

Abstract
Objectives
Coroglaucigenin (CGN), a natural product isolated from Calotropis gigantean by our research group, has been identified as a potential anti‐cancer agent. However, the molecular mechanisms involved remain poorly understood.
Materials and methods
Cell viability and cell proliferation were detected by MTT and BrdU assays. Flow cytometry, SA‐β‐gal assay, western blotting and immunofluorescence were performed to determine CGN‐induced apoptosis, senescence and autophagy. Western blotting, siRNA transfection and coimmunoprecipitation were carried out to investigate the mechanisms of CGN‐induced senescence and autophagy. The anti‐tumour activities of combination therapy with CGN and chloroquine were observed in mice tumour models.
Results
We demonstrated that CGN inhibits the proliferation of colorectal cancer cells both in vitro and in vivo. We showed that the inhibition of cell proliferation by CGN is independent of apoptosis, but is associated with cell‐cycle arrest and senescence in colorectal cancer cells. Notably, CGN induces protective autophagy that attenuates CGN‐mediated cell proliferation. Functional studies revealed that CGN disrupts the association of Hsp90 with both CDK4 and Akt, leading to CDK4 degradation and Akt dephosphorylation, eventually resulting in senescence and autophagy, respectively. Combination therapy with CGN and chloroquine resulted in enhanced anti‐tumour effects in vivo.
Conclusions
Our results demonstrate that CGN induces senescence and autophagy in colorectal cancer cells and indicate that combining it with an autophagy inhibitor may be a novel strategy suitable for CGN‐mediated anti‐cancer therapy.
1. INTRODUCTION
Coroglaucigenin (CGN) is a natural product isolated from the roots of Calotropis gigantean by our research group. CGN is a cardenolide with a special structure as shown in Figure 1A. Traditionally, cardenolides have been used in the treatment of congestive heart failure and arrhythmia.1, 2 Recently, cardenolides have attracted more attention due to their anti‐cancer activities.3, 4, 5 Consistent with these findings, CGN exhibits significant cytotoxicity against hepatoma carcinoma cells, gastric cancer cells and lung cancer cells.6, 7 However, the underlying mechanisms by which CGN inhibits tumour growth remain largely unknown.
Figure 1.
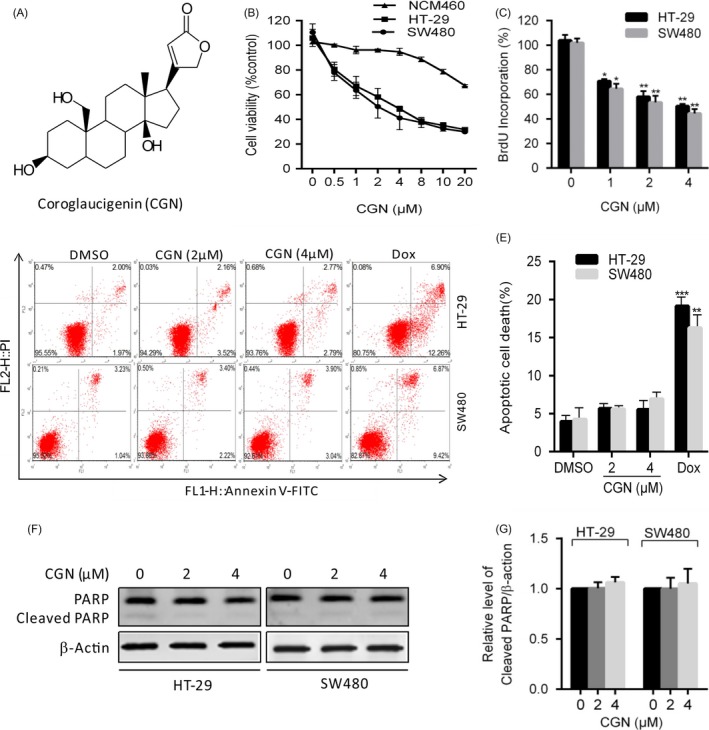
Coroglaucigenin inhibits cell proliferation independent of apoptosis in colorectal cancer cells. (A) The structure of coroglaucigenin (CGN). (B) Cell viability was measured using MTT assays in HT‐29, SW480 and NCM460 cells treated with the indicated concentrations of CGN for 24 hours. (C) Cells were treated with the indicated concentrations of CGN for 24 hours, and the degree to which CGN inhibited cell proliferation was measured using BrdU labelling. (D and E) Cells were treated with the indicated concentrations of GCN for 24 hours, and the level of apoptosis was determined using an Annexin‐V‐FITC/PI double staining assay. (F) The level of cleavage of PARP was determined using western blotting in HT‐29 and SW480 cells treated as in D. (G) ImageJ densitometric analysis of the cleaved PARP/β‐actin ratios from immunoblots. *P < 0.05, **P < .01, ***P < .001
Cellular senescence is defined as a stable cell‐cycle arrest that limits unrestricted cell proliferation and prevents tumorigenesis and tumour progression, which can be triggered by stress signals such as DNA damage response, acute oncogene activation, reactive oxygen species (ROS), ionizing radiation and exposure to chemotherapeutic drugs.8, 9, 10 A wide variety of anti‐cancer agents induce cellular senescence in tumour cells that exhibit senescence‐like morphological changes and senescence‐associated SA‐β‐gal activity through modulation of CDK activities, inhibition of telomerase activity and suppression of c‐MYC.8, 11, 12, 13 Activating programmed senescence in tumour cells may be an attractive approach to cancer treatment.
Autophagy is a conserved catabolic process important for the degradation of protein aggregates and damaged organelles. During autophagy, protein aggregates and damaged organelles are sequestered into a double‐membrane autophagosome that fuses with a lysosome to form an autolysosome, in which enclosed materials are degraded.14 Autophagy can be activated in response to various cellular stresses, such as nutrient stress, hypoxia and cytotoxic therapies.15 Autophagy is induced in many cancer cell models challenged with anti‐cancer therapies, and the Akt/mTOR pathway is considered a classic negative regulator of autophagy.16, 17 The role of autophagy activated by anti‐cancer agents is paradoxical. On the one hand, autophagy is required for tumour progression. Autophagy activation can enable tumour cell survival against chemotherapy and lead to drug resistance.18, 19 On the other hand, autophagy activation inhibits tumour growth and/or induces tumour cell death, thus playing a tumour‐suppressive role.20, 21
In this study, we demonstrated that CGN inhibits cell proliferation in colorectal cancer cells independent of apoptosis, but associated with senescence and autophagy. Our findings suggest that a combination of CGN and an autophagy inhibitor may be a potential alternative approach for the treatment of colorectal cancer.
2. MATERIALS AND METHODS
2.1. Cell culture, agents and antibodies
Human colorectal cancer cell lines HT‐29 and SW480 cells were purchased from the American Type Culture Collection. The NCM460 cell line was a kind gift from Professor Rong‐Hua Xu (Hainan Medical College, Haikou, China). All cell lines were cultured in RPMI‐1640 supplemented with 10% foetal bovine serum (Gibco, Carlsbad, CA), 100 U/mL penicillin (Sigma, St. Louis, MO) and 100 μg/mL streptomycin (Sigma, St. Louis, MO) at 37°C in an atmosphere containing 5% CO2. Coroglaucigenin (CGN) was isolated and purified from C. gigantean in our laboratory. The purity of CGN was proved to be ≥95% by chromatographic analysis. CGN was dissolved in DMSO and stored at −20°C for experimental use in this study. CQ and 3‐MA were obtained from Sigma‐Aldrich (St. Louis, MO). MG132 was obtained from MedChemExpress (Monmouth Junction, NJ). The following antibodies were used in this study: phosphorylated and total forms of Akt were purchased from Cell Signaling Technology (Boston,MA); LC3 was from Sigma‐Aldrich (St. Louis, MO); PARP, Hsp90, CDK4, Goat Anti‐Rabbit IgG H&L (Alexa Fluor® 488), and Donkey Anti‐Mouse IgG H&L (Alexa Fluor® 594) preadsorbed were purchased from Abcam (Cambridge, UK).
2.2. Cell viability assay
Cells were cultured in 96‐well plates overnight and then exposed to the tested compounds for 24 hours. The cell viabilities were determined by 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyltetrazolium bromide (MTT) assay.
2.3. BrdU assay
Cells were cultured in 96‐well plates and exposed to the tested compounds for 24 hours, and proliferation was assayed using a BrdU Cell Proliferation ELISA Kit (Abcam, ab126556).
2.4. Flow cytometry
Cells (approximately 2 × 105) were cultured and exposed to the tested compounds for 24 hours. For apoptosis analysis, cells were harvested and washed once with PBS and then resuspended in PI/Annexin‐V solution (KeyGEN Biotech, Jiangsu, China). At least 10 000 live cells were analysed on a Flow Cytometer (sysmex, CyFlow@ Cube 6, Munster, Nordrhein Westfalen, Germany). For cell‐cycle analysis, cells were harvested and washed once with PBS, fixed with 75% cold ethanol at −20°C overnight, stained with propidium iodide and analysed using a Flow Cytometer (sysmex, CyFlow@ Cube 6) to determine cell‐cycle distribution of DNA content.
2.5. Western blotting and coimmunoprecipitation
Cells were lysed with RIPA buffer. Protein concentrations were quantified by a BCA protein assay kit (23227, Thermo, Rockford, lL). Proteins were resolved on 10% SDS–PAGE and transferred to PVDF (IPVH00010, Merck Millipore, Billerica, MA) membranes. The membranes were incubated with primary antibodies at 4°C overnight after blocking, and then incubated with secondary antibodies at room temperature for 2 hours. Target proteins were examined using Enhanced Chemiluminescence reagents (Merck Millipore, WBKLS0100). Coimmunoprecipitation assays were determined by an Immunoprecipitation Kit (C600689, Sangon, Shanghai, China).
2.6. SA‐β‐gal assay
Cells were grown for 24 hours in 6‐well plates at a density of approximately 20 000 cells/well and treated with CGN for 5 days, and then were fixed and stained using a Senescence β‐Galactosidase Staining Kit (Cell Signaling Technology, #9860).
2.7. Small RNA interference
CDK4 and negative control small interfering RNA (siRNA) were synthesized by Genephama (Nanchang, China). The CDK4 siRNA was designed to target nucleotides 1062 to 1082 according to GenBank accession NM_000075. HT‐29 cells were transfected with siRNA using Lipofectamine 2000 reagent (11668027, Invitrogen, Carlsbad, CA) according to the manufacturer's protocol.
2.8. Immunofluorescence
Cells were fixed with 4% paraformaldehyde at room temperature for 30 minutes, washed 3 times with PBS and then incubated with 0.1% Triton X‐100 for permeabilization; non‐specific receptors on cells were blocked for 30 minutes with 5% BSA. Cells were stained with rabbit anti‐LC3B polyclonal antibody (Sigma, L7543) overnight at 4°C and then incubated with Alexa Fluor 488‐conjugated goat anti‐rabbit IgG (Abcam, ab150077) at room temperature for 1 hour. For confocal imaging of fixed cells, mouse anti‐Hsp90 monoclonal antibody (Abcam, ab13492) and rabbit anti‐CDK4 monoclonal antibody were used for immunostaining. Alexa Fluor 488‐conjugated goat anti‐rabbit IgG (Abcam, ab150077) and donkey anti‐mouse IgG H&L (Alexa Fluor® 594) (Abcam, ab150108) were used as secondary antibodies. Nuclei were stained with Hoechst. Images were captured using confocal laser scanning microscopy (Olympus, FV1000, Tokyo, Japan).
2.9. Animal models
HT‐29 and SW480 tumour models were established in BALB/c immunodeficient nude mice at 6‐8 weeks of age. The mice were injected with 1 × 106 corresponding tumour cells. When the tumour volume was palpable (~50 mm3 at day 6), mice were randomly divided into 3 groups for each tumour model and injected with DMSO (Ctrl, CGN 0 mg/kg), CGN (50 mg/kg) or CGN (50 mg/kg) + CQ (80 mg/kg), respectively, through the tail vein once every 2 days. Treatment was continued for 5 total times (10 days), and mice were observed to 18 days. The tumour volumes were monitored every 3 days using a handheld imaging device (TM900; Peira, Belgium).
2.10. Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0. Statistical differences were determined using a 2‐sample equal variance Student's t test. Statistical significance was defined as *P < .05. Error bars indicate SEM unless otherwise indicated.
3. RESULTS
3.1. Coroglaucigenin inhibits cell proliferation independent of apoptosis in colorectal cancer cells
To examine the effect of CGN on colorectal cancer cells, human colorectal cancer cell lines HT‐29, SW480 and NCM460 were treated with different concentrations of CGN for 24 hours, and the cell viability was measured by MTT assay. As shown in Figure 1B, CGN treatment markedly decreased cell viability in a dose‐dependent manner in both HT‐29 and W480 cell lines, but had less cytotoxic effect in NCM460 cells. Consistently, BrdU assays showed that there were a significantly lower percentage of BrdU‐positive cells in CGN‐treated cells compared with controls (Figure 1C). Collectively, these results indicated that CGN inhibits cell proliferation in colorectal cancer cells. To further evaluate whether CGN‐mediated inhibition of cell proliferation was associated with apoptosis, cells were analysed by flow cytometry following Annexin V‐FITC and propidium iodide (PI) staining. As shown in Figure 1D,E, CGN treatment for 24 hours did not obviously increase the percentage of apoptosis in both HT‐29 and SW480 cells, respectively. This was further supported by equivalent levels of cleaved PARP in CGN‐treated cells and control cells (Figure 1F,G). Collectively, these results demonstrated that CGN did not induce apoptosis in colorectal cancer cells. Taken together, these findings suggested that CGN inhibits cell proliferation independent of apoptosis in colorectal cancer cells.
3.2. Coroglaucigenin induces cell‐cycle arrest and senescence in colorectal cancer cells
To investigate the mechanism underlying CGN‐mediated cell proliferation inhibition, we analysed the effect of CGN on cell‐cycle distribution using flow cytometry. As shown in Figure 2A,B, CGN treatment altered cell‐cycle distribution profiles in HT‐29 cells. Colorectal cancer cells accumulated in the G2/M phase and decreased in the G0/G1 phase upon CGN treatment, indicating that CGN induces G2/M arrest. Senescence is defined as a stable cell‐cycle arrest, and we next examined whether cellular senescence is involved in CGN‐mediated cell growth inhibition. As shown in Figure 2C,D, CGN treatment for 7 days inhibited long‐term colony formation by 70%, indicating that upon CGN treatment, cells lost their proliferative capacity. Consistently, SA‐β‐gal assays showed that CGN significantly enhanced senescence as indicated by a significant increase in the percentage of SA‐β‐gal‐positive cells (Figure 2E,F). Taken together, these findings indicated that CGN inhibits cell growth in colorectal cancer cells via induction of G2/M cell‐cycle arrest and cellular senescence.
Figure 2.
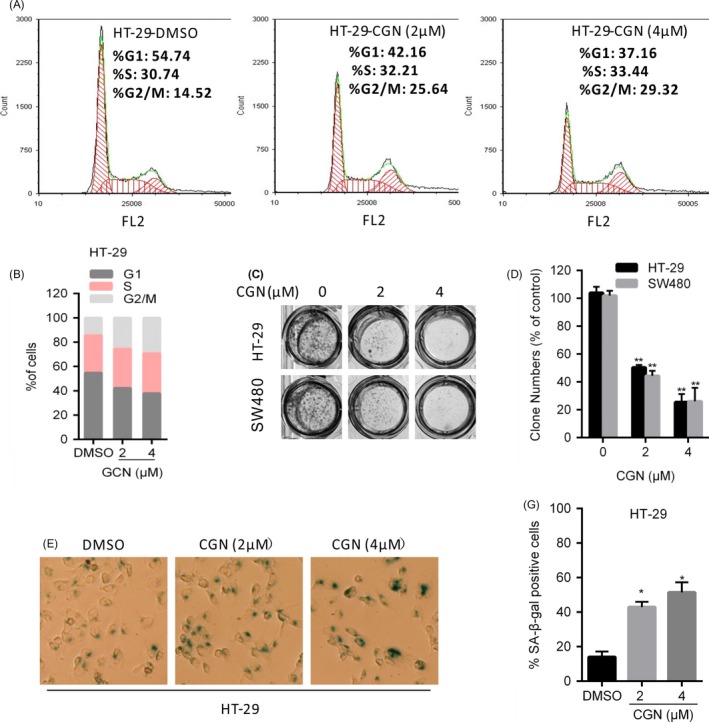
Coroglaucigenin induces cell‐cycle arrest and senescence in colorectal cancer cells. (A and B) HT‐29 cells were treated with the indicated concentrations of CGN for 24 hours, and cell‐cycle distribution was determined by flow cytometry. (C and D) Cell proliferation was measured by colony formation assay in HT‐29 and SW480 cells treated with the indicated concentrations of CGN and grown for 7 d. (E and F) HT‐29 cells were treated with the indicated concentrations of CGN for 5 d and then stained for SA‐β‐gal activity 2 d after treatment. *P < .05, **P < .01
3.3. Coroglaucigenin induces protective autophagy in colorectal cancer cells
Because autophagy is considered a target for anti‐cancer therapy,22, 23 we next explored whether autophagy is induced in CGN‐treated colorectal cancer cells. We first detected the conversion of LC3‐I to lapidated LC3‐II, one classical marker of autophagy. As shown in Figure 3A, CGN treatment markedly increased LC3‐II conversion in colorectal cancer cells. Next, we detected the distribution of endogenous LC3 puncta, another classical marker of autophagy. As shown in Figure 3C,D, CGN treatment markedly increased LC3 puncta in colorectal cancer cells. Moreover, combinatorial treatment with 3‐methyladenine (3‐MA, an autophagy early stage inhibitor) markedly decreased LC3‐II conversion in CGN‐treated cells (Figure 3B). These findings indicated that CGN induces autophagy in colorectal cancer cells. To determine whether autophagy is involved in CGN‐mediated inhibition of cell growth, we performed MTT and BrdU assays to detect cell viability and cell proliferation under inhibition of autophagy by chloroquine (CQ). As shown in Figure 3E, inhibition of autophagy by CQ, a lysosomal inhibitor, resulted in significant lower cell viability than CGN treatment alone. Consistently, similar results were observed by BrdU assay (Figure 3F). Taken together, these results indicated that CGN induces protective autophagy against CGN‐mediated inhibition of cell growth.
Figure 3.
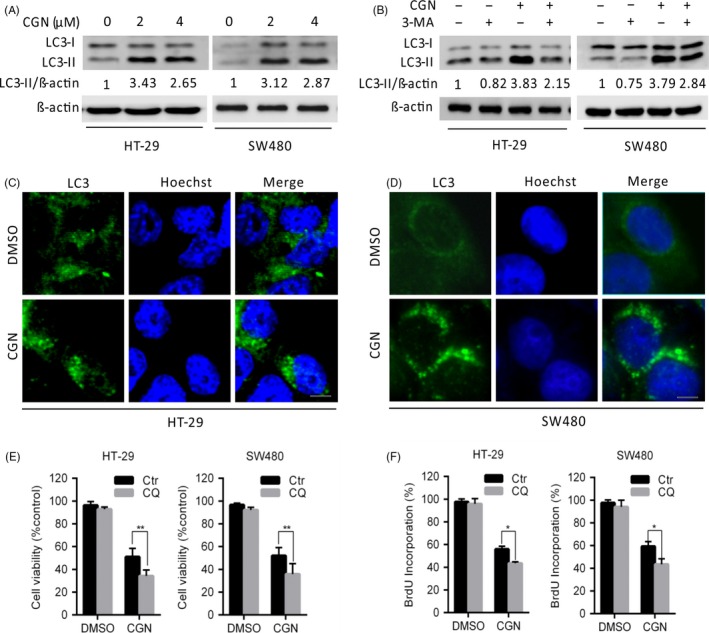
Coroglaucigenin induces protective autophagy in colorectal cancer cells. (A) Immunoblot analysis of LC3 in HT‐29 and SW480 cells treated with the indicated concentrations of CGN for 24 hours. Protein ratios were calculated following ImageJ densitometric analysis. (B) Immunoblot analysis of LC3 in cells treated with the indicated concentrations of CGN for 24 hours in the absence or presence of 3‐methyladenine (3‐MA). (C and D) HT‐29 and SW480 cells were treated with DMSO or 4 μM of CGN for 24 hours, and the formation of endogenous LC3 puncta (arrows) was visualized under a fluorescent microscope. Scale bars, 10 μm. (E) Cell viability was measured by MTT assays in cells treated with DMSO or 4 μM of CGN for 24 hours in the presence and absence of chloroquine (CQ). (F) Cell proliferation was measured using BrdU labelling in cells treated with DMSO or 4 μM of CGN for 24 hours in the presence or absence of CQ. *P < .05, **P < .01
3.4. Coroglaucigenin induces senescence through downregulation of CDK4
Cellular division is carefully monitored by cell‐cycle checkpoints, which are regulated at different stages by various cyclins, cyclin‐dependent kinases (CDK) and CDK inhibitors.24, 25 Recent studies suggest that targeting specific CDK and CDKI in the appropriate genetic context can result in cellular senescence.12, 26 Accumulating evidence has demonstrated that inhibition of CDK4/6 induces senescence in a variety of tumour cells;27, 28, 29 therefore, we investigated whether CDK4/6 is involved in CGN‐induced cellular senescence. First, we examined the expression of CDK4/6 in colorectal cells upon CGN treatment. As shown in Figure 4A, CDK4 expression was significantly decreased, but CDK6 expression was not clearly affected upon CGN treatment (data not shown), suggesting that CGN selectively inhibits CDK4 by downregulation of CDK4 expression. In agreement with previous observations that inhibition of CDK4 by chemotherapeutic drugs leads to the induction of cellular senescence,27, 29 CGN treatment markedly induced senescence in colorectal cancer cells, as evidenced by increased SA‐β‐gal activity (Figure 4B,C). Moreover, similar to CGN treatment, siRNA‐mediated knockdown of CDK4 led to a significant increase in senescence in colorectal cancer cells (Figure 4B,C). Collectively, these results indicated that CGN‐induced senescence in colorectal cells is associated with downregulation of CDK4. To determine the mechanism underlying the regulation of CDK4 by CGN, we performed reverse transcription PCR (RT‐PCR) analysis after 24 hours of CGN treatment. The results showed that there were no significant changes in mRNA levels of CDK4, suggesting that transcription regulation might not account for the decreased CDK4 expression upon CGN treatment (Figure 4D). Thus, we next examined whether CDK4 was degraded through the ubiquitination/proteasome pathway. The results showed that combinatorial treatment with proteasome inhibitor MG132 was able to rescue the CDK4 protein levels after CGN treatment (Figure 4E). Collectively, these results indicated that CGN promoted CDK4 degradation through the ubiquitination/proteasome pathway. Hsp90 is an ubiquitous molecular chaperone, and inhibition of Hsp90 prevents the stabilization of its client proteins and leads to their degradation or dephosphorylation. Since CDK4 is an Hsp90 client protein, we thus investigated whether Hsp90 is involved in CGN‐mediated downregulation of CDK4. Immunofluorescence confocal microscopy analysis revealed that CGN treatment markedly reduced the colocalization of Hsp90 with CDK4 (Figure 4F). Consistently, coimmunoprecipitation analysis showed that CGN treatment significantly decreased the association of Hsp90 with CDK4 (Figure 4G). Taken together, these results indicated that CGN disrupts the interaction of Hsp90 with CDk4, leading to downregulated CDK4, and then induces senescence in colorectal cancer cells.
Figure 4.
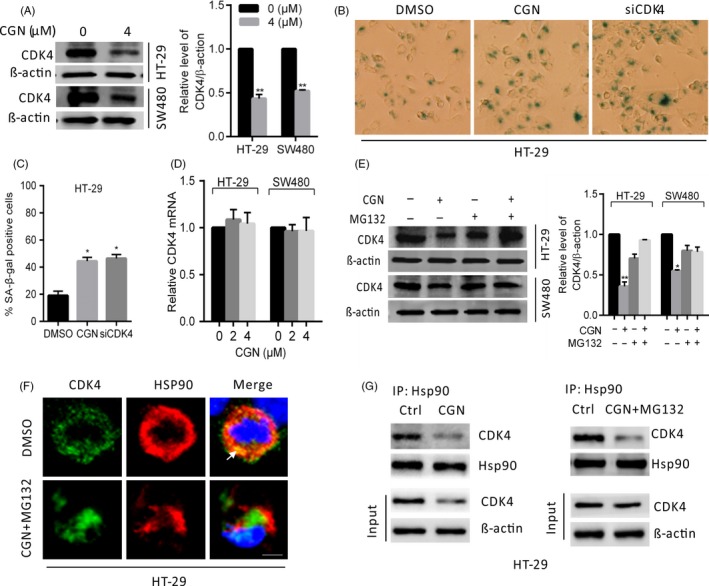
Coroglaucigenin induces senescence through the downregulation of CDK4. (A) Immunoblot analysis of CDK4 in HT‐29 and SW480 cells treated with 4 μM of CGN for 24 hours. ImageJ densitometric analysis of the CDK4/β‐actin ratios from immunoblots. (B and C) HT‐29 cells were transfected with siCDK4 or treated with 4 μM of CGN, and then stained for SA‐β‐gal activity 2 d after treatment. (D) CDK4 mRNA expression in cells treated with the indicated concentrations of CGN for 24 hours was evaluated by RT‐PCR. (E) Immunoblot analysis of CDK4 in cells treated with 4 μM of CGN for 24 hours in the presence and absence of MG132. ImageJ densitometric analysis of the CDK4/β actin ratios from immunoblots. (F) Confocal microscopy of HT‐29 cells treated with 4 μM of CGN for 24 hours in the presence and absence of MG132, then immunostained for Hsp90 (red) and CDK4 (green). Arrows indicate colocalization of green (CDK4) and red (Hsp90) signals. Scale bars, 10 μm. (G) Hsp90 interaction with CDK4 was determined by coimmunoprecipitation assays in HT‐29 cells treated with 4 μM of CGN for 24 hours in the presence and absence of MG132. *P < .05, **P < .01
3.5. Coroglaucigenin induces autophagy through disrupting the association of Hsp90 with Akt
The Akt/mTOR pathway is a major negative regulator of autophagy.16, 17 Therefore, we first investigated the phosphorylation status of Akt in CGN‐treated colorectal cancer cells. As shown in Figure 5A,B, CGN treatment significantly decreased the phosphorylation levels of Akt (Ser473). To determine the role of Akt dephosphorylation in CGN‐induced autophagy, cells were treated with insulin (an Akt activator) to reactivate Akt and rescue its phosphorylation levels. The results showed that Akt activation by insulin markedly attenuated CGN‐induced autophagy, as evidenced by the decreased LC3‐II conversion and LC3 puncta accumulation in CGN‐treated cells (Figure 5C‐G). Furthermore, we transfected a constitutively active form of Akt (CA‐Akt, the active form of Akt) to restore CGN‐induced Akt/mTOR inhibition. As shown in Figure 5E,F, over‐expressing CA‐Akt leads to decreased levels of LC3‐II conversion. Collectively, these results suggested that CGN‐induced autophagy is associated with the inactivation of Akt, which leads to blockage of the Akt/mTOR pathway. Since Akt is a Hsp90 client protein, we, thus, investigated whether Hsp90 is involved in the CGN‐mediated dephosphorylation of Akt. Coimmunoprecipitation analysis showed that CGN treatment markedly decreased the interaction of Hsp90 with Akt (Figure 5H). Taken together, these results indicated that CGN disrupts the association of Hsp90 with Akt, leading to Akt dephosphorylation, which blocks the Akt/mTOR pathway and induces autophagy in colorectal cancer cells.
Figure 5.
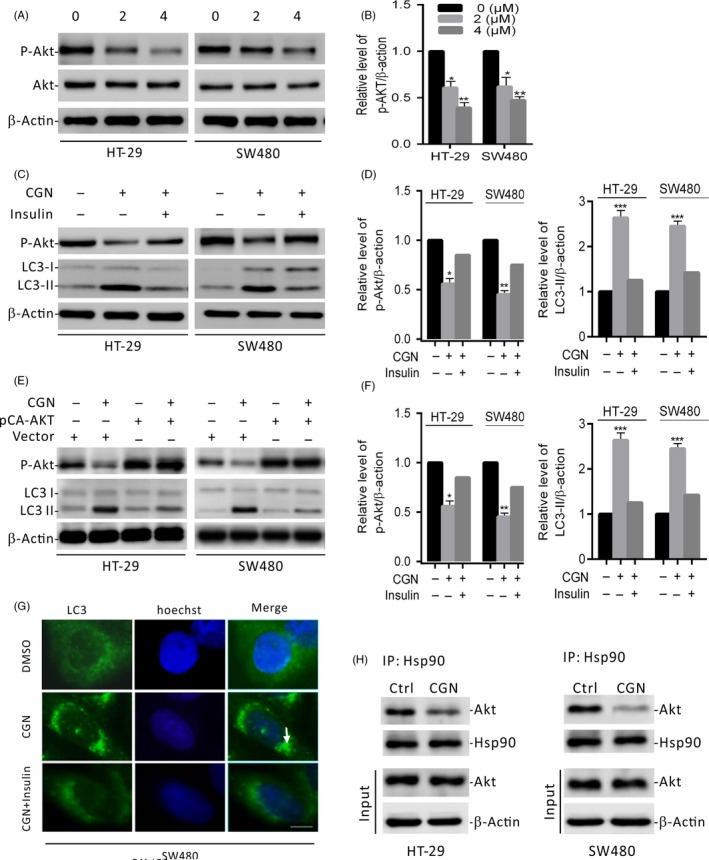
Coroglaucigenin induces autophagy through disrupting the interaction of Hsp90 with Akt. (A) Immunoblot analysis of phosphorylation of Akt (Ser473) in HT‐29 and SW480 cells treated with the indicated concentrations of CGN for 24 hours. Total Akt expression served as an internal control. (B) ImageJ densitometric analysis of the p‐AKT/β‐actin ratios from immunoblots. (C) Immunoblot analysis of LC3 and p‐Akt in cells treated with 4 μM of CGN for 24 hours in the presence and absence of insulin. (D) ImageJ densitometric analysis of the p‐AKT/β‐actin and the LC3‐II/β‐actin ratios from immunoblots. (E) Cells were transfected with an empty vector or with constitutively active CA‐Akt for 48 hours, and then cells were treated with 4 μM of CGN for another 24 hours. p‐Akt and LC3 were determined by immunoblotting. (F) ImageJ densitometric analysis of the p‐AKT/β‐actin and the LC3‐II/β‐actin ratios from immunoblots. (G) SW480 cells were treated as in C. The formation of endogenous LC3 puncta (arrows) was visualized under a fluorescent microscope. Scale bars, 10 μm. (H) Hsp90 interaction with Akt was determined by coimmunoprecipitation assays in cells treated with 4 μM of CGN for 24 hours. *P < .05, **P < .01, ***P < .001
3.6. Combination therapy with coroglaucigenin and chloroquine enhances anti‐tumour activities in vivo
HT‐29 and SW480 cells were subcutaneously implanted into BALB/c Nu/Nu mice. The mice were treated with DMSO (control), CGN, or a combination of CGN and CQ, respectively, in intervals of 2 days for 5 total treatments and observed to 18 days. Figure 6A shows the images of tumour masses (5 in each group) that were collected at day 18. Tumour volumes at different time points are shown in Figure 6B. Compared with the control group, the tumours in the CGN and CGN + CQ groups grew significantly slower, and the CGN + CQ group grew the slowest. These data strongly indicate that the disruption of CGN‐induced autophagy by CQ enhances the anti‐tumour activities of CGN.
Figure 6.
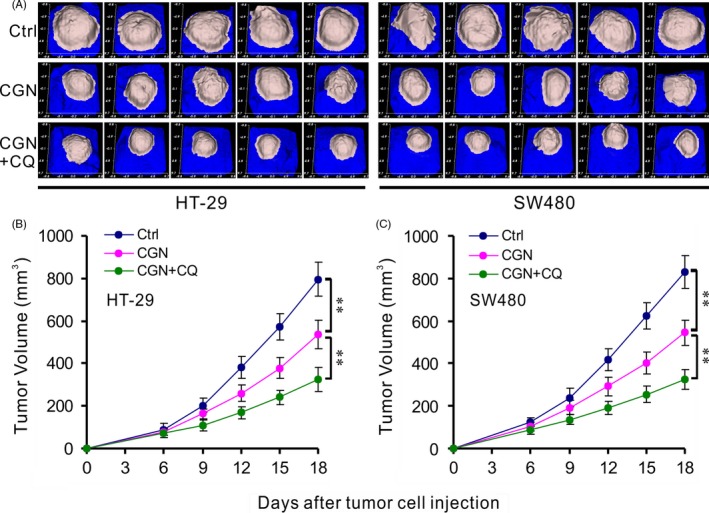
Combination therapy with coroglaucigenin and chloroquine enhances anti‐tumour activities in vivo. HT‐29 and SW480 tumour cells were injected into the right flanks of Nu/Nu mice (5 mice/group). When tumour volumes reached 50 mm3, mice were treated with indicated formulations every 2 d for 5 total treatments. (A) Images of tumour masses at day 18 in each mouse. (B) Tumour volumes at different time points. **P < .01
4. DISCUSSION
Natural products have been the major resource of compounds of value to medicine. Several natural products derived from plants are currently successfully employed in cancer treatment, such as vincristine, irinotecan, etoposide and paclitaxel.30, 31, 32 At present, cardenolides, a class of natural products, especially those derived from plants such as digitoxin, oleandrin and ouabain, have attracted much attention due to their anti‐cancer activities.3, 33 Coroglaucigenin (CGN), a cardenolide with a special structure (Figure 1A), isolated from the roots of C. gigantean by our research group, has shown potential anti‐cancer activities. CGN significantly inhibits cell proliferation in SMMC‐77210, SGC‐7901, A549, NCI‐H460 and NCI‐H446 cells. CGN is also a promising sensitizer for cancer radiotherapy in lung cancer cells.6, 7 In this study, we demonstrated that CGN markedly suppresses cell proliferation in colorectal cancer cells independent of apoptosis, but associated with cell‐cycle arrest and senescence. CGN also induces protective autophagy against CGN‐mediated inhibition of cell proliferation. Combination therapy with CGN and chloroquine resulted in enhanced anti‐tumour effects in vivo. Furthermore, we revealed that CGN targets the interaction of Hsp90 with both CDK4 and Akt to modulate the CDK4 protein levels and Akt phosphorylation status, thus resulting in senescence and autophagy, respectively. Our findings suggest that senescence and autophagy are involved in CGN‐mediated inhibition of cell proliferation in colorectal cancer cells.
Recently, several studies have linked autophagy with senescence,34, 35 but the role of autophagy on senescence is still subject to debate. Young et al showed that autophagy mediates the mitotic senescence transition,36 but Garcia‐Prat et al revealed that autophagy is essential to maintain the stem‐cell quiescent state by preventing senescence.37 Since many of the same stimuli that induce autophagy also induce senescence,38, 39 autophagy and senescence may be viewed as a continuum or 2 related phases of the same overall biological process. In this study, we showed that CGN induces both senescence and autophagy in colorectal cancer cells, but the possible role of autophagy activation in CGN‐induced senescence needs further study.
Cyclin‐dependent kinases (CDKs) regulate major cell‐cycle transitions in eukaryotic cells.24, 25 CDK4 and CDK6 (CDK4/6) mediate the transition from G0/G1‐phase to S‐phase of the cell cycle. Previous research has demonstrated that inhibition of CDK4/6 induces G1 arrest and/or senescence in a variety of tumour cells.27, 40 But in this study, we showed that downregulation of CDK4 in CGN‐treated colorectal cancer cells was not required for G1 arrest. Although CDK4 and CDK6 have similar properties in mammalian cell proliferation, they exhibit distinct functions depending on different cellular conditions and diverse cancer types.41, 42 Yu et al indicated that CDK4, but not CDK6, is required for the development of breast tumours induced by ErbB‐2.43 Puyol et al also demonstrated that ablation of CDK4, but not CDK6, induces an immediate senescence response only in lung cells that express an endogenous K‐Ras oncogene.12 In addition, Molenaar et al reported that CDK4, but not CDK6, contributes to the undifferentiated phenotype in neuroblastoma.44 Consistent with these observations, in this study, we demonstrated that CGN selectively targets CDK4, but not CDK6, to induce senescence in colorectal cancer cells, suggesting that CDK4 probably plays a more pronounced role upon CGN treatment in colorectal cancer cells compared with CDK6.
Heat shock protein 90 (Hsp90) is a ubiquitous molecular chaperone essential for the folding, maturation, activation and assembly of a variety of proteins, which are generally called “clients” of Hsp90. Inhibition of Hsp90 prevents the stabilization of these client proteins and leads to their degradation.45, 46, 47 Considering that both CDK4 and Akt are Hsp90 client proteins, we, thus, investigated whether Hsp90 is involved in CGN‐mediated downregulation of CDK4 and dephosphorylation of Akt in colorectal cancer cells. In this study, we indicated that CGN disrupts the association of Hsp90 with both CDK4 and Akt, leading to CDK4 downregulation and Akt dephosphorylation, respectively. The sustained downregulation of CDK4 induces senescence in CGN‐treated cells. Likewise, the dephosphorylation of Akt blocks the Akt/mTOR pathway, resulting in autophagy. Our findings demonstrated that CGN targets the interaction of Hsp90 with both CDK4 and Akt to modulate CDK4 expression and Akt dephosphorylation, suggesting that Hsp90 plays a critical role in CGN‐mediated cell proliferation in colorectal cancer cells.
In summary, we demonstrated that CGN inhibits the proliferation of colorectal cancer cells both in vitro and in vivo. We showed that CGN inhibits cell proliferation independent of apoptosis, but is associated with cell‐cycle arrest, senescence and autophagy in colorectal cancer cells. Mechanistically, CGN disrupts the association of Hsp90 with both CDK4 and Akt, leading to CDK4 degradation and Akt dephosphorylation, eventually resulting in senescence and autophagy, respectively. Moreover, combination therapy with CGN and chloroquine resulted in enhanced anti‐tumour effects in vivo. Our findings suggest that CGN is a potential anti‐cancer agent against colorectal cancer, and the combined use of autophagic inhibitors such as CQ may be a promising strategy for CGN‐mediated anti‐cancer therapy.
CONFLICTS OF INTEREST
There is no competing interest.
ACKNOWLEDGEMENTS
This work was funded by the National Natural Science Foundation of China (81460557, 81560599 and 81460020).
Huang Y‐H, Lei J, Yi G‐H, et al. Coroglaucigenin induces senescence and autophagy in colorectal cancer cells. Cell Prolif. 2018;51:e12451 10.1111/cpr.12451
Yong‐Hao Huang, Jing Lei and Guo‐Hui Yi contributed equally to the work.
Contributor Information
Yan Sun, Email: 18712798207@163.com.
Hao‐Fu Dai, Email: daihaofu@itbb.org.cn.
Guang‐Hong Tan, Email: tanhoho@163.com.
REFERENCES
- 1. Gheorghiade M, van Veldhuisen DJ, Colucci WS. Contemporary use of digoxin in the management of cardiovascular disorders. Circulation. 2006;113:2556‐2564. [DOI] [PubMed] [Google Scholar]
- 2. Shi LS, Liao YR, Su MJ, et al. Cardiac glycosides from Antiaris toxicaria with potent cardiotonic activity. J Nat Prod. 2010;73:1214‐1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Newman RA, Yang P, Pawlus AD, et al. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv. 2008;8:36‐49. [DOI] [PubMed] [Google Scholar]
- 4. Cerella C, Dicato M, Diederich M. Assembling the puzzle of anti‐cancer mechanisms triggered by cardiac glycosides. Mitochondrion. 2013;13:225‐234. [DOI] [PubMed] [Google Scholar]
- 5. McConkey DJ, Lin Y, Nutt LK, et al. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen‐independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 2000;60:3807‐3812. [PubMed] [Google Scholar]
- 6. Wang MY, Mei WL, Deng YY, et al. Cytotoxic cardenolides from the roots of Calotropis gigantea. Modern Pharm Res. 2008;1:4‐9. [Google Scholar]
- 7. Sun M, Pan D, Chen Y, et al. Coroglaucigenin enhances the radiosensitivity of human lung cancer cells through Nrf2/ROS pathway. Oncotarget. 2017;8:32807‐32820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Acosta JC, Gil J. Senescence: a new weapon for cancer therapy. Trends Cell Biol. 2012;22:211‐219. [DOI] [PubMed] [Google Scholar]
- 9. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705‐2715. [PubMed] [Google Scholar]
- 11. Campaner S, Doni M, Hydbring P, et al. Cdk2 suppresses cellular senescence induced by the c‐myc oncogene. Nat Cell Biol. 2010;12:54‐59. [DOI] [PubMed] [Google Scholar]
- 12. Puyol M, Martin A, Dubus P, et al. A synthetic lethal interaction between K‐Ras oncogenes and Cdk4 unveils a therapeutic strategy for non‐small cell lung carcinoma. Cancer Cell. 2010;18:63‐73. [DOI] [PubMed] [Google Scholar]
- 13. Wu CH, van Riggelen J, Yetil A, et al. Cellular senescence is an important mechanism of tumor regression upon c‐Myc inactivation. Proc Natl Acad Sci U S A. 2007;104:13028‐13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Janku F, McConkey DJ, Hong DS, et al. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528‐539. [DOI] [PubMed] [Google Scholar]
- 15. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jung CH, Ro SH, Cao J, et al. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marino G, Niso‐Santano M, Baehrecke EH, et al. Self‐consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elgendy M, Sheridan C, Brumatti G, et al. Oncogenic Ras‐induced expression of Noxa and Beclin‐1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23‐35. [DOI] [PubMed] [Google Scholar]
- 21. Liu R, Li J, Zhang T, et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: involvement of abnormal cholesterol trafficking. Autophagy. 2014;10:1241‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014;74:647‐651. [DOI] [PubMed] [Google Scholar]
- 23. Yang PM, Liu YL, Lin YC, et al. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res. 2010;70:7699‐7709. [DOI] [PubMed] [Google Scholar]
- 24. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153‐166. [DOI] [PubMed] [Google Scholar]
- 25. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zou X, Ray D, Aziyu A, et al. Cdk4 disruption renders primary mouse cells resistant to oncogenic transformation, leading to Arf/p53‐independent senescence. Genes Dev. 2002;16:2923‐2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rader J, Russell MR, Hart LS, et al. Dual CDK4/CDK6 inhibition induces cell‐cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013;19:6173‐6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. VanArsdale T, Boshoff C, Arndt KT, et al. Molecular pathways: targeting the cyclin D‐CDK4/6 axis for cancer treatment. Clin Cancer Res. 2015;21:2905‐2910. [DOI] [PubMed] [Google Scholar]
- 30. Gordaliza M. Natural products as leads to anticancer drugs. Clin Transl Oncol. 2007;9:767‐776. [DOI] [PubMed] [Google Scholar]
- 31. Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005;4:206‐220. [DOI] [PubMed] [Google Scholar]
- 32. Mishra BB, Tiwari VK. Natural products: an evolving role in future drug discovery. Eur J Med Chem. 2011;46:4769‐4807. [DOI] [PubMed] [Google Scholar]
- 33. Prassas I, Diamandis EP. Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov. 2008;7:926‐935. [DOI] [PubMed] [Google Scholar]
- 34. Kang C, Elledge SJ. How autophagy both activates and inhibits cellular senescence. Autophagy. 2016;12:898‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang H, Puleston DJ, Simon AK. Autophagy and immune senescence. Trends Mol Med. 2016;22:671‐686. [DOI] [PubMed] [Google Scholar]
- 36. Young AR, Narita M, Ferreira M, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garcia‐Prat L, Martinez‐Vicente M, Perdiguero E, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37‐42. [DOI] [PubMed] [Google Scholar]
- 38. Tian LM, Xie HF, Xiao X, et al. Study on the roles of beta‐catenin in hydrogen peroxide‐induced senescence in human skin fibroblasts. Exp Dermatol. 2011;20:836‐838. [DOI] [PubMed] [Google Scholar]
- 39. Liu DH, Chen YM, Liu Y, et al. Rb1 protects endothelial cells from hydrogen peroxide‐induced cell senescence by modulating redox status. Biol Pharm Bull. 2011;34:1072‐1077. [DOI] [PubMed] [Google Scholar]
- 40. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from Discovery to Therapy. Cancer Discov. 2016;6:353‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee Y, Dominy JE, Choi YJ, et al. Cyclin D1‐Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510:547‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang H, Nicolay BN, Chick JM, et al. The metabolic function of cyclin D3‐CDK6 kinase in cancer cell survival. Nature. 2017;546:426‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23‐32. [DOI] [PubMed] [Google Scholar]
- 44. Molenaar JJ, Ebus ME, Koster J, et al. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res. 2008;68:2599‐2609. [DOI] [PubMed] [Google Scholar]
- 45. Mahalingam D, Swords R, Carew JS, et al. Targeting HSP90 for cancer therapy. Br J Cancer. 2009;100:1523‐1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl). 2004;82:488‐499. [DOI] [PubMed] [Google Scholar]
- 47. Lorenz OR, Freiburger L, Rutz DA, et al. Modulation of the Hsp90 chaperone cycle by a stringent client protein. Mol Cell. 2014;53:941‐953. [DOI] [PubMed] [Google Scholar]