

Abstract
Apoptosis is a mode of regulated cell death that is indispensable for the morphogenesis, development and homeostasis of multicellular organisms. Caspases are cysteine‐dependent aspartate‐specific proteases, which function as initiators and executors of apoptosis. Caspases are cytosolic proteins that can cleave substrates located in different intracellular compartments during apoptosis. Many years ago, the involvement of caspases in the regulation of nuclear changes, a hallmark of apoptosis, was documented. Accumulated data suggest that apoptosis‐associated alterations in nucleocytoplasmic transport are also linked to caspase activity. Here, we aim to discuss the current state of knowledge regarding this process. Particular attention will be focused on caspase nuclear entry and their functions in the demolition of the nucleus upon apoptotic stimuli.
1. INTRODUCTION
Apoptosis is a caspase‐dependent mode of regulated cell death that is indispensable for the morphogenesis, development and homeostasis of multicellular organisms; the dysregulation of apoptosis is often linked to cancer and neurodegenerative disorders.1 Caspases are cysteine‐dependent aspartate‐specific proteases, which function as both the initiators (caspase‐2, ‐8, ‐9, ‐10) and executors (caspase‐3, ‐6, ‐7) of apoptotic cell death. The executioner caspases are also considered the main proteases responsible for the dismantling of intracellular compartments during apoptosis. Over 520 caspase substrates have been experimentally determined, and more than 2000 proteins have also been shown to be cleaved by caspases in recent mass spectrometry studies.2, 3, 4 Notably, whereas procaspases are normally located in the cytosol, their substrates seem to be evenly distributed among all of the intracellular compartments.3 However, the mechanisms by which these cytosolic proteases perform the cleavage of proteins located inside the isolated cellular organelles remain largely elusive.
Apoptotic changes in the nuclear morphology were described as early as 1972.5 Some of them, namely chromatin condensation and DNA fragmentation, were generally accepted as the hallmarks of apoptosis and are widely used for the detection of this cell death mode. However, some of the apoptotic changes, such as alterations of the nuclear envelope (NE) structure that precede the breakdown of the nucleus, have been much less thoroughly investigated. Later, the significance of the NE rearrangements, as well as of the changes in the nucleocytoplasmic transport system, in the course of apoptotic cell death was re‐evaluated.6 In this way, the redistribution of pro‐apoptotic and some anti‐apoptotic factors was considered an important and, in some cases, even essential step for the amplification of the death signal. Since then, the amount of data showing that apoptotic function of proteins depends on their localization has increased, further supporting the aforementioned notion. Additionally, the regulation of post‐translational modifications not only of cargo molecules, but also of transport factors, was shown to play an important role.
Here, we aim to present the current state of knowledge regarding the alterations in nucleocytoplasmic transport during apoptosis. As caspases, the major players in apoptosis, not only translocate to the nucleus in the course of this type of cell death, but also alter the NE structure and directly regulate the transport of other apoptosis‐related factors, their role in this process will be discussed in detail. Our particular attention will be focused on caspase nuclear entry and functions in response to apoptotic stimuli.
2. APOPTOSIS‐INDUCED ALTERATIONS IN THE NUCLEOCYTOPLASMIC TRANSPORT SYSTEM
The exchange of factors between the cytoplasm and the nucleus of eukaryotic cells is indispensable for their normal functions and the coordination of cellular stress responses.7 Two mechanisms, namely passive diffusion and active translocation, facilitate nucleocytoplasmic exchange and both are predominantly carried out through the nuclear pore complexes (NPCs) embedded in the NE.
The NPC is a large assembly of ∼500 copies of ∼30 protein molecules known as nucleoporins (Nups).8 Nups can be divided into three classes: (i) transmembrane Nups that anchor the NPC in the NE, (ii) structural Nups that stabilize the NE and the NPCs, (iii) Nups that are rich in natively disordered phenylalanine‐glycine (FG) repeats, which coordinate both passive and active transport through the NPC. The control of passive diffusion is achieved by the formation of a filter that inhibits the translocation of molecules larger than ∼40 kDa.9, 10, 11 The active transport allowing the translocation of large macromolecules is accomplished via direct interactions between FG repeats and nuclear transport receptors termed karyopherins (importins and exportins).12 Thus, to actively pass through the NPC, a protein must contain a nuclear localization signal (NLS) or nuclear exports signal (NES) and form complexes with importins or exportins, respectively. The energy and directionality of the process is provided by the small GTPase, Ras‐related nuclear protein, Ran (see reviews10, 11 for the general steps of the nucleocytoplasmic transport).
The induction of apoptosis leads to multiple alterations in the nucleocytoplasmic transport system. These changes, as well as chromatin condensation and DNA fragmentation, are mediated by both caspase‐dependent and ‐independent mechanisms.13, 14, 15 Early alterations in the system of nucleocytoplasmic transport include the redistribution of transport factors, primarily Ran.16, 17 Normally, the cytoplasmic Ran is bound to GDP, which is achieved through the cytoplasmic Ran‐GTPase‐activating protein (RanGAP). On the contrary, in the nucleus, due to the activity of Ran guanine nucleotide exchange factor (RanGEF or RCC1), Ran is predominantly present as Ran‐GTP. In response to different stress stimuli, including apoptosis, Ran translocates to the cytoplasm, where it is rapidly converted to Ran‐GDP by RanGAP. Consequently, nuclear and cellular pools of Ran‐GTP are reduced, leading to perturbations in the distribution of other nucleocytoplasmic transport factors.
Another feature of apoptosis induction is a change in the composition of importin complexes. As the dissociation of import complexes seems to be inhibited due to reduced level of Ran‐GTP, the nuclear accumulation of importin α and β bound to the NLS‐containing proteins is observed. Thus, importin α was shown to migrate from the cytoplasm into the nucleus under oxidative stress and heat shock,18, 19 and most of the importin family members translocate to the nucleus at least in response to oxidative stress.20 The physiological outcomes of the sequestration of importins in the nucleus vary depending on which of the isoforms is involved and which substrate is failed to be transported.21, 22, 23 For example, while the reduced nuclear uptake of pro‐apoptotic factors with a classical NLS, such as apoptosis‐inducing factor (AIF)24 or DEDD,25 should promote apoptotic cell death, the impaired movement of nuclear factor‐kappa B (NF‐κB) to the nucleus, on the contrary, accelerates apoptosis.26, 27, 28 At the same time, a protracted and substantial inhibition of nuclear import also leads to the induction of cell death.23
Not only NLS‐dependent protein import, but all the pathways of regulated nuclear transport are generally suppressed upon the apoptotic‐mediated disruption of the Ran‐GTP gradient.16, 29, 30, 31, 32 At the same time, during apoptosis nonspecific permeability of the nuclear membrane increases, which makes the entry of at least 70 kDa molecules into the nucleus possible.17 The described mechanism by which Ran‐GTP can directly control NPC size‐cutoff involves Ran‐GTP‐mediated regulation of the interaction between importin‐β and Nup153.33
Initially, early apoptotic changes in the distribution of nuclear transport factors were reported to occur prior to caspase activation and shown to be insensitive to pan‐caspase inhibitors.16, 17 However, caspase‐9 activity was later shown to be required for the release of Ran upon TNF‐α treatment.34 Moreover, caspase‐9 mediates a breakdown of the hydrophobicity‐based barrier inside NPC channel, increasing NE leakiness.15 On the other hand, in another study, the disruption of Ran‐mediated nuclear transport after 5‐flurouracil treatment was shown to precede caspase‐9 activation, indicating the caspase‐independent increase in NPC permeability.35 Consequently, the role of caspases in early apoptosis‐mediated alterations of nucleocytoplasmic transport remains controversial and demands further investigation.
Additionally, caspases could indirectly change the Ran‐GTP gradient. For example, the caspase‐dependent translocation of mammalian Sterile 20‐like kinase 1 (Mst1) to the nucleus prior to NE rearrangements was shown to result in a decrease in nuclear Ran‐GTP level upon etoposide treatment.26 When in the nucleus, Mst1 catalyses the phosphorylation of histone H2B at Ser14. This modification causes the immobilization of RanGEF on chromosomes and leads to a significant decrease in the nuclear levels of Ran‐GTP, which is generated only when the RanGEF‐Ran binary complex is dissociated from the nucleosome.36 Importantly, according to the findings of Wong et al, Mst1 function is also essential for the prevention of NF‐κB entry into the nucleus, which downregulates NF‐κB‐mediated anti‐apoptotic pathway.26
The later steps of apoptotic cell death include irreversible caspase‐mediated alterations of the nuclear structure, namely the cleavage of components of the NPC, although the NPCs appear to be morphologically intact.37, 38, 39 Apart from changes in the NPC structure, caspase‐mediated proteolysis of the nuclear lamina and a number of nuclear matrix proteins constitutes another important step that promotes nuclear disruption following the induction of apoptosis.40, 41, 42 Yet again, despite the degradation of both internal and external components of the NPC and nuclear lamina during apoptosis, the general structure of the NE was reported to remain preserved.14 As components of the NE associate with each other and the filaments of the nuclear lamina, Nups and some other proteins of the nuclear membrane on the nucleoplasmic side,43 alterations in the NE structure affect nucleocytoplasmic transport, and the further proteolysis of NE components finally disrupts the connection between the NE and chromatin.
Taken together, disturbances in both the distribution of nuclear factors and the NE structure following apoptosis, as well as other stress stimuli, lead to the inhibition of the regulated nucleocytoplasmic transport. The changes observed during apoptosis are implemented via both caspase‐dependent and ‐independent mechanisms. Caspase‐mediated proteolysis of the NPC components is thought to push the nucleocytoplasmic transport from strictly controlled exchange of the nuclear and cytoplasmic components to a less regulated traffic. Although caspase‐dependent mechanisms undoubtedly play a key role in the latter stages of apoptosis, their functions in the early rearrangements of the transport system are not fully understood.
3. APOPTOTIC CASPASES TARGET NUCLEOCYTOPLASMIC TRANSPORT COMPONENTS AND PROTEINS MAINTAINING NUCLEAR INTEGRITY
A number of nuclear proteins have been reported to be cleaved by caspases during apoptosis (Table 1). Many of these nuclear targets of caspases are components of the NE; therefore, its proteolysis contributes to NE breakdown during apoptotic cell death (Figure 1). The nuclear substrates of caspases can be classified into three groups: (i) components of the NPC and nuclear export‐import system (eg, Nups and importins), (ii) structural proteins maintaining nuclear integrity, (iii) intranuclear substrates (eg, poly ADP‐ribose polymerase (PARP‐1)44). Below, we discuss the substrates from groups 1 and 2, the cleavage of which disrupts barrier function of the nuclear membrane and nuclear structure (Table 1).
Table 1.
The caspase substrates of groups 1 and 2
| Substrates | Result | References |
|---|---|---|
| Nup50 | Disrupting interaction with other Nups | 45 |
| Nup96 | Disrupting interaction with other Nups | 45 |
| Nup93 | Disrupting interaction with other Nups | 45 |
| Nup153 | Impaired nuclear transport, redistribution of importins | 47, 136 |
| Nup214 | Impaired nuclear transport, redistribution of importins | 47 |
| POM121 | Disrupting interactions with other NPC proteins affecting NPC stability | 137 |
| RanBP2 (Nup358) | Increase of nuclear membrane permeability | 17 |
| Tpr | Increase of nuclear membrane permeability | 17 |
| Importin α | Downregulation of the nucleocytoplasmic protein transport and DNA synthesis | 16, 138 |
| Importin β | Inhibition of classical nuclear import | 16 |
| Lamins A/C ‐type | Disruption of nuclear integrity | 40, 41 |
| Lamins B‐type | Disruption of nuclear integrity | 40, 41 |
| Nuclear Mitotic Apparatus protein (NuMA) | Disruption of nuclear integrity | 51, 139 |
| LAP2β | Detachment of NE from chromatin | 38 |
| LBR | Detachment of NE from chromatin | 38 |
| C53/LZAP | Rupture of the NE | 54 |
Figure 1.
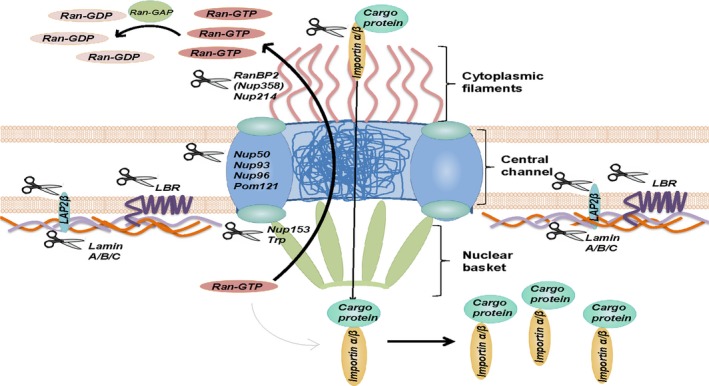
Caspases target components of the nucleocytoplasmic transport and proteins maintaining nuclear integrity. Caspases have been found to cleave at least 8 of 30 proteins of the NPC and importins (Table 1), which leads to the destabilization of active transport. Caspases are depicted as a pair of scissors
The cleavage of a substrate from the first group leads to the deregulation of nucleoplasmic transport and an increase in nuclear membrane leakiness. In particular, out of thirty Nups of the NPC, eight are known to be caspase targets (Figure 1). The destruction of the central core Nup96 and Nup93 was proposed to be a major cause of the impairment of the NPC function.45 Of particular note, the caspase‐mediated cleavage of Nups can also impair protein‐protein interactions. Thus, a few proteolytic events might be sufficient to compromise the NPC function. For example, proteolysis of the structural Nup93 and Nup96 results in the removal of the binding domains for FG Nup62 and Nup98, respectively,45, 46 both of which are required for the establishment of transport‐competent channels. Furthermore, caspase‐mediated cleavage of Nup153 and Nup214 results in the redistribution of importins, which inhibits nucleocytoplasmic transport.47, 48 Additionally, Nup50, which is involved in nuclear protein export and import, is also cleaved in apoptotic cells. The cleavage of Nup50 blocks interaction with Nup153, causing a decrease in nuclear import.45, 49
On the other hand, nucleocytoplasmic transport factors, including importins α and β, are subjected to cleavage by caspases during apoptosis (Figure 1).16 These factors can be linked to NPC components. For example, Nup153 interacts with importin β, which regulates the permeability of NPC in a Ran‐dependent manner.33 Moreover, Nup153 is a binding partner of importin α, interaction with which is important for NLS‐mediated import.50 Consequently, after the induction of apoptosis, the caspase‐dependent decrease in Nup153 or/and importin levels might enhance passive diffusion across the NPC, although active transport is impaired.
Notably, several dozens of potential caspase substrates were found in the nucleus.2 The essential part of them is involved in nucleocytoplasmic transport, but these proteins are not discussed here because the biological significance of their caspase‐dependent cleavage has not yet been determined.
In the later stages of apoptosis, caspases target structural proteins of the NE, including lamins, which presents one of the classical features of the demolition phase of apoptosis. A/C‐ and B‐type lamins are preferentially cleaved by caspase‐6 (Figure 1).40, 41 Moreover, caspase‐dependent proteolysis of the nuclear matrix proteins (eg, cleavage of NuMA, hnRNP proteins C1 and C2, the 70 kDa component of U1 small ribonucleoprotein (snRNP), the scaffold‐attachment factor A and the special AT‐rich sequence‐binding protein 1 (SATB1)) is an important step towards the breakdown of the nucleus.51, 52 Notably, the preservation of intact lamina can at least delay the execution of apoptosis, preventing chromatin condensation and fragmentation,53 which underlines the importance of caspase nuclear function. Additionally, caspases have been reported to cleave protein C53/LZAP, which is indirectly bound to the microtubules and implicated in various signalling pathways. Its cleavage results in the formation of C3 fragment, which disrupts the connection between the NE and the surrounding microtubule system, and, therefore, promotes NE disruption early after the initiation of apoptosis.54 Another key event in the NE breakdown is the rearrangement of the cytoskeleton. Many cytoskeletal proteins, including actins, tubulins, vimentins and actin‐binding proteins are caspase substrates. Moreover, a well‐described mechanism in the demolition phase of apoptosis involves cleavage of the rho‐associated kinase 1 (ROCK1), leading to its activation, which in turn drives the contraction of the actin‐myosin cytoskeleton and promotes the breakdown of the NE.55
In summary, during the early steps of apoptosis, disruption of the NPCs and the nuclear permeability barrier allows caspases to enter the nuclei and then cleave the components of the nuclear lamina and matrix. Hence, caspases play a crucial role not only in chromatin condensation and DNA fragmentation during the demolition phase of apoptosis, but also in the alterations of the nucleocytoplasmic transport system during early apoptotic stages.
4. NUCLEAR TRANSPORT OF CASPASES DURING APOPTOSIS
Nuclear localization of caspase‐3 was observed in early studies of apoptotic processes.56, 57 However, 20 years later, due to the inconsistency and scarcity of the accumulated data on the subject, the redistribution of caspase‐3 and other caspases during apoptosis is still a matter of debate.
Initially, caspase‐3 was suggested to translocate into the nucleus using active transport.58, 59, 60 In confirmation of this, nuclear entry of the p17/p12 caspase‐3 fragments before the disruption of the nuclear and mitochondrial membranes was demonstrated.61 However, another study described the nuclear appearance of active caspase‐3 only after the breakdown of the nucleocytoplasmic barrier, suggesting that the translocation occurs by means of simple diffusion.34 Nevertheless, the nuclear entry of active caspase‐3 was thought to be a necessary step during apoptosis.58, 59, 60, 62, 63 Thus, for example, the impaired nuclear transport of active caspase‐3 was associated with the decreased sensitivity of non‐small cell lung carcinoma cells to radiation and chemotherapy.63
Fluorescence resonance energy transfer (FRET) experiments showed that in response to apoptotic stimuli caspase‐3 activation initiates in the cytoplasm and subsequently proceeds in the nucleus.64 Importantly, caspase‐3 nuclear activation was detected before apoptotic changes in the nuclear morphology, suggesting the implementation of active transport mechanisms during the nuclear entry of caspase‐3. Another study observed the nuclear translocation of caspase‐3 exclusively after its two‐step proteolysis and proposed a carrier protein for the nuclear transport of caspase‐3.65 Moreover, in microglia cells, the formation of p17/p12 heterotetramers in the cytoplasm and its subsequent nuclear entry were critical steps for the promotion of apoptosis.65 The inhibition of the conversion of p19 caspase‐3 subunit to p17 was facilitated by cellular IAP2, which led to the retention of p19/p12 caspase‐3 in the cytoplasm and the development of a pro‐inflammatory response. Taken together, the question of whether caspase‐3 translocates into the nucleus as a proform or active form is still under debate. It is not inconceivable that the status of caspase‐3 during the nuclear translocation depends on the cell type and that both forms of caspase‐3 could translocate into the nucleus. Undoubtedly, the nuclear entry of caspase‐3 is a critical step for the promotion of apoptosis promotion and the inhibition of its cellular redistribution results in a loss of sensitivity to cell death induction.
Interestingly, a functional Crm‐1‐independent NES was identified in the small subunit of caspase‐3.66 The proteolytic activity of caspase‐3 was found to be necessary for blocking its NES, and, therefore, its export from the nucleus.66 The inhibition of caspase‐3 NES might be carried out by different mechanisms, for example, via the binding of caspase‐3 to a nuclear protein. However, if caspase‐3 was transported by a carrier, this protein would have to dissociate from caspase‐3 to unblock its proteolytic activity. In this case, the NES of caspase‐3 would again be exposed, leading to the export of caspase‐3 back to the cytoplasm. More likely, the export of caspase‐3 is impaired due to the degradation of the components, for instance, the related exportin and Nups.66 The latter might serve as an explanation of the molecular mechanism of the amplification of caspase‐3 activity in the nucleus during the demolition phase of apoptosis. Indeed, in the early stages of apoptosis, the nuclear export system is not yet impaired, and if caspase‐3 penetrates into the nucleus it will be immediately exported due to its NES. However, upon the deregulation of the export system, caspase‐3 accumulates in the nucleus, leading to its destruction at the later stages of apoptosis.
Nuclear localization of other active effector caspases, caspase‐667, 68 and ‐7,69, 70 in apoptotic cells was also reported. Notably, correlation between the nuclear localization of caspase‐7 and DNA degradation was observed.69, 70 SUMOylation of caspases plays an important role in the control of their nuclear localization/translocation. With respect to caspase‐7, it possesses a SUMO‐1 site, which is located in the large subunit of caspase‐7 and was identified as a key regulator of its nuclear localization.71 The data describing the role of caspase‐6 in nuclear events are scarce; however, caspase‐6 was found to cleave a number of nuclear substrates including lamina and huntingtin.68 The inhibition of caspase‐6‐mediated proteolysis of the latter prevents the development of behavioural, motor and neuropathological features in a mouse model of Huntington disease. Interestingly, in the mantle cell lymphoma cells, the nuclear activity of caspase‐6 can be blocked by Sox11.72 It might be that a loss or mutation of the Sox11 gene results in the hyperactivation of caspase‐6 in the nucleus and the development of Huntington disease. In addition, the role of Sox11 in suppressing the nuclear activity of other effector caspases was proposed, although the physiological significance of this effect is still unknown.
The nuclear translocation of initiator caspases following apoptosis induction was also shown. In particular, the nuclear entrance of caspase‐8 was reported in a number of studies,61, 73 although there is some controversy in the literature as there are also reports arguing only the cytoplasmic localization of caspase‐8 and ‐10.74 The described mechanisms of caspase‐8 nuclear translocation also include a post‐translational SUMOylation75 and binding to the DED‐containing proteins DEDD and DEDD2.76 The detailed mechanism of the DEDD‐ and DEDD2‐mediated translocation of caspase‐8 remains elusive, but it was shown that caspase‐8 nuclear accumulation preceded caspase‐3 activation, suggesting carrier‐mediated translocation before disruption of the nuclear barrier.76 After its nuclear translocation, caspase‐8 has been shown to amplify nuclear demolition via cleavage of the centromere protein (CENP)‐C and inner CENP (INCENP), resulting in the severe disruption of the centromere structure.77
Nuclear activity of another initiator caspase, caspase‐2, was shown as early as 1994.78 Among all caspases, only caspase‐2 was described as a NLS‐containing protein which is constitutively localized and supposedly activated in the nuclei.74, 79, 80, 81 The presence of caspase‐2 in the cytosol was reported,61, 79 but active caspase‐2 was detected in the nuclear fraction upon the induction of apoptosis.61 As for other caspases, SUMO‐1 modification of the CARD domain of procaspase‐2 was shown to be essential for its nuclear localization.82 Moreover, caspase‐2 was found to co‐localize with promyelocytic leukemia protein nuclear bodies (PML‐NBs).83, 84 Therefore, it was proposed that PML‐NBs could serve as the platforms for caspase‐2 activation, and that SP100, one of the components of PML‐NB that presumably contains a CARD domain, might directly interact with caspase‐2.83
The use of bimolecular fluorescence complementation (BiFC) to measure caspase‐2 induced proximity in real‐time demonstrated caspase‐2 activation exclusively in the cytosol.85 However, importantly, only the fragment of caspase‐2 was used in this study that did not possess NLS. The obtained results were in accordance with the previous data, demonstrating that the disruption of caspase‐2 NLS and abrogating its nuclear localization did not prevent caspase‐2 from activation.80, 81 Thus, the authors postulated that not the nuclear localization of caspase‐2, but rather its binding to macromolecular complexes, is necessary for caspase‐2 activation.80, 85 However, the later study showed the appearance of two distinct caspase‐2 activation complexes—one of them in the cytoplasm and the second in the nucleolus.86 Given that caspase‐2 is not only involved in the induction of apoptosis in response to DNA damage and other stimuli, but also functions as a tumour suppressor and in a number of other processes,87, 88 its nuclear localization might be important for maintaining the normal metabolism and the genetic stability of the cell, as well as playing a key role in ageing.
Thus, data on the mechanisms of caspase redistribution during apoptosis remain controversial and contradictory. Even the molecular mechanisms of the nuclear translocation of the most‐studied caspase‐3 are still largely unknown. The discrepancy of the data might be caused by the use of different cell lines, stress stimuli and tools (ie, antibodies). Other problems are inherent to the methods employed, such as the ectopic expression of caspases linked to a fluorescent protein or the isolation of subcellular fractions.
Initially, the controversial issue of whether caspases are imported actively or passively enter the nucleus was addressed by Ferrando‐May et al.6 It is generally thought that cargos smaller than ~40 kDa can slowly diffuse into the nucleus, and only larger cargos are actively imported by karyopherins.9, 10, 11 However, there has been a recent study supporting another model. According to Timney et al, no rigorous size threshold exists, but rather the NPC forms a soft barrier that gradually intensifies with increasing molecular mass (or volume).89 The soft barrier results from the highly dynamic FG repeat domains of Nups and the diffusion of macromolecules is controlled by the volume available in the interior of the NPC, setting up entropic repulsion forces. Indeed, proteins larger than 40‐60 kDa are often equally partitioned between the nucleus and the cytoplasm, similar to smaller proteins.90 For example, in spite of molecular sizes of ∼100 kDa, karyopherins can pass through the NPC even together with their specific cargos. In particular, karyopherins, as well as other HEAT (a pair of amphiphilic α‐helices)‐rich proteins were demonstrated to undergo flexible conformational changes that enable them to pass through the hydrophobic milieu of the pore.91, 92 Moreover, some of the smallest proteins, including Hiv‐1 Rev, PTHrP and histone H1, use karyopherin‐mediated import pathways,93 suggesting that their physicochemical features do not allow passive nuclear entry and require active import. Thus, there is no strict correlation between the molecular size of the protein and its capability to passively diffuse through the NE. Cargo properties, eg, size and hydrophobicity, are other important factors that determine the influx and efflux rates.
Independent of the mechanism, the issue of active nuclear uptake of caspases must be discussed in the context of apoptosis‐induced alterations of the nucleocytoplasmic barrier. If the size exclusion limit of the NPC augments in the initial stage of cell death in such a way as to transmit molecules of at least 70 kDa, the active tetrameric forms of caspases may not be efficiently retained outside of the nucleus as they have a molecular weight of about 60 kDa. Given that the NPC is an early caspase target, an implication of active import for caspase translocation seems questionable. Nups that are crucially involved in cargo translocation and NPC selectivity are efficiently cleaved by caspases, which leads to the destruction of the selective NPC barrier.45 Consequently, the NPC size exclusion limit increases,17, 34 allowing the entry of larger proteins, such as caspases, into the nucleus. Nucleocytoplasmic barrier disruption also allows the leakage of resident nuclear proteins into the apoptotic cytoplasm,34 although the contribution of such a release mechanism to apoptotic cell dynamics or execution has not been assessed.
5. ROLE OF CASPASES IN THE DEMOLITION OF THE NUCLEUS DURING APOPTOSIS
One of the most important functions of caspases during apoptosis is promoting the degradation of genetic material. Caspases were found to contribute to the activation or nuclear translocation of a set of proteins promoting chromatin condensation and DNA fragmentation a long time ago. The most important mechanism for oligonucleosomal DNA fragmentation represents the caspase‐3 or ‐7‐mediated activation of caspase‐activated DNase (CAD)/DFF40 via cleavage of its inhibitor ICAD/DFF45 (Figure 2).94, 95, 96, 97 Once activated, CAD endonuclease hydrolyzes the DNA into oligonucleosomal‐size pieces, facilitating the chromatin package. Another factor activated by caspases and directly involved in chromatin fragmentation is a CARD‐containing helicase, Helicard. The processing of Helicard by caspases generates a C‐terminal fragment with helicase activity which translocates to the nucleus and accelerates DNA degradation (Figure 2).98 Moreover, the role of caspases in the nuclear translocation of endonuclease DNAS1L3 for inter‐nucleosomal DNA fragmentation was also described.99
Figure 2.
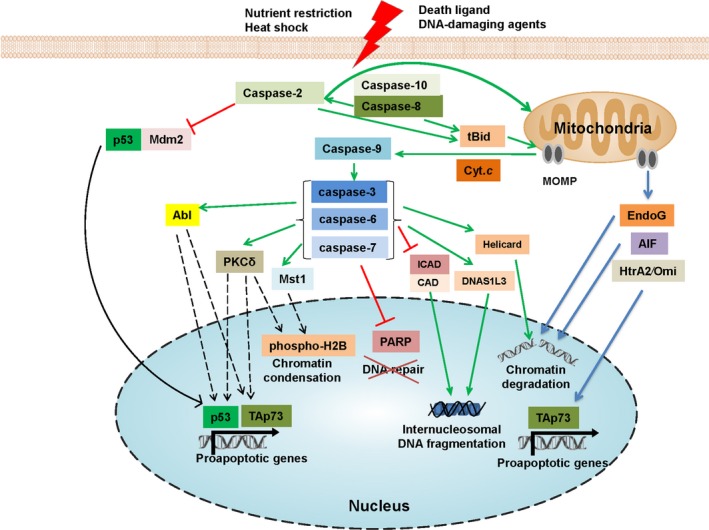
Caspases indirectly promote the demolition of the nucleus during apoptosis. The caspase‐dependent cleavage of a target can lead to its inactivation (red line) or the formation of an active product (green line). The phosphorylation of a target protein (dashed line) by caspase‐activated kinases results in chromatin condensation and the transcription of pro‐apoptotic genes
It is important to note that apoptotic chromatin condensation and DNA fragmentation might occur in a caspase‐independent manner. Thus, upon mitochondrial outer membrane permeabilization (MOMP), at least three intra‐mitochondrial proteins, AIF, endonuclease G (EndoG) and HtrA2/Omi, are released and translocated into the nucleus in order to promote chromatin condensation and DNA fragmentation.100, 101 EndoG is a DNase/RNase that induces inter‐ and intra‐nucleosomal DNA cleavage.102 AIF is a flavoprotein that indirectly triggers peripheral chromatin condensation and large‐scale DNA fragmentation after its translocation to the nucleus.24 Serine protease HtrA2⁄Omi after its release from the mitochondria upon MOMP also accumulates in the nucleus, leading to TAp73‐mediated up‐regulation of several pro‐apoptotic genes, including bax and bak.103 Thus, as proteases, caspases are not able to cleave DNA themselves; nevertheless, these proteases play an indispensable role in DNA fragmentation via proteolysis of their substrates and translocation of DNases into the nucleus.
Furthermore, nuclear targets of caspases include proteins that are important for various cellular processes. For instance, once the decision to die is made, there is no reason to spend further energy on DNA repair. Thus, proteins involved in DNA repair, eg, PARP‐1, one of the first reported caspase substrates, are cleaved by caspases. In the case of PARP‐1, its degradation is suggested to also promote apoptosis (Figure 2).44 Notably, virtually all pathways of macromolecular synthesis, including DNA replication, RNA and protein synthesis, are impaired by caspases. In addition, various chromatin‐bound proteins that have structural or transcriptional functions, and a number of RNA‐binding proteins, are cleaved.
Many cellular processes are controlled by enzymes providing post‐translational modifications: phosphorylation, ubiquitination, acetylation, methylation, etc. The amount of data confirming that caspases induce apoptotic nuclear changes via the proteolysis of protein kinases, phosphatases and other enzymes has increased. Recently, it has been shown that among the caspase‐targeted protein kinases, the cleavage of Mst1, protein kinase C delta (PKCδ) and PAK2 results in the generation of catalytically active fragments that translocate from the cytosol to the nucleus and promote chromatin condensation (Figure 2).104 The phosphorylation of histone H2B at Ser14 by Mst1 and PKCδ is the best known modification involved in chromatin condensation.105, 106, 107 Additionally, caspase‐mediated activation of acinus, a protein provoking chromatin condensation in the nucleus, leads to the enhanced kinase activity of PKCδ.106 Consequently, the cleavage of acinus by caspases and its PKCδ binding acts as an amplification loop for chromatin condensation. Moreover, nuclear PKCδ was suggested to facilitate apoptosis, not only promoting chromatin condensation but also via the phosphorylation of lamin B, DNA‐PK, p53, TAp73 and Rad9.108
In addition to the aforementioned kinases, several pro‐apoptotic kinases, including Abelson murine leukemia viral oncogene homolog 1 (Abl) and dual‐specificity tyrosine phosphorylation‐regulated kinase 2 (DYRK2), undergo nucleocytoplasmic shuttling and contribute to the apoptotic response following genotoxic stress.109 Normally, Abl is sequestered in the cytosol by 14‐3‐3 proteins and promotes cell survival. The caspase‐mediated cleavage of Abl results in the separation of the kinase domain from the NES, which leads to its nuclear accumulation.110, 111 Additionally, in response to DNA damage, Abl is phosphorylated by ATM and DNA‐PK, whereas activated JNK phosphorylates 14‐3‐3, resulting in the entry of Abl into the nucleus.112 Nuclear localization of full‐length Abl is critical for DNA damage‐induced apoptosis.112 In the nucleus, Abl phosphorylates p53, TAp73, and other targets, promoting apoptosis (Figure 2).113 Additionally, caspase‐mediated cleavage of the retinoblastoma (Rb) protein, a negative regulator of Abl,114 may robustly amplify the pro‐apoptotic Abl pathway in the nucleus. The oncogenic fusion protein Bcr‐Abl, the major genetic lesion in chronic myelogeneous leukemia and a target for the drug imatinib, is exclusively localized to the cytoplasm, where it promotes proliferation. Interestingly, the forced translocation of Bcr‐Abl to the nucleus promotes caspase activation115, 116 and cell death,109 suggesting that mimicking caspase activity to induce Abl relocalization could be a potent anti‐cancer therapy.117
Upon exposure to genotoxic stress, one of the main tumour suppressors—the p53 protein—undergoes post‐translational modifications, stabilization and nuclear translocation, resulting in the transcription of a huge number of p53 targets. Caspases can regulate p53 status via cleavage of the enzymes involved in post‐translational modifications of p53. In normal cells, Mdm2 regulates p53 by promoting its nuclear export and degradation through the ubiquitin‐proteasome pathway.118 DNA damage and some other stimuli promote a series of reversible post‐translational modifications of p53, including multisite phosphorylation of the transactivation domain, and the accumulation of p53 in the nucleus. A set of proteins regulates this cascade, including ATM, DYRK2, PKCδ, etc.109, 119, 120 For example, caspase‐activated PKCδ phosphorylates p53 on Ser46, which potentiates p53‐dependent apoptosis in response to genotoxic stress DNA damage (Figure 2).120 Moreover, PKCδ has been shown to transactivate p53 expression. On the other hand, kinase‐mediated regulation of p53 status could require additional partners such as an adaptor or scaffold protein. Thus, during apoptosis, caspases cleave Golgi protein p115, generating 205 amino acid C‐terminal fragment. This fragment translocates into the nucleus and promotes p53‐ERK1 interaction for the amplification of apoptotic cell death.121 Notably, caspases could play a role in the regulation of p53, not only via the cleavage of kinases and following phosphorylation, but also via the alteration of p53 ubiquitination. Mdm2 is known to target p53 degradation through its E3 ubiquitin‐protein ligase activity. The disruption of the interaction between Mdm2 and p53 is one of the most promising strategies for the improvement of anti‐cancer therapy.122 Upon genotoxic stress, caspase‐2 has been shown to directly cleave Mdm2 at Asp367 (Figure 2), leading to the disconnection of its C‐terminal RING domain which is responsible for p53 ubiquitination. N‐terminally truncated Mdm2 promotes p53 stability.123 Furthermore, this role of caspase‐2 in the inhibition of Mdm2 and reinforcing p53 stability and activity is essential for the prevention of chromosomal instability and the suppression of tumours.88, 124 Taken together, despite the common view that p53 triggers caspase activation, vice versa, caspases can regulate p53 stabilization and activation, amplifying apoptotic processes.
Additionally, the Cdk inhibitors p21Cip1/Waf1, p27Kip1 and Wee1 were demonstrated to be proteolytically inactivated by caspases in cells undergoing apoptosis, consequently triggering an increase in Cdk2 activity.104 In contrast, Cdc25A, an activator of Cdk2, is activated by caspase‐3 cleavage, generating NLS‐containing fragment with enhanced phosphatase activity that is retained in the nucleus to induce the activation of Cdk2.125 Similarly, another activator of Cdk2, Cyclin E, is also a caspase target126; the cleavage of the fragment sequesters the Bax‐binding protein Ku70, allowing the release of Bax from its inactive complex.127
These findings collectively indicate that the caspase‐mediated nuclear targeting of pro‐apoptotic proteins might be a pivotal regulatory mechanism for cell fate that directly influences the induction of apoptosis. Caspases could control the nuclear level of apoptosis execution directly and indirectly, which underlies the importance of further analysis of the role of caspases as apoptosis regulators on a genetic level. Importantly, in many cases, caspase‐mediated proteolysis results in the generation of proteins that not only lose their normal function, but also acquire a pro‐apoptotic activity, and therefore, further facilitate the development of apoptotic cell death. Hence, further investigations of the caspase functions in the nucleus will undoubtedly provide new views on the execution of apoptosis and pave the way for the development of new therapies against diseases with apoptosis deregulation.
6. CONCLUSIONS AND OUTLOOK
The degradation of the nucleus represents an important step and hallmark of apoptotic cell death that is crucial for impeding the release of potentially immunogenic nuclear proteins and nucleic acids into the extracellular space.128, 129 Taking into account the key role of caspases in its accomplishment, their entry into the nucleus might constitute an important regulatory step during apoptosis. However, the mechanisms by which these cytosolic proteases perform the cleavage of proteins located in the nucleus are not clear and demand further investigation. Even the changes in localization patterns of caspases during apoptosis remain contradictory. Advancing our knowledge of apoptotic mechanisms is not only of great significance for improving our general understanding of apoptosis signalling, but also for the development of novel therapeutic strategies, as the deregulation of apoptotic pathways is associated with various pathological conditions, such as cancer, neurodegenerative disorders and viral infections. Caspases, being the key players in apoptosis, are especially promising targets for pharmacological modulation, and the stimulation or inhibition of their functions in the nucleus represents an important way to regulate its activity. Therefore, further studies are needed to improve our understanding of their nuclear translocation and function.
Nucleocytoplasmic transport is regulated by cellular signalling systems via cargo protein modifications and the transport machinery, including the transport receptors, the NPC and the Ran system. Targeting nucleocytoplasmic transport represents a new therapeutic avenue to treat cancers that display an aberrant protein subcellular localization.130, 131 Given the broad effect of Ran‐GTP levels on apoptosis, the Ran system could be a potential target for developing therapeutic reagents that promote or suppress cell death. For instance, when Ran expression was suppressed specifically in neuroblastoma cells by siRNA in mice, both tumour growth and apoptosis were reduced.132 Therapeutic agents that attempt to normalize or target protein localization are aimed at various regulatory components within the transport process, including the upstream regulatory components, cargo proteins, transport receptors, Ran regulators and the NPC itself. In some cases, compounds have been developed to successfully influence subcellular protein distribution in disease states, including CRM1 and importin α/ß inhibitors. Further research aimed at better understanding the nuclear transport mechanisms will likely reveal novel targets for the treatment of cancer and viral infections. The ability to avoid apoptosis by regulating the cellular localization of key effector enzymes, rather than preventing activation of the apoptotic cascade, brings the possibility for novel therapeutic targets as well as other disease states where apoptosis is a key pathogenic factor.
In the course of apoptosis, caspases obtain access to the external nuclear components as they are located in the cytoplasm. An important part of promoting apoptosis is the active disruption of nuclear trafficking before the NE breakdown. It seems that apoptotic disturbances in the nucleocytoplasmic transport might allow the entrance of caspases to the nucleus. Importantly, the redistribution of Ran and other nucleocytoplasmic transport factors is not limited to the initiation of apoptosis, but is also observed in response to various stresses, both physiological and pathological.
The apoptotic changes of the Ran gradient might also play an essential role in the last stage of nuclear demolition. It is known that actin–myosin‐II activity is crucial for the nuclear remodeling observed during the execution phase of apoptosis.55, 133 During the latter stages of the execution phase, actin–myosin‐II drives the movement of apoptotic nuclear fragments into surface blebs.133 Given the close association between microtubules and nuclear fragments in apoptotic cells,134 it is possible that factor(s) associated with or released from the apoptotic nucleus contribute to microtubule assembly. Thus, Ran‐GTP is released from the apoptotic nucleus and retains its GTP status in the cytoplasm that might stimulate apoptotic microtubule assembly. Due to the continued association of RanGEF with apoptotic chromatin and the rapid loss of the NPC selectivity towards soluble components,17, 135 a gradient of Ran‐GTP would be established in the vicinity of the apoptotic nucleus, promoting microtubule growth and stabilization towards apoptotic nuclear fragments. Moreover, proteolytic cleavage of the C‐terminus of α‐tubulin may also contribute to apoptotic microtubule stabilization. These non‐centrosomal microtubule arrays subsequently assist in the fragmentation of the apoptotic cell.
Thus, it became clear that cell death pathways may be regulated by nuclear transport at different levels and with different outcomes depending on the cellular context and the type of insult. Of particular note, an additional layer of regulation is post‐translational modifications of the nucleocytoplasmic transport factors and NE components, although the available data are scarce, and further research is needed to form a more general picture. Understanding how post‐translational modifications affect the nucleocytoplasmic transport is a comprehensive task, and it is very likely that there are many cargos regulated by post‐translational modifications.
Caspases are involved not only in alterations of nuclear trafficking but also in the activation of pro‐apoptotic factors which regulate post‐translational modifications and transcription of apoptosis‐related proteins, as well as chromatin condensation and DNA fragmentation. Intriguingly, together with the disruption of protein structures and functions via cleavage, caspases can target multiple proteins to form active products with stably folded functional domains. This suggests that rather than the complete degradation of proteins, the apoptotic proteolytic cascades aim at the generation of new forms of proteins that may acquire new functions that further contribute to the self‐killing event.
ACKNOWLEDGEMENTS
This work was supported by the Grant from the Russian Science Foundation (17‐75‐20102). The work in the authors’ laboratories is also supported by the Grants from the Russian Foundation for Basic Research, the Swedish Research Foundation, the Swedish and Stockholm Cancer Societies and Swedish Childhood Cancer Foundation. We apologize to those authors whose primary works could not be cited owing to space limitations.
Kopeina GS, Prokhorova EA, Lavrik IN, Zhivotovsky B. Alterations in the nucleocytoplasmic transport in apoptosis: Caspases lead the way. Cell Prolif. 2018;51:e12467 10.1111/cpr.12467
Gelina S. Kopeina and Evgeniia A. Prokhorova are the authors who contributed equally to this work.
REFERENCES
- 1. Galluzzi L, Bravo‐San Pedro JM, Vitale I, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crawford ED, Seaman JE, Agard N, et al. The DegraBase: a database of proteolysis in healthy and apoptotic human cells. Mol Cell Proteomics. 2013;12:813‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Julien O, Zhuang M, Wiita AP, et al. Quantitative MS‐based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles. Proc Natl Acad Sci. 2016;113:E2001‐E2010. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang C, Ye M, Wei X, Bian Y, Cheng K, Zou H. A bead‐based cleavage method for large‐scale identification of protease substrates. Sci Rep. 2016;6:22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kerr JFR, Wyllie AH, Currie ARD. Apoptosis: a basic biological phenomenon with wide‐ranging implications in tissue kinetics. Br J Cancer. 1972;26:239‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferrando‐May E. Nucleocytoplasmic transport in apoptosis. Cell Death Differ. 2005;12:1263‐1276. [DOI] [PubMed] [Google Scholar]
- 7. Tran EJ, Wente SR. Dynamic nuclear pore complexes: life on the edge. Cell. 2006;125:1041‐1053. [DOI] [PubMed] [Google Scholar]
- 8. Alber F, Dokudovskaya S, Veenhoff LM, et al. The molecular architecture of the nuclear pore complex. Nature. 2007;450:695‐701. [DOI] [PubMed] [Google Scholar]
- 9. Ribbeck K, Görlich D. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J. 2002;21:2664‐2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kabachinski G, Schwartz TU. The nuclear pore complex ‐ structure and function at a glance. J Cell Sci. 2015;128:423‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Knockenhauer KE, Schwartz TU. The nuclear pore complex as a flexible and dynamic gate. Cell. 2016;164:1162‐1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Conti E, Müller CW, Stewart M. Karyopherin flexibility in nucleocytoplasmic transport. Curr Opin Struct Biol. 2006;16:237‐244. [DOI] [PubMed] [Google Scholar]
- 13. Prokhorova EA, Zamaraev AV, Kopeina GS, Zhivotovsky B, Lavrik IN. Role of the nucleus in apoptosis: signaling and execution. Cell Mol Life Sci. 2015;72:4593‐4612. 10.1007/s00018-015-2031-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kramer A, Liashkovich I, Oberleithner H, Ludwig S, Mazur I, Shahin V. Apoptosis leads to a degradation of vital components of active nuclear transport and a dissociation of the nuclear lamina. Proc Natl Acad Sci U S A. 2008;105:11236‐11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kramer A, Liashkovich I, Oberleithner H, Shahin V. Caspase‐9‐dependent decrease of nuclear pore channel hydrophobicity is accompanied by nuclear envelope leakiness. Nanomedicine. 2010;6:605‐611. [DOI] [PubMed] [Google Scholar]
- 16. Kodiha M, Chu A, Matusiewicz N, Stochaj U. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 2004;11:862‐874. [DOI] [PubMed] [Google Scholar]
- 17. Ferrando‐May E, Cordes VC, Biller‐Ckovric I, Mirkovic J, Görlich D, Nicotera P. Caspases mediate nucleoporin cleavage, but not early redistribution of nuclear transport factors and modulation of nuclear permeability in apoptosis. Cell Death Differ. 2001;8:495‐505. [DOI] [PubMed] [Google Scholar]
- 18. Tachibana T, Sakaguchi N, Miyamoto Y, Sekimoto T, Yoneda Y, Azuma M. Generation and characterization of a monoclonal antibody against NPI‐1 subfamily of importin alpha. Hybridoma (Larchmt). 2008;27:285‐289. [DOI] [PubMed] [Google Scholar]
- 19. Kodiha M, Bański P, Ho‐Wo‐Cheong D, Stochaj U. Dissection of the molecular mechanisms that control the nuclear accumulation of transport factors importin‐α and CAS in stressed cells. Cell Mol Life Sci. 2008;65:1756‐1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu M, Zak J, Chen S, et al. A code for RanGDP binding in ankyrin repeats defines a nuclear import pathway. Cell. 2014;157:1130‐1145. [DOI] [PubMed] [Google Scholar]
- 21. Yasuda Y, Miyamoto Y, Yamashiro T, et al. Nuclear retention of importin α coordinates cell fate through changes in gene expression. EMBO J. 2012;31:83‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miyamoto Y, Loveland KL, Yoneda Y. Nuclear importin alpha and its physiological importance. Commun Integr Biol. 2012;5:220‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grote P, Schaeuble K, Ferrando‐May E. Commuting (to) suicide: an update on nucleocytoplasmic transport in apoptosis. Arch Biochem Biophys. 2007;462:156‐161. [DOI] [PubMed] [Google Scholar]
- 24. Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis‐inducing factor. Nature. 1999;397:441‐446. [DOI] [PubMed] [Google Scholar]
- 25. Stegh AH, Schickling O, Ehret A, et al. DEDD, a novel death effector domain‐containing protein, targeted to the nucleolus. EMBO J. 1998;17:5974‐5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wong C‐H, Chan H, Ho C‐Y, et al. Apoptotic histone modification inhibits nuclear transport by regulating RCC1. Nat Cell Biol. 2009;11:36‐45. [DOI] [PubMed] [Google Scholar]
- 27. Wilde A, Zheng Y. Ran out of the nucleus for apoptosis. Nat Cell Biol. 2009;11:11‐12. [DOI] [PubMed] [Google Scholar]
- 28. Tsubaki M, Ogawa N, Takeda T, et al. Dimethyl fumarate induces apoptosis of hematopoietic tumor cells via inhibition of NF‐κB nuclear translocation and down‐regulation of Bcl‐xL and XIAP. Biomed Pharmacother. 2014;68:999‐1005. [DOI] [PubMed] [Google Scholar]
- 29. Miyamoto Y, Saiwaki T, Yamashita J, et al. Cellular stresses induce the nuclear accumulation of importin alpha and cause a conventional nuclear import block. J Cell Biol. 2004;165:617‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stochaj U, Rassadi R, Chiu J. Stress‐mediated inhibition of the classical nuclear protein import pathway and nuclear accumulation of the small GTPase Gsp1p. FASEB J. 2000;14:2130‐2132. [DOI] [PubMed] [Google Scholar]
- 31. Schwoebel ED, Ho TH, Moore MS. The mechanism of inhibition of Ran‐dependent nuclear transport by cellular ATP depletion. J Cell Biol. 2002;157:963‐974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Furuta M, Kose S, Koike M, et al. Heat‐shock induced nuclear retention and recycling inhibition of importin alpha Genes Cells. 2004;9:429‐441. [DOI] [PubMed] [Google Scholar]
- 33. Lowe AR, Tang JH, Yassif J, et al. Importin‐β modulates the permeability of the nuclear pore complex in a Ran‐dependent manner. Elife. 2015;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Faleiro L, Lazebnik Y. Caspases disrupt the nuclear‐cytoplasmic barrier. J Cell Biol. 2000;151:951‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Higby KJ, Bischak MM, Campbell CA, et al. 5‐Flurouracil disrupts nuclear export and nuclear pore permeability in a calcium dependent manner. Apoptosis. 2017;22:393‐405. [DOI] [PubMed] [Google Scholar]
- 36. Li HY, Wirtz D, Zheng Y. A mechanism of coupling RCC1 mobility to RanGTP production on the chromatin in vivo. J Cell Biol. 2003;160:635‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lazebnik YA, Cole S, Cooke CA, Nelson WG, Earnshaw WC. Nuclear events of apoptosis in vitro in cell‐free mitotic extracts: a model system for analysis of the active phase of apoptosis. J Cell Biol. 1993;123:7‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buendia B, Santa‐Maria A, Courvalin JC. Caspase‐dependent proteolysis of integral and peripheral proteins of nuclear membranes and nuclear pore complex proteins during apoptosis. J Cell Sci. 1999;112:1743‐1753. [DOI] [PubMed] [Google Scholar]
- 39. Falcieri E, Gobbi P, Cataldi A, Zamai L, Faenza I, Vitale M. Nuclear pores in the apoptotic cell. Histochem J. 1994;26:754‐763. [DOI] [PubMed] [Google Scholar]
- 40. Slee EA, Adrain C, Martin SJ. Executioner caspase‐3, ‐6, and ‐7 perform distinct, non‐redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320‐7326. [DOI] [PubMed] [Google Scholar]
- 41. Ruchaud S, Korfali N, Villa P, et al. Caspase‐6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 2002;21:1967‐1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cuvillier O, Rosenthal DS, Smulson ME, Spiegel S. Sphingosine 1‐phosphate inhibits activation of caspases that cleave poly(ADP‐ribose) polymerase and lamins during Fas‐ and ceramide‐mediated apoptosis in Jurkat T lymphocytes. J Biol Chem. 1998;273:2910‐2916. [DOI] [PubMed] [Google Scholar]
- 43. Stewart CL, Roux KJ, Burke B. Blurring the boundary: the nuclear envelope extends its reach. Science. 2007;318:1408‐1412. [DOI] [PubMed] [Google Scholar]
- 44. Boulares AH, Yakovlev AG, Ivanova V, et al. Role of poly(ADP‐ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3‐resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932‐22940. [DOI] [PubMed] [Google Scholar]
- 45. Patre M, Tabbert A, Hermann D, et al. Caspases target only two architectural components within the core structure of the nuclear pore complex. J Biol Chem. 2006;281:1296‐1304. [DOI] [PubMed] [Google Scholar]
- 46. Sachdev R, Sieverding C, Flötenmeyer M, Antonin W. The C‐terminal domain of Nup93 is essential for assembly of the structural backbone of nuclear pore complexes. Mol Biol Cell. 2012;23:740‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fischer U, Jänicke RU, Schulze‐Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fornerod M, van Deursen J, van Baal S, et al. The human homologue of yeast CRM1 is in a dynamic subcomplex with CAN/Nup214 and a novel nuclear pore component Nup88. EMBO J. 1997;16:807‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Makise M, Mackay DR, Elgort S, Shankaran SS, Adam SA, Ullman KS. The Nup153‐Nup50 protein interface and its role in nuclear import. J Biol Chem. 2012;287:38515‐38522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ogawa Y, Miyamoto Y, Oka M, Yoneda Y. The interaction between importin‐alpha and Nup153 promotes importin‐alpha/beta‐mediated nuclear import. Traffic. 2012;13:934‐946. [DOI] [PubMed] [Google Scholar]
- 51. Taimen P, Kallajoki M. NuMA and nuclear lamins behave differently in Fas‐mediated apoptosis. J Cell Sci. 2003;116:571‐583. [DOI] [PubMed] [Google Scholar]
- 52. Gerner C, Gotzmann J, Frohwein U, Schamberger C, Ellinger A, Sauermann G. Proteome analysis of nuclear matrix proteins during apoptotic chromatin condensation. Cell Death Differ. 2002;9:671‐681. [DOI] [PubMed] [Google Scholar]
- 53. Broers JLV, Ramaekers FCS. The role of the nuclear lamina in cancer and apoptosis. Adv Exp Med Biol. 2014;773:27‐48. [DOI] [PubMed] [Google Scholar]
- 54. Wu J, Jiang H, Luo S, et al. Caspase‐mediated cleavage of C53/LZAP protein causes abnormal microtubule bundling and rupture of the nuclear envelope. Cell Res. 2013;23:691‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Croft DR, Coleman ML, Li S, et al. Actin‐myosin‐based contraction is responsible for apoptotic nuclear disintegration. J Cell Biol. 2005;168:245‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martins LM, Kottke T, Mesner PW, et al. Activation of multiple interleukin‐1beta converting enzyme homologues in cytosol and nuclei of HL‐60 cells during etoposide‐induced apoptosis. J Biol Chem. 1997;272:7421‐7430. [DOI] [PubMed] [Google Scholar]
- 57. Nakagawara A, Nakamura Y, Ikeda H, et al. High levels of expression and nuclear localization of interleukin‐1 beta converting enzyme (ICE) and CPP32 in favorable human neuroblastomas. Cancer Res. 1997;57:4578‐4584. [PubMed] [Google Scholar]
- 58. Yasuhara N, Eguchi Y, Tachibana T, Imamoto N, Yoneda Y, Tsujimoto Y. Essential role of active nuclear transport in apoptosis. Genes Cells. 1997;2:55‐64. [DOI] [PubMed] [Google Scholar]
- 59. Kuwana T, Smith JJ, Muzio M, Dixit V, Newmeyer DD, Kornbluth S. Apoptosis induction by caspase‐8 is amplified through the mitochondrial release of cytochrome c. J Biol Chem. 1998;273:16589‐16594. [DOI] [PubMed] [Google Scholar]
- 60. Köhler C, Håkansson A, Svanborg C, Orrenius S, Zhivotovsky B. Protease activation in apoptosis induced by MAL. Exp Cell Res. 1999;249:260‐268. [DOI] [PubMed] [Google Scholar]
- 61. Zhivotovsky B, Samali A, Gahm A, Orrenius S. Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ. 1999;6:644‐651. [DOI] [PubMed] [Google Scholar]
- 62. Cao G, Pei W, Lan J, et al. Caspase‐activated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci. 2001;21:4678‐4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Joseph B, Ekedahl J, Lewensohn R, Marchetti P, Formstecher P, Zhivotovsky B. Defective caspase‐3 relocalization in non‐small cell lung carcinoma. Oncogene. 2001;20:2877‐2888. [DOI] [PubMed] [Google Scholar]
- 64. Takemoto K, Nagai T, Miyawaki A, Miura M. Spatio‐temporal activation of caspase revealed by indicator that is insensitive to environmental effects. J Cell Biol. 2003;160:235‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. Nuclear translocation of caspase‐3 is dependent on its proteolytic activation and recognition of a substrate‐like protein(s). J Biol Chem. 2005;280:857‐860. [DOI] [PubMed] [Google Scholar]
- 66. Luo M, Lu Z, Sun H, et al. Nuclear entry of active caspase‐3 is facilitated by its p3‐recognition‐based specific cleavage activity. Cell Res. 2010;20:211‐222. [DOI] [PubMed] [Google Scholar]
- 67. Romero MJ, Yao L, Sridhar S, et al. I‐Citrulline protects from kidney damage in type 1 diabetic mice. Front Immunol. 2013;4:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Warby SC, Doty CN, Graham RK, et al. Activated caspase‐6 and caspase‐6‐cleaved fragments of huntingtin specifically colocalize in the nucleus. Hum Mol Genet. 2008;17:2390‐2404. [DOI] [PubMed] [Google Scholar]
- 69. Svandova E, Lesot H, Vanden Berghe T, et al. Non‐apoptotic functions of caspase‐7 during osteogenesis. Cell Death Dis. 2014;5:e1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Matalova E, Vanden Berghe T, Svandova E, et al. Caspase‐7 in molar tooth development. Arch Oral Biol. 2012;57:1474‐1481. [DOI] [PubMed] [Google Scholar]
- 71. Hayashi N, Shirakura H, Uehara T, Nomura Y. Relationship between SUMO‐1 modification of caspase‐7 and its nuclear localization in human neuronal cells. Neurosci Lett. 2006;397:5‐9. 10.1016/j.neulet.2005.11.057. [DOI] [PubMed] [Google Scholar]
- 72. Waldron‐Roby E, Hoerauf J, Arbez N, Zhu S, Kulcsar K, Ross CA. Sox11 reduces Caspase‐6 cleavage and activity. PLoS ONE. 2015;10:e0141439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Benchoua A, Couriaud C, Guégan C, et al. Active caspase‐8 translocates into the nucleus of apoptotic cells to inactivate poly(ADP‐ribose) polymerase‐2. J Biol Chem. 2002;277:34217‐34222. [DOI] [PubMed] [Google Scholar]
- 74. Shikama Y, Mami U, Miyashita T, Yamada M. Comprehensive studies on subcellular localizations and cell death‐inducing activities of eight GFP‐tagged apoptosis‐related caspases. Exp Cell Res. 2001;264:315‐325. [DOI] [PubMed] [Google Scholar]
- 75. Besnault‐Mascard L, Leprince C, Auffredou MT, et al. Caspase‐8 sumoylation is associated with nuclear localization. Oncogene. 2005;24:3268‐3273. 10.1038/sj.onc.1208448. [DOI] [PubMed] [Google Scholar]
- 76. Alcivar A, Hu S, Tang J, Yang X. DEDD and DEDD2 associate with caspase‐8/10 and signal cell death. Oncogene. 2003;22:291‐297. [DOI] [PubMed] [Google Scholar]
- 77. Faragher AJ, Sun XM, Butterworth M, et al. Death receptor‐induced apoptosis reveals a novel interplay between the chromosomal passenger complex and CENP‐C during interphase. Mol Biol Cell. 2007;18:1337‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP‐ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346‐347. [DOI] [PubMed] [Google Scholar]
- 79. Colussi PA, Harvey NL, Kumar S. Prodomain‐dependent nuclear localization of the caspase‐2 (Nedd2) precursor: a novel function for a caspase prodomain. J Biol Chem. 1998;273:24535‐24542. [DOI] [PubMed] [Google Scholar]
- 80. Baliga BC, Colussi PA, Read SH, Dias MM, Jans DA, Kumar S. Role of prodomain in importin‐mediated nuclear localization and activation of caspase‐2. J Biol Chem. 2003;278:4899‐4905. [DOI] [PubMed] [Google Scholar]
- 81. Paroni G, Henderson C, Schneider C, Brancolini C. Caspase‐2 can trigger cytochrome C release and apoptosis from the nucleus. J Biol Chem. 2002;277:15147‐15161. [DOI] [PubMed] [Google Scholar]
- 82. Shirakura H, Hayashi N, Ogino S, Tsuruma K, Uehara T, Nomura Y. Caspase recruitment domain of procaspase‐2 could be a target for SUMO‐1 modification through Ubc9. Biochem Biophys Res Commun. 2005;331:1007‐1015. 10.1016/j.bbrc.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 83. Sanchez‐Pulido L, Valencia A, Rojas AM. Are promyelocytic leukaemia protein nuclear bodies a scaffold for caspase‐2 programmed cell death? Trends Biochem Sci. 2007;32:400‐406. [DOI] [PubMed] [Google Scholar]
- 84. Tang J, Xie W, Yang X. Association of caspase‐2 with the promyelocytic leukemia protein nuclear bodies. Cancer Biol Ther. 2005;4:645‐649. [DOI] [PubMed] [Google Scholar]
- 85. Bouchier‐Hayes L, Oberst A, McStay GP, et al. Characterization of cytoplasmic caspase‐2 activation by induced proximity. Mol Cell. 2009;35:830‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ando K, Kernan JL, Liu PH, et al. PIDD death‐domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling. Mol Cell. 2012;47:681‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, Kumar S. A tumor suppressor function for caspase‐2. Proc Natl Acad Sci U S A. 2009;106:5336‐5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. López‐García C, Sansregret L, Domingo E, et al. BCL9L dysfunction impairs caspase‐2 expression permitting aneuploidy tolerance in colorectal cancer. Cancer Cell. 2017;31:79‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Timney BL, Raveh B, Mironska R, et al. Simple rules for passive diffusion through the nuclear pore complex. J Cell Biol. 2016;215:57‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wühr M, Güttler T, Peshkin L, et al. The nuclear proteome of a vertebrate. Curr Biol. 2015;25:2663‐2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kumeta M, Yamaguchi H, Yoshimura SH, Takeyasu K. Karyopherin‐independent spontaneous transport of amphiphilic proteins through the nuclear pore. J Cell Sci. 2012;125:4979‐4984. [DOI] [PubMed] [Google Scholar]
- 92. Yoshimura SH, Kumeta M, Takeyasu K. Structural mechanism of nuclear transport mediated by importin β and flexible amphiphilic proteins. Structure. 2014;22:1699‐1710. [DOI] [PubMed] [Google Scholar]
- 93. Nardozzi JD, Lott K, Cingolani G. Phosphorylation meets nuclear import: a review. Cell Commun Signal CCS. 2010;8:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liu X, Li P, Widlak P, et al. The 40‐kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc Natl Acad Sci U S A. 1998;95:8461‐8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase‐activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43‐50. [DOI] [PubMed] [Google Scholar]
- 96. Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96‐99. [DOI] [PubMed] [Google Scholar]
- 97. Nagata S, Nagase H, Kawane K, Mukae N, Fukuyama H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003;10:108‐116. [DOI] [PubMed] [Google Scholar]
- 98. Kovacsovics M, Martinon F, Micheau O, Bodmer JL, Hofmann K, Tschopp J. Overexpression of Helicard, a CARD‐containing helicase cleaved during apoptosis, accelerates DNA degradation. Curr Biol. 2002;12:838‐843. [DOI] [PubMed] [Google Scholar]
- 99. Errami Y, Naura AS, Kim H, et al. Apoptotic DNA fragmentation may be a cooperative activity between caspase‐activated deoxyribonuclease and the Poly(ADP‐ribose) Polymerase‐regulated DNAS1L3, an endoplasmic reticulum‐localized endonuclease that translocates to the nucleus during apoptosis. J Biol Chem. 2013;288:3460‐3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861‐2874. [DOI] [PubMed] [Google Scholar]
- 101. Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak‐mediated permeabilization. EMBO J. 2003;22:4385‐4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kalinowska M, Garncarz W, Pietrowska M, Garrard WT, Widlak P. Regulation of the human apoptotic DNase/RNase Endonuclease G: involvement of Hsp70 and ATP. Apoptosis. 2005;10:821‐830. [DOI] [PubMed] [Google Scholar]
- 103. Marabese M, Mazzoletti M, Vikhanskaya F, Broggini M. HtrA2 enhances the apoptotic functions of p73 on bax. Cell Death Differ. 2008;15:849‐858. [DOI] [PubMed] [Google Scholar]
- 104. Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell. 2009;138:838‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Graves JD, Draves KE, Gotoh Y, Krebs EG, Clark EA. Both phosphorylation and caspase‐mediated cleavage contribute to regulation of the Ste20‐like protein kinase Mst1 during CD95/Fas‐induced apoptosis. J Biol Chem. 2001;276:14909‐14915. [DOI] [PubMed] [Google Scholar]
- 106. Hu Y, Liu Z, Yang S‐J, Ye K. Acinus‐provoked protein kinase C delta isoform activation is essential for apoptotic chromatin condensation. Cell Death Differ. 2007;14:2035‐2046. [DOI] [PubMed] [Google Scholar]
- 107. Cheung WL, Ajiro K, Samejima K, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113:507‐517. [DOI] [PubMed] [Google Scholar]
- 108. Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281‐294. [DOI] [PubMed] [Google Scholar]
- 109. Yoshida K. Nuclear trafficking of pro‐apoptotic kinases in response to DNA damage. Trends Mol Med. 2008;14:305‐313. [DOI] [PubMed] [Google Scholar]
- 110. Barilà D, Rufini A, Condò I, et al. Caspase‐dependent cleavage of c‐Abl contributes to apoptosis. Mol Cell Biol. 2003;23:2790‐2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Machuy N, Rajalingam K, Rudel T. Requirement of caspase‐mediated cleavage of c‐Abl during stress‐induced apoptosis. Cell Death Differ. 2004;11:290‐300. [DOI] [PubMed] [Google Scholar]
- 112. Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14‐3‐3 proteins regulates nuclear targeting of c‐Abl in the apoptotic response to DNA damage. Nat Cell Biol. 2005;7:278‐285. [DOI] [PubMed] [Google Scholar]
- 113. Wang JY. Regulation of cell death by the Abl tyrosine kinase. Oncogene. 2000;19:5643‐5650. [DOI] [PubMed] [Google Scholar]
- 114. Jänicke RU, Walker PA, Lin XY, Porter AG. Specific cleavage of the retinoblastoma protein by an ICE‐like protease in apoptosis. EMBO J. 1996;15:6969‐6978. [PMC free article] [PubMed] [Google Scholar]
- 115. Dixon AS, Kakar M, Schneider KMH, Constance JE, Paullin BC, Lim CS. Controlling subcellular localization to alter function: sending oncogenic Bcr‐Abl to the nucleus causes apoptosis. J Control Release. 2009;140:245‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Vigneri P, Wang JY. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR‐ABL tyrosine kinase. Nat Med. 2001;7:228‐234. [DOI] [PubMed] [Google Scholar]
- 117. Crawford ED, Wells JA. Caspase substrates and cellular remodeling. Annu Rev Biochem. 2011;80:1055‐1087. [DOI] [PubMed] [Google Scholar]
- 118. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296‐299. [DOI] [PubMed] [Google Scholar]
- 119. Taira N, Yamamoto H, Yamaguchi T, Miki Y, Yoshida K. ATM augments nuclear stabilization of DYRK2 by inhibiting MDM2 in the apoptotic response to DNA damage. J Biol Chem. 2010;285:4909‐4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yoshida K, Liu H, Miki Y. Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J Biol Chem. 2006;281:5734‐5740. [DOI] [PubMed] [Google Scholar]
- 121. How PC, Shields D. Tethering function of the caspase cleavage fragment of golgi protein p115 promotes apoptosis via a p53‐dependent pathway. J Biol Chem. 2011;286:8565‐8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science. 2004;303:844‐848. [DOI] [PubMed] [Google Scholar]
- 123. Oliver TG, Meylan E, Chang GP, et al. Caspase‐2‐mediated cleavage of Mdm2 creates a p53‐induced positive feedback loop. Mol Cell. 2011;43:57‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Fava LL, Schuler F, Sladky V, et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017;31:34‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mazars A, Fernandez‐Vidal A, Mondesert O, et al. A caspase‐dependent cleavage of CDC25A generates an active fragment activating cyclin‐dependent kinase 2 during apoptosis. Cell Death Differ. 2009;16:208‐218. [DOI] [PubMed] [Google Scholar]
- 126. Mazumder S, Gong B, Chen Q, Drazba JA, Buchsbaum JC, Almasan A. Proteolytic cleavage of cyclin E leads to inactivation of associated kinase activity and amplification of apoptosis in hematopoietic cells. Mol Cell Biol. 2002;22:2398‐2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Mazumder S, Plesca D, Kinter M, Almasan A. Interaction of a cyclin E fragment with Ku70 regulates Bax‐mediated apoptosis. Mol Cell Biol. 2007;27:3511‐3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1‐deficient mice. Nat Genet. 2000;25:177‐181. [DOI] [PubMed] [Google Scholar]
- 129. Kawane K, Fukuyama H, Yoshida H, et al. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol. 2003;4:138‐144. [DOI] [PubMed] [Google Scholar]
- 130. Hill R, Cautain B, de Pedro N, Link W. Targeting nucleocytoplasmic transport in cancer therapy. Oncotarget. 2014;5:11‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Cautain B, de Pedro N, Murillo Garzón V, et al. High‐content screening of natural products reveals novel nuclear export inhibitors. J Biomol Screen. 2014;19:57‐65. [DOI] [PubMed] [Google Scholar]
- 132. Tietze N, Pelisek J, Philipp A, et al. Induction of apoptosis in murine neuroblastoma by systemic delivery of transferrin‐shielded siRNA polyplexes for downregulation of Ran. Oligonucleotides. 2008;18:161‐174. [DOI] [PubMed] [Google Scholar]
- 133. Lane JD, Allan VJ, Woodman PG. Active relocation of chromatin and endoplasmic reticulum into blebs in late apoptotic cells. J Cell Sci. 2005;118:4059‐4071. [DOI] [PubMed] [Google Scholar]
- 134. Moss DK, Betin VM, Malesinski SD, Lane JD. A novel role for microtubules in apoptotic chromatin dynamics and cellular fragmentation. J Cell Sci. 2006;119:2362‐2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Moss DK, Wilde A, Lane JD. Dynamic release of nuclear RanGTP triggers TPX2‐dependent microtubule assembly during the apoptotic execution phase. J Cell Sci. 2009;122:644‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Roehrig S, Tabbert A, Ferrando‐May E. In vitro measurement of nuclear permeability changes in apoptosis. Anal Biochem. 2003;318:244‐253. [DOI] [PubMed] [Google Scholar]
- 137. Kihlmark M, Imreh G, Hallberg E. Sequential degradation of proteins from the nuclear envelope during apoptosis. 2001;114:3643‐3653. [DOI] [PubMed] [Google Scholar]
- 138. Kim BJ, Lee H. Caspase‐mediated cleavage of importin‐α increases its affinity for MCM and downregulates DNA synthesis by interrupting the binding of MCM to chromatin. Biochim Biophys Acta ‐ Mol Cell Res. 2008;1783:2287‐2293. [DOI] [PubMed] [Google Scholar]
- 139. Sun QY, Schatten H. Role of NuMA in vertebrate cells: review of an intriguing multifunctional protein. Front Biosci. 2006;11:1137‐1146. [DOI] [PubMed] [Google Scholar]