

Abstract
Objectives
Methamphetamine (MA) abuse evokes pulmonary toxicity. The aim of our study is to investigate if autophagy is induced by MA and if autophagy‐initiated apoptosis in alveolar epithelial cells is involved in MA‐induced chronic pulmonary toxicity.
Materials and Methods
The rats in Control group and MA group were tested by Doppler and HE staining. The alveolar epithelial cells were treated with MA, following by western blot, RT‐PCR and immunofluorescence assay.
Results
Chronic exposure to MA resulted in lower growth ratio of weight and in higher heart rate and peak blood flow velocity of the main pulmonary artery of rats. MA induced infiltration of inflammatory cells in lungs, more compact lung parenchyma, thickened alveolar septum and reduction in the number of alveolar sacs. In alveolar epithelial cells, the autophagy marker LC3 and per cent of cells containing LC3‐positive autophagosome were significantly increased. MA dose dependently suppressed the phosphorylation of mTOR to inactivate mTOR, elicited autophagy regulatory proteins LC3 and Beclin‐1, accelerated the transformation from LC3 I to LC3 II and initiated apoptosis by decreasing Bcl‐2 and increasing Bax, Bax/Bcl‐2 and cleaved Caspase 3. The above results suggest that sustained autophagy was induced by long‐term exposure to MA and that the increased Beclin‐1 autophagy initiated apoptosis in alveolar epithelial cells.
Conclusions
Concurrence of autophagy with apoptosis in alveolar epithelial cells contributes to chronic pulmonary toxicity induced by MA.
Keywords: apoptosis, autophagy, aveolar epithelial cells, Beclin‐1, LC3, methamphetamine
1. INTRODUCTION
Methamphetamine (MA) is a kind of highly addictive drug.1 Long‐term abuse of MA can cause pulmonary toxicity with clinical manifestations: pulmonary oedema, inflammation and pulmonary hypertension.2, 3 A growing body of evidence indicates that alveolar epithelial cells play an important role in the protecting against the toxic stimulants to maintain pulmonary homeostasis.4 Therefore, disruption of alveolar epithelium may be the key contribution to chronic pulmonary toxicity induced by MA.
Autophagy is a process that degrades proteins and organelles in lysosomes to maintain the cellular homeostasis.5 Autophagy dysfunction is involved in the progression of a number of diseases including diabetes, cardiovascular disease and pulmonary disease.6 Mechanistic target of rapamycin (mTOR) is a prosurvival factor and is involved in the regulation of autophagy.7, 8 It is reported that autophagy can be induced by rapamycin through inhibiting mTOR and then activating autophagy regulatory proteins (ie, LC3, Beclin 1) in the downstream of mTOR.9, 10
There are some proteins encoded by ATG genes which can tightly regulate autophagy.11 LC3 is called the microtubule‐associated protein 1 light‐chain 3, which is the ortholog of yeast ATG8.11, 12 LC3 has two forms: LC3‐ I and LC3‐ II.12 LC3 (cytosolic) is cleaved at its C‐terminus by ATG4 to form LC3‐ I which is covalently conjugated to phosphatidylethanolamine (PE) to form LC3‐ II.11 LC3 II is localized to autophagosomes, so LC3‐II levels are correlated with the amount of autophagosome.11, 12, 13 LC3 is the only known protein specifically associated with autophagosomes but not with other vesicular structures, so that LC3 becomes the marker of autophagy.12, 14
Beclin‐1 is the mammalian ortholog of yeast ATG6 and is involved in autophagosome formation by interacting with PI3KC3/VPS34.15, 16 The autophagy protein, Beclin‐1 can interact with anti‐apoptotic protein Bcl‐2 or Bcl‐XL to represent a potentially important point of convergence of the autophagic and apoptotic machinery.17, 18 When the autophagy occurs, Beclin‐1 is dissociated with Bcl‐2 and Bcl‐XL, and constituted Beclin‐1‐VPS34 complex.15 ATG14 also interact with Beclin‐1‐VPS34 complex to enhance its activity and induce the formation of autophagosome structure.15, 19 Thus, autophagy and apoptosis may be co‐regulated at the same direction.14
However, it is always controversial between autophagy and apoptosis. On one hand, autophagy can counteract apoptosis,20 and on the other hand increased autophagy can be found in dying cells, although the relationship between autophagy and programmed cell death remains unclear.10 Therefore, some experts postulated that severe and persistent autophagy may initiate apoptosis or cell death.8 A growing body of evidence indicates that the psychostimulant MA can induce the cellular autophagy in nerve system, for example, dopaminergic neuronal cells, cortical cells and astrocytes.21, 22, 23, 24, 25, 26 However, it has been not detected if autophagy in alveolar epithelial cells can be induced by MA. In our previous studies, it was proved that endoplasmic reticulum stress (ERS) is induced in chronic exposure to MA in rats.2, 27 ERS and unfolded protein response can activate the autophagy signalling pathways.28, 29, 30, 31 Taken together, it is hypothesized that MA may induce autophagy in alveolar epithelial cells.
On basis of our hypothesis, this study is designed to investigate if autophagy can be induced by MA in lungs, and if concurrence of autophagy with apoptosis in alveolar epithelial cells contributes to chronic pulmonary toxicity induced by MA.
2. MATERIALS AND METHODS
2.1. Animal models
A total of 20 male Wistar rats (200 ± 10 g) were obtained from Animal Resource Center, China Medical University (Certificate number: Liaoning SCSK 2012‐0005) and were randomly divided into control group and MA group. Establishment of the rat models is referred to our previous study.2 In briefly, at the 1st week, rats in MA group were intraperitoneally injected with MA (China Criminal Police University, China) at 10 mg/kg for 1 week, and then additional 1 mg/kg was added into daily dosage per week. At the 6th week, a daily dosage was increased to 15 mg/kg (twice per day for 6 weeks). Meanwhile, the rats in control group were intraperitoneally injected with an equal volume of 0.9% normal saline. All animals were maintained in animal facility with controlled temperature (18‐22°C) and humidity (50%‐70%) and were fed with solid food and water in an alternating 12 hours light and 12 hours dark cycle. All procedures were complied with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH), and all protocols were approved by the Institutional Animal Care and Use Committee of China Medical University.
2.2. Cells culture and drug treatment
Alveolar epithelial cell lines A549 were obtained from the Experimental Center of China Medical University (Shenyang, China). Cells were cultured in RPMI‐1640 medium containing 10% foetal bovine serum and 1% penicillin/streptomycin at 37°C in 5% CO2. Cells were treated by MA (China Criminal Police University, Shenyang, China) with the dosage of 0.1, 0.5, 1 and 5 mmol L−1 for 6, 12 and 24 hours.25 Cells in positive control were treated with 1 mg/L rapamycin (Dalian Meilun Biotech Co., Ltd, China) in 6, 12 and 24 hours.
2.3. Doppler ultrasonic measurement
After 6 weeks, five rats from each group selected randomly were intraperitoneally anaesthetized with 3% pentobarbital sodium (45 mg/kg; Sigma‐Aldrich; Merck KGaA, Darmstadt, Germany). Thoroughly shaving the hair in rat abdomen to expose the skin to completely contact with the probe, Acoustic gel (Skintact; Leonard Lang GmbH, Innsbruck, Austria) was applied to hold the animal dander and improve the resolution of the ultrasound. The rats were examined in the supine position using the Philips IE33 cardiovascular ultrasound (Philips Healthcare, Andover, MA, USA) with a linear transducer probe S12‐4. The frequency of 4.5 MHz and one focal zone set at a depth of 0.5‐2 cm were used to achieve detailed imaging of the rats.
2.4. Haematoxylin and eosin (H&E) staining
The right lower lobes were separated, fixed with paraformaldehyde and embedded in paraffin for H&E staining. Sections (5 μm) were performed for observation and analysis under light microscopy (Olympus BX 51, Japan). Lung injury was calculated by the thickness of alveolar septum and the number of alveolar sacs (three visual fields selected randomly were analysed in each section; magnification, ×200).
2.5. Western blot
After the drugs treatment, A549 cells were lysed in RIPA buffer with PMSF on ice. Total protein was extracted and the concentration was determined by BCA kit (Beyotime Biotechnology). After electrophoresis, protein was transferred to a PVDF membrane. The PVDF membrane was blocking for 2 hours and then incubated with corresponding primary antibodies overnight at 4°C and was incubated with secondary antibodies for 2 hours in room temperature, followed by detecting using enhanced chemiluminescence (Proteintech, USA). The primary antibodies of correlative proteins were listed in Table 1. The relevant protein was represented by the relative yield to the β‐actin.
Table 1.
Primary antibodies for western blot in this study
| Primary antibody | Dilution | Company | Catalogue |
|---|---|---|---|
| mTOR | 1:1000 | Proteintech, USA | 20657‐1‐AP |
| p‐mTOR | 1:500 | Bioworld Technology, USA | BS4706 |
| LC3 | 1:1000 | Cell Signaling Technology, USA | #3868 |
| Beclin‐1 | 1:1000 | Proteintech, USA | 11306‐1‐AP |
| Bax | 1:1000 | Proteintech, USA | 50599‐2‐lg |
| Bcl‐2 | 1:1000 | Proteintech, USA | 12789‐1‐AP |
| Cleaved Caspase 3 | 1:1000 | Proteintech, USA | 25546‐1‐AP |
| β‐actin | 1:2000 | Proteintech, USA | 60008‐1‐lg |
2.6. Real‐time polymerase chain reaction
Total RNA was extracted from cells using the TRIzol® reagent (Invitrogen Life Technologies, Paisley, Scotland) after drug treatment. The concentration of total RNA was detected by Ultra microultraviolet spectrophotometer (Thermo NanoDrop 2000, USA). Total RNA was reverse transcribed to cDNA by PrimeScriptTM RT Reagent Kit (Perfect Real Time) (TAKARA, Japan). For the semi‐quantitative analysis, cDNA was incubated with SYBR® Premix Ex TaqTM II (Tli RNase Plus) (TAKARA, Japan) and primers for LC3 and Beclin‐1(Proteintech, USA) (Table 2). We performed 40 cycles (95°C for 5 seconds and 55°C for 30 seconds) on CFX96 Touch™ Real‐Time PCR Detection System (Bio‐Rad, USA). The levels of LC3 and Beclin‐1 mRNA were normalized to β‐actin and were then expressed as values relative to the control using the comparative threshold cycle (Ct) method.
Table 2.
Primers of LC3 and Beclin‐1 for Real‐time PCR
| Forward | Reversed | |
|---|---|---|
| LC3 | 5′‐GGCGCTTACAGCTCAATGCT‐3′ | 5′‐GATTGGTGTGGAGACGCTGA‐3′ |
| Beclin‐1 | 5′‐GGCTGAGAGACTGGATCAGG‐3′ | 5′‐CTGCGTCTGGGCATAACG‐3′ |
| β‐actin | 5′‐GGAGATTACTGCCCTGGCTCCTA‐3′ | 5′‐GACTCATCGTACTCCTGCTTGCTG‐3′ |
2.7. Immunofluorescence assay
The cells were cultured on the cover slips and treated with drugs for 6, 12 and 24 hours. And then cover the cells with ice‐cold 100% methanol for 15 minutes at −20°C. Remove the fixative and block the specimens in Blocking Buffer for 1 hour at room temperature. The cells were incubated with primary antibody LC3 (Cell Signaling Technology, USA) overnight at 4°C. The FITC‐conjugated secondary antibody (Proteintech, USA) was incubated for 1.5 hours at room temperature in dark. The nuclear was counterstained with DAPI (blue) (Beyotime Biotechnology, China) for 5 minutes in dark. All the specimen were observed in 100× oil immersion lens of Nikon Eclipse Ni epifluorescence microscope (Nikon Instruments Inc., Tokyo, Japan) equipped with a DS‐Qi2 camera immediately. The quantification of immunofluorescence was analysed using Image J to calculate as the average intensity of the fluorescent signal at three visual fields randomly selected in each section respectively.
2.8. Statistical analysis
All the data are expressed as mean ± standard deviation (SD). Statistical analysis was performed by GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA). Statistical comparisons were performed using t‐test, and two‐way ANOVA followed by Bonferroni post‐test. Value of P < .05 is considered to be statistically significant.
3. RESULTS
3.1. Changes in weight and Doppler ultrasonic indexes from different groups
Changes in weight of rats were calculated as the growth ratio of weight by the following equation:
Although the rat weights were substantially increased in both control group and MA group, but the growth ratios of weight were different between them after 6 weeks, and even two rats in MA group were dead at the 4th and 5th week respectively. Compared with control group, the growth ratio of weight was significantly lower in MA group (*** P < .001, vs control; Figure 1A).
Figure 1.
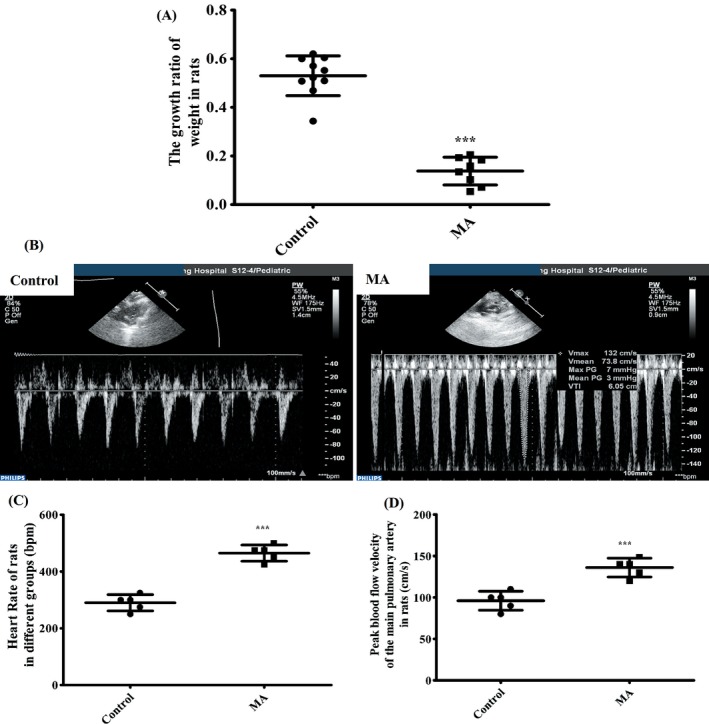
Alteration of weight and Doppler ultrasonic indexes from different groups in rats. A, The growth ratio of weight in rats in different groups (n = 8‐10). B, Doppler ultrasonic imaging in different groups (n = 5). C, Heart rate of rats in different groups (bpm). D, Peak blood flow velocity of the main pulmonary artery in rats (cm/s). Data were shown as means ± SD, ***P < .01 vs Control; Control, Control group; MA, methamphetamine group
Doppler ultrasonic imaging showed that compared with control group, heart rate (bpm) and peak blood flow velocity of the main pulmonary artery (cm/s) in MA group were significantly increased (Figure 1B). Heart rate was remarkably increased from 290 ± 12.75 bpm in the control group to 465 ± 12.75 bpm in MA group (***P < .001, vs control; Figure 1C). Peak blood flow velocity of the main pulmonary artery was significantly accelerated from 96 ± 5.1 cm/s in the control group to 136 ± 5.1 cm/s in MA group (***P < .001, vs control; Figure 1D).
3.2. Pulmonary injury induced by chronic exposure to MA
The results from H&E staining suggested that the pulmonary injury was significantly induced by chronic exposure to MA (Figure 2A). We observed that the lungs of rats were infiltrated by inflammatory cells in the MA group (black arrows), the lung parenchyma became more compact and the alveolar septum was thickened (***P < .001, vs control; Figure 2B), alveoli was fused and the number of alveolar sacs was significantly reduced in the MA group, compared with the control group (***P < .001, vs control; Figure 2C).
Figure 2.
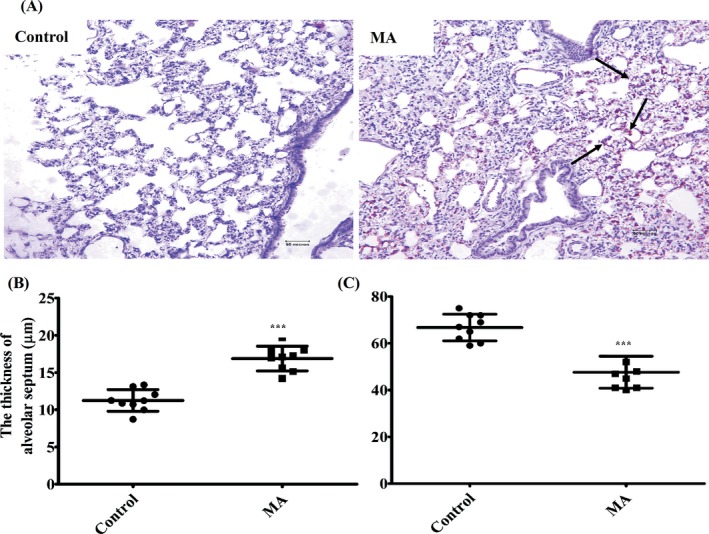
Microscopic analysis of pulmonary injury induced by chronic exposure of MA. A, MA‐induced pulmonary injury by H&E staining (Olympus BX 51, Japan, ×200): infiltration by inflammatory cells in the MA group, more compact lung parenchyma, thickened alveolar septum, alveoli fusion and reduced number of alveolar sacs in the MA group. B, The thickness of alveolar septum (μm) in different groups. C, The number of alveolar sacs in different groups. The quantification of Figure 2B,C was analysed in three visual fields randomly selected in each section, respectively. Data were shown as means ± SD, ***P < .01 vs Control; Control, Control group; MA, methamphetamine group
3.3. MA induced autophagy in alveolar epithelial cells
LC3 is the only known protein specifically associated with autophagosomes and becomes the marker of autophagy.12, 14 LC3 protein was detected with Immunofluorescence assay. It was found that compared with control group, LC3 fluorescent expression (green) was partly increased at 0.5 and 1 m mol L−1 MA, and was most densely expressed in cytoplasm at 5 m mol L−1 MA for 24 hours incubation with statistical significance (Figure 3A). Immunofluorescence results indicate that autophagy in alveolar epithelial cells was induced by MA with dose‐dependent and time‐dependent enhancement (Figure 3B).
Figure 3.
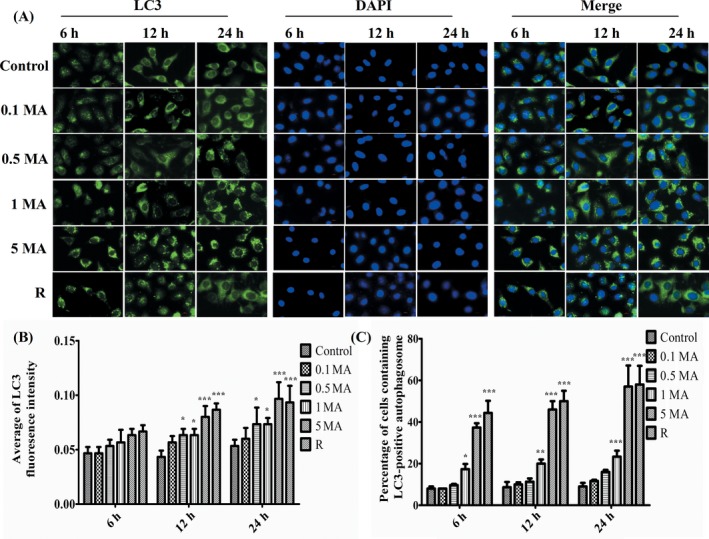
The effect of MA on autophagy marker LC3 in alveolar epithelial cells. A, Immunofluorescence assay of LC3 in alveolar epithelial cells (AECs). LC3 fluorescent expression was in cytoplasm (green), and the nuclear of AECs was counterstained by DAPI (blue). All the specimen were immediately observed in 100× oil immersion lens of Nikon Eclipse Ni epifluorescence microscope (Nikon Instruments Inc., Tokyo, Japan) equipped with a DS‐Qi2 camera. B, Average of LC3 fluorescent intensity in different groups. C, The percentage of cell containing LC3‐positive autophagosome. The quantification of Figure 3,C was analysed using Image J from three visual fields randomly selected in each section respectively. Data were shown as means ± SD, *P < .05, **P < .01, ***P < .01 vs Control group. Control, Control group; 0.1 MA, 0.1 m mol L−1 MA group; 0.5 MA, 0.5 m mol L−1 MA group; 1 MA, 1 m mol L−1 MA group; 5 MA: 5 m mol L−1 MA group; R, 1 mg/L rapamycin group; MA, methamphetamine
Quantitative analysis of the number of cells with LC3‐positive autophagosome structures relative to the total number of cells is expressed as the percentage of cells containing LC3‐positive autophagosome. Three areas of microscopic images were captured and analysed by counting the stained cells. Figure 3C showed that the percentage of cells containing LC3‐positive autophagosome is significantly increased at 1 and 5 m mol L−1 MA, and gradually increased from 6 to 24 hours in comparison with control group.
3.4. The effect of MA on mTOR phosphorylation
The protein expression of mTOR is noticeably decreased at 1 and 5 m mol L−1 MA for 6, 12 and 24 hours, compared with control group as shown in Figure 4A,B. However, it was found that the expression of p‐mTOR was gradually augmented in correlation with time and was significantly inhibited with the increasing concentration of MA in comparison with control group (Figure 4C). Therefore, the phosphorylation of mTOR was dose dependently suppressed by MA to inactivate mTOR in alveolar epithelial cells.
Figure 4.
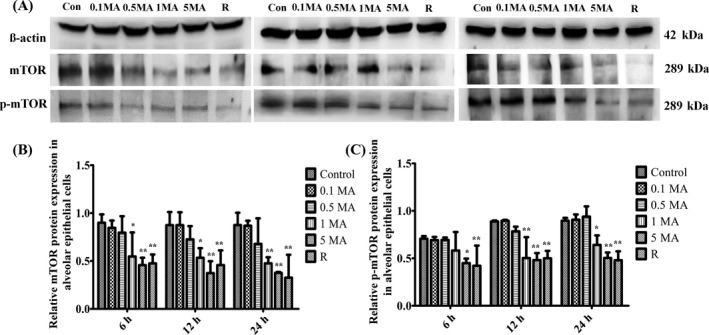
The effect of MA on mTOR phosphorylation. A, Western blot analysis of mTOR and p‐mTOR in different groups. B, Relative mTOR protein expression in alveolar epithelial cells. C, Relative p‐mTOR protein expression in alveolar epithelial cells. Data were shown as means ± SD (n = 5), * P < .05, ** P < .01, *** P < .01 vs Control group. Control, Control group; 0.1 MA, 0.1 m mol L−1 MA group; 0.5 MA, 0.5 m mol L−1 MA group; 1 MA, 1 m mol L−1 MA group; 5 MA: 5 m mol L−1 MA group; R, 1 mg/L rapamycin group; MA, methamphetamine
3.5. Activation of autophagy regulatory proteins by MA
LC3 and Beclin‐1 in the downstream of mTOR are the autophagy regulatory proteins.9, 10 The protein and mRNA levels of LC3 and Beclin‐1 in alveolar epithelial cells were detected by western blot and Real‐time PCR respectively.
Western blot analysis showed that MA improved the transformation from LC3 I to LC3 II, and that LC3 II levels were significantly higher in 5 m mol L−1 MA group than those in other groups (Figure 5A). Compared with Control group, the ratio of LC3 II and LC3 I (LC3 II/LC3 I) was evidently increased at 5 m mol L−1 MA for 6, 12 and 24 hours (Figure 5B). LC3 II/LC3 I was also dose dependently increased by MA for 24 hours with statistical significance (Figure 5B). The LC3 mRNA was obviously increased at 5 m mol L−1 MA for 12 and 24 hours. Particularly, for 24 hours, LC3 mRNA levels were upregulated in dose‐dependent manner. (Figure 5D), which was in consistency with the protein level of LC3.
Figure 5.
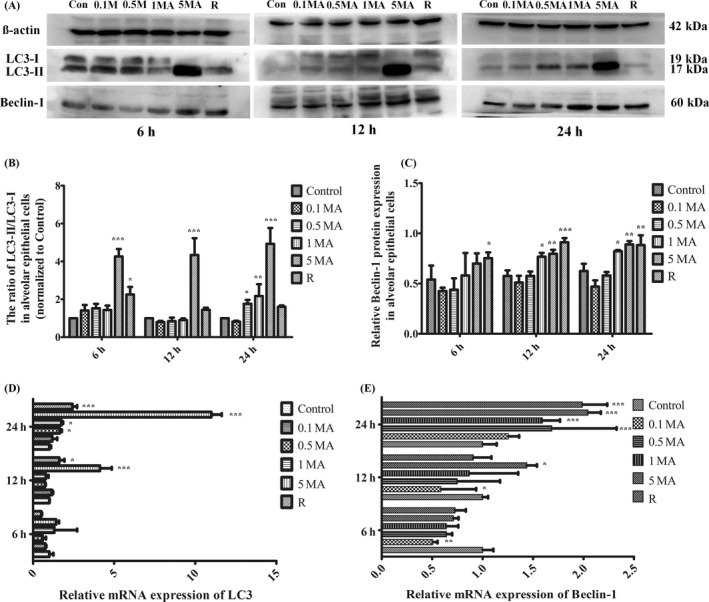
Activation of autophagic regulator proteins by MA. A, Western blot analysis of LC3 and Beclin‐1 in different groups. B, The ratio of LC3‐ II/LC3‐ I in alveolar epithelial cells (normalized to control). C, Relative Beclin‐1 protein expression in alveolar epithelial cells. D, Relative mRNA expression of LC3 by real‐time PCR. E, Relative mRNA expression of Beclin‐1 by real‐time PCR. Data were shown as means ± SD (n = 5), * P < .05, ** P < .01, *** P < .01 vs Control group. Control, Control group; 0.1 MA, 0.1 m mol L−1 MA group; 0.5 MA, 0.5 m mol L−1 MA group; 1 MA, 1 m mol L−1 MA group; 5 MA: 5 m mol L−1 MA group; R, 1 mg/L rapamycin group; MA, methamphetamine
Beclin‐1 is another crucial regulator of the autophagy. It was found that compared with control group, Beclin‐1 protein was slightly decreased at 0.1 and 0.5 m mol L−1 without statistical significance, but was significantly increased at 1 and 5 m mol L−1 (Figure 5A). MA of 1 and 5 m mol L−1 remarkably induced overexpression of Beclin‐1 for 12 and 24 hours, which indicated that Beclin‐1 protein was dose‐dependently and time‐dependently increased by MA (Figure 5C). Figure 5E showed that the mRNA levels of Beclin‐1 were lower in 0.1, 0.5 and 1 m mol L−1 MA groups than those in control group for 6 and 12 hours. However, 5 m mol L−1 MA significantly induced Beclin‐1 mRNA for 12 and 24 hours. Meanwhile, for 24 hours, the mRNA levels of Beclin‐1 were increased dose dependently by MA.
3.6. Increasing Beclin‐1 suppressed Bcl‐2‐mediated anti‐apoptosis
As shown in Figure 5, the overexpression of Beclin‐1 stimulated by MA induced the autophagy in alveolar epithelial cells, but in order to investigate if the autophagy‐stimulatory effect can cause the apoptosis in alveolar epithelial cells, anti‐apoptotic factor Bcl‐2, apoptotic factor BAX and apoptotic executive protein Caspase 3 were detected by western blot (Figure 6A).
Figure 6.
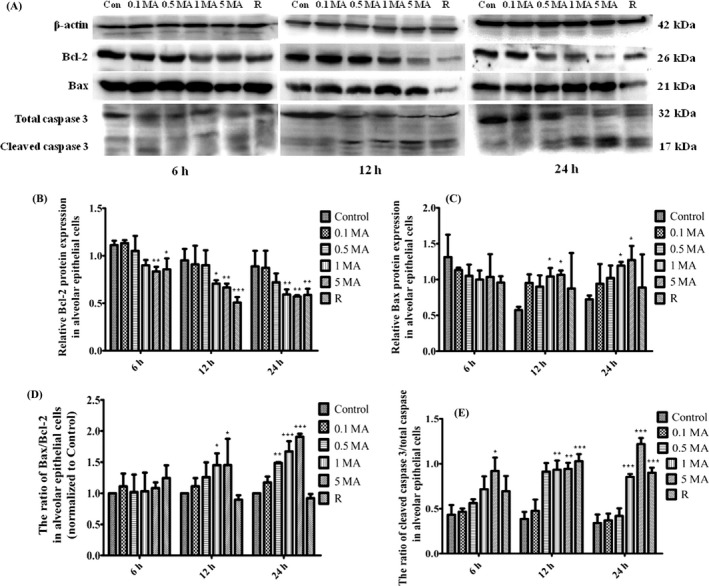
The effect of MA on apoptosis in alveolar epithelial cells. A, Western blot analysis of anti‐apoptotic and apoptotic‐relevant factors. B, Relative anti‐apoptotic protein Bcl‐2 expression in alveolar epithelial cells. C, Relative apoptotic protein Bax expression in alveolar epithelial cells. D, The ratio of Bax/Bcl‐2 in alveolar epithelial cells (normalized to control). E, The ratio of cleaved Caspase 3/total Caspase 3 in alveolar epithelial cells. Data were shown as means ± SD (n = 5), * P < .05, ** P < .01, *** P < .01 vs Control group. Control, Control group; 0.1 MA, 0.1 m mol L−1 MA group; 0.5 MA, 0.5 m mol L−1 MA group; 1 MA, 1 m mol L−1 MA group; 5 MA: 5 m mol L−1 MA group; R, 1 mg/L rapamycin group; MA, methamphetamine
Bcl‐2, an anti‐apoptotic protein, can interact with Beclin‐1 to form complex to represent a crosstalk between the autophagic and apoptotic mechanism.17, 18 It was found in our current study that Beclin1 overexpression in protein and mRNA levels when MA stimulated the autophagy in alveolar epithelial cells, which is probably associated with that MA disrupted the interaction between Beclin‐1 and Bcl‐2 and then released Beclin‐1. Western blot analysis suggested that Bcl‐2 protein level was time dependently downregulated at 5 m mol L−1 MA for 6, 12 and 24 hours, and that it was significantly suppressed at 1 m mol L−1 MA for 12 and 24 hours (Figure 6B). The reduced Bcl‐2 may be associated with depletion of Bcl‐2 caused by Beclin‐1 overexpression‐related autophagy.17 Our results also showed that apoptotic protein Bax level was not obviously changed after MA stimulation for 6 hours, but that, for 12 and 24 hours, Bax protein expression was heightened dose dependently by MA, particularly, in 1 and 5 m mol L−1 groups, compared with control group (Figure 6C). As shown in Figure 6D that the ratio between Bax and Bcl‐2 was partly increased at 1 and 5 m mol L−1 MA for 12 hours, especially, for 24 hours, Bax/Bcl‐2 level was significantly elevated dose dependently, compared with control group. Apoptotic executor Caspase 3 is also obviously elevated at 1 and 5 m mol L−1 MA for 12 and 24 hours in comparison with control group (Figure 6E).
4. DISCUSSION
Chronic exposure of MA resulted in the slower growth of weight and in the higher heart rate and peak blood flow velocity of rats. The toxicity of lung tissue in rats was included by infiltration of inflammatory cells, more compact lung parenchyma, thickened alveolar septum, alveoli fusion and reduction in the number of alveolar sac. In alveolar epithelial cells, the autophagy marker LC3 was overexpressed and per cent of cells containing LC3‐positive autophagosome was significantly increased, which suggested autophagy was induced by MA. The phosphorylation of mTOR was dose dependently depressed by MA at 24 hours to inactivate mTOR, and then elicited its downstream autophagic regulator proteins LC3 and Beclin‐1. MA accelerated the transformation from LC3 I to LC3 II, and upregulated the protein levels and mRNA level of LC3 and Beclin‐1. Increasing Beclin‐1‐autophagy suppressed Bcl‐2‐mediated anti‐apoptosis by decreasing Bcl‐2 and increasing Bax, Bax/Bcl‐2 and cleaved Caspase 3, especially stimulated by 12 or 24 hours exposure to MA. The above results suggested that sustained autophagy was induced by long‐term exposure to MA and that the increased Beclin‐1 autophagy initiated apoptosis in alveolar epithelial cells. Therefore, concurrence of autophagy with apoptosis in alveolar epithelial cells contributes to chronic pulmonary toxicity induced by MA.
Methamphetamine is an addictive stimulant, which caused the disorder of nervous system, cardiovascular system and endocrine system, and even individual death.32, 33, 34 There is a growing evidence shown that pulmonary toxicity is one of the main causes of death by MA addiction. In our rat models, pulmonary toxicity was prominent after 6‐week exposure to MA. In MA group, rats had a slower growth in weight, and even individual death. MA caused the acceleration of heart rate and peak blood flow velocity of the main pulmonary artery. The velocity of blood flow can change with respiration.35 Alteration of respiratory blood flow velocity is beneficial to assess the pulmonary injury.35, 36 In addition, our morphological analysis showed that the lungs of rats were infiltrated by inflammatory cells, the lung parenchyma became more compact, the alveolar septum was thickened, alveoli was fused and the number of alveolar sacs was significantly reduced in the MA group, compared with the control group, which hinted that long‐term exposed to MA induces chronic pulmonary toxicity.
Pulmonary epithelium acts as a barrier to limit the access of various toxic to the respiratory system to maintain lung homeostasis, so that alveolar epithelial cells are also considered to take effects on the defence and infection.4 Hence, it is the key to study the mechanism of MA‐induced pulmonary toxicity using alveolar epithelial cells. In this study, it was found that in alveolar epithelial cells, autophagy marker LC3 was overexpressed and per cent of cells containing LC3‐positive autophagosome was significantly increased, which suggested autophagy in alveolar epithelial cells was involved in MA‐induced pulmonary toxicity. Autophagy is a vacuolar process of cytoplasmic degradation by lysosome to maintain cell homeostasis and organelle self‐renew. Some studies were proved that autophagy can be induced through activating some protein kinases and mTOR‐Beclin‐1 pathway.37, 38 Autophagy is suppressed by the factor mTOR, thus, the mTOR inhibitor rapamycin‐induced autophagy.39 It was reported that the substrates of mTOR are involved in the regulation of autophagy and formation of autophagosome.40, 41, 42 In this study, it was found that mTOR expression was noticeably decreased in 1 and 5 m mol L−1 MA groups at 6, 12 and 24 hours, and that p‐mTOR expression was significantly inhibited with the increasing concentration of MA in comparison with control group. Therefore, the phosphorylation of mTOR was dose dependently depressed by MA and inactivate mTOR to induce autophagy in alveolar epithelial cells.
LC3 and Beclin‐1 in the downstream of mTOR are the autophagy regulatory proteins.9, 10 LC3 is the marker of autophagy and the modification of LC3 is necessary for autophagosome formation.12, 13 By multiple modification, LC3 I is activated to be transformed into LC3 II.43 LC3 II is located on the membrane of autophagosome, and is degraded after the fusion of autophagosomes with lysosomes.13 Thus, LC3 II become the marker of autophagosome in mammals.12 Beclin‐1 also plays a crucial role in autophagic process, such as autophagosome formation and autophagy protein localization.16, 44 When the autophagy is evoked, Beclin1 is overexpressed and interacted with PI3K to extend lipid membrane and to facilitate the maturation of autophagosome.44 Results from this study showed that MA increased the transformation from LC3 I to LC3 II, and that LC3 II levels were significantly higher in 5 m mol L−1 MA group than those in other groups. Compared with Control group, the ratio of LC3 II /LC3 I is also dose dependently increased by MA in 24 hours with statistical significance. The LC3 mRNA was obviously increased in dose‐dependent manner, which was in consistency with the protein level of LC3 protein. In addition, Beclin‐1 protein and mRNA were both increased by long‐term stimulation of MA. Taken together with suppressed phosphorylation of mTOR, MA‐induced autophagy in alveolar epithelial cells is probably through mTOR/LC3/Beclin‐1 signalling.
The relation between autophagy and apoptosis remains debatable, but the interaction between them is closely associated with many diseases.45 The autophagy protein, Beclin‐1, can interact with anti‐apoptotic protein Bcl‐2 or Bcl‐XL to form complex to inhibit autophagy.46 When the autophagy appears, Beclin‐1 is dissociated with Bcl‐2 to take a part in the formation of autophagasomes.15 Our results in this study showed that Beclin‐1 was overexpressed but that anti‐apoptotic Bcl‐2 expression is decreased by MA, which is probably associated with that MA disrupted the interaction between Beclin‐1 and Bcl‐2, released Beclin‐1 and then depleted Bcl‐2.17 Meanwhile, our results also showed that compared with control group, apoptotic protein Bax level was enhanced dose dependently by MA, and that the ratio of Bax/Bcl‐2 level was significantly elevated dose dependently in 1 and 5 m mol L−1 groups. Active Caspase 3, cleaved Caspase 3, is also markedly elevated at 1 and 5 m mol L−1 MA for 12 and 24 hours in comparison with control group. Taken together, it was indicated that increasing Beclin‐1 autophagy suppressed Bcl‐2‐mediated anti‐apoptosis.
In summary, sustained autophagy was induced by long‐term exposure to MA. And the increased Beclin‐1 autophagy initiated apoptosis in alveolar epithelial cells, which indicated the concurrence of autophagy with apoptosis in alveolar epithelial cells contributes to chronic pulmonary toxicity induced by MA.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
ACKNOWLEDGEMENTS
Authors thank to Zai‐Xing Chen and Wei‐Fan Yao for providing technical assistance. This work was supported by National Natural Science Foundation of China [no. 81503058] and Natural Science foundation of Liaoning Province [no. 2014021065]. Study design: Yun and Yu‐Han; Contribution of new reagents or analytical tools: Ming and Bin; Data Collection: Yu‐Han, Li‐Ye, Bin, Mei‐Jia, Xin and Lin; Data analysis: Yun and Yu‐Han; Manuscript preparation: Yun.
Wang Y, Gu Y‐H, Liang L‐Y, et al. Concurrence of autophagy with apoptosis in alveolar epithelial cells contributes to chronic pulmonary toxicity induced by methamphetamine. Cell Prolif. 2018;51:e12476 10.1111/cpr.12476
REFERENCES
- 1. Yu S, Zhu L, Shen Q, Bai X, Di X. Recent advances in methamphetamine neurotoxicity mechanisms and its molecular pathophysiology. Behav Neurol. 2015;2015:103969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang Y, Gu YH, Liu M, Bai Y, Liang LY, Wang HL. TBHQ alleviated endoplasmic reticulum stress‐apoptosis and oxidative stress by PERK‐Nrf2 crosstalk in methamphetamine‐induced chronic pulmonary toxicity. Oxid Med Cell Longev. 2017;2017:4310475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y, Gu YH, Liu M, Bai Y, Wang HL. Fluoxetine protects against methamphetamine‐induced lung inflammation by suppressing oxidative stress through the SERT/p38 MAPK/Nrf2 pathway in rats. Mol Med Rep. 2017;15:673‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang L, Chen X, Simet SM, et al. Reactive oxygen species/hypoxia‐inducible factor‐1a/platelet‐derived growth factor‐BB autocrine loop contributes to cocaine‐mediated alveolar epithelial barrier damage. Am J Resp Cell Mol. 2016;55:736‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383‐1435. [DOI] [PubMed] [Google Scholar]
- 6. de la Vega MR, Dodson M, Gross C, et al. Role of Nrf2 and autophagy in acute lung injury. Curr Pharmacol Rep. 2016;2:91‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu G, Pei F, Yang F, et al. Role of autophagy and apoptosis in non‐small‐cell lung cancer. Int J Mol Sci. 2017;18:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Levine B, Klionsky DJ. Development by self‐digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463‐477. [DOI] [PubMed] [Google Scholar]
- 10. Ryter SW, Choi AM. Autophagy in the lung. Proc Am Thorac Soc. 2010;7:13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee YK, Lee JA. Role of the mammalian ATG8/LC3 family in autophagy: differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016;49:424‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503‐2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720‐5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1‐dependent autophagosome synthesis: an effect rescued by Bcl‐xL. Cell Death Differ. 2010;17:268‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu H, He L. Beclin 1 biology and its role in heart disease. Curr Cardiol Rev. 2015;11:229‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maejima Y, Isobe M, Sadoshima J. Regulation of autophagy by Beclin 1 in the heart. J Mol Cell Cardiol. 2016;95:19‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl‐X (L) and a BH3‐like domain in Beclin‐1. EMBO J. 2007;26:2527‐2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672‐676. [DOI] [PubMed] [Google Scholar]
- 19. Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy‐specific factor for Beclin 1 and class III phosphatidylinositol 3‐kinase. Proc Natl Acad Sci. 2008;105:19211‐19216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu GY, Jiang XX, Zhu X, et al. ROS activates JNK‐mediated autophagy to counteract apoptosis in mouse mesenchymal stem cells in vitro. Acta Pharmacol Sin. 2015;36:1473‐1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine‐induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951‐8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fornai F, Lenzi P, Gesi M, et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J Neurochem. 2014;88:114‐123. [DOI] [PubMed] [Google Scholar]
- 23. Kanthasamy A, Anantharam V, Ali SF, Kanthasamy AG. Methamphetamine induces autophagy and apoptosis in a mesencephalic dopaminergic neuronal culture model: role of cathepsin‐D in methamphetamine‐induced apoptotic cell death. Ann N Y Acad Sci. 2006;1074:234‐244. [DOI] [PubMed] [Google Scholar]
- 24. Funakoshi‐Hirose I, Aki T, Unuma K, Funakoshi T, Noritake K, Uemura K. Distinct effects of methamphetamine on autophagy‐lysosome and ubiquitin‐proteasome systems in HL‐1 cultured mouse atrial cardiomyocytes. Toxicology. 2013;312:74‐82. [DOI] [PubMed] [Google Scholar]
- 25. Huang YN, Yang LY, Wang JY, Lai CC, Chiu CT, Wang JY. L‐Ascorbate protects against methamphetamine‐induced neurotoxicity of cortical cells via inhibiting oxidative stress, autophagy, and apoptosis. Mol Neurobiol. 2017;54:125‐136. [DOI] [PubMed] [Google Scholar]
- 26. Cao L, Fu M, Kumar S, Kumar A. Methamphetamine potentiates HIV‐1 gp120‐mediated autophagy via Beclin‐1 and Atg5/7 as a pro‐survival response in astrocytes. Cell Death Dis. 2016;7:e2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gu YH, Wang Y, Bai Y, Liu M, Wang HL. Endoplasmic reticulum stress and apoptosis via PERK‐eIF2α‐CHOP signaling in the methamphetamine‐induced chronic pulmonary injury. Environ Toxicol Pharmacol. 2017;49:194‐201. [DOI] [PubMed] [Google Scholar]
- 28. Xie WY, Zhou XD, Yang J, Chen LX, Ran DH. Inhibition of autophagy enhances heat‐induced apoptosis in human non‐small cell lung cancer cells through ER stress pathways. Arch Biochem Biophys. 2016;607:55‐66. [DOI] [PubMed] [Google Scholar]
- 29. Shen M, Wang L, Wang B, et al. Activation of volume‐sensitive outwardly rectifying chloride channel by ROS contributes to ER stress and cardiac contractile dysfunction: involvement of CHOP through Wnt. Cell Death Dis. 2014;5:e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo ML, Liao K, Periyasamy P, et al. Cocaine‐mediated microglial activation involves the ER stress‐autophagy axis. Autophagy. 2015;11:995‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Høyer‐Hansen M, Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14:1576‐1582. [DOI] [PubMed] [Google Scholar]
- 32. Koriem KM, Soliman RE. Chlorogenic and caftaric acids in liver toxicityand oxidative stress induced by methamphetamine. J Toxicol. 2014;2014:583494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramirez SH, Potula R, Fan S, et al. Methamphetamine disrupts blood‐brain barrier function by induction ofoxidative stress in brain endothelial cells. J Cereb Blood Flow Metab. 2009;29:1933‐1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quinton MS, Yamamoto BK. Causes and consequences of methamphetamine and MDMA toxicity. AAPS J. 2006;8:E337‐E347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kudo K, Terae S, Ishii A, et al. Physiologic change in flow velocity and direction of dural venous sinuses with respiration: MR venography and flow analysis. AJNR Am J Neuroradiol. 2004;25:551‐557. [PMC free article] [PubMed] [Google Scholar]
- 36. Halimi KE, Negadi M, Bouguetof H, Zemour L, Boumendil D, Chentouf MZ. Respiratory variations in aortic blood flow velocity and inferior vena cava diameter as predictors of fluid responsiveness in mechanically ventilated children using transthoracic echocardiography in a pediatric PICU. Crit Care. 2015;19(Suppl 1):S181. [Google Scholar]
- 37. Hernández‐Gea V, Hilscher M, Rozenfeld R, et al. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J Hepatol. 2013;59:98‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141‐150. [DOI] [PubMed] [Google Scholar]
- 39. Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963‐3966. [DOI] [PubMed] [Google Scholar]
- 40. Hosokawa N, Hara T, Kaizuka T, et al. Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981‐1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang M, Su L, Xiao Z, Liu X, Liu X. Methyl jasmonate induces apoptosis and pro‐apoptotic autophagy via the ROS pathway in human non‐small cell lung cancer. Am J Cancer Res. 2016;6:187‐199. [PMC free article] [PubMed] [Google Scholar]
- 42. Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1 ATG13 FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297‐12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanida I, Tanida‐Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiaeApg7p is a Protein‐activating enzyme for multiple substrates including human Apg12p, GATE‐16, GABARAP, and MAP–LC3. J Biol Chem. 2001;276:1701‐1706. [DOI] [PubMed] [Google Scholar]
- 44. Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3‐kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ou L, Lin S, Song B, Liu J, Lai R, Shao L. The mechanisms of graphene‐based materials‐induced programmed cell death: a review of apoptosis, autophagy, and programmed necrosis. Int J Nanomedicine. 2017;12:6633‐6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pattingre S, Tassa A, Qu X, et al. Bcl‐2 antiapoptotic proteins inhibit Beclin 1‐dependent autophagy. Cell. 2005;122:927‐939. [DOI] [PubMed] [Google Scholar]