

Abstract
Objectives
Restoring a functional beta‐cell mass is a fundamental goal in treating diabetes. A complex signalling pathway network coordinates the regulation of beta‐cell proliferation, although a role for ERK5 in this network has not been reported. This question was addressed in this study.
Materials and methods
We studied the activation of extracellular‐signal‐regulated kinase 5 (ERK5) in pregnant mice, a well‐known mouse model of increased beta‐cell proliferation. A specific inhibitor of ERK5 activation, BIX02189, was intraperitoneally injected into the pregnant mice to suppress ERK5 signalling. Beta‐cell proliferation was determined by quantification of Ki‐67+ beta cells. Beta‐cell apoptosis was determined by TUNEL assay. The extent of beta‐cell proliferation was determined by beta‐cell mass. The alteration of ERK5 activation and CyclinD1 levels in purified mouse islets was examined by Western blotting.
Results
Extracellular‐signal‐regulated kinase 5 phosphorylation, which represents ERK5 activation, was significantly upregulated in islets from pregnant mice. Suppression of ERK5 activation by BIX02189 in pregnant mice significantly reduced beta‐cell proliferation, without affecting beta‐cell apoptosis, resulting in increases in random blood glucose levels and impairment of glucose response of the mice. ERK5 seemed to activate CyclinD1 to promote gestational beta‐cell proliferation.
Conclusions
Extracellular‐signal‐regulated kinase 5 plays an essential role in the gestational augmentation of beta‐cell proliferation. ERK5 may be a promising target for increasing beta‐cell mass in diabetes patients.
1. INTRODUCTION
Diabetes, often referred to as diabetes mellitus, describes a group of prevalent metabolic diseases in which the patient has high blood sugar, due to inadequate insulin production by pancreatic beta cells or improper body responses to insulin.1 There are two types of diabetes, type 1 diabetes and type 2 diabetes, which are different in disease prevalence and pathogenesis; however, both share a gradual loss‐of‐functional beta‐cell mass.1 Indeed, past studies have demonstrated that restoring functional beta‐cell mass is a fundamental goal in treating diabetic patients.1
We and others have shown that increases in adult beta‐cell number preliminarily stem from beta‐cell self‐replication.2, 3, 4, 5, 6 An established opinion upon the control of beta‐cell proliferation highlights the coordinating role of a complex signalling pathway network.7, 8, 9, 10, 11, 12, 13 Among the many molecules that participate in this sophisticated regulation, the mitogen‐activated protein kinases (MAPKs) and their downstream targets appear to be essential signalling modules. There are four major subfamilies of MAPKs: the extracellular‐signal‐regulated kinases (ERK1/2), the p38 kinases, the Jun amino‐terminal kinases (JNKs) and extracellular‐signal‐regulated kinase 5 (ERK5). ERK1/2 are the most frequently studied MAPK in pancreatic beta cells, and platelet‐derived growth factor (PDGF)14 and insulin‐like growth factor 1 (IGF‐1)15, 16, 17 have independently been shown to activate Ras/c‐Raf/ERK1/2 to promote beta‐cell proliferation. Moreover, JNKs were found to be mainly involved in regulating beta‐cell stress and apoptosis,18, 19, 20, 21, 22, 23 while p38 kinases appeared to be involved in control of both beta‐cell proliferation and apoptosis.24, 25, 26, 27, 28 However, a role of ERK5 in the control of beta‐cell proliferation or beta‐cell apoptosis has not been examined.
Extracellular‐signal‐regulated kinase 5 is the largest MAPK and is ubiquitously expressed in mammalian tissues. ERK5 is activated by the upstream kinase MEK5 in response to several growth factors (eg, nerve growth factor (NGF), granulocyte colony‐stimulating factor (G‐CSF), fibroblast growth factor (FGF), or PDGF and oxidative and hyperosmotic stress stimulation.29 The MEK5/ERK5 pathway plays a crucial role in cell proliferation in a variety of normal and cancer cells.29 MEK5 phosphorylates ERK5 at the C‐terminal, resulting in its dissociation from Hsp90 and Cdc37, which allows phosphorylated ERK5 (pERK5) to translocate into the nuclei to exert its function as a transcriptional activator in a canonical mechanism.29 So far, the role of ERK5 signalling in pancreatic beta‐cell proliferation remains unknown. Very recently, it was reported that suppression of ERK5‐SUMOylation by constitutive MEK5 expression inhibited diabetic cardiac dysfunction in mice.30 In another study, elevated blood glucose and compromised insulin sensitivity were detected in adipocyte‐specific ERK5‐deficient mice.31 In addition, ERK5 was found to protect against pancreatic beta‐cell apoptosis and hyperglycaemia in mice by interrupting the ER stress‐mediated apoptotic pathway.32 Since cellular apoptosis and proliferation often share regulatory components,33 we hypothesized that ERK5 may play a role in beta‐cell proliferation in certain settings and thus studied it in pregnant mice, a well‐accepted model for beta‐cell proliferation.34, 35, 36, 37, 38
Here, we found that ERK5 phosphorylation, which represents ERK5 activation, was significantly upregulated in islets from pregnant mice. Suppression of ERK5 activation by a specific inhibitor of ERK5 activation, BIX02189, in pregnant mice significantly reduced beta‐cell proliferation, without affecting beta‐cell apoptosis, resulting in increases in random blood glucose levels and impairment of glucose responses of the mice. Moreover, ERK5 seemed to activate CyclinD1 to promote gestational beta‐cell proliferation. These data suggest that ERK5 may play an essential role in the gestational augmentation of beta‐cell proliferation and could be a target for increasing beta‐cell mass in diabetes patients.
2. MATERIALS AND METHODS
2.1. Protocol and mouse treatment
All animal experiments were approved by the Animal Research and Care Committee at Wenzhou Medical University. C57BL/6 mice were purchased from the SLAC Laboratory Animal Co. Ltd (Shanghai, China). Pregnancy was induced in female mice at 12 weeks of age by a timed breeding, and the pregnant mice were analysed at gestational day 15. The ERK5 inhibitor BIX02189 (Selleck Chemicals, Houston, TX, USA) was intraperitoneally injected into the pregnant mice from gestational day 1 to day 15 (time of analysis) at a dose of 10 mg/kg body weight in 100 mL saline once per day. The control mice received the same volume of saline and at the same frequency.
2.2. Random blood glucose and glucose tolerance test
Random blood glucose and glucose tolerance were measured as has been previously described.6, 11, 13, 39 Briefly, the mice were not fasted and blood samples were obtained at around 10 am by tail tip puncture using a glucometer (Accu‐Chek, Roche, Nutley, NJ, USA). To determine the glucose tolerance, after a 16‐hour fasting, the mice received an intraperitoneal injection of dextrose (2 mg/g body weight) and blood samples were obtained by tail clipping at 0, 15, 30, 60 and 120 minutes.
2.3. Ki‐67 staining, TUNEL staining, quantification of proliferating/apoptotic beta cells and beta‐cell mass
Tissues were fixed in 4% paraformaldehyde at 4°C for 6 hours, followed by cryoprotection in 30% sucrose for 16 hours before snap‐freezing. Sectioning was performed to prepare slides of 6 μm thickness. All slides were incubated with primary antibodies at 4°C overnight, then incubated with secondary antibodies for 2 hours at room temperature. Primary antibodies used in this study were Guinea pig anti‐insulin (Dako, Carpinteria, CA, USA) and Rat anti‐Ki‐67 (Abcam, Cambridge, MA, USA). The secondary antibodies were Cy2 or Cy3 conjugated anti‐guinea pig or anti‐rat secondary antibodies. Nucleus staining was performed with Hoechst 33342 (Becton‐Dickinson Biosciences, San Jose, CA, USA). Quantification of beta‐cell proliferation was based on the percentage of ki‐67+ beta cells. At least 2000 beta cells were counted for each experimental repeat, as has been described before.11, 13 Terminal deoxynucleotidyltransferase‐mediated dUTP‐biotin nick end‐labelling (TUNEL) staining was performed with an ApopTag® Peroxidase In Situ Apoptosis Detection Kit (Millipore, Billerica, MA, USA), according to the manufacturer's instruction, as previously described.12, 40, 41 Beta‐cell mass was analysed as described before.11, 13, 39, 40, 42
2.4. Pancreatic digestion and islet isolation
Mouse pancreas digestion and islet isolation were performed as described before.42, 43, 44, 45 Briefly, the pancreas was infused with 0.25 mg/mL collagenase P (Sigma‐Aldrich, St. Louis, MO, USA) for 40 minutes to obtain a single‐cell population. Islets were handpicked at least five times to avoid contamination of non‐islets.
2.5. Western blotting
Western blotting was performed as previously described.11, 39, 40 Primary antibodies for Western blot are rabbit anti‐GAPDH (Cell Signaling, San Jose, CA, USA), rabbit anti‐ERK5 (Cell Signaling), rabbit anti‐pERK5 (Cell Signaling) and rabbit anti‐CyclinD1 (Santa Cruz Biotechnology, Dallas, TX, USA). Secondary antibody is HRP‐conjugated anti‐rabbit antibody (Dako).
2.6. Data Analysis
GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA) was used for statistical analyses. All values are depicted as mean ± SD. Five repeats were analysed in each condition. All data were statistically analysed using one‐way ANOVA with a Bonferroni correction, followed by Fisher's exact test to compare two groups. Significance was considered when P < .05.
3. RESULTS
3.1. Significantly increased beta‐cell proliferation and elevated beta‐cell mass are detected in pregnant mice at gestational day 15
During pregnancy, beta‐cell proliferation significantly increases in response to augmented metabolic need.34, 35, 36, 37, 38 Based on previous findings from us6 and from others,46 the peak of beta‐cell proliferation appears to be from gestational day 13 to 16. Thus, in this study, we focused on gestational day 15. We found that at gestational day 15, beta‐cell mass was significantly increased, compared with control non‐pregnant littermates (Figure 1A). This significant increase in beta‐cell mass appeared to stem from the significant increases in beta‐cell proliferation, shown by quantification of Ki67+ beta cells (Figure 1B) and representative Ki‐67 staining images (Figure 1C), rather than from an alteration in beta‐cell apoptosis, shown by quantification of TUNEL + beta cells (Figure 1D) and representative TUNEL staining images (Figure 1E).
Figure 1.
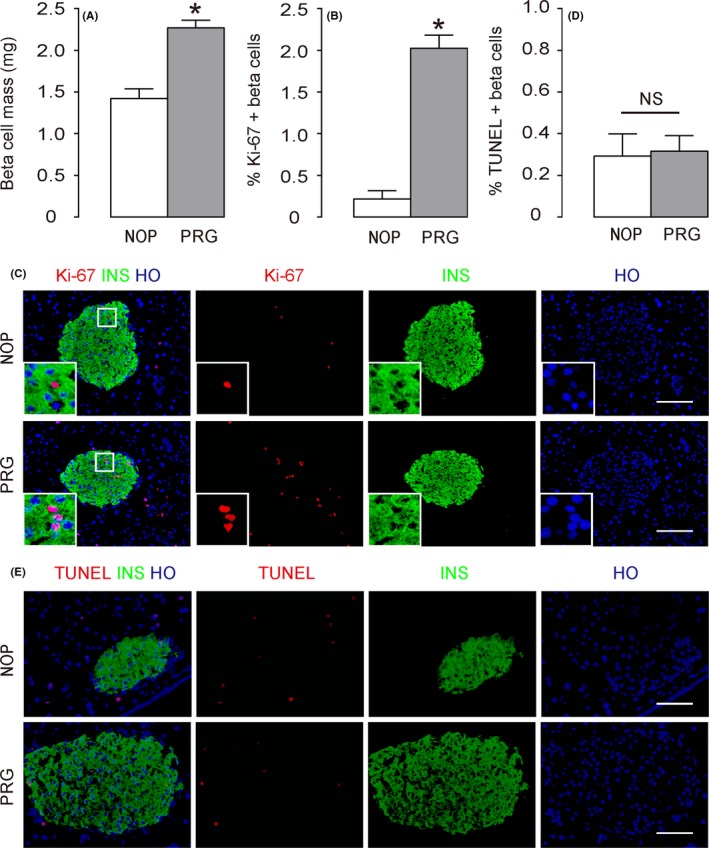
Significantly increased beta‐cell proliferation and elevated beta‐cell mass are detected in pregnant mice at gestational day 15. (A) Beta cell mass was determined at gestational day 15 in pregnant mice (PRG), compared with control non‐pregnant littermates (NOP). (B‐C) Beta cell proliferation was determined by Ki‐67+ beta cells, shown by quantification of Ki67+ insulin (INS)+ cells (B) and representative images (C). A square inset in white was shown for each panel in C. (D‐E) Beta cell apoptosis was determined by Terminal deoxynucleotidyltransferase‐mediated dUTP‐biotin nick end labelling (TUNEL) + beta cells, shown by quantification of TUNEL + INS + cells (D) and representative images (E). *P < .05. NS, no significance, n = 5. Scale bars are 50 μm.
3.2. ERK5 is activated in beta cells from pregnant mice
Mouse islets were, thus, isolated from pregnant mice at gestational day 15, and compared with those from non‐pregnant littermates (Figure 2A). We found that the total ERK5 in islets from pregnant mice remained unaltered (Figure 2B,C), while the phosphorylated ERK5 was significantly increased in islets from pregnant mice, compared with those from non‐pregnant littermates (Figure 2B,D). Thus, ERK5 activation in beta cells occurs concomitantly with pregnancy.
Figure 2.
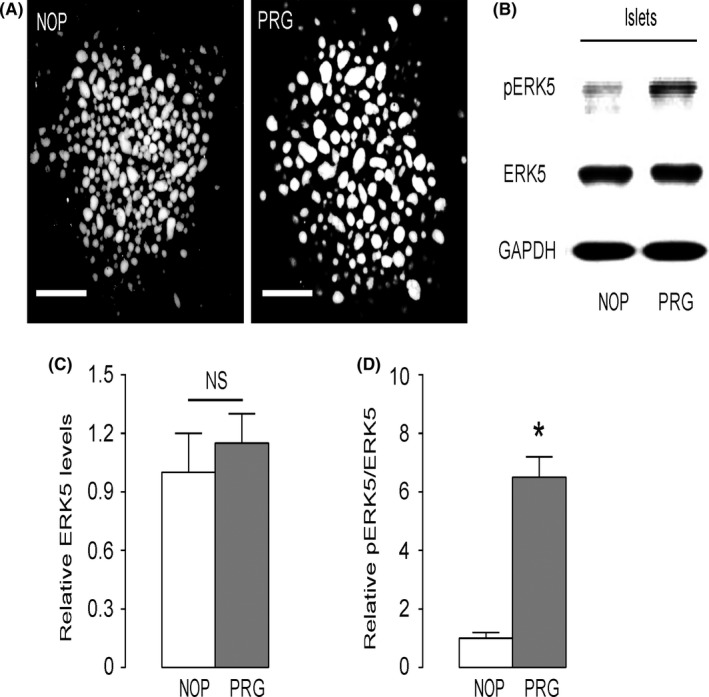
Extracellular‐signal‐regulated kinase 5 (ERK5) is activated in beta cells from pregnant mice. A, Images of purified mouse islets isolated from control non‐pregnant littermates (NOP) or from pregnant mice at gestational day 15 (PRG). B, Representative Western blotting for pERK5, ERK5 and GAPDH. C, Quantification of ERK5 levels (normalized to GAPDH). D, Quantification of pERK5 levels (normalized to ERK5). *P < .05. NS, no significance, n = 5.
3.3. Activation of ERK5 in beta cells is suppressed by BIX02189 in pregnant mice
In order to figure out whether the activation of ERK5 in beta cells may be necessary for the augmentation of beta‐cell proliferation during pregnancy, we injected a specific inhibitor of ERK5 activation, BIX02189, into the pregnant mice to suppress ERK5 signalling. At gestational day 15, mouse islets were isolated and examined for the levels of ERK5 and pERK5. We found that the total ERK5 in islets from the pregnant mice that had received either BIX02189 or control saline did not alter (Figure 3A,B), while the phosphorylated ERK5 was significantly decreased in islets from the pregnant mice that had received BIX02189, compared with those from the pregnant mice that had received saline (Figure 3A,C). Thus, activation of ERK5 in beta cells in pregnant mice is suppressed by BIX02189 injections.
Figure 3.
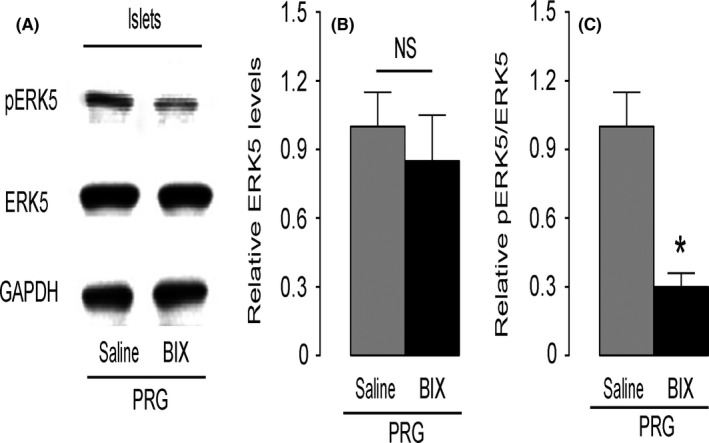
Activation of extracellular‐signal‐regulated kinase 5 (ERK5) in beta cells is suppressed by BIX02189 in pregnant mice. A specific inhibitor of ERK5 activation, BIX02189 (BIX), was injected into the pregnant mice once per day from gestational day 1 to day 15 to suppress ERK5 signalling. The control pregnant mice received the same volume of saline and at the same frequency (saline). At gestational day 15, mouse islets were isolated and examined for levels of ERK5 and pERK5. A, Representative Western blotting for pERK5, ERK5 and GAPDH. B, Quantification of ERK5 levels (normalized to GAPDH). C, Quantification of pERK5 levels (normalized to ERK5). *P < .05. NS, no significance, n = 5.
3.4. Suppression of ERK5 activation impairs gestational beta‐cell proliferation
We found that at gestational day 15, the random blood glucose in pregnant mice that had received BIX02189 was significantly higher, compared with the measurements from pregnant mice that had received saline (Figure 4A). Moreover, the glucose tolerance in the mice that had received BIX02189 was also significantly poorer, compared with pregnant mice that had received saline or control non‐pregnant mice (Figure 4B). Next, beta‐cell proliferation, beta‐cell apoptosis and beta‐cell mass were quantified. We found that at gestational day 15, increases in beta‐cell mass in mice that had received BIX02189 were significantly compromised, compared with the pregnant mice that had received saline (Figure 4C). The attenuated increases in beta‐cell mass by BIX02189 injections appeared to stem from the significant attenuation in beta‐cell proliferation, shown by quantification of Ki67+ beta cells (Figure 4D) and representative Ki‐67 staining images (Figure 4E), rather than from an alteration in beta‐cell apoptosis, shown by quantification of TUNEL + beta cells (Figure 4F) and representative TUNEL staining images (Figure 4G). Together, these data suggest that suppression of ERK5 activation impairs gestational beta‐cell proliferation and increases in beta‐cell mass.
Figure 4.
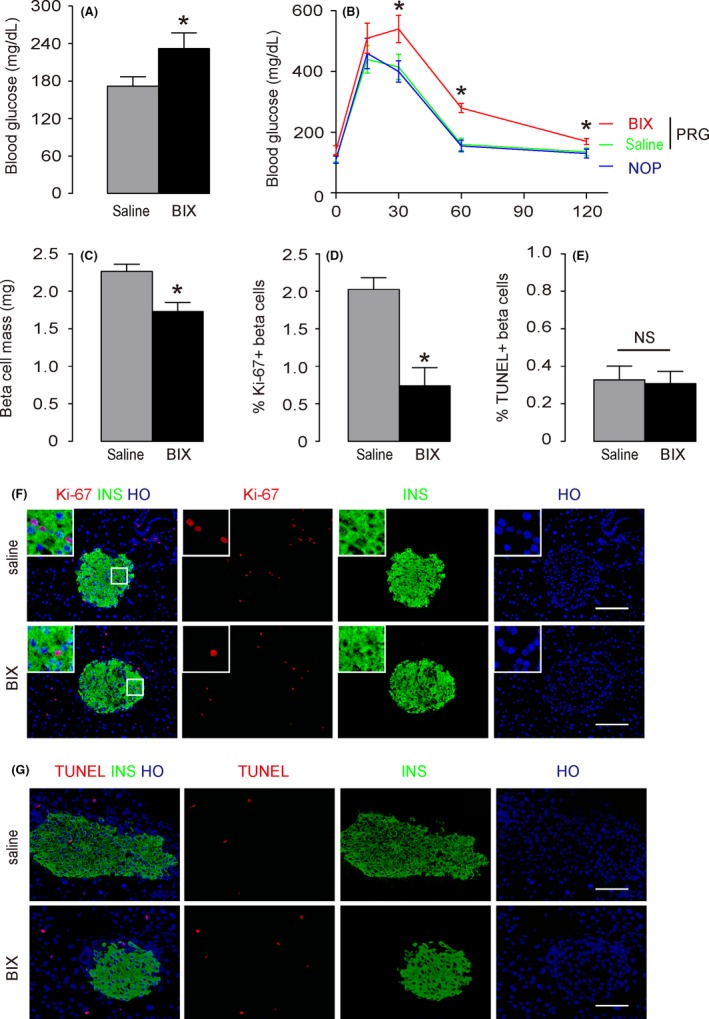
Suppression of extracellular‐signal‐regulated kinase 5 (ERK5) activation impairs gestational beta‐cell proliferation. At gestational day 15, the mice were analysed. A, The random blood glucose in pregnant mice that had received BIX02189 (BIX) and in the pregnant mice that had received saline (saline) was determined. B, The glucose tolerance in BIX and saline‐treated from pregnant mice at gestational day 15 (PRG) and control non‐pregnant littermates (NOP) was determined by IPGTT. C, Beta‐cell mass. D‐E, Beta‐cell proliferation was determined by Ki‐67+ beta cells, shown by quantification of Ki67+ INS + cells (D) and representative images (E). A square inset in white was shown for each panel in E. F‐G, Beta‐cell apoptosis was determined by terminal deoxynucleotidyltransferase‐mediated dUTP‐biotin nick end labelling (TUNEL) + beta cells, shown by quantification of TUNEL + INS + cells (F) and representative images (G). *P < .05. NS, no significance, n = 5. Scale bars are 50 μm.
3.5. ERK5 seems to activate CyclinD1 to promote gestational beta‐cell proliferation
Finally, we investigated the downstream signalling molecules for ERK5‐mediated upregulation of beta‐cell proliferation. A previous study has shown that ERK5 activation promotes cell proliferation through CyclinD1, a key cell‐cycle activator that promotes G1/s phase transition, in a fibroblast cell line.47 Thus, we examined the levels of CyclinD1 in beta cells from non‐pregnant mice, pregnant mice that had received BIX02189 and pregnant mice that had received saline. We found that CyclinD1 levels were significantly increased in beta cells from pregnant mice, compared with non‐pregnant mice, but this increase in CyclinD1 levels was abolished by BIX02189 injections (Figure 5A). Hence, ERK5 seems to promote gestational beta‐cell proliferation at least partially through upregulating CyclinD1 (Figure 5B).
Figure 5.
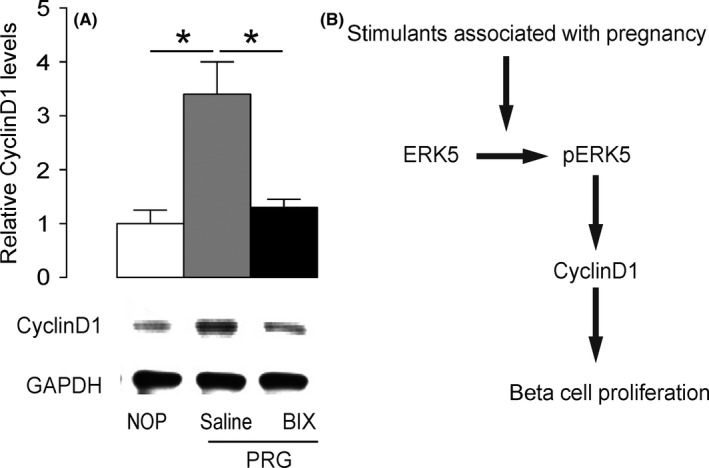
Extracellular‐signal‐regulated kinase 5 (ERK5) seems to activate CyclinD1 to promote gestational beta‐cell proliferation. A, The levels of CyclinD1 in beta cells from non‐pregnant mice (NOP), pregnant mice that had received BIX02189 (BIX) and pregnant mice that had received saline (saline) were determined by Western blotting. B, Schematic of the model: ERK5 seems to promote gestational beta‐cell proliferation through upregulating CyclinD1. *P < .05; n = 5.
4. DISCUSSION
Accumulating evidence in the past years shows that adult beta‐cell proliferation is controlled by a number of signalling pathways that share certain molecular cascades.10 For example, we have investigated the role of transforming growth factor receptor (TGF‐beta) signalling and epidermal growth factor (EGF) receptor signalling in beta‐cell proliferation and proposed that beta‐cell proliferation can be activated either actively in response to a metabolic need/beta‐cell workload or passively in response to inflammatory stimulants.11, 12, 13, 42 Interestingly, although these 2 ways to induce beta‐cell proliferation may mainly use different signalling pathways, some part of the pathway may share overlapping regulatory molecules. For example, we have shown that the activation of both TGF‐beta signalling and EGF receptor signalling results in suppression of the nuclear translocation of the phosphorylated form of SMAD2, but allows SMAD7‐mediated beta‐cell proliferation through CyclinD1 activation and p27 nuclear exclusion.11 These signalling molecules that could be activated by different growth factors through the same or different receptors appear to be essential factors for understanding the molecular control of pancreatic beta‐cell proliferation.
Among all signalling molecules, MAPKs are factors that are connected to different pathways. As a focus of this study, ERK5 can be activated through NGF and its receptor TrkA at the nerve terminal,48 or be activated through G‐CSF/G‐CSF receptor in the regulation of granulopoiesis,49 or be activated through TGF‐beta/TGF‐beta receptor in the control of function and survival of podocytes.50 However, the association of ERK5 with Hsp90 and Cdc37 super‐chaperone renders ERK5 a regulator of other signalling pathways that include Hsp90 or Cdc37 as involved molecules.29 For example, Hsp90 is a chaperone required for the conformation of several signalling proteins including Raf, cdk4 and steroid receptors.51 Hsp90 can stabilize TGF‐beta receptors and prevent ubiquitin‐mediated degradation of TGF‐beta receptors.52, 53 Moreover, like ERK5, intracellular Akt is also associated with Hsp90 and Cdc37 in a complex that is regulated by phosphatidylinositol 3‐kinase.54 Thus, ERK5 appears to be an important molecule that may affect many signalling pathways controlling beta‐cell proliferation and apoptosis.
Here, we investigated the changes in ERK5 activation in primary mouse islets during pregnancy. To the best of our knowledge, it is the first study to show that ERK5 plays an essential role in postnatal beta‐cell proliferation. Since beta cells are the major proliferating cells in response to the gestational metabolic need, our use of primary mouse islets should be well representative of beta cells, taking into account that beta cells represent more than 90% of islet cells in the mouse, and that this percentage further increases during pregnancy. BIX02189 is a specific inhibitor of phosphorylation of ERK5 by MEK5.55 BIX02189 inhibits the catalytic function of the MEK5 enzyme, and MEK5 mediates phosphorylation of ERK5, without affecting phosphorylation of ERK1/2.29 Thus, the effects of BIX02189 on suppression of ERK5 are specific and this loss‐of‐function experiment does not have off‐target effects on ERK1/2.
CyclinD1 was identified as a target for ERK5 in beta cells, similar to a fibroblast cell line in a previous report.47 In our past studies, we have shown that CyclinD1 is an important molecule involved in both SMAD7‐mediated11 and EGFR signalling‐activated beta‐cell proliferation.12 Future studies may apply proper pathway inhibitors in knock‐out animals to further investigate the interaction among pathways that may be affected by ERK5, in order to increase our knowledge on the coordination of beta‐cell proliferation. This study suggests that ERK5 may be a promising target for increasing beta‐cell mass in diabetes patients.
CONFLICT OF INTEREST
None disclosed.
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: XX. Performed the experiments and analysed the data: CC, SW, XL, DW, SF and XX. Wrote the paper: XX.
Chen C, Wu S, Lin X, Wu D, Fischbach S, Xiao X. ERK5 plays an essential role in gestational beta‐cell proliferation. Cell Prolif. 2018;51:e12410 10.1111/cpr.12410
Contributor Information
Congde Chen, Email: pedsurg@163.com.
Xiangwei Xiao, Email: Xiangwei.xiao@chp.edu.
REFERENCES
- 1. Pipeleers D, Chintinne M, Denys B, Martens G, Keymeulen B, Gorus F. Restoring a functional beta‐cell mass in diabetes. Diabetes Obes Metab. 2008;10(suppl 4):54‐62. [DOI] [PubMed] [Google Scholar]
- 2. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta‐cells are formed by self‐duplication rather than stem‐cell differentiation. Nature. 2004;429:41‐46. [DOI] [PubMed] [Google Scholar]
- 3. Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12:817‐826. [DOI] [PubMed] [Google Scholar]
- 4. Meier JJ, Butler AE, Saisho Y, et al. Beta‐cell replication is the primary mechanism subserving the postnatal expansion of beta‐cell mass in humans. Diabetes. 2008;57:1584‐1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963‐968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiao X, Chen Z, Shiota C, et al. No evidence for beta cell neogenesis in murine adult pancreas. J Clin Invest. 2013;123:2207‐2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiao X, Gittes GK. Concise review: new insights into the role of macrophages in beta‐cell proliferation. Stem Cells Transl Med. 2015;4:655‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saunders D, Powers AC. Replicative capacity of beta‐cells and type 1 diabetes. J Autoimmun. 2016;71:59‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging. 2011;3:565‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stewart AF, Hussain MA, Garcia‐Ocana A, et al. Human beta‐cell proliferation and intracellular signaling: part 3. Diabetes. 2015;64:1872‐1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiao X, Gaffar I, Guo P, et al. M2 macrophages promote beta‐cell proliferation by up‐regulation of SMAD7. Proc Natl Acad Sci USA. 2014;111:E1211‐E1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song Z, Fusco J, Zimmerman R, et al. Epidermal growth factor receptor signaling regulates beta cell proliferation in adult mice. J Biol Chem. 2016;291:22630‐22637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiao X, Wiersch J, El‐Gohary Y, et al. TGFbeta receptor signaling is essential for inflammation‐induced but not beta‐cell workload‐induced beta‐cell proliferation. Diabetes. 2013;62:1217‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen H, Gu X, Liu Y, et al. PDGF signalling controls age‐dependent proliferation in pancreatic beta‐cells. Nature. 2011;478:349‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shirakawa J, Okuyama T, Yoshida E, et al. Effects of the antitumor drug OSI‐906, a dual inhibitor of IGF‐1 receptor and insulin receptor, on the glycemic control, beta‐cell functions, and beta‐cell proliferation in male mice. Endocrinology. 2014;155:2102‐2111. [DOI] [PubMed] [Google Scholar]
- 16. Lingohr MK, Dickson LM, McCuaig JF, Hugl SR, Twardzik DR, Rhodes CJ. Activation of IRS‐2‐mediated signal transduction by IGF‐1, but not TGF‐alpha or EGF, augments pancreatic beta‐cell proliferation. Diabetes. 2002;51:966‐976. [DOI] [PubMed] [Google Scholar]
- 17. Shen S, Alt A, Wertheimer E, et al. PKCdelta activation: a divergence point in the signaling of insulin and IGF‐1‐induced proliferation of skin keratinocytes. Diabetes. 2001;50:255‐264. [DOI] [PubMed] [Google Scholar]
- 18. Chan JY, Luzuriaga J, Maxwell EL, West PK, Bensellam M, Laybutt DR. The balance between adaptive and apoptotic unfolded protein responses regulates beta‐cell death under ER stress conditions through XBP1, CHOP and JNK. Mol Cell Endocrinol. 2015;413:189‐201. [DOI] [PubMed] [Google Scholar]
- 19. Humphrey RK, Ray A, Gonuguntla S, Hao E, Jhala US. Loss of TRB3 alters dynamics of MLK3‐JNK signaling and inhibits cytokine‐activated pancreatic beta cell death. J Biol Chem. 2014;289:29994‐30004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Subramanian SL, Hull RL, Zraika S, Aston‐Mourney K, Udayasankar J, Kahn SE. cJUN N‐terminal kinase (JNK) activation mediates islet amyloid‐induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia. 2012;55:166‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheon H, Cho JM, Kim S, et al. Role of JNK activation in pancreatic beta‐cell death by streptozotocin. Mol Cell Endocrinol. 2010;321:131‐137. [DOI] [PubMed] [Google Scholar]
- 22. Fornoni A, Pileggi A, Molano RD, et al. Inhibition of c‐jun N terminal kinase (JNK) improves functional beta cell mass in human islets and leads to AKT and glycogen synthase kinase‐3 (GSK‐3) phosphorylation. Diabetologia. 2008;51:298‐308. [DOI] [PubMed] [Google Scholar]
- 23. Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell‐permeable peptide inhibitors of JNK: novel blockers of beta‐cell death. Diabetes. 2001;50:77‐82. [DOI] [PubMed] [Google Scholar]
- 24. Khan S, Jena GB. Protective role of sodium butyrate, a HDAC inhibitor on beta‐cell proliferation, function and glucose homeostasis through modulation of p38/ERK MAPK and apoptotic pathways: study in juvenile diabetic rat. Chem Biol Interact. 2014;213:1‐12. [DOI] [PubMed] [Google Scholar]
- 25. Widenmaier SB, Ao Z, Kim SJ, Warnock G, McIntosh CH. Suppression of p38 MAPK and JNK via Akt‐mediated inhibition of apoptosis signal‐regulating kinase 1 constitutes a core component of the beta‐cell pro‐survival effects of glucose‐dependent insulinotropic polypeptide. J Biol Chem. 2009;284:30372‐30382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hou N, Torii S, Saito N, Hosaka M, Takeuchi T. Reactive oxygen species‐mediated pancreatic beta‐cell death is regulated by interactions between stress‐activated protein kinases, p38 and c‐Jun N‐terminal kinase, and mitogen‐activated protein kinase phosphatases. Endocrinology. 2008;149:1654‐1665. [DOI] [PubMed] [Google Scholar]
- 27. Schrader J, Rennekamp W, Niebergall U, et al. Cytokine‐induced osteoprotegerin expression protects pancreatic beta cells through p38 mitogen‐activated protein kinase signalling against cell death. Diabetologia. 2007;50:1243‐1247. [DOI] [PubMed] [Google Scholar]
- 28. Burns CJ, Squires PE, Persaud SJ. Signaling through the p38 and p42/44 mitogen‐activated families of protein kinases in pancreatic beta‐cell proliferation. Biochem Biophys Res Commun. 2000;268:541‐546. [DOI] [PubMed] [Google Scholar]
- 29. Gomez N, Erazo T, Lizcano JM. ERK5 and cell proliferation: nuclear localization is what matters. Front Cell Dev Biol. 2016;4:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shishido T, Woo CH, Ding B, et al. Effects of MEK5/ERK5 association on small ubiquitin‐related modification of ERK5: implications for diabetic ventricular dysfunction after myocardial infarction. Circ Res. 2008;102:1416‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu H, Guariglia S, Li W, et al. Role of extracellular signal‐regulated kinase 5 in adipocyte signaling. J Biol Chem. 2014;289:6311‐6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nam DH, Han JH, Lim JH, Park KM, Woo CH. CHOP deficiency ameliorates ERK5 inhibition‐mediated exacerbation of streptozotocin‐induced hyperglycemia and pancreatic beta‐cell apoptosis. Mol Cells. 2017;40:457‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guo M, Hay BA. Cell proliferation and apoptosis. Curr Opin Cell Biol. 1999;11:745‐752. [DOI] [PubMed] [Google Scholar]
- 34. Hakonen E, Ustinov J, Palgi J, Miettinen PJ, Otonkoski T. EGFR signaling promotes beta‐cell proliferation and survivin expression during pregnancy. PLoS ONE. 2014;9:e93651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hughes E, Huang C. Participation of Akt, menin, and p21 in pregnancy‐induced beta‐cell proliferation. Endocrinology. 2011;152:847‐855. [DOI] [PubMed] [Google Scholar]
- 36. Chen G, Liu C, Xue Y, Mao X, Xu K, Liu C. Molecular mechanism of pancreatic beta‐cell adaptive proliferation: studies during pregnancy in rats and in vitro. Endocrine. 2011;39:118‐127. [DOI] [PubMed] [Google Scholar]
- 37. Avril I, Blondeau B, Duchene B, Czernichow P, Breant B. Decreased beta‐cell proliferation impairs the adaptation to pregnancy in rats malnourished during perinatal life. J Endocrinol. 2002;174:215‐223. [DOI] [PubMed] [Google Scholar]
- 38. Butler AE, Cao‐Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010;53:2167‐2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xiao X, Fischbach S, Song Z, et al. Transient Suppression of TGFbeta Receptor Signaling Facilitates Human Islet Transplantation. Endocrinology. 2016;157:1348‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao X, Fischbach S, Zhang T, et al. SMAD3/Stat3 signaling mediates beta‐cell epithelial‐mesenchymal transition in chronic pancreatitis‐related diabetes. Diabetes. 2017;66:2646‐2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xiao X, Chen C, Guo P, et al. Forkhead Box Protein 1 (FoxO1) inhibits accelerated beta cell aging in pancreas‐specific SMAD7 mutant mice. J Biol Chem. 2017;292:3456‐3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xiao X, Guo P, Chen Z, et al. Hypoglycemia reduces vascular endothelial growth factor a production by pancreatic Beta cells as a regulator of Beta cell mass. J Biol Chem. 2013;288:8636‐8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiao X, Prasadan K, Guo P, et al. Pancreatic duct cells as a source of VEGF in mice. Diabetologia. 2014;57:991‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xiao X, Guo P, Shiota C, et al. Neurogenin3 activation is not sufficient to direct duct‐to‐beta cell transdifferentiation in the adult pancreas. J Biol Chem. 2013;288:25297‐25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolation from mouse pancreas. Nat Protoc. 2009;4:1649‐1652. [DOI] [PubMed] [Google Scholar]
- 46. Zhao X. Increase of beta cell mass by beta cell replication, but not neogenesis, in the maternal pancreas in mice. Endocr J. 2014;61:623‐628. [DOI] [PubMed] [Google Scholar]
- 47. Mulloy R, Salinas S, Philips A, Hipskind RA. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene. 2003;22:5387‐5398. [DOI] [PubMed] [Google Scholar]
- 48. Shao Y, Akmentin W, Toledo‐Aral JJ, et al. Pincher, a pinocytic chaperone for nerve growth factor/TrkA signaling endosomes. J Cell Biol. 2002;157:679‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dong F, Gutkind JS, Larner AC. Granulocyte colony‐stimulating factor induces ERK5 activation, which is differentially regulated by protein‐tyrosine kinases and protein kinase C. Regulation of cell proliferation and survival. J Biol Chem. 2001;276:10811‐10816. [DOI] [PubMed] [Google Scholar]
- 50. Badshah II, Baines DL, Dockrell ME. Erk5 is a mediator to TGFbeta1‐induced loss of phenotype and function in human podocytes. Front Pharmacol. 2014;5:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol. 2003;15:419‐424. [DOI] [PubMed] [Google Scholar]
- 52. Wrighton KH, Lin X, Feng XH. Critical regulation of TGFbeta signaling by Hsp90. Proc Natl Acad Sci USA. 2008;105:9244‐9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kavsak P, Rasmussen RK, Causing CG, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365‐1375. [DOI] [PubMed] [Google Scholar]
- 54. Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858‐39866. [DOI] [PubMed] [Google Scholar]
- 55. Tatake RJ, O'Neill MM, Kennedy CA, et al. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun. 2008;377:120‐125. [DOI] [PubMed] [Google Scholar]