) were not affected by PHD1 silencing. In addition, the basal levels of inflammatory cytokine expression were not changed after PHD1 and PHD2 knockdown.
Figure 5.
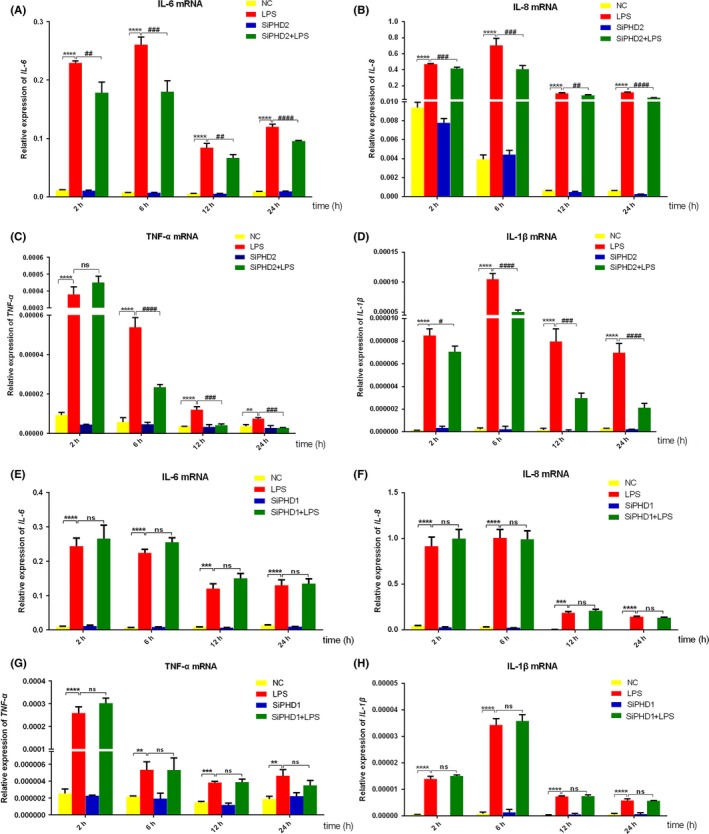
Effects of PHD1/2 knockdown on LPS‐induced IL‐6, IL‐8, TNF‐α and IL‐1β mRNA expression. HGFs were respectively transfected with siPHD1 (E, F, G, H) and siPHD2 (A, B, C, D) and stimulated with LPS for different timings. The mRNA expression of IL‐6 (A, E), IL‐8 (B, F), TNF‐α (C, G) and IL‐1β (D, H) was detected by qRT‐PCR. Histograms represented means ± SD of relative quantification from three independent experiments. ****P < 0.0001, ***P < 0.001 and **P < 0.01 compared with negative control group; ####P < 0.0001, ###P < 0.001, ##P < 0.01, and #P < 0.05 compared with LPS alone group. NC: negative control
Figure 6.
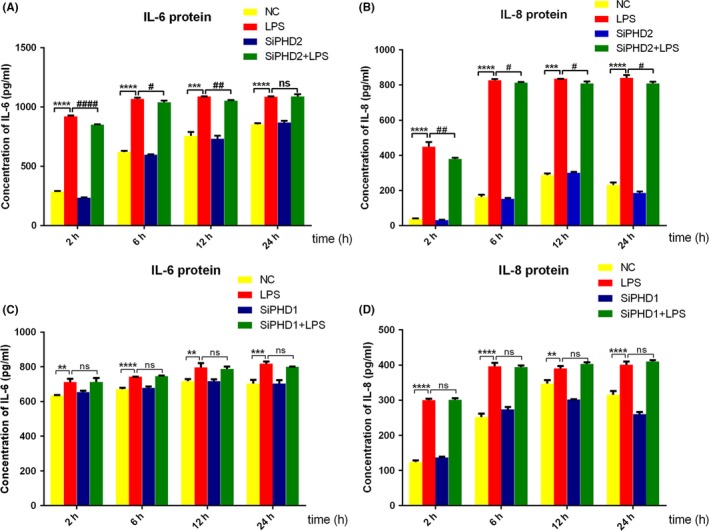
Effects of PHD1/2 knockdown on LPS‐induced IL‐6 and IL‐8 protein expression. HGFs were respectively transfected with siPHD1 (C, D) and siPHD2 (A, B) and stimulated with LPS for different timings. The culture supernatants were harvested to evaluate the secretion protein level of IL‐6 (A, C) and IL‐8 (B, D) by ELISA. Data represented means ± SD from three independent experiments. ****P < 0.0001, ***P < 0.001 and **P < 0.01 compared with negative control group; ####P < 0.0001, ##P < 0.01, and #P < 0.05 compared with LPS alone group. NC: negative control
4. DISCUSSION
Prolyl hydroxylases inhibitor DMOG has been reported to possess extensive application prospects owing to its comprehensive pharmacological effects such as the potential of anti‐inflammation, antimicrobial and bone regeneration.17, 31, 32 In our study, we demonstrated that the expression of IL‐6, IL‐8, TNF‐α and IL‐1β was significantly upregulated in LPS‐stimulated HGFs while DMOG markedly inhibited upregulation of these inflammatory cytokines. The inhibitory effect of DMOG on LPS‐induced inflammatory response was predominantly mediated by PHD2 among the three isoforms. These results suggested that PHD2 may be a novel therapeutic target for periodontal diseases.
TLR4 functions as a crucial signal‐transducing receptor that recognizes pathogen‐associated molecular patterns (PAMPs) such as LPS thereby inducing the secretion of inflammatory cytokines and activating the innate immune system.33, 34 In general, TLR4 activates the downstream signalling cascades via MyD88‐dependent pathways.35 The decreased activation of TLR4/MyD88 could attenuate the binding of LPS and thereby down‐regulated the expression of downstream messenger molecules and related inflammatory genes.9 In our study, DMOG pretreatment dose‐dependently suppressed LPS‐induced expression of TLR4 and MyD88, which indicate that the anti‐inflammatory effect of DMOG on LPS stimulation is associated with TLR4/MyD88 signalling.
Transcription factor NF‐κB pathway has long been considered as a prototypical proinflammatory signalling pathway, largely based on its role in upregulating the expression of proinflammatory genes.36 RelA/p65 and p50 subunit compose a heterodimer, of which activation triggered by stimulus such as LPS is generally defined as the canonical NF‐κB pathway. NF‐κB p65/p50 heterodimer is normally sequestered in the cytoplasm by inhibitors of NF‐κB (IκBs), and the best characterized member of IκBs is IκBα. Upon LPS stimulation, IκBα is phosphorylated and degraded, which liberates p65/p50 heterodimer to translocate to the nucleus and bind to NF‐κB consensus sites. Ultimately, the transcription of target genes is activated.37 In addition to stimulus‐induced nuclear translocation of NF‐κB, the phosphorylation of NF‐κB p65 is essential for the transcriptional activation of the prototypical p65/p50 complex after the nuclear translocation.38 Thus, we speculated that the anti‐inflammation mechanism of DMOG may be relative to its suppressive effects on NF‐κB activation. The results indicated that DMOG inhibited LPS‐induced phosphorylation of IκB and p65, as well as the degradation of IκB. The immunofluorescence staining demonstrated that increased p65 nuclear translocation with LPS stimulation was also declined by DMOG treatment. These results were supported by a recent study in LPS‐stimulated mouse peritoneal macrophages.26
As the upstream kinase of IκB, IκB kinase‐β (IKK‐β) regulates the activation of canonical NF‐κB pathway and mediates LPS‐induced IκB phosphorylation, degradation and NF‐κB liberation.36 It has been reported that LPS‐activated IKK was inhibited by DMOG in mouse peritoneal macrophages, which contributed to the attenuation of NF‐κB activation.26 PI3K/AKT signalling pathway is a critical regulator of NF‐κB activation by targeting the trans‐activation domain of NF‐κB p65 in an IKKβ‐dependent manner.39 However, not only did AKT activate the IKK complex but also IKKβ induced AKT activation and potentiated the phosphorylation of AKT thereby establishing a feed forward control.40 The present study showed that LPS elevated the phosphorylation of PI3K and AKT in HGFs, which was consistent with a previous study41 and DMOG inhibited the activation of AKT, but did not affect the phosphorylation of PI3K. The downregulated AKT phosphorylation by DMOG might be due to the interaction between AKT and IKKβ but not PI3K activation.
TLR4/MyD88‐mediated activation of MAPK is also responsible for LPS‐induced proinflammatory mediator production and regulation of NF‐κB, which is crucial for the progression of periodontal diseases.42, 43 Therefore, the effects of DMOG on LPS‐induced MAPK phosphorylation were also investigated and the results demonstrated that enhanced phosphorylation of p38, JNK and ERK were inhibited by DMOG. Takeda and his colleagues found that the activation of these kinases was not reduced by DMOG pretreatment in LPS‐induced RAW264.7 macrophages.22 In addition, it has been reported that PHD inhibitors regulated inflammatory cytokine production via activating MAPK signalling in endothelial cells and epithelial cells.44, 45 The exact reason for the contradiction between their studies and ours is unknown, and one possibility is that the inflammatory modulation mechanisms of PHDs involvement may be cell type‐specific. Further study needs to be done to confirm the inference.
Prolyl hydroxylases isoforms display special tissue‐specific expression patterns. Accordingly, the relative expression of three isoforms was detected and isoform‐specific knockdown was performed to investigate which isoform contributed to the suppressive effect. The results revealed that PHD2, the most abundant isoform, was predominantly responsible for the inhibitory effects of DMOG. Li and colleagues reported that silencing of PHD2 downregulated the production of inflammatory mediators in TNF‐α‐induced NP cells, because PHD2 promoted NF‐κB transcriptional activity through interaction and co‐localization with p65 in HIF‐1‐independent manner.24 PHD2 is the most critical oxygen sensor among the three isoforms for HIF degradation in normoxia.46 Previous studies have reported that the increased HIF‐1 by PHD inhibition was protective in several inflammatory models.26, 27, 28 Nevertheless, numerous studies have indicated that TLR4‐dependent activation of HIF by LPS stimulation markedly promoted proinflammatory cytokine production, which amplified LPS‐induced inflammatory response47, 48 and TLR4‐mediated activation of HIF‐1α was also observed in LPS‐induced HGFs.49 Hence, we speculated that PHD2 might regulate the inflammatory response via HIF‐independent pathway and further study is needed to elucidate the molecular mechanism by which PHD2 regulates LPS‐induced inflammation. In addition, our study demonstrated that PHD1 was not involved in anti‐inflammation function of DMOG. Previous studies have shown that PHD1 deficiency attenuated NF‐κB‐dependent inflammatory response to LPS in macrophages.22, 23 The reason for the contradictory results is unclear and might be due to the diverse biological effects of PHD1 in different cell types. We demonstrated that PHD3 was almost not expressed in HGFs and the effect of PHD3 was accordingly not investigated in our study.
In conclusion, our study demonstrated that PHD inhibitor DMOG inhibited proinflammatory cytokine expression in LPS‐induced HGFs. The proposed mechanism illustration was presented in Figure 7. Additionally, PHD2 may contribute to the inhibitory effect of DMOG. These results suggest that PHDs positively regulated LPS‐induced inflammation in HGFs via TLR4/MyD88‐mediated AKT/NF‐κB and MAPK pathways and PHD inhibition may provide a novel strategy for controlling the initiation and progression of periodontal diseases. However, in vivo experiments should be performed to evaluate the efficacy and safety for its clinical application in treatment of periodontitis.
Figure 7.
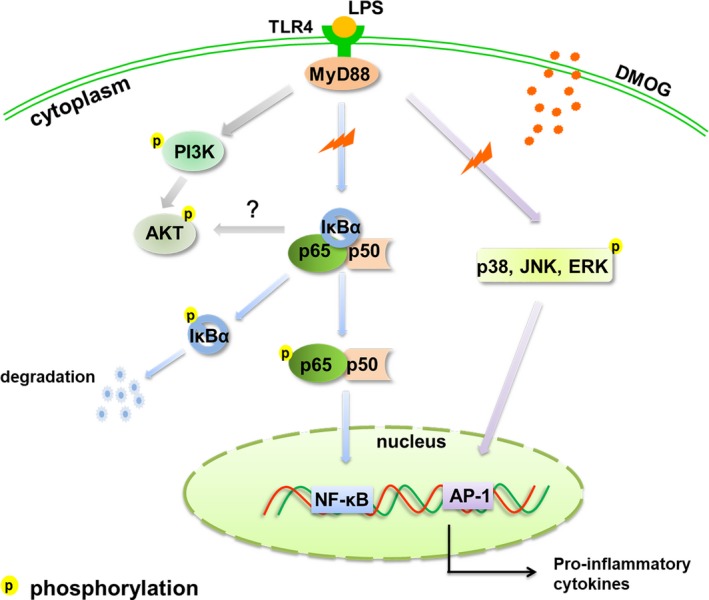
Schematic diagram of the targets for DMOG. LPS promoted the activation of PI3K/AKT, NF‐κB and MAPK signalling pathway via TLR4/MyD88‐dependent manner to upregulate proinflammatory cytokine expression. Our study demonstrated that DMOG inhibited LPS‐upregulated TLR4 and MyD88 expression and subsequently suppressed the phosphorylation and degradation of IκBα. Afterwards, the phosphorylation and nuclear translocation of NF‐κB p65 were reduced. Moreover, DMOG inhibited AKT phosphorylation, which might interact with IKKs, the upstream kinase of NF‐κB. Likewise, the activation of MAPK which was also implicated in LPS‐induced inflammation was downregulated by DMOG. Consequently, LPS‐upregulated inflammatory cytokine expression was attenuated. LPS, lipopolysaccharide; TLR4, Toll‐like receptor 4; MyD88, myeloid differentiation primary response protein 88; DMOG, dimethyloxallyl glycine; IκBs, inhibitors of NF‐κB; PI3K, phosphoinositide‐3‐kinase; AKT/ PKB, protein kinase B; NF‐κB, nuclear factor kappa B; MAPK, mitogen‐activated protein kinase; JNK, c‐Jun N‐terminal kinase; ERK, extracellular signal regulated kinase; AP‐1, Activator protein 1
ACKNOWLEDGEMENT
This research was supported by the National Natural Science Foundation of China (nos. 81670993 and 81371157), Key Research and Development Program of Shandong Province (no. 2018GSF118065), Medical and Health Science and Technology Development Program of Shandong Province (no. 2016WS0339), The National Key Research and Development Program of China (No.2017YFA0104604) and The Construction Engineering Special Fund of “Taishan Scholars” of Shandong Province (no. ts201511106, no. tsqn20161068).
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
Lingling Shang performed all the experiments and wrote the manuscript. Ting Wang and Dongdong Tong participated in collecting the gingiva tissues. Wenyan Kang and Qianyu Liang analysed data and prepare figures. Shaohua Ge designed the study and revised the manuscript.
Shang L, Wang T, Tong D, Kang W, Liang Q, Ge S. Prolyl hydroxylases positively regulated LPS‐induced inflammation in human gingival fibroblasts via TLR4/MyD88‐mediated AKT/NF‐κB and MAPK pathways. Cell Prolif. 2018;51:e12516 10.1111/cpr.12516
REFERENCES
- 1. Ji S, Choi YS, Choi Y. Bacterial invasion and persistence: critical events in the pathogenesis of periodontitis? J Periodontal Res. 2015;50:570‐585. [DOI] [PubMed] [Google Scholar]
- 2. Seneviratne CJ, Zhang CF, Samaranayake LP. Dental plaque biofilm in oral health and disease. Chin J Dent Res. 2011;14:87‐94. [PubMed] [Google Scholar]
- 3. Trindade F, Oppenheim FG, Helmerhorst EJ, Amado F, Gomes PS, Vitorino R. Uncovering the molecular networks in periodontitis. Proteomics Clin Appl. 2014;8:748‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang HWHY, Chou MY. Occurrence of Porphyromonas gingivalis and Tannerella forsythensis in periodontally diseased and healthy subjects. J Periodontol. 2004;75:1077‐1083. [DOI] [PubMed] [Google Scholar]
- 5. Rafiei MKF, Sayehmiri F, Sayehmiri K, Sheikhi A, Zamanian Azodi M. Study of Porphyromonas gingivalis in periodontal diseases: a systematic review and meta‐analysis. Med J Islam Repub Iran. 2017;31:62‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suryono KJ, Hayashi N, Kataoka M, Nagata T. Effect of Porphyromonas gingivalis lipopolysaccharide, tumor necrosis factor‐α, and interleukin‐1β on calprotectin release in human monocytes. J Periodontol. 2003;74:1719‐1724. [DOI] [PubMed] [Google Scholar]
- 7. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll‐like receptors: significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol. 2018;37:20‐36. [DOI] [PubMed] [Google Scholar]
- 8. Ara T, Kurata K, Hirai K, et al. Human gingival fibroblasts are critical in sustaining inflammation in periodontal disease. J Periodontal Res. 2009;44:21‐27. [DOI] [PubMed] [Google Scholar]
- 9. Wang PL, Azuma Y, Shinohara M, Ohura K. Toll‐like receptor 4‐mediated signal pathway induced by Porphyromonas gingivalis lipopolysaccharide in human gingival fibroblasts. Biochem Biophys Res Commun. 2000;273:1161‐1167. [DOI] [PubMed] [Google Scholar]
- 10. Wei C, Tan CK, Xiaoping H, Junqiang J. Acanthoic acid inhibits LPS‐induced inflammatory response in human gingival fibroblasts. Inflammation. 2015;38:896‐901. [DOI] [PubMed] [Google Scholar]
- 11. Gutierrez‐Venegas G, Contreras‐Sanchez A, Ventura‐Arroyo JA. Anti‐inflammatory activity of fisetin in human gingival fibroblasts treated with lipopolysaccharide. J Asian Nat Prod Res. 2014;16:1009‐1017. [DOI] [PubMed] [Google Scholar]
- 12. Brinson CW, Lu Z, Li Y, Lopes‐Virella MF, Huang Y. Lipopolysaccharide and IL‐1beta coordinate a synergy on cytokine production by upregulating MyD88 expression in human gingival fibroblasts. Mol Immunol. 2016;79:47‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chu AJ. Antagonism by bioactive polyphenols against inflammation a systematic view. Inflamm Allergy Drug Targets. 2014;13:34‐64. [DOI] [PubMed] [Google Scholar]
- 14. Gemmell E, Marshall RI, Seymour GJ. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontol. 2000;1997(14):112‐143. [DOI] [PubMed] [Google Scholar]
- 15. Scholz CC, Taylor C. Hydroxylase‐dependent regulation of the NF‐κB pathway. Biol Chem. 2013;394:479‐493. [DOI] [PubMed] [Google Scholar]
- 16. Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393‐402. [DOI] [PubMed] [Google Scholar]
- 17. Fan L, Li J, Yu Z, Dang X, Wang K. The hypoxia‐inducible factor pathway, prolyl hydroxylase domain protein inhibitors, and their roles in bone repair and regeneration. Biomed Res Int. 2014;2014:239356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wenger RH. Cellular adaptation to hypoxia: O2‐sensing protein hydroxylases, hypoxia‐inducible transcription factors, and O2‐regulated gene expression. FASEB J. 2002;16:1151‐1162. [DOI] [PubMed] [Google Scholar]
- 19. Oliver KM, Taylor CT, Cummins EP. Hypoxia. Regulation of NFκB signalling during inflammation: the role of hydroxylases. Arthritis Res Ther. 2009;11:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase‐1 negatively regulates IkappaB kinase‐beta, giving insight into hypoxia‐induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154‐18159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu J, Taubman MB. EGLN3 inhibition of NF‐kappaB is mediated by prolyl hydroxylase‐independent inhibition of IkappaB kinase gamma ubiquitination. Mol Cell Biol. 2013;33:3050‐3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda K, Ichiki T, Narabayashi E, et al. Inhibition of prolyl hydroxylase domain‐containing protein suppressed lipopolysaccharide‐induced TNF‐alpha expression. Arterioscler Thromb Vasc Biol. 2009;29:2132‐2137. [DOI] [PubMed] [Google Scholar]
- 23. Van Welden S, De Vos M, Wielockx B, et al. Haematopoietic prolyl hydroxylase‐1 deficiency promotes M2 macrophage polarization and is both necessary and sufficient to protect against experimental colitis. J Pathol. 2017;241:547‐558. [DOI] [PubMed] [Google Scholar]
- 24. Li J, Yuan W, Jiang S, et al. Prolyl‐4‐hydroxylase domain protein 2 controls NF‐kappaB/p65 transactivation and enhances the catabolic effects of inflammatory cytokines on cells of the nucleus pulposus. J Biol Chem. 2015;290:7195‐7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita N, Gogate SS, Chiba K, Toyama Y, Shapiro IM, Risbud MV. Prolyl hydroxylase 3 (PHD3) modulates catabolic effects of tumor necrosis factor‐alpha (TNF‐alpha) on cells of the nucleus pulposus through co‐activation of nuclear factor kappaB (NF‐kappaB)/p65 signaling. J Biol Chem. 2012;287:39942‐39953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirai K, Furusho H, Hirota K, Sasaki H. Activation of hypoxia‐inducible factor 1 attenuates periapical inflammation and bone loss. Int J Oral Sci. 2018;10:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cummins EP, Seeballuck F, Keely SJ, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156‐165. [DOI] [PubMed] [Google Scholar]
- 28. Natarajan R, Salloum FN, Fisher BJ, Ownby ED, Kukreja RC, Fowler AA. Activation of hypoxia‐inducible factor‐1 via prolyl‐4 hydoxylase‐2 gene silencing attenuates acute inflammatory responses in postischemic myocardium. Am J Physiol Heart Circ Physiol. 2007;293:1571‐1580. [DOI] [PubMed] [Google Scholar]
- 29. Hams E, Saunders SP, Cummins EP, et al. The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL‐10 production by B1 cells. Shock. 2011;36:295‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kang W, Wang T, Hu Z, Liu F, Sun Y, Ge S. Metformin inhibits Porphyromonas gingivalis lipopolysaccharide‐influenced inflammatory response in human gingival fibroblasts via regulating activating transcription factor‐3 expression. J Periodontol. 2017;88:e169‐e178. [DOI] [PubMed] [Google Scholar]
- 31. Harnoss JM, Strowitzki MJ, Radhakrishnan P, et al. Therapeutic inhibition of prolyl hydroxylase domain‐containing enzymes in surgery: putative applications and challenges. Hypoxia. 2015;3:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karhausen J, Schaible B, McClean S, et al. Hypoxia modulates infection of epithelial cells by Pseudomonas aeruginosa . PLoS ONE. 2013;8:e56491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lim KH, Staudt LM. Toll‐like receptor signaling. Cold Spring Harb Perspect Bio. 2013;5:a011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Medzhitov R. Toll‐like receptors and innate immunity. Nat Rev Immunol. 2001;1:135‐145. [DOI] [PubMed] [Google Scholar]
- 35. De Nardo D. Toll‐like receptors: activation, signalling and transcriptional modulation. Cytokine. 2015;74:181‐189. [DOI] [PubMed] [Google Scholar]
- 36. Lawrence T. The nuclear factor NF‐kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gasparini CFM. NF‐κB as a target for modulating inflammatory responses. Curr Pharm Des. 2012;18:5735‐5745. [DOI] [PubMed] [Google Scholar]
- 38. Sakurai H, Suzuki S, Kawasaki N, et al. Tumor necrosis factor‐alpha‐induced IKK phosphorylation of NF‐kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916‐36923. [DOI] [PubMed] [Google Scholar]
- 39. Madrid LV, Mayo MW, Reuther JY, Baldwin AS Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF‐kappa B through utilization of the Ikappa B kinase and activation of the mitogen‐activated protein kinase p38. J Biol Chem. 2001;276:18934‐18940. [DOI] [PubMed] [Google Scholar]
- 40. You P, Fu S, Yu K, et al. Scutellarin suppresses neuroinflammation via the inhibition of the AKT/NF‐kappaB and p38/JNK pathway in LPS‐induced BV‐2 microglial cells. Naunyn Schmiedebergs Arch Pharmacol. 2018;391:743‐751. [DOI] [PubMed] [Google Scholar]
- 41. Wang Q, Zhang B, Yu JL. Farrerol inhibits IL‐6 and IL‐8 production in LPS‐stimulated human gingival fibroblasts by suppressing PI3K/AKT/NF‐kappaB signaling pathway. Arch Oral Biol. 2016;62:28‐32. [DOI] [PubMed] [Google Scholar]
- 42. Kim HG, Shrestha B, Lim SY, et al. Cordycepin inhibits lipopolysaccharide‐induced inflammation by the suppression of NF‐kappaB through Akt and p38 inhibition in RAW 264.7 macrophage cells. Eur J Pharmacol. 2006;545:192‐199. [DOI] [PubMed] [Google Scholar]
- 43. Li Q, Valerio MS, Kirkwood KL. MAPK usage in periodontal disease progression. J Signal Transduct. 2012;2012:308943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. A novel role of hypoxia‐inducible factor in cobalt chloride‐ and hypoxia‐mediated expression of IL‐8 chemokine in human endothelial cells. J Immunol. 2006;177:7211‐7224. [DOI] [PubMed] [Google Scholar]
- 45. Markel TA, Crisostomo PR, Wang M, et al. Iron chelation acutely stimulates fetal human intestinal cell production of IL‐6 and VEGF while decreasing HGF: the roles of p38, ERK, and JNK MAPK signaling. Am J Physiol Gastrointest Liver Physiol. 2007;292:G958‐G963. [DOI] [PubMed] [Google Scholar]
- 46. Berra EBE, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl‐hydroxylase 2 is the key oxygen sensor setting low steady‐state levels of HIF‐1alpha in normoxia. EMBO J. 2003;22:4082‐4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peyssonnaux C, Cejudo‐Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: essential role of hypoxia inducible factor‐1 in development of lipopolysaccharide‐induced sepsis. J Immunol. 2007;178:7516‐7519. [DOI] [PubMed] [Google Scholar]
- 48. Scholz CC, Taylor CT. Targeting the HIF pathway in inflammation and immunity. Curr Opin Pharmacol. 2013;13:646‐653. [DOI] [PubMed] [Google Scholar]
- 49. Li JP, Li FY, Xu A, et al. Lipopolysaccharide and hypoxia‐induced HIF‐1 activation in human gingival fibroblasts. J Periodontol. 2012;83:816‐824. [DOI] [PubMed] [Google Scholar]