

Abstract
Objectives
Yes‐associated protein (YAP) has been reported to regulate cell proliferation and differentiation. We aimed to characterize the role of YAP in angiotensin II (Ang II)‐induced hypertensive vascular remodelling (HVR) and vascular smooth muscle cells (VSMCs) phenotypic modulation and to explore the underlying mechanisms.
Materials and methods
An HVR rat model was established by continuous Ang II infusion for 2 weeks. Western blotting, qRT‐PCR, and confocal microscopy were conducted to assess YAP expression. YAP‐shRNA interfering plasmid and adenovirus were constructed to knock down YAP. We used cell proliferation and migration assays, accompanied by pathway inhibitors, to evaluate the biological function and underlying mechanisms.
Results
Ang II upregulated YAP expression in the media of carotid artery; however, in vivo YAP silencing significantly mitigated HVR, independent of the blood pressure level. Ang II upregulated YAP expression and promoted YAP nuclear accumulation in a dose‐ and time‐dependent manner in rat VSMCs. YAP knockdown ameliorated Ang II‐induced VSMCs phenotypic modulation. The regulation of YAP by Ang II could be blocked by pretreatment with angiotensin receptor type 1 antagonist losartan or F‐actin depolymerizing agent latrunculin B but not the AT2R antagonist PD 123319. Disrupting the YAP‐TEA domain (TEAD) interaction with verteporfin inhibited Ang II‐induced VSMCs phenotypic modulation.
Conclusions
Yes‐associated protein mediated angiotensin II‐induced VSMCs phenotypic modulation and vascular remodelling. YAP is a potential therapeutic target for HVR beyond blood pressure control.
1. INTRODUCTION
As the leading cause of morbidity and mortality worldwide, hypertension has become an epidemic in the 21st century.1, 2 Structural changes of the vascular wall during hypertension were termed “hypertensive vascular remodelling” (HVR),3 which was commonly characterized as media hyperplasia and an increase in the media thickness. HVR was generally accepted as an important determinant in vascular pathologies of hypertension.4 However, the mechanism of HVR remains elusive. Previous researches in hypertensive patients and animal models have provided evidence that the mechanism of HVR is at least partially independent of the blood pressure level.5 Vascular smooth muscle cells (VSMCs) within the media of blood vessels retain remarkable phenotypic plasticity.6 A growing body of evidence has demonstrated that abnormal VSMC proliferation and migration are key events underlying many VSMC‐related pathological conditions, such as hypertension and postangioplasty restenosis.7
The Hippo/Yes‐associated protein (YAP) signalling pathway is an evolutionarily conserved molecular mechanism that has been reported to be closely related to vascular pathophysiological regulation and dysregulation.8 YAP‐null mice die at embryonic day 8.5 due to defects in yolk sac vasculogenesis, suggesting that YAP plays a fundamental role in vasculogenesis.9 Moreover, Wang et al10 reported that YAP expression was upregulated after vascular injury. However, the role of the Hippo/YAP pathway in HVR still remains unknown. Angiotensin II (Ang II) has been reported to induce HVR and VSMCs phenotypic modulation.11, 12 However, the effect of Ang II on the Hippo/YAP pathway in VSMCs has not been reported. Ang II exerts its biological function via two receptors, angiotensin receptor type 1 (AT1R) and AT2R, both of which belong to the G protein‐coupled receptor (GPCR) family. Recently, GPCRs were reported to be able to serve as upstream regulators of Hippo/YAP signalling.13, 14, 15
Based on these findings, this study aimed to characterize the role of the Hippo/YAP pathway in Ang II‐induced HVR and VSMCs phenotypic modulation and to explore the underlying mechanisms.
2. METHODS
2.1. Animals
Twenty male 9‐ to 10‐week‐old Sprague‐Dawley rats were purchased from the Laboratory Animal Research Center of Southern Medical University (Guangzhou, China). All animals were maintained in a 12:12 h light‐dark cycle at an ambient temperature of 23‐25°C and 40%‐60% humidity with free access to standard rodent chow and water.
After a 1‐week acclimatization period, the animals were randomly assigned to the following four groups (n = 5 for each group): the control group received a continuous infusion of phosphate‐buffered saline (PBS); the Ad‐YAP‐shRNA group received continuous PBS infusion and Ad‐YAP‐shRNA transfer in the right common carotid artery (RCCA); the Ang II + Ad‐GFP group and the Ang II + Ad‐YAP‐shRNA group received Ad‐GFP and Ad‐YAP‐shRNA RCCA transfer, respectively, in addition to continuous Ang II infusion.
This protocol was approved by the Academic Administration Committee of Sun Yat‐sen Memorial Hospital of Sun Yat‐sen University, and all animal experiments were performed in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals.
2.2. Construction of the YAP‐shRNA adenovirus and carotid artery adventitial gene transfer
The YAP‐shRNA adenovirus was designed and constructed by Hanbio Company (Shanghai, China) based on the pHBAd‐U6‐Scramble‐CMV‐GFP vector. According to Wang et al,10 the sequence of shRNA targeting rat YAP 3′‐UTR was 5′‐GCTGCCACCAAGTT‐3′. Prior to use, Ad‐GFP or Ad‐YAP‐shRNA was suspended in Pluronic F127 gel (20% w/v; Sigma‐Aldrich, St. Louis, MO, USA) at a 1:9 ratio with a final concentration of 3.5 × 109 PFU/mL.
Application of the adenovirus was performed as previously described by Liu et al.16 Briefly, rats were anaesthetized by intraperitoneal administration of sodium pentobarbital (40 mg/kg; Sigma‐Aldrich), and then the RCCA was exposed. Two hundred microlitre of Pluronic F127 gel containing the adenovirus was carefully applied to the adventitial surface of the RCCA, whereas the control group received gel alone. After coagulation of the gel, the incision was closed, and rats were allowed to recover from anaesthesia.
2.3. Osmotic minipump implantation and systolic blood pressure measurements
An osmotic minipump (2002; Alzet Osmotic Pumps, Cupertino, CA, USA) was used to ensure that the drug was continually and constantly delivered. The pumps were filled with either Ang II (Sigma‐Aldrich) or PBS and implanted according to the manufacturer's instructions. The minipump was inserted into the abdominal cavity. Ang II was continually infused at a rate of 0.5 μg/kg/min, whereas the two control groups received PBS infusion at the same rate and volume.
The systolic blood pressure (SBP), heart rate, and body weight were measured in conscious rats using the computerized caudal artery tail‐cuff system (BP‐98A; Softron, Tokyo, Japan) on days 0, 3, 7, and 14.
2.4. Histological assays and morphometric analyses
Two weeks after the intervention, the rats were euthanized, and the RCCAs were harvested. All RCCAs were fixed, paraffin‐embedded, sectioned, and stained with haematoxylin and eosin, Masson's Trichrome or Vitoria blue (to detect elastic fibres) following standard protocols. Cross‐sectional images were captured using a light microscope (Olympus, Tokyo, Japan) and measured using the Image‐Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA). The media thickness was determined by measuring the distance between the internal elastic lamina (IEL) and the external elastic lamina (EEL). For each slide, measurements from four points (the 3, 6, 9, and 12o'clock positions) were averaged. The media area was determined by measuring the area between the IEL and EEL. The media:lumen ratio was calculated based on the measured lumen and media areas. Five slides per group were randomly selected for the statistical analyses.
For immunohistochemistry, the sections were deparaffinized with xylene and then rehydrated with a graded series of ethanol before incubation with 0.3% hydrogen peroxide for 20 minutes and then blocked with 10% goat serum. The sections were subsequently incubated with primary antibodies against the following proteins: YAP (1:100; sc‐101199; Santa Cruz Biotechnology, Santa Cruz, CA, USA); connective tissue growth factor (CTGF, 1:50; sc‐101586; Santa Cruz Biotechnology); transforming growth factor β1 (TGF β1, 1:100; ab64715; Abcam); cleaved caspase‐3 (1:250; #9664, Cell Signalling Technology, Danvers, MA, USA); collagen I (1:100; ab6308, Abcam) for 2 hours at 37°C, followed by horseradish peroxidase‐conjugated polyclonal goat anti‐mouse (1:5000; ab6789, Abcam) and goat anti‐rabbit (1:3000; #7074, Cell Signalling Technology) secondary antibody. The reaction was visualized with diaminobenzidine, and the sections were counterstained with haematoxylin. The mean optical density of each group was determined using the Image‐Pro Plus software (Media Cybernetics).
2.5. Culture and stimulation of rat VSMCs
Rat VSMCs were isolated from the thoracic aortas as previously described.17 The VSMCs were cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Scientific, Waltham, MA, USA) supplemented with 10% foetal bovine serum (FBS; Thermo Scientific), 100 U/mL of penicillin and 100 μg/mL of streptomycin (Thermo Scientific) in a humidified atmosphere of 95% air/5% CO2 at 37°C. Cells between passages 3 and 8 were used for the experiments. Upon reaching 70%‐80% confluence, the rat VSMCs were made quiescent by incubation in DMEM supplemented with 0.1% FBS for 48 hours prior to treatment.
The rat VSMCs were treated with Ang II at different concentrations and for different lengths of time. For the mechanistic experiments, the rat VSMCs were pretreated with the following compounds: losartan (1 μmol/L; Sigma‐Aldrich) for 30 minutes; PD 123319 (1 μmol/L; Sigma‐Aldrich) for 30 minutes; latrunculin B (500 nmol/L; Thermo Scientific) for 20 minutes; or verteporfin (2 μg/mL, protected from light; Thermo Scientific) for 48 hours.
2.6. Extraction of cytoplasmic/nuclear proteins and Western blot analysis
Proteins from cultured cells or rat carotid arteries were extracted and prepared following standard protocols and as previously described by our laboratory.18 Nuclear and cytoplasmic extracts were prepared using NE‐PER Nuclear and Cytoplasmic Extraction Reagents (#78833, ThermoFisher, Waltham, MA, USA), supplemented with protease inhibitor (#36978; ThermoFisher). Proteins were separated by 12% sodium dodecyl sulphate polyacrylamide gel and electrophoretically transferred onto polyvinylidene fluoride (Millipore Corporation, Billerica, MA, USA) membranes. The membranes were blocked in TBST buffer containing 5% (w/v) bovine serum albumin (BSA) for 1 hour at room temperature and then incubated with following primary antibodies: YAP (1:500; sc‐101199, Santa Cruz Biotechnology); phospho‐YAP1 (S127) (1:5000; ab76252, Abcam); large tumour suppressor kinase 1 (LATS1, 1:2000; ab70562, Abcam); phospho‐LATS1 (T1079) (1:2000; #8654, Cell Signalling Technology); SM α‐actin (1:4000; ab32575, Abcam); SM22‐α (1:2000; ab14106, Abcam); cyclin D1 (1:10,000; ab134175, Abcam); proliferating cell nuclear antigen (PCNA; 1:1000; sc‐56, Santa Cruz Biotechnology); histone H3 (1:2000; #4499, Cell Signalling Technology); glyceraldehyde‐3‐phosphate dehydrogenase (GADPH, 1:5000, ab8245; Abcam); and β‐tubulin (1:1000; ab6046, Abcam). Next, the membranes were incubated with the appropriate horseradish peroxidase‐conjugated secondary antibodies for 1 hour at room temperature.
All proteins were visualized and detected using an enhanced chemiluminescence detection system (Bio‐Rad, Hercules, CA, USA). The band density was captured and quantified using the ImageJ software (http://rsbweb.nih.gov/ij/).
2.7. RNA extraction and quantitative real‐time PCR
Total RNA was extracted from tissue or VSMCs using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and cDNA was synthesized with the PrimeScript® RT Master Mix (Takara Bio, Kusatsu, Japan). The mRNA levels were quantified in triplicate using quantitative real‐time PCR (qRT‐PCR) on LightCycler 480 Real‐Time PCR System (Roche, Basel, Switzerland) with the SYBR Premix EX Taq (Tli RNase H Plus) kit (Takara Bio). The primers used for qRT‐PCR were listed in Supplementary Table S1. All primers were designed using Primer Premier 6 or adopted as previously reported,19, 20 and were synthesized by Invitrogen. The PCR cycling conditions were as follows: one cycle at 95°C for 10 minutes, followed by 40 cycles at 95°C for 5 seconds, 60°C for 20 seconds and 72°C for 15 seconds. At the end of the reaction, the samples were subjected to additional reaction steps at 95°C for 5 seconds, 60°C for 60 seconds, 95°C for 5 seconds, and 60°C for 20 seconds to generate a dissociation curve. The dissociation curve analysis confirmed that the signals corresponded to unique amplicons. All reactions were performed in triplicate. Specific mRNA expression levels were normalized to the GAPDH mRNA levels using the comparative ΔΔCt method.21
2.8. Immunofluorescence staining
Vascular smooth muscle cells were seeded onto coverslips and treated. Afterwards, the VSMCs were fixed with 4% paraformaldehyde for 30 minutes and then permeabilized with 0.1% Triton X‐100. After washing with PBST, the cells were incubated with 1% BSA for 30 minutes. Then, the cells were incubated with a primary anti‐YAP antibody (1:100, sc‐101199, Santa Cruz Biotechnology) overnight at 4°C, followed by an Alexa Fluor 594‐conjugated goat anti‐mouse secondary antibody (1:300, #8890, Cell Signalling Technology) and Alexa Fluor® 488 Phalloidin to stain polymerized actin (1:200, A12379, Thermo Scientific) for 1 hour at room temperature. The cell nuclei were stained with DAPI (1 μg/mL). Then, the slides were rinsed with PBST and mounted. Immunofluorescence was detected using confocal microscopy (LSM 710; Zeiss, Oberkochen, Germany).
2.9. Construction of the YAP‐shRNA plasmid and transfection into VSMCs
Three YAP1‐shRNA sequences were designed using Primer Premier 6.0 as follows: 5′‐CCATAAGAACAAGACCACA‐3′; 5′‐GGTCAGAGATACTTCTTAA‐3′ and 5′‐GGCAGGCAATACGGAATAT‐3′. A scrambled sequence served as a control. The YAP‐shRNA plasmids were constructed based on the pRNAT‐U6.1/Neo backbone system (RioBo Bio, Guangzhou, China). VSMCs were transfected with the YAP‐shRNA plasmids using the Lipofectamine 3000 Transfection kit (Thermo Scientific) following the manufacturer's protocol.
2.10. VSMC proliferation assay
Vascular smooth muscle cells proliferation was determined using the 5‐ethynyl‐2′‐deoxyuridine (EdU) Cell Proliferation kit (RiboBio, Guangzhou, China) according to the manufacturer's instructions. Briefly, VSMCs were seeded into 96‐well plates at a density of 3 × 104 cells/well and cultured to 70%‐80% confluence. After pretreatment, the cells were made quiescent with 48‐h incubation in 0.1% FBS‐DMEM. Then, the VSMCs were treated with either Ang II (10−7 mol/L) or PBS in serum‐free medium for an additional 24 hours, followed by incubation for 2 hours with an EdU labelling solution (50 μmol/L). Thereafter, the cells were fixed and incubated with 100 μL of Apollo® 488 (anti‐EdU) working solution for 30 minutes at room temperature. After permeabilization, the cells were incubated with Hoechst 33342 for 30 minutes to stain the nuclei. VSMC proliferation was quantified by determining the percentages of EdU‐positive cells from three independent experiments using five randomly selected fields per experiment. Each condition was assayed in triplicate.
2.11. VSMC scratch migration assay
Vascular smooth muscle cells were seeded into 12‐well plates and grown to a confluent density. After serum starvation and pretreatment, the cell monolayer was carefully scratched with the fine end of a sterile 1‐mL pipette tip. The cells were washed twice with PBS to remove any cell debris and incubated at 37°C in serum‐free medium supplemented with either Ang II (10−7 mol/L) or PBS. Photographs of each wound were captured in the same fields at 0, 8, and 16 hours postwound induction. VSMC migration was quantified by measuring the change in distance between the wound edges using the ImageJ software.
2.12. VSMC transwell migration assay
Pretreated VSMCs were seeded into Transwell chambers with 8.0‐μm pores (BD Falcon, NY, USA) at a density of 2 × 105 cells/well and cultured in serum‐free DMEM. DMEM supplemented with either Ang II (10−7 mol/L) or PBS was added to the bottom chamber. The VSMCs were allowed to migrate at 37°C in 5% CO2 for 12 hours, after which the upper surface of the membrane was cleaned with a cotton swab. The migrated cells on the bottom surface of the membrane were fixed and stained with DAPI to visualize the nuclei. DAPI‐positive cells were counted in five random fields under a fluorescence microscope. Each experiment was conducted in triplicate.
2.13. Statistical analysis
Continuous variables were presented as the mean ± standard error of the mean, whereas categorical variables were expressed as percentages. Differences between two mean values were analysed using Student's t‐test. Multiple mean values were compared using either one‐way ANOVA or the Kruskal‐Wallis test depending on the variable type. A two‐tailed P value <0.05 was considered significant. All analyses were performed using the SPSS 13.0 software (SPSS, Inc., Chicago, IL , USA), and figures were generated using the GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA).
3. RESULTS
3.1. In vivo silencing of YAP ameliorated Ang II‐induced vascular remodelling through a blood pressure‐independent mechanism
The Ang II‐infused rats displayed significantly higher SBPs, heart rates, and YAP expression in the media of RCCAs, but lower p‐YAP/YAP and slightly lower body weights than the control rats. Adventitial application of the Ad‐YAP‐shRNA was effective in attenuating Ang II‐regulated YAP expression and p‐YAP/YAP level in the RCCAs media. However, neither SBPs, heart rates, and baseline YAP expression, nor baseline and Ang II‐regulated LATS1 and p‐LATS1/LATS1 level was affected by in vivo silencing of YAP (Figure 1 and Supplementary Table S2).
Figure 1.
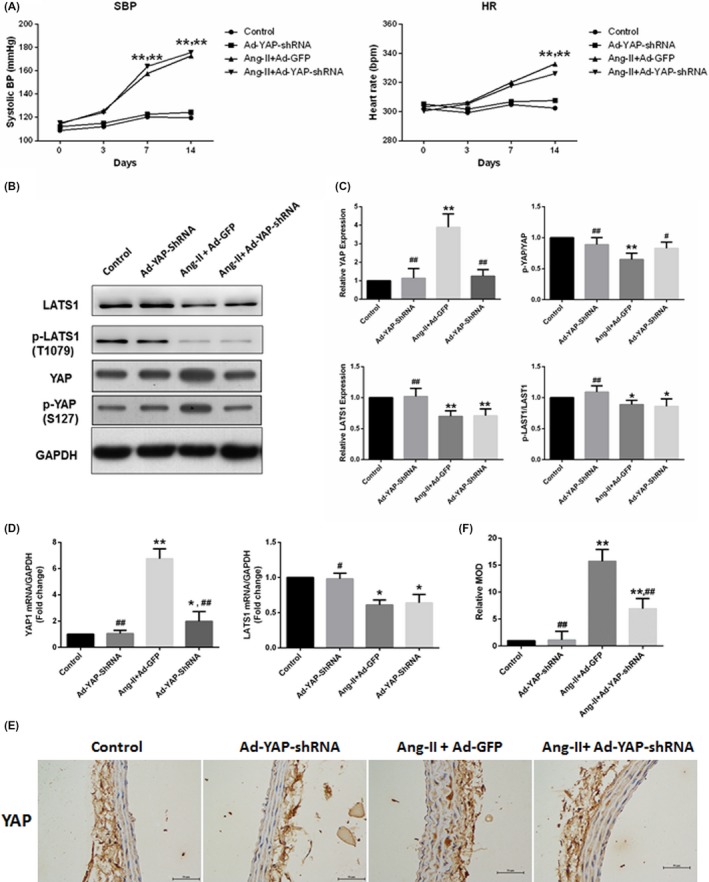
YAP upregulation by Ang II. An HVR rat model was established by continuous Ang II infusion for 2 weeks. YAP was knockdown by adventitial application of the Ad‐YAP‐shRNA. Values were presented as the mean ± SEM. A, Temporal trend of SBP and heart rate which were measured using a caudal artery tail‐cuff system on days 0, 3, 7, and 14 after treatment. B and C, Ang II regulated YAP and LATS1 expression and phosphorylation in the RCCAs. Densitometric quantification and representative western blots of total protein lysates for YAP and LATS1 were shown. Data were normalized to the housekeeping proteins GAPDH. D, Ang II regulated the gene expression of Yap1 and Last1 were analysed using quantitative real‐time polymerase chain reaction. Data were expressed relative to the control (set to 1) after normalization to GAPDH (served as an internal control). E, Immunohistochemical staining of YAP in RCCAs. F, Quantitative analysis of the relative MOD (related to the control group) shown in (E). *P < 0.05 compared with the control group, **P < 0.01 compared with the control group; #P < 0.05 compared with the Ang II + Ad‐GFP group, ##P < 0.01 compared with the Ang II + Ad‐GFP group. HVR, hypertensive vascular remodelling; RCCA, right common carotid artery; MOD, mean optical density; SBP, systolic blood pressure; LATS1, large tumour suppressor kinase 1; GAPDH, glyceraldehyde phosphate dehydrogenase (n = 5)
The media thickness and area and the media:lumen ratio of the RCCAs were used to assess vascular remodelling. Compared with the control group (PBS infusion alone), the Ang II‐infused rats exhibited a significant increase in the media thickness, media area and media:lumen ratio, indicating that vascular remodelling had occurred. However, this effect was significantly attenuated by YAP silencing (Figure 2). CTGF and TGF‐β1 have been reported to be involved in vascular remodelling. In vivo YAP knockdown also ameliorated Ang II‐induced CTGF expression, while TGF‐β1 and cleaved caspase‐3 expression did not differ among groups (Supplementary Figure S1).
Figure 2.
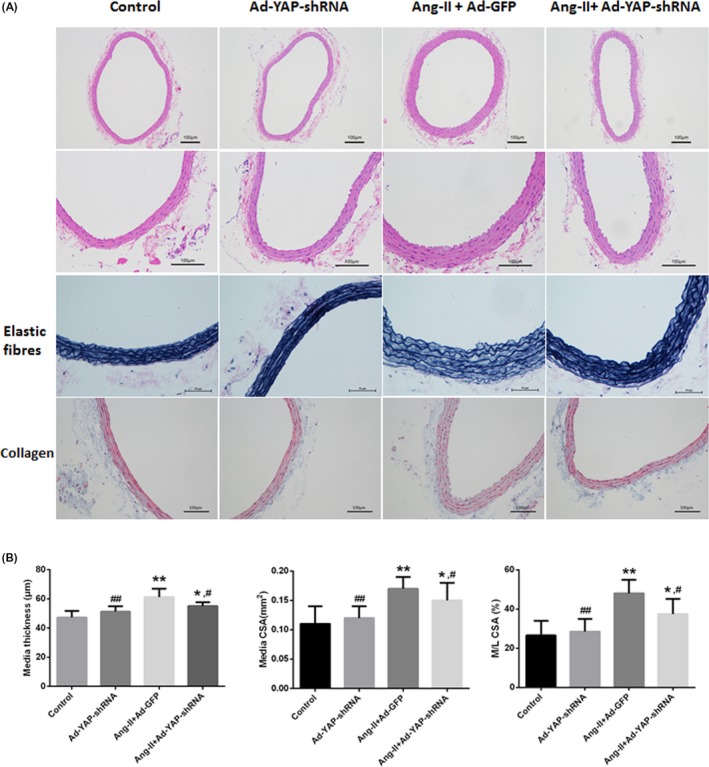
YAP mediated Ang II‐induced hypertensive vascular remodelling independent of the blood pressure levels. Two weeks after continuous infusion with either Ang II or PBS, and adventitial gene transfer with either adenovirus (Ad‐YAP‐shRNA or Ad‐GFP) or gel on the RCCAs, RCCAs were harvested for histological assessments and morphometric analyses. A, H&E staining, Vitoria blue staining for elastic fibres and Masson's Trichrome staining for collagen. Intact elastic arrangement of the RCCAs media was noted whereas no obvious collagen deposition had been demonstrated by Masson staining among groups. B, Quantitative analysis of the vascular remodelling parameters, including medial thickness, medial CSA and the M/L CSA ratio calculated from (A). *P < 0.05 compared with the control group, **P < 0.01 compared with the control group; #P < 0.05 compared with the Ang II + Ad‐GFP group, ##P < 0.01 compared with the Ang II + Ad‐GFP group. PBS, phosphate‐buffered saline (n = 5)
Furthermore, intact elastic arrangement of the RCCAs media was noted during the process of Ang II challenge and adenovirus application. No obvious collagen deposition had been demonstrated by Masson staining and collagen I immunochemistry (Figure 2 and Supplementary Figure S1).
Taken together, these data indicated that YAP mediated Ang II‐induced vascular remodelling independent of changes in blood pressure.
3.2. Ang II regulated YAP expression and promoted YAP nuclear accumulation in rat VSMCs
Ang II administration has been shown to increase YAP expression in the RCCAs media; however, determining whether Ang II could regulate YAP in VSMCs required in vitro investigation. Quantitative Western blotting analysis of rat VSMCs stimulated with Ang II revealed that YAP expression increased in a dose‐ and time‐dependent manner, with maximal increases observed at 10−7 mol/L and 24 hours, respectively. Ang II also increased YAP phosphorylation at serine 127 (S127), a direct LATS phosphorylation site resulting in YAP cytoplasmic localization, to a lesser extent than total YAP level. Consequently, the p‐YAP/YAP ratio was reduced in a dose‐ and time‐dependent manner. Maximal decreases in the ratio were elicited by Ang II treatment at a concentration of 10−7 mol/L and for 24 hours. Thus, Ang II dose‐ and time‐dependently decreased YAP phosphorylation (Figure 3A,B). In contrast, Ang II reduced LATS1 expression and the p‐LATS1/LATST ratio (Figure 3A,B). qRT‐PCR analysis showed that Ang II increased YAP mRNA expression but decreased LATS1 mRNA expression (Figure 3C).
Figure 3.
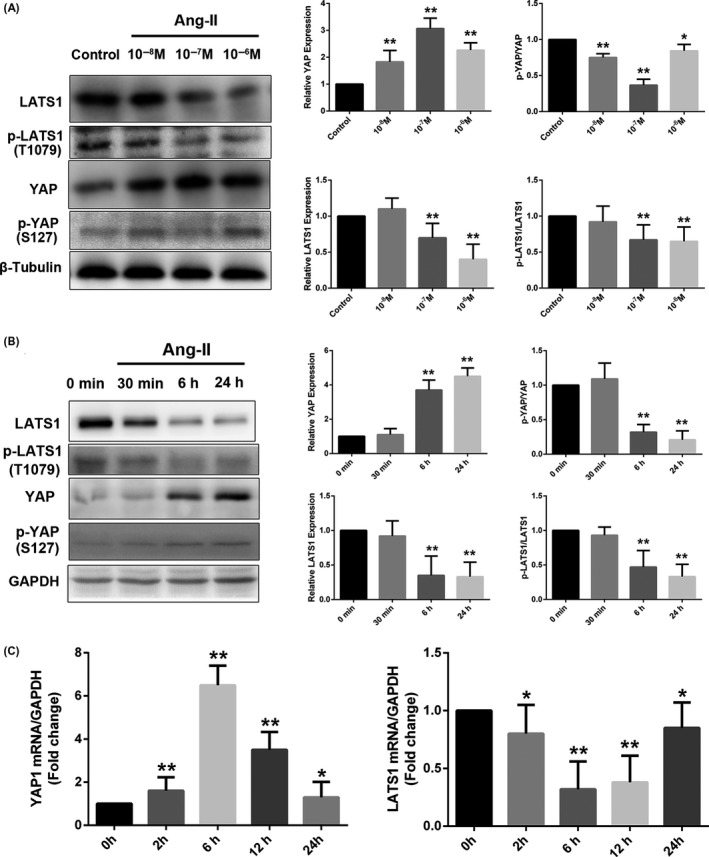
Regulation of YAP and LATS1 by Ang II. Rat VSMCs (70%‐80% confluence) underwent mitogenic quiescence by incubation in DMEM supplemented with 0.1% FBS for 48 h and were subsequently stimulated with Ang II at either the indicated concentrations for 24 h or 10−7 mol/L Ang II for the indicated times. A and B, Ang II regulated YAP and LATS1 expression and phosphorylation in a dose‐ and time‐dependent manner. Densitometric quantification and representative Western blots of total protein lysates for YAP and LATS1 were shown. The data were normalized to the housekeeping proteins β‐tubulin or GAPDH as indicated. C, Temporal changes in the mRNA of Yap1 and Lats1 were analysed using quantitative real‐time polymerase chain reaction. The data were expressed relative to the control (0 h, set to 1) after normalization to GAPDH (served as an internal control). All results were presented as the mean ± SEM of three independent experiments performed in triplicate. *P < 0.05 compared with the control group, **P < 0.01 compared with the control group. GAPDH, glyceraldehyde phosphate dehydrogenase
YAP cytoplasmic/nuclear shuttling plays a critical role in YAP function. Only nuclear YAP can regulate its target genes and thus exert its biological functions. Consistent with decreased YAP phosphorylation with Ang II, a 30‐minute stimulation with Ang II augmented the localization of YAP in the nucleus, as demonstrated on immunofluorescence staining of YAP (Figure 4A). However, this effect was lost after prolonged stimulation for 24 hours. Subcellular fractionation assays confirmed the Ang II‐induced YAP nuclear accumulation (Figure 4B,C).
Figure 4.
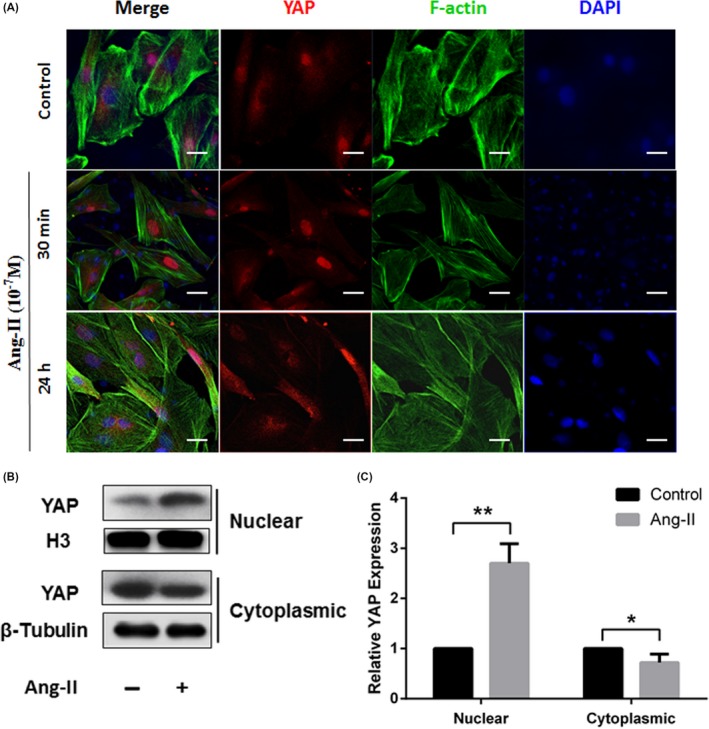
Ang II promoted the nuclear accumulation of YAP. A, Cultured rat VSMCs were incubated with 10−7 mol/L Ang II for the indicated times. The cells were subjected to immunofluorescence staining for YAP (red) and F‐actin (Alexa Fluor® 488 Phalloidin, green). All sections were counterstained with DAPI to visualize the nuclei (blue). Immunofluorescence images were acquired using a confocal microscope (Zeiss). Scale bars represent 10 μm. B, Rat VSMCs were treated with or without Ang II (10−7 mol/L) for 30 min. Nuclear and cytoplasmic fractions were extracted, and the protein levels of YAP in different fractions were measured by Western blot analysis. C, Quantification of YAP level in nuclear and cytoplasmic fractions in (B). The protein levels of YAP in nuclear or cytoplasmic fractions were normalized to H3 or GAPDH, respectively, and compared to that in the PBS control (set as 1). All results were presented as the mean ± SEM of three independent experiments performed in triplicate. *P < 0.05 compared with the control group, **P < 0.01 compared with the control group. GAPDH, glyceraldehyde phosphate dehydrogenase
3.3. YAP mediated Ang II‐induced VSMC phenotypic modulation
Vascular smooth muscle cells phenotypic modulation is an important mechanism that contributes to vascular remodelling. Given the above results, we elected to investigate whether Ang II induced vascular remodelling via YAP‐mediated VSMC phenotypic modulation. We constructed YAP‐shRNA plasmid to knockdown YAP expression in rat VSMCs (Supplementary Figure S2). Ang II induced VSMC proliferation (Figure 5A,B) and migration (Figure 5C‐F). Ang II also upregulated cyclin D1 and PCNA expression (synthesis markers), but downregulated SM α‐actin and SM22‐α expression (contractile markers) (Figure 5G,H), indicating that Ang II induced VSMC phenotypic modulation. Treatment with the YAP‐shRNA alone had no impact on VSMC proliferation, migration, or phenotypic marker expression. However, shRNA‐mediated YAP knockdown attenuated the abovementioned effects of Ang II on VSMCs (Figure 5A‐H). These findings suggested that YAP mediated Ang II‐induced phenotypic modulation of VSMCs.
Figure 5.
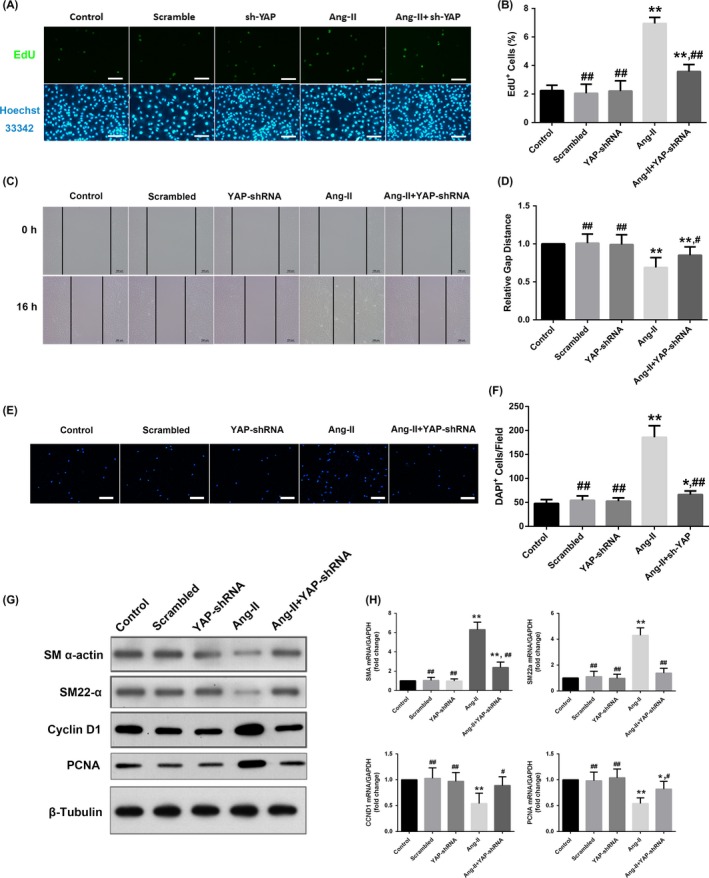
YAP mediated Ang II‐induced VSMCs phenotypic modulation. Rat VSMCs were transfected with either the YAP‐shRNA or scrambled control plasmids. A, YAP mediated Ang II‐induced VSMC proliferation. Quiescent VSMCs were stimulated with either 10−7 mol/L Ang II or PBS for 24 h and then incubated with an EdU labelling solution (50 μmol/L) diluted in DMEM for another 2 h. The cells were subjected to immunofluorescence staining for EdU (green) and counterstained with Hoechst 33342 to visualize the nuclei (blue). B, Quantification of the percentages of proliferating cells (EdU‐positive cells) shown in (A). C, YAP mediated Ang II‐induced VSMC migration. VSMCs were seeded into 12‐well plates and grown to a confluent density. Cell migration was assayed 16 h after a wound was scratched across the cell monolayer under serum‐free conditions, and the relative closure distance was measured. D, Quantification of the gap distance relative to the control (set to 1). E, VSMCs were seeded into Transwell chambers with 8.0‐μm pores and allowed to migrate for 12 h. The migrated cells on the bottom surface of the membrane were stained with DAPI to visualize the nuclei (blue) and detected using fluorescence microscopy. F, Quantification of the migrated cells (DAPI‐positive) in low‐power field shown in (E). G, Representative Western blotting analysis of total protein lysates from the cultured rat VSMCs treated with either 10−7 mol/L Ang II or PBS for 24 h after serum starvation. H, the mRNA levels of different phenotypic markers were detected by quantitative real‐time polymerase chain reaction. The data were expressed relative to the control (set to 1) after normalization to GAPDH (served as an internal control). Scale bars in A and E represent 80 μm. All results from B, D, and F were presented as the mean ± SEM from three independent experiments with five random fields counted per experiment. *P < 0.05 compared with the control group, **P < 0.01 compared with the control group; #P < 0.05 compared with the Ang II group, ##P < 0.01 compared with the Ang II group. CCND1, cyclin D1; PCNA, proliferating cell nuclear antigen
3.4. Ang II regulated YAP via an AT1R‐ and F‐actin‐dependent mechanism
We further explored the underlying mechanism by which Ang II regulated YAP. F‐actin has been shown to be an important upstream regulator of Hippo/YAP signalling.22 To further delineate the role of F‐actin in Ang II‐mediated regulation of YAP, we pretreated the VSMCs with the F‐actin depolymerizing agent latrunculin B (500 nmol/L) for 20 minutes before exposing the cells to Ang II. The quantitative Western blotting analysis demonstrated that losartan (AT1R antagonist), PD 123319 (AT2R antagonist), and latrunculin B alone had no impact on LATS1 and YAP expression in the corresponding groups compared with the control group. However, the effect of Ang II on YAP and LATS1 expression could be partially blocked by pretreatment with either losartan or latrunculin B but not PD 123319 (Figure 6A,B). The confocal microscopy data also revealed that Ang II‐induced nuclear accumulation of YAP was prevented by latrunculin B‐mediated F‐actin cytoskeleton disruption (Figure 6C). These results suggested that AT1R and a functional F‐actin cytoskeleton were basic prerequisites for the normal function of the Ang II/YAP signalling axis.
Figure 6.
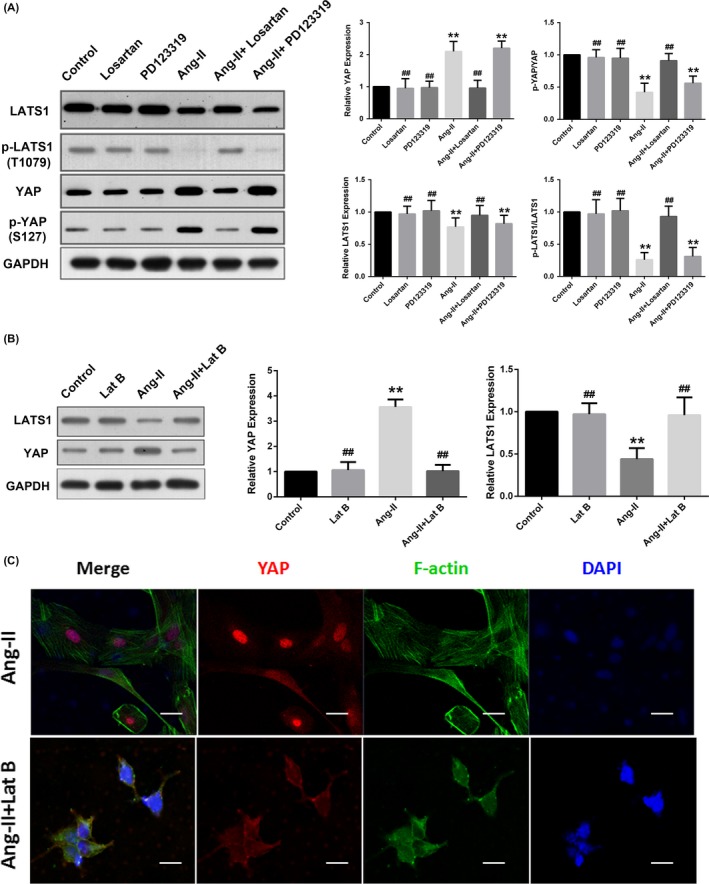
Ang II regulated YAP activity via AT1R‐ and F‐actin‐dependent mechanisms. Quiescent rat VSMCs were pretreated under the following conditions: 1 μmol/L losartan for 30 min; 1 μmol/L PD 123319 for 30 min; or 500 nmol/L latrunculin B for 20 min. Then, all cells were stimulated with Ang II (10−7 mol/L for 24 h) and harvested for the assay. A and B, Densitometric quantification and representative Western blotting for YAP and LATS1 were shown. The data were normalized to GAPDH and expressed relative to the control values (set to 1). C, Representative immunofluorescence staining of YAP (red) and F‐actin (Alexa Fluor® 488 Phalloidin, green). All sections were counterstained with DAPI to visualize the nuclei (blue). Images were acquired using a Zeiss confocal microscope. Scale bars represent 10 μm. *P < 0.05 compared with the control group, **P < 0.01 compared with the control group; #P < 0.05 compared with the Ang II group, ##P < 0.01 compared with the Ang II group. Lat B, latrunculin B. GAPDH, glyceraldehyde phosphate dehydrogenase
3.5. The YAP‐TEAD interaction mediated the VSMC synthetic phenotype induced by Ang II
As a transcriptional coactivator, YAP does not directly bind to DNA but instead mediates the major physiological functions of the Hippo/YAP pathway after partnering with TEA domain (TEAD) transcription factors. Verteporfin disrupts this YAP‐TEAD interaction and therefore inhibits YAP activity. As shown in Figure 7, verteporfin alone did not significantly impact on VSMC proliferation and migration, nor did it affect the cyclin D1 and PCNA expression levels. However, the phenotypic modulatory effect of Ang II on VSMCs could be partially abrogated by verteporfin (Figure 7A‐I), suggesting that the YAP‐TEAD interaction was involved in the formation of the VSMC synthetic phenotype induced by Ang II.
Figure 7.
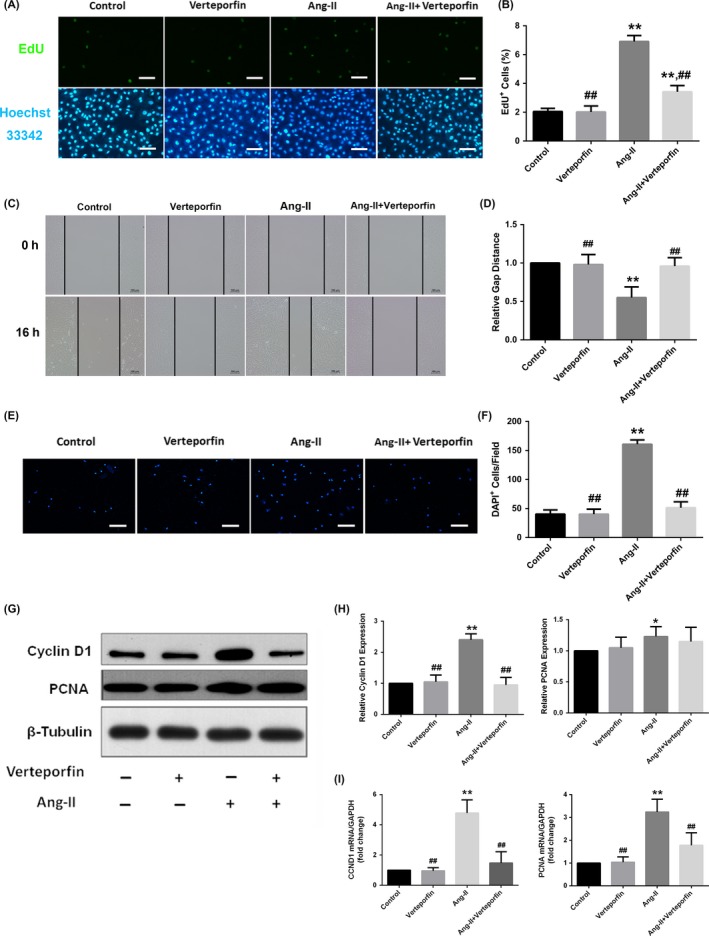
The YAP‐TEAD interaction mediated the induction of the VSMC synthetic phenotype by Ang II. Quiescent rat VSMCs were pretreated with 2 μg/mL of verteporfin (protected from light) or buffer (DMSO) for 48 h prior to Ang II stimulation (10−7 mol/L for different times as indicated). A and B, Representative EdU proliferation assay images and quantification of the percentages of proliferating cells (EdU‐positive cells). The cells were subjected to immunofluorescence staining for EdU (green) and counterstained with Hoechst 33342 to visualize the nuclei (blue). C and D, Representative scratch migration assay images and quantification of the gap distance relative to the control (set to 1). VSMCs were seeded into 12‐well plates at a confluent density. The relative closure distance was measured 16 h after a wound was scratched across the cell monolayer under serum‐free conditions. E and F, Representative Transwell migration assay images and quantification. The migrated cells were visualized by staining with DAPI (blue) and detected via fluorescence microscopy. G and H, Representative Western blotting analysis and densitometric quantification of total protein lysates from cultured rat VSMCs to detect the expression of synthetic markers cyclin D1 and PCNA. The data were normalized to β‐tubulin and expressed relative to the control values (set to 1). I, the mRNA level of cyclin D1 and PCNA were detected by quantitative real‐time polymerase chain reaction. The data were expressed relative to the control (set to 1) after normalization to GAPDH (served as an internal control). Scale bars in A and E represent 80 μm. All results from B, D, and F were presented as the mean ± SEM from three independent experiments with five randomly selected fields counted per experiment. *P < 0.05 compared with the control group, **P < 0.01 compared with the control group; #P < 0.05 compared with the Ang II group, ##P < 0.01 compared with the Ang II group. DMSO, dimethyl sulphoxide; CCND1, cyclin D1; PCNA, proliferating cell nuclear antigen
4. DISCUSSION
Unravelling the mechanisms involved in HVR is an important step towards better understanding of the pathology of hypertension‐induced target organ damage and ultimately defining therapeutic targets for treatment beyond blood pressure control. In this study, we showed that Ang II upregulated YAP expression and promoted its nuclear accumulation via AT1R‐ and F‐actin‐dependent mechanism. More importantly, YAP acted as a crucial molecule in mediating Ang II‐induced VSMCs phenotypic modulation and vascular remodelling. The most important finding of our work was that by targeting on YAP signalling, HVR could be ameliorated independently of blood pressure level. Despite a 53‐56 mmHg increase in SBP level compared with the control group, in vivo YAP knockdown significantly mitigated HVR in Ang II‐infused rats.
In consistent with previous results in mice,10 baseline expression of YAP was low in arteries under physiological condition. However, a 15‐30 minutes short‐term stimulation with Ang II23, 24 or platelet‐derived growth factor‐BB10 was ample to elicit YAP activation and target genes expression. These data suggest that YAP might act as a rapid reaction molecule in mediating pathological signals. In contrast to results from rodent, recent report in normal human aortic samples obtained from heart transplantation donors showed abundant YAP expression.25 However, the demographic and haemodynamic data of these donors were unclear. Moreover, low YAP expression in human aorta was associated with aortic aneurysm formation. Since both over expression and insufficient expression of YAP were related to vascular pathologies, further studies are warranted to explore how and to what extent to maintain appropriate YAP expression in order to preserve normal vascular morphology and function.
Adventitial fibroblasts have been reported to participate in vascular remodelling.26, 27 In response to stimulation, adventitial fibroblasts migrate from the adventitia into the media and intima. During this process, fibroblasts display phenotypic modulation with a switch to myofibroblasts that possess greater secretion and contraction capabilities. YAP has been reported to be a driver of myofibroblast differentiation from dermal fibroblasts.28 However, no obvious collagen deposition had been demonstrated both by Masson staining and collagen I immunochemistry in the RCCAs media among groups. Moreover, the expression of TGF‐β1, a potent inducer of interstitial fibrosis, was significantly low in the media of RCCAs and did not differ among groups. Consequently, these data suggested that YAP might induce vascular remodelling via regulation of VSMCs, the most majority of component in the media, but not interstitial fibrosis.
The Hippo/YAP signalling pathway has been reported to promote cell proliferation and inhibit apoptosis.22 For example, downregulation of YAP by extracellular matrix disorders in human aorta would provoke VSMCs apoptosis and contribute to aortic aneurysms formation eventually.25 However, no obvious cleaved caspase‐3 expression had been detected in the RCCAs media in YAP knockdown rats. Differences in experimental models and methods might be responsible for this discrepancy. First, as discussed above, baseline YAP expression was low in the media under normal condition. In vivo YAP knockdown did not further reduce YAP expression and induce VSMCs apoptosis in the control rats. Second, in the Ang II + Ad‐YAP‐shRNA group, the proapoptotic effect of VSMCs by YAP knockdown would be offset by the promitotic effect of Ang II infusion. The low apoptosis level of VSMCs in the present study would raise another concern. The ability of YAP knockdown to reverse HVR that has already formed would not be as effective as that seen when YAP knockdown was applied during the formation of HVR.
The core Hippo/YAP pathway has been well established in both Drosophila and mammals; however, the upstream and downstream mechanisms by which this signalling pathway was regulated have not been fully elucidated.22 Proteins involved in cell contacts (eg, Mer/NF2, Crb1‐3, and α/β‐Catenin), cell‐cell adhesion, apical‐basal polarity (eg, Fat1‐4 and Fjx1) and extracellular matrix rigidness as well as several growth factors have been reported to serve as upstream regulators of Hippo/YAP signalling.29 In 2012, two laboratories13, 14, 15 independently demonstrated that GPCRs on the eukaryotic cell surface could serve as upstream regulators of Hippo/YAP signalling. Activation of different GPCRs elicited a stimulatory or inhibitory effect on Hippo/YAP signalling depending on the coupled heterotrimeric G proteins. Specifically, Gα12/13‐, Gαq/11‐ or Gαi/o‐coupled signals induced YAP activity, whereas Gαs‐coupled signals repressed YAP activity.13 In 2014, Yu et al30 provided the first in vivo evidence that mutant Gαq/11 activated YAP, thereby promoting uveal melanoma tumourigenesis. AT1R coupled with Gα12/13, Gαq/11, and Gαi/o, although the AT2R‐coupled G proteins remained elusive.31 In this study, we demonstrated that Ang II regulated YAP expression and dephosphorylation in VSMCs via AT1R rather than AT2R (Figure 5A).
Consistent with the findings of previous studies,32, 33 we also showed that F‐actin was a crucial upstream mediator of YAP activation (Figure 5B). F‐actin can regulate YAP either directly or indirectly. As mentioned above, the Hippo/YAP pathway was regulated by cell contact, cell adhesion, apical‐basal polarity, and GPCRs signalling. One common feature of these regulatory mechanisms is involvement of the actin cytoskeleton. F‐actin can act as a signal convergence for these upstream stimulators.22 Additionally, both mammalian STE20‐like protein kinase 1/2 (MST1/2) and LATS1 have been shown to bind or colocalize with F‐actin,34 suggesting that F‐actin can either function as a platform to facilitate more efficient signal transmission for the Hippo/YAP pathway or directly regulate the activity of Hippo/YAP pathway kinases. However, further investigations are required to clarify the underlying mechanism.
YAP does not contain any intrinsic DNA‐binding domains; instead, YAP binds to the promoters of target genes by interacting with DNA‐binding transcription factors (primarily TEAD) to regulate transcription of target genes. Verteporfin, which is a drug approved by the Food and Drug Administration for photodynamic therapy to eliminate abnormal blood vessels in the eyes, has been shown to disrupt the YAP‐TEAD interaction. Consistent with the results of previous in vitro and in vivo studies,30, 35, 36 our results demonstrated that verteporfin significantly ameliorated the effects of YAP activation (Figure 6). Therefore, the YAP‐TEAD interaction played a critical role in mediating the functions of the Hippo/YAP pathway and could be a target for Hippo/YAP pathway manipulation.
This study also had important implication for clinical practice. A convincing body of evidence has demonstrated that Ang II elevation plays an essential role in the pathogenesis of hypertension and its complications. As we could not manipulate blood pressure level without affecting any other signal systems of the subject, it is difficult to determine to what extent the YAP‐dependent and the BP‐dependent mechanism may play a role in the formation of HVR. However, this dilemma could be solved by the use of BP lowering agent, the AT1R blockers such as losartan. Numerous clinical trials have demonstrated that AT1R blockers are effective in reducing BP level. Furthermore, our in vitro study showed that losartan was also effective in inhibited Ang II‐upregulated YAP expression. Consequently, it was suggestive that the use of AT1R blockers could produce synergic effect on the inhibition of HVR formation. In LIFE trial,37 losartan has been shown to be more efficient in reversing left ventricular hypertrophy than other BP lowering drugs in hypertensive patients. Therefore, AT1R blocker might be the treatment of choice in the inhibition of HVR.
Two limitations of the present study are noteworthy. First, our results have not been validated in a vascular‐specific YAP‐knockout animal model. Second, an investigation of the crosstalk of the Hippo/YAP and other signalling pathways downstream of Ang II is beyond the scope of this study but requires further evaluation. Notwithstanding these limitations, this study has provided new insights into how Hippo/YAP signalling mediates Ang II‐induced VSMC phenotypic modulation and HVR.
In conclusion, this study provided evidence that YAP played a critical role in Ang II‐induced HVR and VSMCs phenotypic modulation and that YAP may be a potential therapeutic target for HVR beyond blood pressure control.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
MHL, YXC, and JFW designed the study and interpreted the data; MHL, WLY, and ZZS performed the experiments and collected the data; CNL and TCH analysed and interpreted the data; MHL and YXC drafted the manuscript; all authors reviewed and approved the manuscript.
Supporting information
ACKNOWLEDGEMENTS
This study was supported in part by grants from the National Natural Science Foundation for Young Scientists of China (grant no. 81600233 to Maohuan Lin), the National Natural Science Foundation of China (grant No. 81570213 to Jingfeng Wang, 81770229 to Yangxin Chen).
Lin M, Yuan W, Su Z, et al. Yes‐associated protein mediates angiotensin II‐induced vascular smooth muscle cell phenotypic modulation and hypertensive vascular remodelling. Cell Prolif. 2018;51:e12517 10.1111/cpr.12517
Maohuan Lin, Woliang Yuan and Zizhuo Su contributed equally to the development of this research study.
Funding information
This study was supported in part by grants from the National Natural Science Foundation for Young Scientists of China (grant No. 81600233 to Maohuan Lin), the National Natural Science Foundation of China (grant No. 81570213 to Jingfeng Wang, 81770229 to Yangxin Chen).
Contributor Information
Yangxin Chen, Email: tjcyx1995@163.com.
Jingfeng Wang, Email: drwjfsums@126.com.
REFERENCES
- 1. Benjamin EJ, Blaha MJ, Chiuve SE; American Heart Association Statistics Committee and Stroke Statistics . Heart disease and stroke statistics‐2017 update: a report from the American heart association. Circulation. 2017;51:e146‐e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Briet M, Schiffrin EL. Treatment of arterial remodeling in essential hypertension. Curr Hypertens Rep. 2013;51:3‐9. [DOI] [PubMed] [Google Scholar]
- 3. Forrester SJ, Elliott KJ, Kawai T, et al. Caveolin‐1 deletion prevents hypertensive vascular remodeling induced by angiotensin ii. Hypertension. 2017;51:79‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pasterkamp G, Galis ZS, de Kleijn DP. Expansive arterial remodeling: location, location, location. Arterioscler Thromb Vasc Biol. 2004;51:650‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schiffrin EL. Vascular remodeling in hypertension: mechanisms and treatment. Hypertension. 2012;51:367‐374. [DOI] [PubMed] [Google Scholar]
- 6. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;51:767‐801. [DOI] [PubMed] [Google Scholar]
- 7. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;51:194‐204. [DOI] [PubMed] [Google Scholar]
- 8. Zhou J. An emerging role for hippo‐yap signaling in cardiovascular development. J Biomed Res. 2014;51:251‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morin‐Kensicki EM, Boone BN, Howell M, et al. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of yap65. Mol Cell Biol. 2006;51:77‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang X, Hu G, Gao X, et al. The induction of yes‐associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation. Arterioscler Thromb Vasc Biol. 2012;51:2662‐2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heeneman S, Sluimer JC, Daemen MJ. Angiotensin‐converting enzyme and vascular remodeling. Circ Res. 2007;51:441‐454. [DOI] [PubMed] [Google Scholar]
- 12. Cui M, Cai Z, Chu S, et al. Orphan nuclear receptor nur77 inhibits angiotensin ii‐induced vascular remodeling via downregulation of beta‐catenin. Hypertension. 2016;51:153‐162. [DOI] [PubMed] [Google Scholar]
- 13. Yu FX, Zhao B, Panupinthu N, et al. Regulation of the hippo‐yap pathway by g‐protein‐coupled receptor signaling. Cell. 2012;51:780‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mo JS, Yu FX, Gong R, Brown JH, Guan KL. Regulation of the hippo‐yap pathway by protease‐activated receptors (pars). Genes Dev. 2012;51:2138‐2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miller E, Yang J, DeRan M, et al. Identification of serum‐derived sphingosine‐1‐phosphate as a small molecule regulator of yap. Chem Biol. 2012;51:955‐962. [DOI] [PubMed] [Google Scholar]
- 16. Liu CF, Zhang J, Shen K, et al. Adventitial gene transfer of catalase attenuates angiotensin ii‐induced vascular remodeling. Mol Med Rep. 2015;51:2608‐2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bond M, Sala‐Newby GB, Wu YJ, Newby AC. Biphasic effect of p21cip1 on smooth muscle cell proliferation: role of pi 3‐kinase and skp2‐mediated degradation. Cardiovasc Res. 2006;51:198‐206. [DOI] [PubMed] [Google Scholar]
- 18. Su Z, Lin R, Chen Y, et al. Oxidized low‐density lipoprotein‐induced cyclophilin a secretion requires rock‐dependent diphosphorylation of myosin light chain. J Vasc Res. 2016;51:206‐215. [DOI] [PubMed] [Google Scholar]
- 19. Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem. 2012;51:14598‐14605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang L, Carlson SG, McBurney D, Horton WE Jr. Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J Biol Chem. 2005;51:31156‐31165. [DOI] [PubMed] [Google Scholar]
- 21. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta c(t)) method. Methods. 2001;51:402‐408. [DOI] [PubMed] [Google Scholar]
- 22. Yu FX, Guan KL. The hippo pathway: regulators and regulations. Genes Dev. 2013;51:355‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J, Sinnett‐Smith J, Stevens JV, Young SH, Rozengurt E. Biphasic regulation of yes‐associated protein (yap) cellular localization, phosphorylation, and activity by g protein‐coupled receptor agonists in intestinal epithelial cells: a novel role for protein kinase d (pkd). J Biol Chem. 2016;51:17988‐18005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wennmann DO, Vollenbroker B, Eckart AK, et al. The hippo pathway is controlled by angiotensin ii signaling and its reactivation induces apoptosis in podocytes. Cell Death Dis. 2014;51:e1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H, Jiang W, Ren W, et al. Downregulation of the yes‐associated protein is associated with extracellular matrix disorders in ascending aortic aneurysms. Stem Cells Int. 2016;51:6786184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Siow RC, Mallawaarachchi CM, Weissberg PL. Migration of adventitial myofibroblasts following vascular balloon injury: insights from in vivo gene transfer to rat carotid arteries. Cardiovasc Res. 2003;51:212‐221. [DOI] [PubMed] [Google Scholar]
- 27. Stenmark KR, Yeager ME, El Kasmi KC, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;51:23‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piersma B, de Rond S, Werker PM, et al. Yap1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am J Pathol. 2015;51:3326‐3337. [DOI] [PubMed] [Google Scholar]
- 29. Piccolo S, Dupont S, Cordenonsi M. The biology of yap/taz: hippo signaling and beyond. Physiol Rev. 2014;51:1287‐1312. [DOI] [PubMed] [Google Scholar]
- 30. Yu FX, Luo J, Mo JS, et al. Mutant gq/11 promote uveal melanoma tumorigenesis by activating yap. Cancer Cell. 2014;51:822‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wettschureck N, Offermanns S. Mammalian g proteins and their cell type specific functions. Physiol Rev. 2005;51:1159‐1204. [DOI] [PubMed] [Google Scholar]
- 32. Dupont S, Morsut L, Aragona M, et al. Role of yap/taz in mechanotransduction. Nature. 2011;51:179‐183. [DOI] [PubMed] [Google Scholar]
- 33. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;51:54‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Visser‐Grieve S, Zhou Z, She YM, et al. Lats1 tumor suppressor is a novel actin‐binding protein and negative regulator of actin polymerization. Cell Res. 2011;51:1513‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu‐Chittenden Y, Huang B, Shim JS, et al. Genetic and pharmacological disruption of the tead‐yap complex suppresses the oncogenic activity of yap. Genes Dev. 2012;51:1300‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brodowska K, Al‐Moujahed A, Marmalidou A, et al. The clinically used photosensitizer verteporfin (vp) inhibits yap‐tead and human retinoblastoma cell growth in vitro without light activation. Exp Eye Res. 2014;51:67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lindholm LH, Ibsen H, Dahlof B; LIFE Study Group . Cardiovascular morbidity and mortality in patients with diabetes in the losartan intervention for endpoint reduction in hypertension study (life): a randomised trial against atenolol. Lancet. 2002;51:1004‐1010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials