

Abstract
Objectives
The transmembrane prostate androgen‐induced protein (TMEPAI) is aberrantly expressed in many cancer and plays a crucial role in tumourigenesis, which makes it a potential cancer therapeutic target for drug discovery.
Materials and methods
Here, we employed a firefly luciferase reporter driven by the TMEPAI gene promoter to screen for compound capable of inhibiting the expression of TMEPAI , and the effects of TMEPAI inhibitor on cancer cell proliferation were evaluated using the colony formation assay, cell cycle analysis, Ki‐67 immunofluorescence assay and EdU incorporation assay.
Results
2‐(2‐nitrobenzylidene) indolin‐3‐one (JHY‐A007‐50) was identified and shown to effectively inhibit the TMEPAI promoter activity. Further studies revealed that JHY‐A007‐50 specifically inhibited the expression of TMEPAI at both the mRNA and protein levels. Moreover, we found that JHY‐A007‐50 could inhibit cell proliferation and induce cell cycle arrest at the G1 phase. Our results showed that overexpression of TMEPAI decreased the inhibitory effects of JHY‐A007‐50 on cancer cell proliferation, and JHY‐A007‐50 did not affect the cell viability of HeLa cells knocked down of TMEPAI.
Conclusions
Taken together, these results suggest that compound JHY‐A007‐50 mediates the downregulation of TMEPAI expression and inhibits cell proliferation in cancer cells.
1. INTRODUCTION
Cancer is one of the major causes of death worldwide.1 Traditional cancer therapies consist of chemotherapy, radiotherapy and surgery. However, these traditional cancer therapies often have a poor prognosis and various side effects.2 Contrast to these traditional cancer therapies, molecular‐targeted therapy has many significant advances and becomes a critical anti‐cancer therapy. The target protein of molecular‐targeted therapy should be essential for the proliferation and survival of cancer cells, enable molecular‐targeted therapy has low cytotoxicity to normal cells.3
Transmembrane prostate androgen‐induced protein (TMEPAI), also named solid tumour‐associated gene 1 (STAG1) or PMEPA1 (prostate transmembrane protein androgen induced‐1), has important roles in tumourigenesis.4, 5 TMEPAI is highly expressed in different kinds of cancer, such as lung,6 breast,7 colon8 and renal cell cancer.9 Previous studies showed that expression of TMEPAI promotes PC‐3 prostate cancer cell proliferation,10 and depletion; the expression of TMEPAI restrains cell growth, migration as well as the invasion of breast cancer cell MDA‐MB‐231.7 Inhibition of the expression of TMEPAI also significantly decreases the growth of tumour xenograft.6, 7, 10 Our studies showed that the expression of TMEPAI promoted lung cancer cell proliferation, migration and invasion, and studies using nude mice models also demonstrated that the expression of TMEPAI promoted cancer growth. Our mechanistic study showed that TMEPAI regulates TGF‐β signalling pathway through the modulation of TβRI protein levels by promoting its lysosomal degradation.11 Our results also showed that expression of TMEPAI improves lysosomal stability against stress‐induced lysosomal rupture and promotes autophagy.12 Moreover, our study indicated that CI‐M6PR and clathrin mediated TMEPAI transport from the Golgi into the endolysosomal pathway, and ubiquitination modification of TMEPAI is an important signal for TMEPAI lysosomal trafficking.13 Our recent research found that the sequence between −298 and +24 includes the basal promoter activity for TMEPAI. Furthermore, Sp1 promotes TMEPAI expression and contributes to cell proliferation.14 All these studies indicated that TMEPAI could be a novel anti‐cancer drug screening target.
In the present study, a firefly luciferase reporter screening system driven by TMEPAI promoter was established, and purified compounds were screened. It was found that JHY‐A007‐50 could effectively inhibit the expression of TMEPAI. Further results have shown that JHY‐A007‐50 could induce G1 phase arrest in cancer cells that expressed high levels of TMEPAI. These results have demonstrated that TMEPAI is a novel anti‐cancer therapeutic target, and JHY‐A007‐50 could be a potential anti‐tumour drug for cancer expressing high levels of TMEPAI.
2. MATERIALS AND METHODS
2.1. Materials and antibodies
All materials were purchased from Sigma (St. Louis, MO, USA) unless otherwise stated. Antibodies were purchased from the following sources: Ki‐67, CDK2, Cyclin E1, p53, Legumain, SM22, Histone and β‐actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Sp1 (Proteintech Group, Rosemont, IL, USA). Fluorophore (D&S488 and D&R647) and HRP‐conjugated secondary antibodies were obtained from Invitrogen (Carlsbad, CA, USA).
2.2. Cell culture
HeLa, MGC‐803, HepG2, L02 cells were grown at 37°C and 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM) medium, supplemented with 10% foetal bovine serum (FBS). All the cells used for experiments were less than 15 generations. The cell line was obtained from the Cell Resource Center, Peking Union Medical College (which is the headquarters of National Infrastructure of Cell Line Resource, NSTI). The cell line was checked free of mycoplasma contamination by PCR and culture. Its species origin was confirmed with PCR. The identity of the cell line was authenticated with STR profiling (FBI, CODIS). All the results can be viewed on the website. (http://cellresource.cn)
2.3. Screening of potential inhibitors of TMEPAI expression
HeLa cells were plated at 2 × 106 cells/well in a 6‐cm‐plate. After 24 h, cells were transfected with 2 μg of pGL3‐TMEPAI promoter plasmid or 2 μg of pGL3 vector plasmid per well plus 0.1 μg of pCMV‐β‐galactosidase plasmid. After 24 h, the transfected cells were plated onto the 96‐well plates 5 × 103 cells/well. Cells were treated with compounds at concentrations of 5 μM in DMEM containing 3% FBS for 24 h, and luciferase activity was detected as described.14
2.4. MTT assay
Cell viability was measured using the MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐ diphenyltetrazolium bromide; thiazolyl blue) assay. Cells were plated (5000 cells/well) with 200 μL of cell growth medium in 96‐well plates and cultured for the indicated time. MTT was added at a final concentration of 0.5 mg/mL in PBS and incubated for 4 h at 37°C. The medium was then removed and the remaining blue precipitate was solubilized in dimethylsulfoxide (DMSO). The absorbance was read at 570 nm in a microplate reader. All the MTT data were averaged from 3 independent experiments. The IC50 value was assessed using probit regression analysis by spss statistical software.
2.5. Western blot
Cultured cells were washed twice using PBS before addition of ice cold lysis buffer (50 mM Tris‐HCl pH 7.5, 150 mM NaCl, 1 mM NaF, 1 mM phenylmethylsulphonyl fluoride, 4 mg/mL leupeptin and 1 mg/mL aprotinin, and 1% Nonidet P‐40). Bradford method was used to measure the protein level, and equal amounts of protein were loaded into each well and separated by 7%, 10% or 12% SDS‐PAGE gel, followed by transfer to PVDF membranes. These PVDF membranes were blocked in PBST buffer containing 5% non‐fat milk for 1 h at room temperature. Blots were then incubated at 4°C with primary antibodies overnight. Secondary antibody incubation was carried out for 1 h at room temperature. Finally, Odyssey infrared laser imaging system (LI‐COR Biosciences, Lincoln, NE, USA) was used to image the results.
2.6. RNA extract and RT‐PCR
Total RNA was isolated from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. RNA was quantified by measuring the absorbance (A260 nm). One microgram of RNA was reverse transcribed with oligo (dT) primers using a reverse transcription system. The single‐stranded cDNA was amplified by PCR using the specific primers. PCR was performed for 30 cycles (each cycle consisting of 94°C for 30 s, 54°C for 30 s and 72°C for 30 s). The PCR products were analysed by electrophoresis on a 1% agarose gel. Primer sequences used were shown in Table 1.
Table 1.
Primer sequences used in the RT‐PCR assay
| Primer | Sequence (5′–3′) |
|---|---|
| TMEPAI‐For | GGAATTCATGCACCGCTTGATGGGGGTC |
| TMEPAI‐Rev | CGGGATCCTTACTTGTCGTCATCGTCTTTG |
| p53‐For | CAGCCAAGTCTGTGACTTGCACGTAC |
| p53‐Rev | CTATGTCGAAAAGTGTTTCTGTCATC |
| p57‐For | AGATCAGCGCCTGAGAAGTC |
| p57‐Rev | GGGACCAGTGTACCTTCTCG |
| GAPDH‐For | CTTTGGTATCGTGGAAGGA |
| GAPDH‐Rev | CACCCTGTTGCTGTAGCC |
2.7. Chromatin immunoprecipitation assays
HeLa cells were cultured in the 10‐cm dishes and treated with 4 μM JHY‐A007‐50 for 24 h, with DMSO as the negative control. Cells were processed for ChIP assay as described previously.14 Briefly, the IgG or anti‐Sp1 was used for the immunoprecipitation. Then, the immunoprecipitated DNAs were purified and used for PCR. The primers for detection of TMEPAI promoter DNA sequence containing Sp1‐binding sites are as follows: forward, 5′‐CGGGTCTACGTGGGCCGCCTAGC‐3′; reverse, 5′‐AGGTTCCCCCGCACCCCCTCC‐3′. Analysis of the PCR products was performed on a 1.2% agarose gel electrophoresis.
2.8. Colony formation assay
Cells were plated (800 cells/well) in triplicate with 4 μM compound JHY‐A007‐50, in six‐well plates. After 20 d, colonies were stained with 5 mg/mL MTT and photographed.14
2.9. Cell cycle analysis
Cell cycle analysis was performed using Cell Cycle Kit (Beyotime, China) following the manufacturer's instructions. The percentages of cells in the different cell cycle phases were measured using a flow cytometer.
2.10. Immunofluorescence assay
Cells were cultured overnight in 96‐well plate and then treated with the compound in 3% FBS medium for 24 h. Cells were fixed in 4% PFA (paraformaldehyde solution) in PBS for 30 min and then washed 3 times using PBS. Cells were blocked with 5% BSA for 30 min at room temperature and probed overnight with Ki‐67 antibodies at 4°C. After washing 3 times with PBS, cells were incubated with fluorochrome‐conjugated secondary antibody for 1 h at room temperature in the dark. Labelled cells were then rinsed with PBS and analysed under a Nikon fluorescence microscope.
2.11. EdU incorporation assay
DNA synthesis was detected by measuring the incorporation of EdU into newly synthesized DNA strands. Cells were plated into 96‐well plates for 24 h and then treated with JHY‐A007‐50 in 3% FBS medium. After incubation for the 24 h, cells were labelled with EdU labelling reagent according to the manufacturer's protocol.
2.12. RNA interference
Lentiviral vectors expressing TMEPAI shRNAs (TRC332: 5′‐CCGGATCACGGAGCTGGAGTTTGTTCTCGAGAACAAACTCCAGCTCCGTGATTTTTT‐3′ or the non‐target shRNA control vector (SHC002) was obtained from Sigma and the knockdown level was tested by Sigma. The TMEPAI shRNA has been tested to inhibit the expression of TMEPAI specifically.11 The lentiviruses were obtained according to the manufacturers’ protocol. Cells were infected with lentiviruses for 3 d before experiments were performed.
2.13. Statistical analysis
All the experiments were repeated at least 3 times, the average values used as results and a set of representative figures shown as illustration. Statistical analysis was performed using the χ 2 test or Fisher's exact test and Spearman's rank correlation coefficient analysis. P < .05 indicated that the difference was significant, and P < .01 indicated that the difference was highly significant.
3. RESULTS
3.1. Compound JHY‐A007‐50 inhibits TMEPAI promoter activity
It has been shown that TMEPAI is highly expressed in many kinds of cancer cells and plays an important role in tumourigenesis, which makes it a potential drug screening target for cancer therapy. To obtain potential candidate inhibitors capable of inhibiting the expression of TMEPAI, a luciferase assay system driven by TMEPAI promoter was established and screened against a compound library (Figure 1A). JHY‐A007‐89 (see structure in Figure 1B) was found to have significant inhibitory effects on the promoter activity of TMEPAI, indicating that JHY‐A007‐89 may be a potential inhibitor of TMEPAI expression. Further analysis also confirmed that JHY‐A007‐89 markedly inhibited the TMEPAI luciferase reporter activity in a dose‐dependent manner (Figure 1C).
Figure 1.
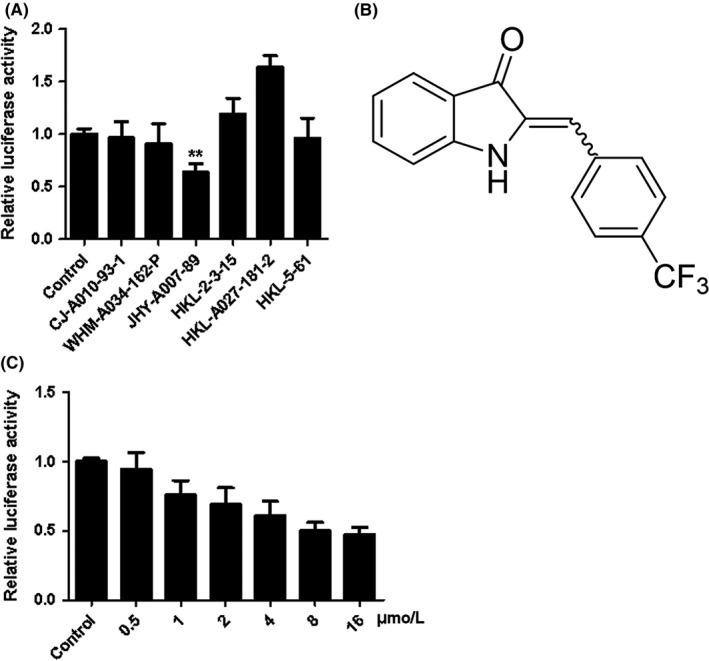
JHY‐A007‐89 inhibits TMEPAI promoter activity. (A) HeLa cells were transfected with pGL3‐TMEPAI promoter plasmid for 24 h, and then treated with the compounds (5 μM each). Cells were treated with 0.1% (v/v) DMSO as the negative control. Results are expressed as the fold activity over the activity of the negative control. The screening assay was carried out in triplicate. Partial data are shown (**P < .01 compared with negative control). (B) Structure of JHY‐A007‐89 (C) HeLa cells were transfected with a pGL3‐TMEPAI promoter plasmid for 24 h, and then were treated with compound JHY‐A007‐89 at the indicated dose
To explore the possibility of obtaining more effective compounds, various derivatives of the lead compound JHY‐A007‐89 (Table S1) were synthesized and their inhibitory effects on TMEPAI gene promoter activity were detected in HeLa cells at a concentration of 4 μM (Figure 2A). It was found that JHY‐A007‐50 (see structure in Figure 2B) could inhibit TMEPAI promoter activity more potently, and the inhibitory concentration (IC50) value was 3.5 μmol/L (Figure 2C).
Figure 2.
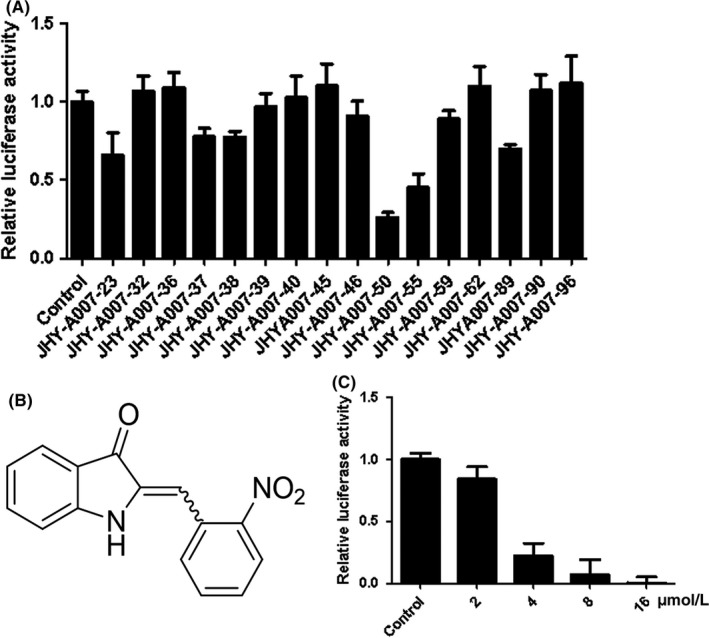
JHY‐A007‐50 inhibits TMEPAI promoter activity. (A) HeLa cells were transfected with pGL3‐TMEPAI promoter plasmid for 24 h, and then were treated with the derivatives of compound JHY‐A007‐89 (5 μM). Cells were treated with 0.1% (v/v) DMSO as the negative control. Results are expressed as the fold activity over the activity of the negative control. Data are presented as the mean ± SD (n = 3). (B) Structure of JHY‐A007‐50 (C) HeLa cells were transfected with a pGL3‐TMEPAI promoter plasmid for 24 h, and then were treated with compound JHY‐A007‐50 at the indicated dose
3.2. Compound JHY‐A007‐50 downregulates TMEPAI expression in cancer
To confirm the inhibitory effect of JHY‐A007‐50 on the expression of TMEPAI, the levels of TMEPAI mRNA and protein in HeLa cells were analysed. The results showed that JHY‐A007‐50 downregulated the expression of both TMEPAI mRNA (Figure 3A,B) and protein (Figure 3C,D). Similarly, JHY‐A007‐50 could inhibit the expression of TMEPAI in MCF‐7 and MGC‐803 cells (Figure 3E,F), which express high levels of endogenous TMEPAI.11, 12 Moreover, we found that JHY‐A007‐50 did not affect the luciferase activity driven by SM22 (smooth muscle 22) promoter (Figure 3G). Legumain (LGMN) is highly expressed in various cancers, and it plays an important role in tumourigenesis.15, 16 Previous result showed that the expression of Legumain is regulated by p53.17 Furthermore, the putative transcriptional factors of LGMN were analysed with TRANSFAC database (http://www.gene-regulation.com) and sabiosciences (http://www.sabiosciences.com), and the results showed that Sp1 is not a transcription factor of LGMN. The expression of LGMN and SM22 in HeLa cells was detected after cells treated with JHY‐A007‐50. As shown in Figure 3H, JHY‐A007‐50 did not affect the expression of LGMN and SM22 (Figure 3H). These results suggest that JHY‐A007‐50 is an efficient and specific inhibitor of TMEPAI expression in cancer cells.
Figure 3.
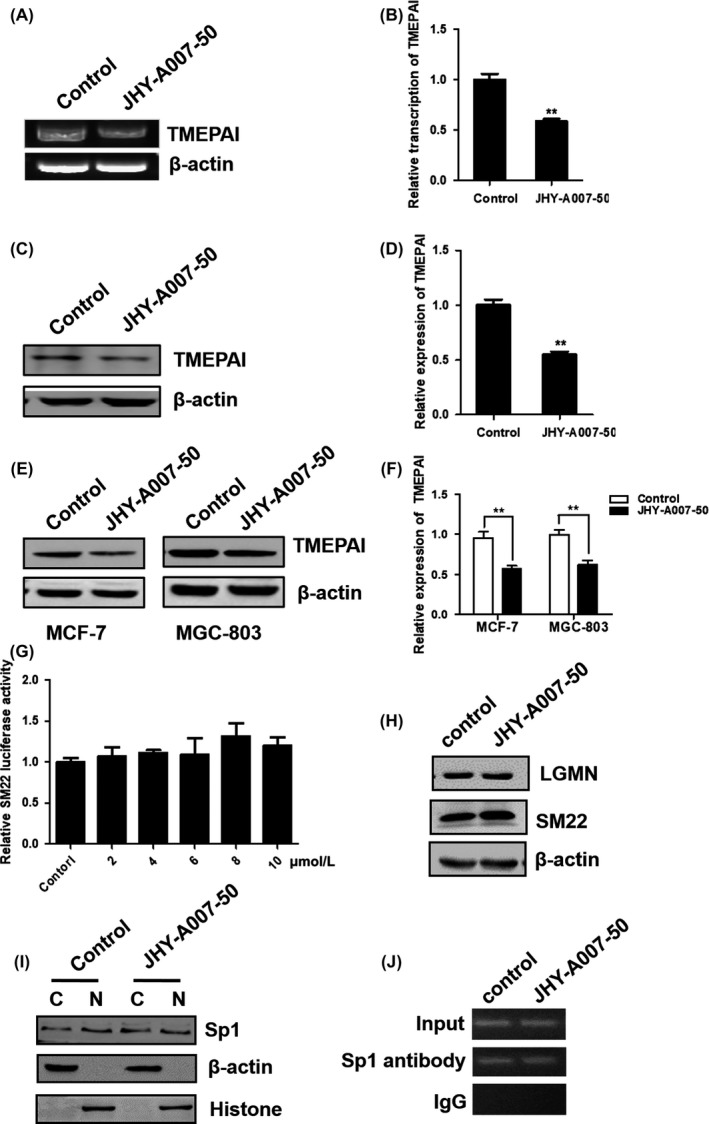
Compound JHY‐A007‐50 downregulates TMEPAI expression in cancer cells. (A) HeLa cells were treated with 4 μM JHY‐A007‐50 for 24 h, with 0.1% (v/v) DMSO being used as a negative control. TMEPAI mRNA levels were analysed by RT‐PCR. β‐actin was used as an internal control to check the efficiency of cDNA synthesis and PCR amplification. (B) TMEPAI mRNA levels were analysed by RT‐PCR, and quantified using Image J software (**P < .01). HeLa (C), MCF‐7 and MGC‐803 (E) cells were treated with 4 μM of JHY‐A007‐50 for 24 h and 0.1% (v/v) DMSO was used as a negative control. Cells were then lysed and TMEPAI expression was analysed by Western blotting. (D) and (F) TMEPAI protein levels were analysed by Western blotting, and quantified using Image J software (**P < .01). (G) HeLa cells were transfected with pGL3‐SM22 promoter plasmid for 24 h, and then were treated with compound JHY‐A007‐50 (5 μM) for 24 h. Cells treated with 0.1% (v/v) DMSO as the negative control. Results are expressed as the fold activity over the activity of the negative control. Data are presented as the mean ± SD (n = 3). (H) HeLa cells were treated with 4 μM of JHY‐A007‐50 for 24 h, and 0.1% (v/v) DMSO was used as a negative control. Cells were then lysed, LGMN and SM22 expression were analysed by Western blot. (I) HeLa cells were treated with 4 μM of JHY‐A007‐50 for 24 h and 0.1% (v/v) DMSO was used as a negative control. The protein level of Sp1 in cytoplasm (C) and nucleus (N) was examined, with β‐actin and Histone as loading control, respectively. (J) Chromatin fragments isolated from HeLa cells treating with 4 μM JHY‐A007‐50 (with DMSO as the negative control) were subjected to immunoprecipitation with an Sp1 antibody and IgG as the negative control, binding of Sp1 to TMEPAI promoter was detected by PCR
Our previous study showed that transcription factor Sp1 could promote the expression of TMEPAI.14 To test whether JHY‐A007‐50 targets Sp1 to regulate the expression of TMEPAI, the protein level of Sp1 in cytoplasm and nucleus with or without this compound treatment was examined. The result showed that the expression or the nucleus translocation of Sp1 was not altered by treating with JHY‐A007‐50 (Figure 3I), which indicates Sp1 is not the target of JHY‐A007‐50. Furthermore, whether Sp1 binding to TMEPAI promoter is affected by compound JHY‐A007‐50 was analysed using CHIP assay. As shown in Figure 3J, Sp1 binding to TMEPAI promoter is not affected by treating with compound JHY‐A007‐50.
3.3. Compound JHY‐A007‐50 inhibits the cell proliferation of HeLa cells
Previous reports found that TMEPAI plays an important role in tumourigenesis, which could be an important target for cancer therapy.11, 12, 18 Firstly, TMEPAI protein expression levels were tested in several cancer cell lines by Western blot. As shown in Figure 4A, TMEPAI protein is highly expressed in HeLa, MCF‐7, MGC‐803 and HepG2 cells, but with low expression in normal liver cell line L02. To determine whether JHY‐A007‐50 has anti‐cancer activity, we quantified the cytotoxicity of JHY‐A007‐50 in cancer cells using a MTT assay. The results showed that JHY‐A007‐50 treatment reduced the cell viability of HeLa, HepG2, MCF‐7, MGC‐803 cancer cells in a dose‐dependent manner (Figure 4B‐E). However, the L02 cells which have a low level of endogenous TMEPAI were not sensitive to JHY‐A007‐50 treatment (Figure 4F). These results suggest that compound JHY‐A007‐50 may have anti‐cancer activity due to its ability to downregulate TMEPAI expression.
Figure 4.
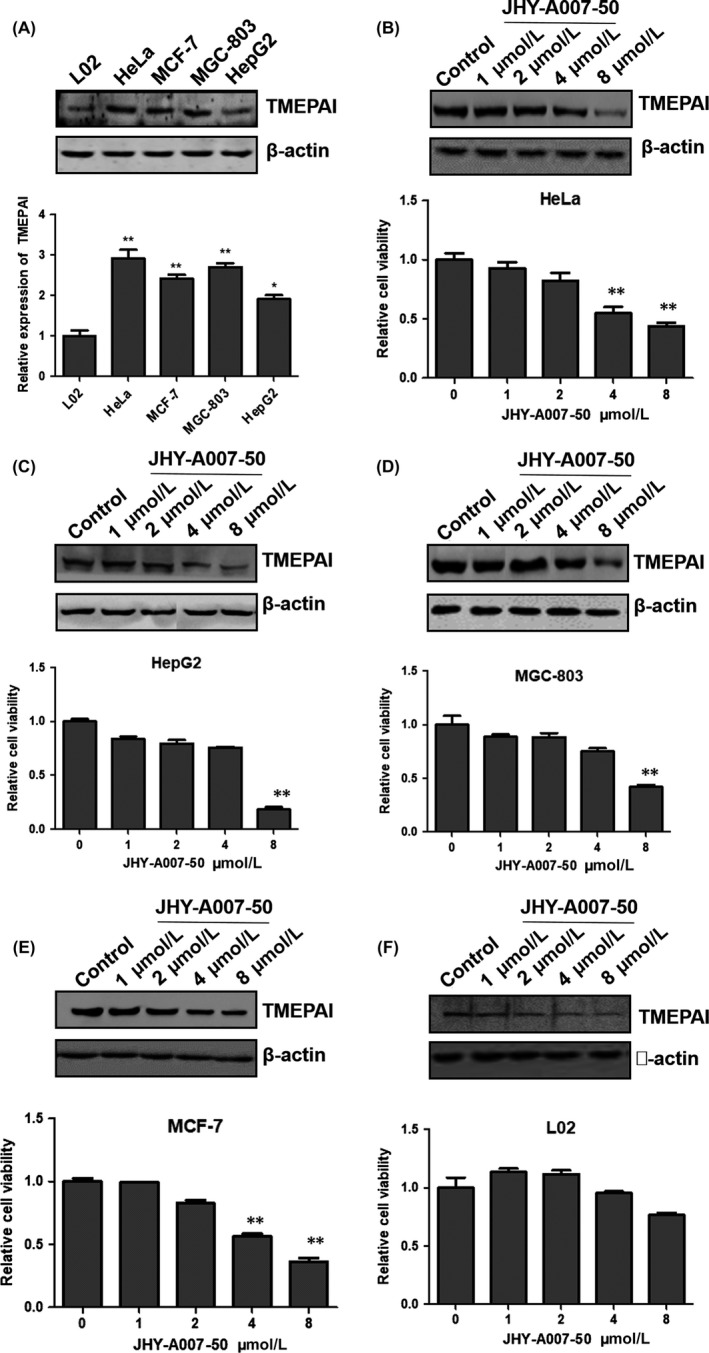
The effect of JHY‐A007‐50 on cell viability. (A) The cells (L02, HeLa, MCF‐7, MGC‐803 and HepG2) were collected and cell lysates were analysed by Western blot with antibodies to TMEPAI and β‐actin as a loading control. The quantified result was shown in the below panel. HeLa (B), HepG2 (C), MGC‐803 (D), MCF‐7 (E) and L02 (F) cells were treated with JHY‐A007‐50 (1, 2, 4, 8 μM) for 48 h. Cells treated with 0.1% (v/v) DMSO were used as the negative control. Cell viability was determined by an MTT assay (*.01 < P < .05, **P < .01). The dose‐dependent effects of JHY‐A007‐50 on TMEPAI expression in HeLa, HepG2, MGC‐803, MCF‐7 and L02 cells were shown in the corresponding up panel
To further detect the mechanism by which JHY‐A007‐50 reduced the viability of cancer cells, we evaluated the effects of JHY‐A007‐50 using a colony formation assay, EdU incorporation assay and Ki‐67 fluorescence assay. The colony formation assay showed that JHY‐A007‐50 reduced the colony formation rate (Figure 5A‐C). The EdU incorporation in HeLa cells treated with JHY‐A007‐50 was also inhibited (Figure 5D‐F). Antigen Ki‐67 is a nuclear protein that is strictly associated with cell proliferation.19 Fluorescence assay data showed that JHY‐A007‐50 reduced the expression of Ki‐67 (Figure 5G‐I). Taken together, these results show that JHY‐A007‐50 has an inhibitory effect on cancer cell proliferation. Moreover, the effect of JHY‐A007‐50 on cell migration was also investigated by Wound‐Healing assay, and the results showed that JHY‐A007‐50 can significantly inhibit HeLa cell migration (Figure 5J).
Figure 5.
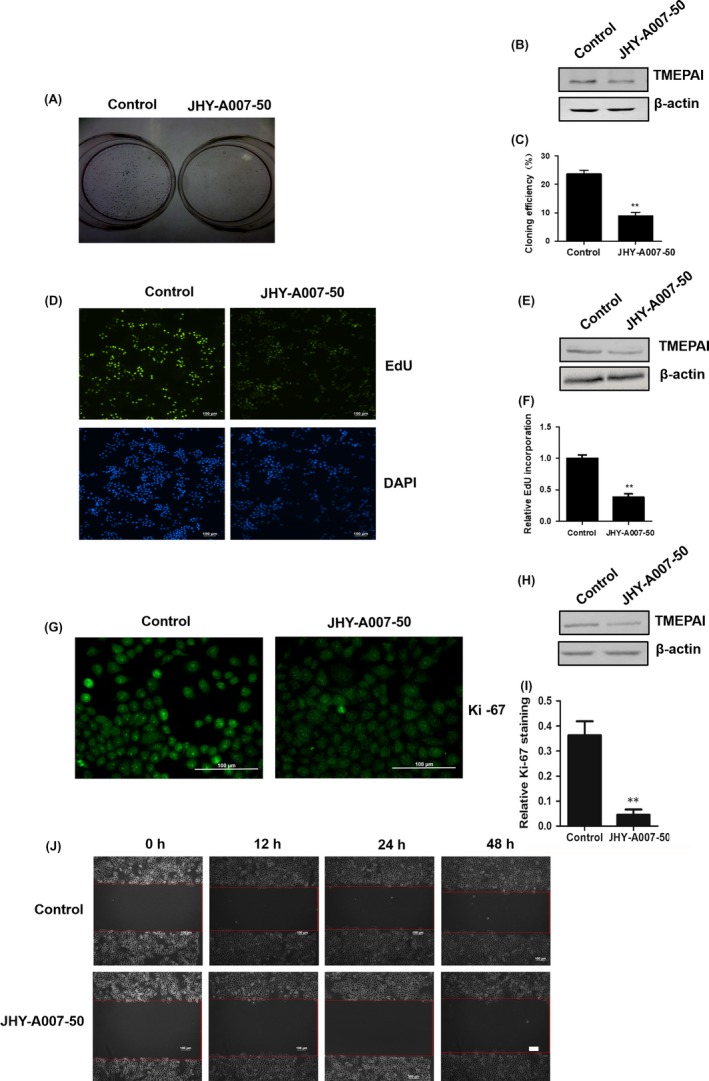
Compound JHY‐A007‐50 inhibits the cell proliferation of HeLa cells. (A) HeLa cells treated with 4 μM JHY‐A007‐50 were used for colony formation assays after 20 d culture, and then plates were photographed. The correlated TMEPAI protein level and the quantified results of colony formation assays were shown in (B) and (C) (**P < .01). (D) HeLa cells were treated with 4 μM JHY‐A007‐50 for 24 h, and DNA synthesis was assessed by EdU incorporation assay. Scale bar, 100 μm. The correlated TMEPAI protein level and the quantified using Image J software results of EdU incorporation assay were shown in (E) and (F) (**P < .01). (G) HeLa cells were treated with 4 μM JHY‐A007‐50 for 24 h, and the amount of Ki‐67 was evaluated by immunofluorescence assay. Scale bar, 100 μm. The correlated TMEPAI protein level and the quantified using Image J software results of immunofluorescence assay were shown in (H) and (I) (**P < .01). (J) HeLa cells were seeded into six‐well plates in DMEM and cultured overnight. Photographs were taken after the wound was made. HeLa cells were treated with 4 μM JHY‐A007‐50 for 12, 24 and 48 h. The experiment was repeated 3 times and similar results were obtained each time. Scale bar, 100 μm
3.4. JHY‐A007‐50 induces G1‐phase arrest in cancer cells
The cell cycle plays an important role in cell proliferation, and many anti‐cancer drugs could inhibit cell proliferation through inducing cell cycle arrest.20, 21 To examine whether the anti‐proliferation effect of JHY‐A007‐50 is associated with cell cycle arrest, we measured the cell cycle distribution in JHY‐A007‐50‐treated cells using flow cytometry. The results showed that in HeLa cells the cell population in the G1 phase was 40.58% in the control group; however, upon treatment with JHY‐A007‐50, the percentage of G1 phase cells increased to 59.16% (Figure 6A). Similar result was also found in MCF‐7 cells, where the percentage of G1 phase cells increased from 54.83% to 64.54% (Figure 6B). This suggested that JHY‐A007‐50 induces cell cycle arrest at the G1 phase.
Figure 6.
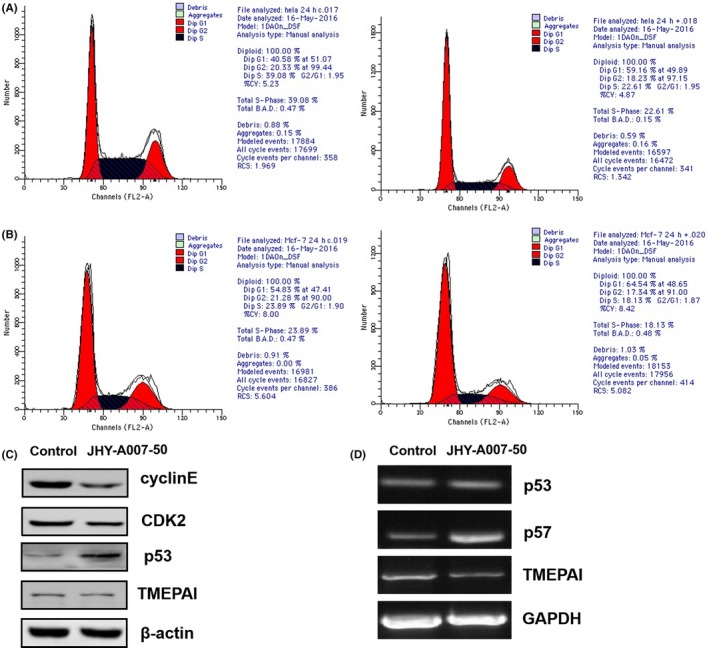
JHY‐A007‐50 induces G1‐phase arrest in cancer cells. HeLa (A) and MCF‐7 (B) cells were treated with 4 μM JHY‐A007‐50 for 24 h, and 0.1% (v/v) DMSO was used as a negative control. The relative number of cells within each cell cycle was determined using flow cytometry. (C) HeLa cells were incubated with 4 μM JHY‐A007‐50, and 0.1% (v/v) DMSO was used as a negative control. Twenty‐four hours after treatment, cell lysates were prepared, and the expression levels of Cyclin E1, CDK2, and p53 were determined using Western blotting. (D) HeLa cells were treated with 4 μM JHY‐A007‐50 for 24 h, and 0.1% (v/v) DMSO was used as a negative control. TMEPAI, p53 and p57 mRNA levels were analysed by RT‐PCR. GAPDH was used as an internal control to check the efficiency of cDNA synthesis and PCR amplification
Since cyclins D, E, and CDK2 have a key role in regulating the progression of G1 and S phase, then the expression levels of these proteins were measured. As shown in Figure 6C, the expression of cyclin E1 and CDK2 was decreased upon treated with JHY‐A007‐50. Furthermore, JHY‐A007‐50 increases the expression of p53 at the mRNA and protein levels (Figure 6C,D). Taken together, these results indicated that JHY‐A007‐50 could induce G1‐phase cell cycle arrest and inhibit the proliferation of cancer cells.
3.5. The inhibitory effect of JHY‐A007‐50 on cell proliferation depends on the downregulation of TMEPAI
Since JHY‐A007‐50 could inhibit TMEPAI expression and reduce cell viability, we next determined if the anti‐proliferative effect of JHY‐A007‐50 is related to the downregulation of TMEPAI expression. HeLa cells were transfected with pEF‐TMEPAI plasmid and treated with JHY‐A007‐50. The results showed that the overexpression of TMEPAI (Figure 7A) partially reversed the inhibitory effects of JHY‐A007‐50 on cell viability as measured by the MTT assay. The Ki‐67 fluorescence assay also showed that overexpression of TMEPAI reversed the inhibitory effects of JHY‐A007‐50 on cell proliferation (Figure 7B). We also found that overexpression of TMEPAI partially reversed the suppression of cyclin E1 and CDK2 as well as promotion of p53 induced by JHY‐A007‐50 treatment (Figure 7C). Furthermore, knockdown of TMEPAI in the JHY‐A007‐50 treated HeLa cells showed that JHY‐A007‐50 did not affect the cell viability of TMEPAI‐depleted HeLa cells (Figure 7D,E). These results suggest that JHY‐A007‐50 inhibits cell proliferation partially through the downregulation of TMEPAI expression.
Figure 7.
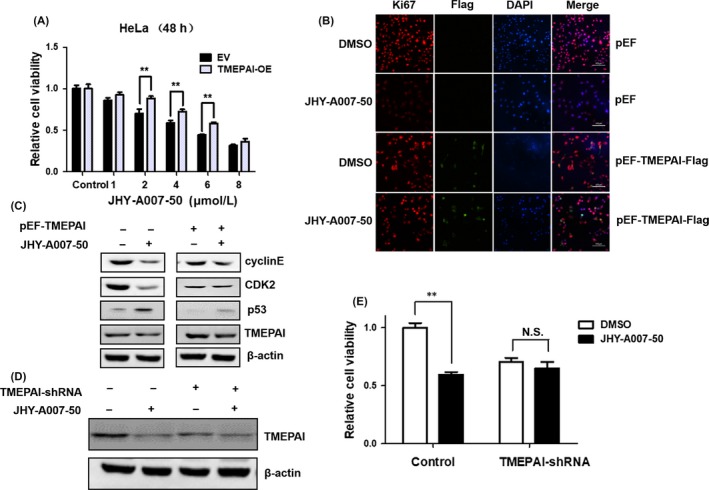
The inhibitory effect of JHY‐A007‐50 on cell proliferation depends on downregulation of TMEPAI. (A) HeLa cells were transfected with pEF‐TMEPAI or empty vector for 48 h, then cells were treated with different concentrations of JHY‐A007‐50, and 0.1% (v/v) DMSO was used as a negative control. MTT assay was used to analyse the cell viability (**P < .01). (B)The amount of Ki‐67 and Flag‐TMEPAI were evaluated by immunofluorescence assay. Scale bar, 100 μm. (C) HeLa cells were transfected with pEF‐TMEPAI or empty vector for 48 h, then the cells were incubated with 4 μM JHY‐A007‐50, and 0.1% (v/v) DMSO was used as a negative control. Twenty‐four hours after treatment, cell lysates were prepared, and the expression levels of Cyclin E1, CDK2 and p53 were determined using Western blot. (D) HeLa cells were infected with TMEPAI‐shRNA lentiviruses 3 d before treated with 4 μM JHY‐A007‐50. TMEPAI protein levels were determined by Western blot. (E) Cell viability was measured 48 h after treated with 4 μM JHY‐A007‐50 (**P < .01, N.S. indicates P > .05)
4. DISCUSSION
TMEPAI is abnormally activated in many cancers and previous studies have suggested that downregulating the expression of TMEPAI could subsequently depress cell proliferation and colony formation. These results indicate that TMEPAI could be a potential novel drug screening target for cancer therapy.
Our previous study showed that 2 Sp1‐binding sites are crucial for maintaining basal transcriptional activity of the TMEPAI promoter, and chromatin immunoprecipitation assays confirmed the direct binding of Sp1 to the TMEPAI promoter. In addition, Sp1 upregulated TMEPAI protein expression, as well as promoted TMEPAI‐induced cell proliferation. Here, a luciferase reporter construct driven by the TMEPAI promoter was used to screen for TMEPAI expression inhibitors in HeLa cells. Compound JHY‐A007‐50 was found that could significantly depress the luciferase reporter activity in comparison with the DMSO control. Further results revealed that compound JHY‐A007‐50 could also inhibit the endogenous expression of TMEPAI mRNA and protein levels in HeLa cells, and that this inhibitory effect was dose‐dependent. These results suggested that compound JHY‐A007‐50 could potently downregulate the expression of endogenous TMEPAI.
TMEPAI is known to be induced by many signalling pathways such as androgen, EGF, TGF‐beta and Wnt signalling. In the downstream of these signalling, many transcriptional factors (Androgen receptor, Smad3, TCF7L2/beta‐catenin, Sp1, ELK‐1, HIF‐1 and c‐fos) are involved in the regulation of TMEPAI gene expression.14, 22, 23, 24 Here, we showed that JHY‐A007‐50 is an efficient and specific inhibitor of TMEPAI expression in cancer cells. As shown in Figure 3G, JHY‐A007‐50 did not affect the luciferase activity driven by SM22 (smooth muscle 22) promoter. We also found that JHY‐A007‐50 did not alter the expression of SM22 and Legumain at protein level, which are not regulated by Sp1 transcriptional factor (Figure 3H). These results proved the specificity of JHY‐A007‐50 on the inhibiting TMEPAI expression. To study the mechanism of JHY‐A007‐50 induced‐inhibiting of TMEPAI expression, we have examined the protein level of Sp1 in cytoplasm and nucleus in this compound treated cells; however, the result indicates that Sp1 is not the target of JHY‐A007‐50 (Figure 3G). Moreover, the CHIP assay showed that Sp1 binding to TMEPAI promoter is not affected by treating with compound JHY‐A007‐50 (Figure 3J). Considering the expression of TMEPAI is induced by many signalling pathways such as androgen, EGF, TGF‐beta and Wnt signalling, we will try to clarify the mechanism of JHY‐A007‐50 inhibiting TMEPAI expression in our further study.
JHY‐A007‐50 seems more effective in inhibiting TMEPAI gene promoter activity (Figures 2 and 3), but less effective in inhibiting the cell viability or cell proliferation detected by MTT, cell cycle analysis, Ki‐67 staining and EdU incorporation assay (Figures 4, 5 and 6). This may be because JHY‐A007‐50 affects the promoter activity more directly. The different sensitivity of these various assays probably is the reason that the dose of the drug to cause an effect appears to be different in different experiments. In spite of these differences, the trends of the results are consistent in these assays.
The cell cycle plays a key role in cell proliferation and cancer progression. Our results indicate that JHY‐A007‐50 induced cell cycle arrest at the G1 phase. Moreover, we showed that this cell cycle arrest was accompanied by the downregulation of Cyclin E and CDK2. The turnover assay showed that overexpression of TMEPAI reversed the inhibitory effects of JHY‐A007‐50 on cell proliferation (Figure 7). As shown in Figure 7B, overexpression of TMEPAI reversed the inhibitory effect of JHY‐A007‐50 on cell proliferation. However, TMEPAI‐expressing cells and Ki‐67‐positive cells are not overlapped in some cells. The paradoxical result may be due to Ki‐67 is periodically expressed in cells. Moreover, the different sensitivities of different antibodies using in IF assay may also lead to this seeming contradiction. Moreover, JHY‐A007‐50 did not affect the cell viability of HeLa cells knocked down of TMEPAI (Figure 7D,E). These results suggest that JHY‐A007‐50 inhibits cell proliferation partially through the downregulation of TMEPAI expression. Our previous study had indicated that TMEPAI could increase lysosome stability and promotes autophagy12 and so a future study will focus on the effects that JHY‐A007‐50 may have on autophagy and its in vivo anti‐tumour activity.
In conclusion, these results indicate that JHY‐A007‐50 is a novel and potent inhibitor of TMEPAI expression. Additionally, the anti‐tumour effect of JHY‐A007‐50 may be achieved through cell cycle arrest at the G1 phase. These results demonstrate that JHY‐A007‐50 could be a very promising anti‐cancer drug candidate for TMEPAI high‐expressing tumours.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGEMENTS
We are grateful to Prof. Edward McKenzie (The University of Manchester, UK) for the critical reading of the manuscript. This research is supported by the National Natural Science Foundation of China grant (31471335).
Li Y, Wang J, Song N, et al. 2‐(2‐nitrobenzylidene) indolin‐3‐one compound inhibits transmembrane prostate androgen‐induced protein (TMEPAI) expression and cancer cell proliferation. Cell Prolif. 2018;51:e12469 10.1111/cpr.12469
Contributor Information
Peng Yu, Email: yupeng@tust.edu.cn.
Aipo Diao, Email: diaoaipo@tust.edu.cn.
REFERENCES
- 1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893‐2917. [DOI] [PubMed] [Google Scholar]
- 2. Xu W, Towers AD, Li P, Collet JP. Traditional Chinese medicine in cancer care: perspectives and experiences of patients and professionals in China. Eur J Cancer Care (Engl). 2006;15:397‐403. [DOI] [PubMed] [Google Scholar]
- 3. Ocana A, Pandiella A, Siu LL, Tannock IF. Preclinical development of molecular‐targeted agents for cancer. Nat Rev Clin Oncol. 2010;8:200‐209. [DOI] [PubMed] [Google Scholar]
- 4. Xu LL, Shanmugam N, Segawa T, et al. A novel androgen‐regulated gene, PMEPA1, located on chromosome 20q13 exhibits high level expression in prostate. Genomics. 2000;66:257‐263. [DOI] [PubMed] [Google Scholar]
- 5. Xu LL, Shi Y, Petrovics G, et al. PMEPA1, an androgen‐regulated NEDD4‐binding protein, exhibits cell growth inhibitory function and decreased expression during prostate cancer progression. Cancer Res. 2003;63:4299‐4304. [PubMed] [Google Scholar]
- 6. Vo Nguyen TT, Watanabe Y, Shiba A, Noguchi M, Itoh S, Kato M. TMEPAI/PMEPA1 enhances tumorigenic activities in lung cancer cells. Cancer Sci. 2014;105:334‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singha PK, Yeh IT, Venkatachalam MA, Saikumar P. Transforming growth factor‐beta (TGF‐beta)‐inducible gene TMEPAI converts TGF‐beta from a tumor suppressor to a tumor promoter in breast cancer. Cancer Res. 2010;70:6377‐6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brunschwig EB, Wilson K, Mack D, et al. PMEPA1, a transforming growth factor‐beta‐induced marker of terminal colonocyte differentiation whose expression is maintained in primary and metastatic colon cancer. Cancer Res. 2003;63:1568‐1575. [PubMed] [Google Scholar]
- 9. Rae FK, Hooper JD, Nicol DL, Clements JA. Characterization of a novel gene, STAG1/PMEPA1, upregulated in renal cell carcinoma and other solid tumors. Mol Carcinog. 2001;32:44‐53. [DOI] [PubMed] [Google Scholar]
- 10. Liu R, Zhou Z, Huang J, Chen C. PMEPA1 promotes androgen receptor‐negative prostate cell proliferation through suppressing the Smad3/4‐c‐Myc‐p21 Cip1 signaling pathway. J Pathol. 2011;223:683‐694. [DOI] [PubMed] [Google Scholar]
- 11. Bai X, Jing L, Li Y, et al. TMEPAI inhibits TGF‐beta signaling by promoting lysosome degradation of TGF‐beta receptor and contributes to lung cancer development. Cell Signal. 2014;26:2030‐2039. [DOI] [PubMed] [Google Scholar]
- 12. Luo S, Yang M, Lv D, et al. TMEPAI increases lysosome stability and promotes autophagy. Int J Biochem Cell Biol. 2016;76:98‐106. [DOI] [PubMed] [Google Scholar]
- 13. Luo S, Jing L, Zhao T, Li Y, Liu Z, Diao A. Ubiquitination and dynactin regulate TMEPAI lysosomal trafficking. Sci Rep. 2017;7:42668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li YY, Guo AL, Feng YJ, et al. Sp1 transcription factor promotes TMEPAI gene expression and contributes to cell proliferation. Cell Proliferat. 2016;49:710‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gawenda J, Traub F, Luck HJ, Kreipe H, von Wasielewski R. Legumain expression as a prognostic factor in breast cancer patients. Breast Cancer Res Tr. 2007;102:1‐6. [DOI] [PubMed] [Google Scholar]
- 16. Liu C, Sun CZ, Huang HN, Janda K, Edgington T. Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Can Res. 2003;63:2957‐2964. [PubMed] [Google Scholar]
- 17. Yamane T, Murao S, Kato‐Ose I, et al. Transcriptional regulation of the legumain gene by p53 in HCT116 cells. Biochem Biophys Res Commun. 2013;438:613‐618. [DOI] [PubMed] [Google Scholar]
- 18. Hu Y, He K, Wang D, et al. TMEPAI regulates EMT in lung cancer cells by modulating the ROS and IRS‐1 signaling pathways. Carcinogenesis. 2013;34:1764‐1772. [DOI] [PubMed] [Google Scholar]
- 19. Kale A, Soylemez F, Ensari A. Expressions of proliferation markers (Ki‐67, proliferating cell nuclear antigen, and silver‐staining nucleolar organizer regions) and of p53 tumor protein in gestational trophoblastic disease. Am J Obstet Gynecol. 2001;184:567‐574. [DOI] [PubMed] [Google Scholar]
- 20. Zang QQ, Zhang L, Gao N, Huang C. Ophiopogonin D inhibits cell proliferation, causes cell cycle arrest at G2/M, and induces apoptosis in human breast carcinoma MCF‐7 cells. J Integr Med. 2016;14:51‐59. [DOI] [PubMed] [Google Scholar]
- 21. Kang N, Wang MM, Wang YH, et al. Tetrahydrocurcumin induces G2/M cell cycle arrest and apoptosis involving p38 MAPK activation in human breast cancer cells. Food Chem Toxicol. 2014;67:193‐200. [DOI] [PubMed] [Google Scholar]
- 22. Masuda K, Werner T, Maheshwari S, et al. Androgen receptor binding sites identified by a GREF_GATA model. J Mol Biol. 2005;353:763‐771. [DOI] [PubMed] [Google Scholar]
- 23. Nakano N, Kato M, Itoh S. Regulation of the TMEPAI promoter by TCF7L2: the C‐terminal tail of TCF7L2 is essential to activate the TMEPAI gene. J Biochem. 2016;159:27‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watanabe Y, Itoh S, Goto T, et al. TMEPAI, a transmembrane TGF‐beta‐inducible protein, sequesters Smad proteins from active participation in TGF‐beta signaling. Mol Cell. 2010;37:123‐134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials