

Abstract
Objectives
Colorectal cancer is one of the most common malignancies both in men and women. Owing to metastasis and resistance, the prognosis of colorectal cancerCRC patients remains extremely poor with chemotherapy. A disintegrin and metalloproteinase 17 (ADAM17) induces the activation of Notch pathway and contributes to the chemoresistance. This study aimed to discover a novel ADAM17 inhibitor and investigate the chemosensitization effect.
Materials and methods
Pharmacophore model, western blot and enzymatic assay were used to discover ZLDI‐8. Cell proliferation was determined by MTT and colony formation assay. Cell migratory and invasive ability were determined by wound healing scratch and transwell assay. Immunofluorescence images and western blot analysed the expression of Notch or epithelial‐mesenchymal transition (EMT) pathway markers. Xenografts were employed to evaluate the chemosensitization effect of ZLDI‐8 in vivo.
Results
We found that ZLDI‐8 cell‐specifically inhibited the proliferation of CRC, and this effect was due to abrogation of ADAM17 and Notch pathway. Meanwhile, we reported for the first time that ZLDI‐8 synergistically improved the anti‐tumour and anti‐metastasis activity of 5‐fluorouracil or irinotecan by reversing Notch and EMT pathways. Interestingly, in vivo studies further demonstrated that ZLDI‐8 promoted the anti‐tumour effect of 5‐fluorouracil through Notch and EMT reversal.
Conclusions
A novel ADAM17 inhibitor ZLDI‐8 may be a potential chemosensitizer which sensitized CRC cells to 5‐fluorouracil or irinotecan by reversing Notch and EMT pathways.
1. INTRODUCTION
Colorectal cancer (CRC) is one of the most common malignancies both in men and women.1 In recent years, CRC has a rising mortality rate mainly due to the liver metastases.2, 3 Though surgery is the preferred option for patients with primary CRC and resectable colorectal liver metastases, 80%‐90% of patients with CRC liver metastases are not resectable.3 Therefore, chemotherapy as the effective regimens increased both patient survival and quality of life. 3 FOLFOX and FOLFIRI, as systemic chemotherapies based on 5‐fluorouracil (5‐FU) and irinotecan, have traditionally been treated as first‐line regimens for metastatic CRC (mCRC).4, 5 However, the prognosis of mCRC patients remains extremely poor due to the chemoresistance.6 Nowadays, combination chemotherapy with agents which inhibit key cellular signaling pathways becoming a potential strategy to improve mCRC therapy.7, 8
Notch pathway is a conserved signalling pathway which contributes to cell proliferation, epithelial‐mesenchymal transition (EMT) and chemoresistance.9, 10 Accumulating evidences indicated that Notch signalling pathway is dysregulated in non‐small cell lung cancer (NSCLC), breast cancer and CRC.11, 12, 13 Our previous study demonstrated that NSCLC cells acquire chemoresistance by activating Notch pathway.14 Notch pathway is activated when ligands bind to related transmembrane receptors and cleaved by ADAM17 and a γ‐secretase complex at S2 and S3 sites, then the cytoplasmic portion known as the Notch intracellular domain (NICD) was released.9, 15 ADAM17, also known as TNF‐α converting enzyme (TACE), is responsible for S2 site cleavage and was important to disclose the S3 site in Notch pathway.16 Chemotherapy, using in clinic for first‐line, acutely induces the expression of ADAM1717 and results in chemoresistance. However, there is a small number of ADAM17 inhibitors reach to clinical trials for malignant tumour patients.18 Thus, it is still a tremendous need for a novel inhibitor of ADAM17 to improve anti‐tumour effects of chemotherapy or overcome cellular drug resistance in mCRC patients.
The early step of metastasis induced by activating Notch receptors in CRC was EMT.19, 20 EMT is a highly coordinated process, in which epithelial cells acquired properties of mesenchymal cells.11 Cancer cells, which undergone EMT involved in migration, invasion, chemoresistance21and linked to poor survival of cancer patients.22 Down‐regulated Notch pathway could reverse these effects and induce drug sensitivity.23 We have found that terfenadine sensitized chemoresistant NSCLC cells to epirubicin by inhibiting Notch and EMT pathways.14 RUI WANG et al reported that TAPI‐2, an inhibitor of ADAM17, regulated cancer stem cells phenotype and chemosensitivity (5‐FU) of CRC cells through Notch1 signaling.24
In this study, we performed a virtual screening based on a pharmacophore model of ADAM17 which arranged atoms or groups of atoms in a molecule that determined its bioactivity against SPECS database.25 Biological activity assays demonstrated that ZLDI‐8, 5‐((1‐(2‐(2,4‐dimethylphenoxy)ethyl)‐2‐methyl‐1H‐indol‐3‐yl)methylene)‐2‐thioxodihydropyrimidine‐4,6(1H,5H)‐dione, was the most potential compound against 6 cancer cell lines especially in CRC. It has been reported that ZLDI‐8 inhibited lymphoid‐specific tyrosine phosphatase which predominantly expressed in immune cells for autoimmune diseases.26 Whereas, its anti‐tumour and underlying molecular mechanisms have not been clarified. In this study, we mainly explored the potential mechanism of anti‐proliferation and chemosensitization effect of ZLDI‐8 in vitro and in vivo.
2. MATERIALS AND METHODS
2.1. Computational methods
All computational experiments were conducted on Dell PowerEdge R900 workstation under Redhat 6.4 platform.
2.2. Reagents and antibodies
The selected compounds (ZLDI‐1‐14) were purchased from SPECS database, and the purity was more than 95%. ZLDI‐8 was dissolved in dimethylsulphoxide (DMSO) and stored in −20°C. RPMI 1640 and DMEM were obtained from Hyclone (Logan, UT, USA). Antibodies for Notch1, 2, 3, E‐cadherin, Vimentin, Hes1, Twist1, Survivin were purchased from CST (Beverly, MA). Antibodies against ADAM17 and Notch4 were purchased from Abcam (Cambridge, UK). β‐actin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The secondary antibodies were purchased from ZSGB‐BIO (Beijing, China).
2.3. Cell cultures
The non‐small cell lung cancer (A549), human breast adenocarcinoma (MCF‐7) and human cervical cancer cell (Hela) were donated by Professor Takashi Ike jima from Shenyang Pharmaceutical University, Shenyang, China. Human carcinoma of mouth floor (KB), Human colorectal adenocarcinoma (LoVo and SW480) and hepatocellular carcinoma (HepG2) cells were obtained from the American Type Culture Collection (ATCC). The cells were cultured in DMEM (A549, Hela, HepG2, KB, MCF‐7 and LoVo) and RPMI‐1640 (SW480) medium with 10% foetal bovine serum (FBS) and incubated in a humidified atmosphere containing 5% CO2 at 37°C.
2.4. Cell viability assay
Cancer cells were seeded in 96‐well plates and then treated with ZLDI‐8 with or without different chemotherapeutics for 24 h, 48 h or 72 h. At the end of treatments, 3‐(4, 5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) (0.5 mg/mL) was added to the medium and incubated for another 4 h in the dark. After adding 150 μL DMSO to dissolve the formed insoluble formazan crystals, the optical density values at 490 nm were measured by a microplate reader (Elx 800 Bio‐Tek, USA).
2.5. ADAM17 enzymatic assay
The ADAM17 enzymatic assay was measured by ADAM17 Activity Assay Kit following the manufacturer's protocol. The human purified ADAM17 protein was treated with ZLDI‐8 at different concentrations in a black 384‐well plate, and the fluorescence intensity was measured by an automatic microplate reader.
2.6. Colony formation assay
LoVo and SW480 cells were seeded in a 6‐well plate with a density of 1 × 103 cells/well and treated with ZLDI‐8 (3.13 μM), 5‐FU (2 μM), irinotecan (8 μM) alone or combination. After 7 days, the colonies were stained with crystal violet and quantified by glacial acetic acid using the microplate reader (Elx 800 Bio‐Tek, USA).
2.7. Wound healing scratch assay
LoVo cells (7 × 105/mL) was seeded in 6‐well plates until they reached 90% confluence and formed a monolayer. A volume of 100 μL pipette tip was used to create scratch wounds and the cells were washed with phosphate‐buffered saline (PBS). The migration of cells was observed after treating with ZLDI‐8, 5‐FU/irinotecan alone or combination. Image J software was used to measure the wound area at 0 h and 72 h. Experiments were repeated at least 3 times.
2.8. Cell invasion assay
The invasion ability of LoVo cells was assessed using 24‐well transwell (Corning Life Sciences, Bedford, MA, USA). Matrigel (Becton Dickinson, San Jose, CA, USA) was coated to the transwell filters. The LoVo cells (5 × 105) suspended in serum‐free medium with or without FU/irinotecan were seeded into the upper chamber and the complete medium containing 10% FBS filled the lower chamber. After 48 h, the cells invaded to the lower surface of the transwell membrane were stained with 0.1% crystal violet and were quantified by counting 3 random fields per transwell. The experiments were performed in triplicate.
2.9. Immunofluorescence images
LoVo cells were seeded into 24‐well plates and treated with ZLDI‐8, FU, irinotecan alone or combination for 72 h. Then, the cells were incubated with immunol staining blocking buffer for 1 h and stained with rabbit anti‐Vimentin at 4°C overnight. Afterwards, the cells were incubated with Cy3‐labelled goat anti‐rabbit IgG (H + L) (Beyotime, Beijing, China) for additional 1 h at 37°C and counter‐stained with DAPI for 10 min. The results were carried out for 2 independent experiments.
2.10. Western blot analysis
Cells and tumour tissues were homogenized in RIPA buffer including protease inhibitor. The BCA Protein Assay Kit was used to determine the protein concentration of extracts. The total protein was separated by SDS‐PAGE and electrophoretically transferred onto PVDF membranes. Membranes were immunoblotted using specific primary antibodies overnight and incubated with HRP‐conjugated secondary antibodies. The immunoreactive bands were visualized by ECL detection kit.
2.11. Animal experiments
All 5‐week‐old male BALB/cA‐nu mice were purchased from Beijing Vital River Laboratory Animal Technology in accordance with institutional guidelines. 3 × 106 cells/mouse LoVo cells were injected into the back next to the left forelimb subcutaneously of a nude mouse. Body weight and tumour volume were measured every 2 days and calculated using the formula V = length × width2/2. When the tumours reach to about 120 mm3, 80 mg/kg ZLDI‐8 (oral administration) and 30 mg/kg 5‐FU (intraperitoneal injection, ip) were used alone or combined, normal saline in the NS group. ZLDI‐8 was taken every day and 5‐FU was injected every other day (Figure 7A). After 25 days, tumours were harvested when nude mice sacrificed, and the weight of the tumours in each mouse was evaluated. The same experiments were performed in twice with 7 mouse/group in all.
Figure 7.
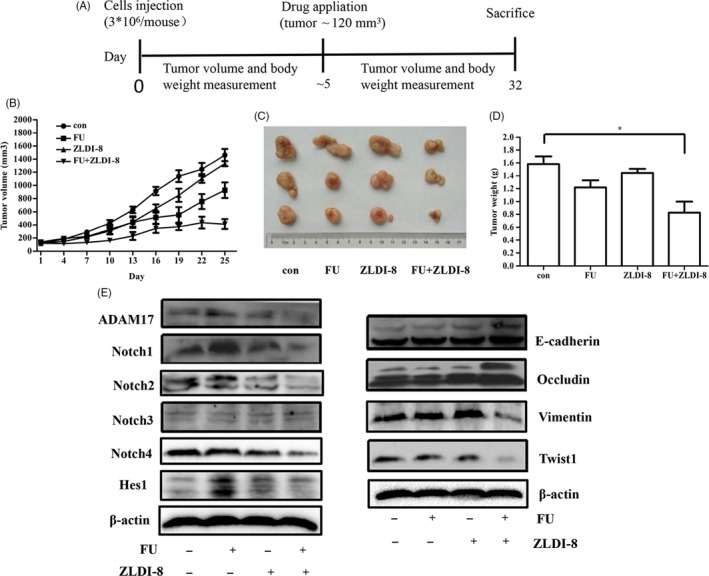
ZLDI‐8 and 5‐FU synergistically inhibited the growth of CRC in xenograft models. A, Schematic diagram of the xenograft model in this study. ZLDI‐8 was suspended in 5‰ CMCNa, 5‐FU was dissolved in normal saline. control (CMCNa,qd×25, Po;normal saline, q4d×25, ip); ZLDI‐8 (qd×25, Po, 80 mg/kg); 5‐FU (q4d×25, ip 30 mg/kg); ZLDI‐8 (qd×25, Po, 80 mg/kg) and 5‐FU (q4d×25, ip 30 mg/kg). B, Tumour volume was measured every other day. (C,D), Tumour weight and imaging of representative tumours from each group were shown at the end of the experiment. E, Tumour lysate come from 3 mice. Noth signalling and EMT markers were analysed by Western blot assay. *P < .05, ***P < .001 vs control
2.12. Statistical analysis
GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis. One‐way analysis of variance (ANOVA) was used to analyse the significance between the groups. All data are expressed as means values ± SEM. P < .05 indicated significance.
2.12.1. Other methods
Please see “Supplementary materials and methods” in Data S1.
3. RESULTS
3.1. Pharmacophore‐based virtual screen to identify a potential ADAM17 inhibitor
Receptor‐Ligand Pharmacophore results showed that the first pharmacophore (FigureS1A) was the most relevant to the Crystal structure of TACE–IK682 complex.27 However, considering the restriction on screening, the secondary features H4 and H5 were deleted manually. Table S1 showed that eighteen co‐crystallized ligands of ADAM1727, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40 could be identified by fitvalues with little change of AbsoluteEnergy and predicted by XSCORE mostly. Therefore, an ideal pharmacophore model (pharmacophore a) was confirmed (Figures 1A and S1B), which including 1 hydrogen bond acceptor (A) mapped on the carbonyl group of the pyrrolidine, 2 hydrogen bond donors (D1 and D2) matched with hydroxamate moiety and 3 hydrophobic‐aliphatic groups (H1, H2 and H3) mapped on the phenyl group and quinolinyl group of IK682. Pharmacophore a was used as a 3D structural query for retrieving SPECS database which was prepared previously.41 As a result, fourteen compounds were selected based on their scaffolds and visual observations (Figure S2).
Figure 1.
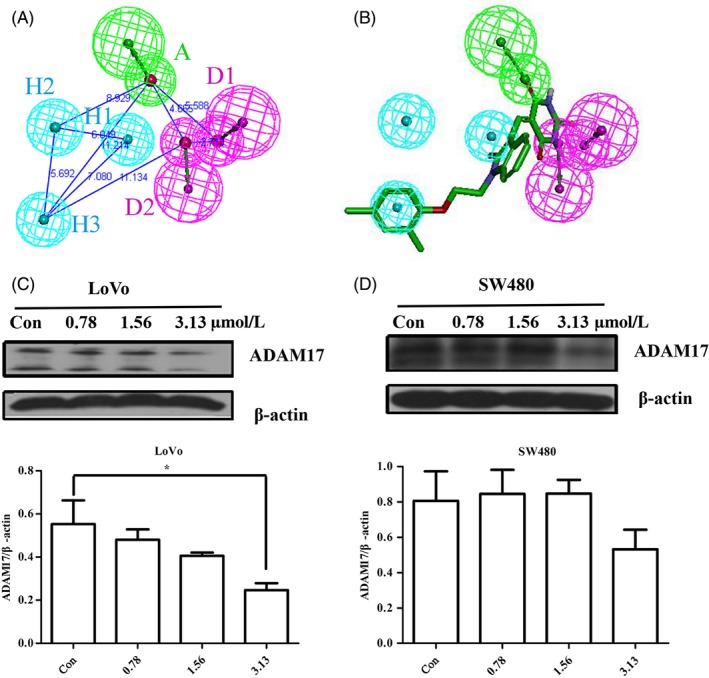
Discovery of a potential ADAM17 inhibitor based on pharmacophore hypothesis. A, Pharmacophore a consisting of 1 hydrogen bond acceptor A (green), 2 hydrogen bond donor D1 and D2 (magenta) and 3 hydrophobic‐aliphatic groups H1, H2 and H3 (cyan). Distances between features were expressed in Å. B, 3D mapping of the pharmacophore features onto ZLDI‐8. (C, D), Western blot assay was used to assess the expression of ADAM17 and the quantitative results of ADAM17 protein level were analysed in LoVo and SW480 cells. β‐actin was used as a loading control. *P < .05 vs control
Six human cancer cell lines including A549, HepG2, LoVo, KB, Hela and MCF‐7 were used to estimate the anti‐proliferative effect of fourteen compounds by MTT assay. Table 1 showed that ZLDI‐8 inhibited the proliferation of 6 cancer cell lines especially in CRC (IC50 value was 9.65 ± 3.3 μM at 24 h). In contrast, other compounds showed no significant effect on 6 human cancer cell lines (data not shown).
Table 1.
Summary of IC50 values of ZLDI‐8 on 6 cancer cell lines
| Cell lines, IC50 ± SD value (μM) | |||||
|---|---|---|---|---|---|
| A549 | HepG2 | LoVo | KB | Hela | MCF‐7 |
| 30.7 ± 3.3 | 22.9 ± 4.8 | 9.65 ± 3.3 | 37.4 ± 9.9 | 25.1 ± 6.8 | 20.0 ± 1.0 |
IC50: Half maximal inhibitory concentration. All experiments were done at least 3 times.
3D mapping and the molecular docking showed that ZLDI‐8 interacts with most pharmacophore features and key amino acids of ADAM17 which including Leu348, Glu406, His405, Ala439, Leu401 and Leu402 (Figure 1B and S1C). Consistent with the results of pharmacophore and docking, enzymatic activity assays showed that ZLDI‐8 inhibited ADAM17 and the IC50 value was approximately 72 μM (shown in Figure S1D). Furthermore, we investigated the effect of ZLDI‐8 on the ADAM17 protein level using western blot assay. Figure 1C,D showed that the protein levels of ADAM17 were decreased both in SW480 and LoVo cells.
3.2. ZLDI‐8 inhibited the effect of Notch pathway
Figure 2A,B showed that ZLDI‐8 inhibited the proliferation of SW480 and LoVo cells in a dose‐ and time‐dependent manner (IC50 value was about 5.57 μM in LoVo cells and 7.42 μM in SW480 cells at 48 h). Meanwhile, Figure 2C,D showed that ZLDI‐8 also effectively attenuated the colony formation of SW480 and LoVo cells in a dose‐dependent manner. Moreover, the protein level of Survivin, an anti‐apoptosis regulator, was suppressed by ZLDI‐8 (Figure 2E).
Figure 2.
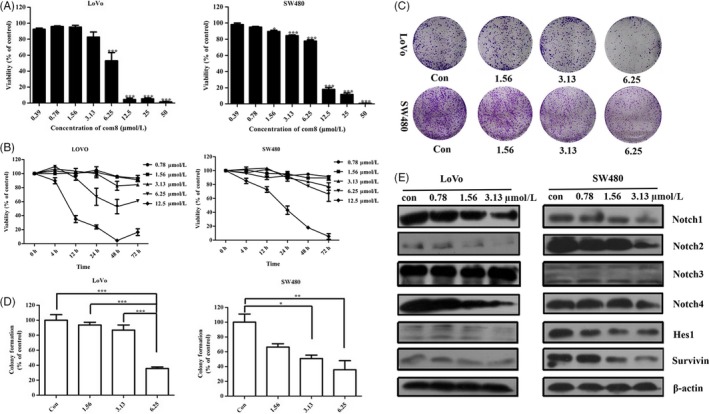
ZLDI‐8 attenuates colorectal cancer cells growth in vitro though Notch pathway. A, The growth inhibition was evaluated after the cells were treated with the indicated concentrations of ZLDI‐8 in SW480 and LoVo cells for 48 h. B, ZLDI‐8 decreased the viability of SW480 and LoVo cells in a time‐dependent manner. C, Cell proliferation was assessed by colony formation assay in SW480 and LoVo cells. D, Bar graphs showed the quantitative results of clonogenicity viability. E, Western blot assay was used to assess the expression of Notch pathway members (N1‐4ICD and Hes1) and anti‐apoptosis protein survivin. β‐actin was used as a loading control. *P < .05. **P < .01, ***P < .001 vs control
It is reported that ADAM17 regulates the activation of Notch signalling pathway.16 In CRC cells (SW480 and LoVo) (Figure 2E), the protein levels of Notch1, 2, 4 intracellular domains (N1ICD, N2ICD, N4ICD) were significantly decreased in a dose‐dependent manner when treated with ZLDI‐8. Whereas, there was no difference in the protein level of N3ICD in any of the treatment (Figure 2E). Moreover, Hes1, a downstream target genes of Notch pathway, was down‐regulated by ZLDI‐8 (Figure 2E). The expression of Survivin was also down‐regulated when treatment with ZLDI‐8. Taken together, these results demonstrated that ZLDI‐8 effectively inhibited the proliferation of CRC cells by inhibiting Notch signalling pathway.
3.3. ZLDI‐8 enhanced the anti‐proliferation effect of 5‐FU or irinotecan in CRC cells
To determine whether ZLDI‐8 sensitizes the CRC cells to different anti‐tumour agents, SW480 and LoVo cells were treated with 5‐FU, irinotecan, ZLDI‐8, and the cell viability was measured. SW480 and LoVo cells were pre‐incubated with ZLDI‐8 for 1 h before treatment with 5‐FU or irinotecan. Table 2 and table 3 showed that the IC50 values decreased from 2.17 ± 0.22 μM to 1.05 ± 0.23 μM (5‐FU in LoVo cells), 12.32 ± 0.62 μM to 7.71 ± 0.47 μM (irinotecan in LoVo cells), 4.71 ± 0.27 μM to 1.86 ± 0.17 μM (5‐FU in SW480 cells) or 10.00 ± 0.44 μM to 6.80 ± 0.39 μM (irinotecan in SW480 cells) when combined with ZLDI‐8, respectively. Moreover, ZLDI‐8 enhanced the anti‐proliferation effect of 5‐FU or irinotecan in a time‐dependent manner (Figure 3A,B).
Table 2.
ZLDI‐8 enhanced the sensitivity of LoVo cells to FU or irinotecan
| Compounds | FU | Irinotecan |
|---|---|---|
| IC50 value (μM) | ||
| Solvent control | 2.17 ± 0.22 | 12.32 ± 0.62 |
| ZLDI‐8 | 1.05 ± 0.23 | 7.71 ± 0.47 |
Table 3.
ZLDI‐8 enhanced the sensitivity of SW480 cells to FU or irinotecan
| Compounds | FU | Irinotecan |
|---|---|---|
| IC50 value (μM) | ||
| Solvent control | 4.71 ± 0.27 | 10.00 ± 0.44 |
| ZLDI‐8 | 1.86 ± 0.17 | 6.80 ± 0.39 |
Figure 3.
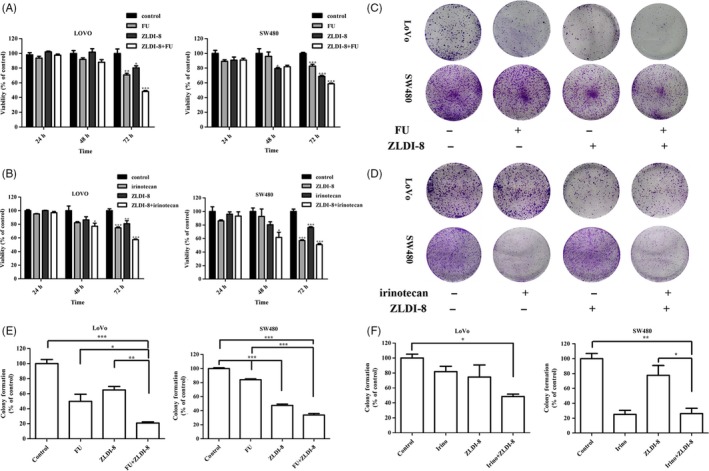
ZLDI‐8 sensitized colorectal cancer cells to 5‐FU and irinotecan. Cells were pre‐incubated with 0.78, 1.56 and 3.13 (μM) ZLDI‐8 for 1 h and then treated with different concentrations of 5‐FU (A) or irinotecan (B) in LoVo and SW480 cells for another 24 h, 48 h or 72 h. Cell proliferation was assessed by colony formation assay. ZLDI‐8 combined with 5‐FU (2 μM) (C) or irinotecan (8 μM) (D) in LoVo and SW480 cells. (E and F) Bar graphs showed the quantitative results of clonogenicity viability. *P < .05. **P < .01, ***P < .001 vs control. Irinotecan (irino)
To determine whether the combination of ZLDI‐8 with 5‐FU or irinotecan results in synergistic effects, the combination index (CI) values were calculated according to the equation as described by Kern et al.42 Table S2A showed that the combination of ZLDI‐8 with 5‐FU exhibited synergistic anti‐proliferation effects in LoVo and SW480 cells (CI > 1.0). However, the combination of ZLDI‐8 with irinotecan only has synergistic effect in LoVo cells (Table S2B).
To further investigate the synergistic anti‐proliferation effect of ZLDI‐8 with 5‐FU or irinotecan, colony formation was performed. Figure 3C,E showed that ZLDI‐8 sensitized LoVo and SW480 cells to 5‐FU in clonogenicity viability. However, ZLDI‐8 only enhanced this inhibition effect of irinotecan in LoVo cells (Figure 3D,F). Together, all results suggested that ZLDI‐8 sensitized both 5‐FU and irinotecan in LoVo cells.
3.4. ZLDI‐8 sensitized CRC cells to 5‐FU or irinotecan through Notch signalling pathway
Colorectal cancer cells respond to chemotherapy by increasing ADAM‐17 activity, which further resulted in resistance.17 Pharmacologic inhibition of ADAM17 in conjunction with chemotherapy may enhance response rates of CRC patients. Figure 4A,C,D showed that 5‐FU or irinotecan increased the expression of ADAM17; however, this effect could be reversed when combined with ZLDI‐8. ADAM17 cleaved the Notch receptors and activated the expression of Notch signalling pathway in CRC. Figure 4B‐D showed that the expression of Notch1 and Hes1 was increased induced by 5‐FU or irinotecan, nonetheless, ZLDI‐8 reversed this effect when combined with 5‐FU or irinotecan. Moreover, the combination of ZLDI‐8 with 5‐FU or irinotecan reduced the protein levels of N2ICD and N4ICD compared with ZLDI‐8 or 5‐FU/irinotecan alone treatment (Figure 4B‐D). Whereas, there was no change in the protein level of N3ICD in any of the treatment (Figure 4B‐D).
Figure 4.
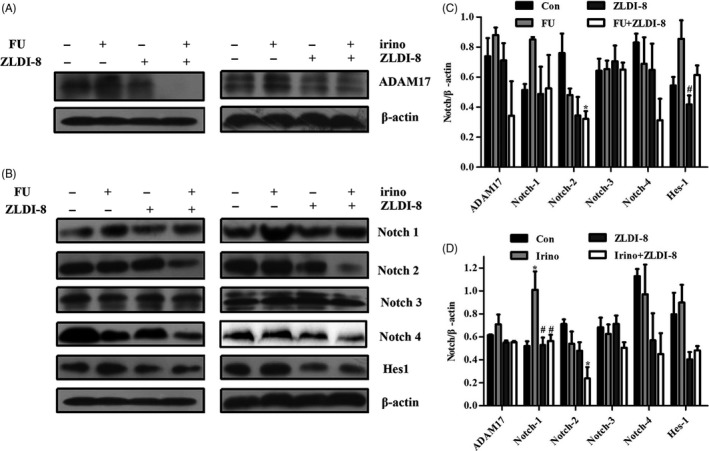
Effect of the combination of ZLDI‐8 with 5‐FU/irinotecan on the Notch pathway. A, Protein level of ADAM17 in LoVo cells was assayed by Western blot. B, Western blot was used to analyse expression of N1‐4ICD and Hes1 when treatment with ZLDI‐8, 5‐FU/irinotecan, alone or combination. β‐actin was used as a loading control. (C,D), Bar graphs showed the quantitative results of ADAM17, N1‐4ICD and Hes1. *P < .05 vs control, # P < .05 vs 5‐FU or irinotecan. Irinotecan (irino)
3.5. ZLDI‐8 sensitized CRC cells to 5‐FU or irinotecan by reversing EMT process
EMT is a major mechanism that involves in migration, invasion and chemoresistance.21 The wound healing scratch assay showed that ZLDI‐8, 5‐FU or irinotecan slightly inhibited the migration ability of LoVo cells, and this effect could be enhanced by combination groups (Figure 5A,B). As shown in Figure 5C,D, ZLDI‐8, 5‐FU or irinotecan alone treatment have no significant influence on the invasion ability, but, the combination groups enhanced the inhibitory effects of 5‐FU or irinotecan on LoVo cells’ invasion.
Figure 5.
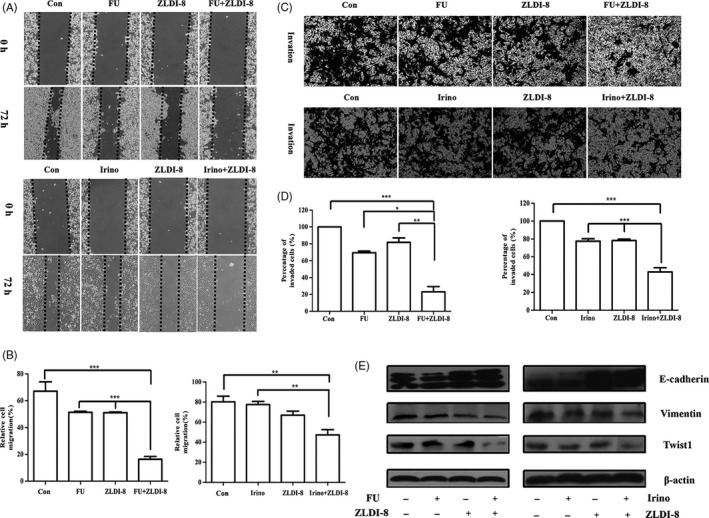
ZLDI‐8 (3.13 μM) combined with 5‐FU or irinotecan reversed the epithelial‐mesenchymal transition. A, Migratory ability was detected by wound healing assays when cells were treated with ZLDI‐8 (3.13 μM), 5‐FU (2 μM)/irinotecan (8 μM), alone or combination. B, Bar graphs showed the quantitative results of the migration. C, Transwell assays was used to measure the invasive ability of ZLDI‐8 (3.13 μM), 5‐FU (2 μM)/irinotecan (8 μM), alone or combination. (magnification, ×40). D, Bar graphs showed the quantitative results of the invasion (right). (E) Western blot assay was used to evaluate the expression of EMT markers (E‐cadherin, Vimentin and Twist1). β‐actin was used as a loading control. *P < .05. **P < .01, ***P < .001. Irinotecan (irino)
Then, whether migration and invasion ability blocked by ZLDI‐8 or 5‐FU/irinotecan involved in changing of EMT markers was evaluated by western blot assay and immunofluorescence staining. Figure 5E showed that the expression of epithelial marker E‐cadherin was slightly increased, in contrast, mesenchymal markers Vimentin and transcription factor Twist1 were significantly decreased when ZLDI‐8 combined with 5‐FU or irinotecan. Figure 6A,B showed that the intensity of Vimentin in comination groups was obviously weaker than ZLDI‐8 or 5‐FU/irinotecan alone treatment. Those results indicated that EMT process may be the key mechanism in synergistic anti‐tumour effect of ZLDI‐8 with 5‐FU or irinotecan.
Figure 6.
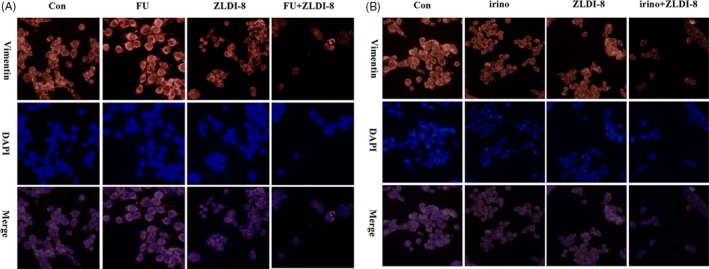
Immunofluorescence staining for the expression of Vimentin: ZLDI‐8 (3.13 μM) combined with 5‐FU (2 μM) (A) or irinotecan (8 μM) (B) in LoVo cells (magnification, ×40). Irinotecan (irino)
3.6. ZLDI‐8 enhanced in vivo anti‐tumour effect of 5‐FU on CRC cells
We next tested the combined therapeutic effect of ZLDI‐8 with 5‐FU in xenografts established with the LoVo cells. Treatment with ZLDI‐8 and 5‐FU alone slightly inhibited the growth of tumour. However, the combination group significantly inhibited the tumour growth compared to the tumours in the vehicle group (Figure 7B). Figure 7C,D showed that the similar trend in tumour weight loss when the mice were sacrificed. To further identify the anti‐tumour mechanism of the combination in vivo, the tumour lysate was measured with western blot analysis. Consistent with the finding in vitro, the epithelial markers E‐cadherin and Occludin were significantly increased, in contrast, the mesenchymal markers Vimentin and EMT transcription factor Twist1 were decreased in the combination group (Figure 7E). At the same time, the protein levels of ADAM17, N1ICD and Hes1 were increased in 5‐FU treatment, and this effect was reversed by combining with ZLDI‐8 (Figure 7E). In addition, the expression of N2ICD and N4ICD was remarkably decreased, while the N3ICD has no significant change in the combination group (Figure 7E). Collectively, all results suggested that ZLDI‐8 might enhance the anti‐proliferation and anti‐metastases effects of 5‐FU by inhibiting Notch signalling pathway and EMT process.
4. DISCUSSION
In mCRC, chemotherapy is the primary option to reduce tumour recurrence and prolong survival.43 However, the resistance of CRC cells to chemotherapy is the major obstacle for clinical application. Though targeted therapies were approved in recent years, the resistance was also an emergent problem.43 A novel inhibitor of potent pathways in conjunction with chemotherapy may be a potential approach for treatment with chemoresistance. In this study, we found a novel ADAM17 inhibitor ZLDI‐8 which cell‐specifically inhibited the proliferation of CRC and sensitized CRC to 5‐FU by reversing Notch and EMT pathway in vitro and in vivo.
ADAM17 was an indispensable regulator of almost all cellular events in ovarian cancer, non‐small cell lung cancer, breast cancer or CRC.44, 45, 46, 47, 48 It involved in a series of substrates, including cytokines, growth factors, TNF‐α and Epidermal Growth Factor Receptor (EGFR), especially in Notch pathway which contributed to chemoresistance.44, 49 Inhibition of ADAM17 is a potent approach to target the activation of Notch signalling and decrease tumourigenicity and metastasis in CRC.6 Therefore, inhibition of ADAM17 and synergistic interaction with existing chemotherapy may improve the outcomes of therapy for CRC patients.17, 50
Computational methods are important tools to design and screen activity compounds in recent years. Moreover, relationships among compounds, scaffolds and biological activities are initiating to be globally explored, beyond individual applications.25 In this study, we generated a pharmacophore model based on ADAM17 (PDB: 2FV5) validated by Fitvalues, AbsoluteEnergy and XSCORE, and gained fourteen potential compounds. Furthermore, biological activity assay showed that ZLDI‐8, a barbiturate acid‐based compound, was the most potent compound against 6 cancer cell lines especially in CRC. At the same time, enzymatic activity assays and western blot assay demonstrated that ZLDI‐8 suppressed the activated ADAM17 protein.
In CRC, accumulating evidences indicated that Notch signalling plays an oncogenic role.51 But, not all signalling components related to the development of CRC.52 In this study, we demonstrated that the expression of N1ICD, N2ICD and N4ICD was down‐regulated by ZLDI‐8. However, there was no significant difference in the protein level of N3ICD treated with ZLDI‐8 or not. In addition, ZLDI‐8 also suppressed the expression of Notch target gene Hes1. Moreover, the protein level of survivin, involved in anti‐apoptosis and chemoresistance followed by overexpressing of Notch1,53 was down‐regulated by ZLDI‐8. These findings suggested that ZLDI‐8 inhibited the proliferation of CRC cells through inhibiting Notch pathway.
FOLFOX (5‐FU + leucovorin + oxaliplatin) and FOLFIRI (5‐FU + leucovorin + irinotecan) are an important therapeutic strategy and remains the mainstay for CRC. In recent years, FOLFOX and FOLFIRI always result in treatment failure due to the chemoresistance. Accumulating evidences demonstrated that 5‐FU or irinotecan resulted in resistance through activating ADAM17, Notch1 and Hes1 in CRC patients.17, 53 Pharmacologic inhibition of ADAM17 could inhibit the activation of Notch pathway in CRC cells.50 Our current study found that ZLDI‐8 reversed the high expressions of ADAM17, Notch1 and Hes1 induced by 5‐FU or irinotecan. Nevertheless, in vitro and in vivo results showed that 5‐FU down‐regulated the expression of N2ICD and N4ICD, furthermore, ZLDI‐8 could increase these effects. But, there was no change in the protein level of N3ICD in any of the treatment. Nowadays, a series of inhibitors targeting Notch pathway for metastatic malignancies as a single agent or combination with chemotherapeutical drugs were studied.54 It is reported that GSI34 or parthenolide sensitized CRC cells to chemotherapy and synergistic with 5‐FU or irinotecan.53, 55 In this study, we demonstrated that ZLDI‐8 synergistically enhanced the anti‐proliferation effect when combined with 5‐FU or irinotecan. Meanwhile, ZLDI‐8 sensitized both 5‐FU and irinotecan in LoVo cells more than SW480.
It has been shown that Notch pathway is implicated in EMT process, in which epithelial cells obtain a mesenchymal phenotype, ultimately leading to migration and invasion.13, 23 Significantly, at present study, we demonstrated that the combination of ZLDI‐8 with 5‐FU or irinotecan dramatically decreased migratory and invasive capabilities of CRC cells compared with control or 5‐FU/irinotecan treatment alone. In addition, ZLDI‐8 enhanced the expression of epithelial cell marker E‐cadherin (a critical cell‐to‐cell adhesion molecule), decreased the expression of mesenchymal molecules Vimentin and EMT transcription factors Twist1 when combined with 5‐FU or irinotecan in vitro and in vivo. Furthermore, in vivo results also showed that the effect of 5‐FU on tumour growth and tumour weight was enhanced when combined with ZLDI‐8. Collectively, ZLDI‐8 sensitized CRC cells to 5‐FU or irinotecan may be through inhibiting Notch and EMT pathway. ZLDI‐8 may be a new adjuvant agent to increase the therapeutic benefits to metastatic CRC patients.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
Dan‐Dan Li, Jian Wang and Qing‐Chun Zhao designed and acquired the data. Chang‐Hao Zhao, Huai‐Wei Ding, Qiong Wu, Tian‐Shu Ren and Cong‐Qin Chen involved in drafting the manuscript or revising it critically for important intellectual content. Qing‐Chun Zhao gave final approval of the version to be published.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by Liaoning Science and technology research projects (No. 20170540969).
Li D‐D, Zhao C‐H, Ding H‐W, et al. A novel inhibitor of ADAM17 sensitizes colorectal cancer cells to 5‐Fluorouracil by reversing Notch and epithelial‐mesenchymal transition in vitro and in vivo. Cell Prolif. 2018;51:e12480 10.1111/cpr.12480
Contributor Information
Jian Wang, jianwang@email.com.
Cong‐Qin Chen, Email: ccq0120@163.com.
Qing‐Chun Zhao, Email: zhaoqingchun1967@163.com.
REFERENCES
- 1. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177‐193. [DOI] [PubMed] [Google Scholar]
- 2. Kranenburg O. Prometastatic NOTCH signaling in colon cancer. Cancer Discov. 2015;5:115‐117. [DOI] [PubMed] [Google Scholar]
- 3. Adam R, Vinet E. Regional treatment of metastasis: surgery of colorectal liver metastases. Ann Oncol. 2004;15:iv103‐iv106. [DOI] [PubMed] [Google Scholar]
- 4. Prenen H, Vecchione L, Van CE. Role of targeted agents in metastatic colorectal cancer. Target Oncol. 2013;8:83‐96. [DOI] [PubMed] [Google Scholar]
- 5. Barton MK. Primary tumor location found to impact prognosis and response to therapy in patients with metastatic colorectal cancer. Ca A Cancer J Clin. 2017;67:3‐4. [DOI] [PubMed] [Google Scholar]
- 6. Dosch J, Ziemke E, Wan S, et al. Targeting ADAM17 inhibits human colorectal adenocarcinoma progression and tumor‐initiating cell frequency. Oncotarget. 2017;8(39):65090‐65099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wei N, Chu E, Wu SY, Wipf P, Schmitz JC. The cytotoxic effects of regorafenib in combination with protein kinase D inhibition in human colorectal cancer cells. Oncotarget. 2015;6:4745‐4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin L, Velazquez OC, Liu ZJ. Notch signaling: emerging molecular targets for cancer therapy. Biochem Pharmacol. 2010;80:690‐701. [DOI] [PubMed] [Google Scholar]
- 9. Kopan R. Notch signaling. Cold Spring Harb Perspect Biol. 2012;4:759‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Km C, SR P. The notch signaling pathway as a mediator of tumor survival. Carcinogenesis. 2013;34:1420‐1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuan X, Wu H, Han N, et al. Notch signaling and EMT in non‐small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014;7:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parr C, Watkins G, Jiang WG. The possible correlation of Notch‐1 and Notch‐2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. 2004;14:779‐786. [DOI] [PubMed] [Google Scholar]
- 13. Vinson KE, George DC, Fender AW, Bertrand FE, Sigounas G. The Notch pathway in colorectal cancer. Int J Cancer. 2016;138:1835‐1842. [DOI] [PubMed] [Google Scholar]
- 14. An L, Li DD, Chu HX, et al. Terfenadine combined with epirubicin impedes the chemo‐resistant human non‐small cell lung cancer both in vitro and in vivo through EMT and Notch reversal. Pharmacol Res. 2017;124: 105‐115. [DOI] [PubMed] [Google Scholar]
- 15. Yuan X, Wu H, Xu H, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20‐27. [DOI] [PubMed] [Google Scholar]
- 16. Mumm JS, Schroeter EH, Saxena MT, et al. A ligand‐induced extracellular cleavage regulates gamma‐secretase‐like proteolytic activation of Notch1. Mol Cell. 2000;5:197‐206. [DOI] [PubMed] [Google Scholar]
- 17. Kyula JN, Schaeybroeck SV, Doherty J, Fenning CS, Longley DB, Johnston PG. Chemotherapy‐induced activation of ADAM‐17: a novel mechanism of drug resistance in colorectal cancer. Clin Cancer Res. 2010;16:3378‐3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saftig P, Reiss K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur J Cell Biol. 2011;90:527‐535. [DOI] [PubMed] [Google Scholar]
- 19. Scheel C, Weinberg RA. Cancer stem cells and epithelial–mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22:396‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fender AW, Nutter JM, Fitzgerald TL, Bertrand FE, Sigounas G. Notch‐1 Promotes Stemness and Epithelial to Mesenchymal Transition in Colorectal Cancer. J Cell Biochem. 2015;116:2517‐2527. [DOI] [PubMed] [Google Scholar]
- 21. Lu Z, Lai ZQ, Leung AWN, Leung PS, Li ZS, Lin ZX. Exploring brusatol as a new anti‐pancreatic cancer adjuvant: biological evaluation and mechanistic studies. Oncotarget. 2017;8(49):84974‐84985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug‐resistance. Oncotarget. 2015;6:10697‐10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Z, Li Y, Ahmad A, et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Curr Drug Targets. 2010;1806:258‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang R, Ye X, Bhattacharya R, et al. A disintegrin and metalloproteinase domain 17 regulates colorectal cancer stem cells and chemosensitivity via Notch1 signaling. Stem Cells Transl Med. 2016;5:331‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu Y, Stumpfe D, Bajorath J. Recent advances in Scaffold Hopping. J Med Chem. 2017;60:1238‐1246. [DOI] [PubMed] [Google Scholar]
- 26. Hou X, Li R, Li K, Yu X, Sun JP, Fang H. Fast identification of novel lymphoid tyrosine phosphatase inhibitors using target‐ligand interaction‐based virtual screening. J Med Chem. 2014;57:9309‐9322. [DOI] [PubMed] [Google Scholar]
- 27. Niu X, Umland S, Ingram R, et al. IK682, a tight binding inhibitor of TACE. Arch Biochem Biophys. 2006;451:43‐50. [DOI] [PubMed] [Google Scholar]
- 28. Mazzola RD Jr, Zhu Z, Sinning L, et al. Discovery of novel hydroxamates as highly potent tumor necrosis factor‐alpha converting enzyme inhibitors. Part II: optimization of the S3’ pocket. Bioorg Med Chem Lett. 2008;18:5809‐5814. [DOI] [PubMed] [Google Scholar]
- 29. Levin JI, Chen JM, Laakso LM, et al. Acetylenic TACE inhibitors. Part 2: SAR of six‐membered cyclic sulfonamide hydroxamates. Bioorg Med Chem Lett. 2005;15:4345‐4349. [DOI] [PubMed] [Google Scholar]
- 30. Li D, Popovicimuller J, Belanger DB, et al. Structure and activity relationships of tartrate‐based TACE inhibitors. Bioorg Med Chem Lett. 2010;20:4812‐4815. [DOI] [PubMed] [Google Scholar]
- 31. Rosner KE, Guo Z, Orth P, et al. The discovery of novel tartrate‐based TNF‐α converting enzyme (TACE) inhibitors. Bioorg Med Chem Lett. 2010;20:1189‐1193. [DOI] [PubMed] [Google Scholar]
- 32. Levin JI, Chen JM, Laakso LM, et al. Acetylenic TACE inhibitors. Part 3: thiomorpholine sulfonamide hydroxamates. Bioorg Med Chem Lett. 2006;16:1605‐1609. [DOI] [PubMed] [Google Scholar]
- 33. Yu W, Tong L, Kim SH, et al. Biaryl substituted hydantoin compounds as TACE inhibitors. Bioorg Med Chem Lett. 2010;20:5286‐5289. [DOI] [PubMed] [Google Scholar]
- 34. Dai C, Li D, Popovicimuller J, et al. 2‐(2‐Aminothiazol‐4‐yl)pyrrolidine‐based tartrate diamides as potent, selective and orally bioavailable TACE inhibitors. Bioorg Med Chem Lett. 2011;21:3172‐3176. [DOI] [PubMed] [Google Scholar]
- 35. Yu W, Guo Z, Orth P, et al. Discovery and SAR of hydantoin TACE inhibitors. Bioorg Med Chem Lett. 2010;20:1877‐1880. [DOI] [PubMed] [Google Scholar]
- 36. Ingram RN, Orth P, Strickland CL, Le HV, Madison V, Beyer BM. Stabilization of the autoproteolysis of TNF‐alpha converting enzyme (TACE) results in a novel crystal form suitable for structure‐based drug design studies. Protein Eng Des Sel. 2006;19:155‐161. [DOI] [PubMed] [Google Scholar]
- 37. Condon JS, Joseph‐McCarthy D, Levin JI, et al. Identification of potent and selective TACE inhibitors via the S1 pocket. Bioorg Med Chem Lett. 2007;17:34‐39. [DOI] [PubMed] [Google Scholar]
- 38. Bandarage UK, Wang T, Come JH, Perola E, Wei Y, Rao BG. Novel thiol‐based TACE inhibitors. Part 2: rational design, synthesis, and SAR of thiol‐containing aryl sulfones. Bioorg Med Chem Lett. 2008;18:44‐48. [DOI] [PubMed] [Google Scholar]
- 39. Govinda RB, Bandarage UK, Wang T, et al. Novel thiol‐based TACE inhibitors: rational design, synthesis, and SAR of thiol‐containing aryl sulfonamides. Bioorg Med Chem Lett. 2007;17:2250‐2253. [DOI] [PubMed] [Google Scholar]
- 40. Park K, Gopalsamy A, Aplasca A, et al. Synthesis and activity of tryptophan sulfonamide derivatives as novel non‐hydroxamate TNF‐alpha converting enzyme (TACE) inhibitors. Bioorg Med Chem. 2009;17:3857‐3865. [DOI] [PubMed] [Google Scholar]
- 41. Li R, Su X, Cheng Z, et al. Structure‐based virtual screening and ADME/T‐based profiling for low molecular weight chemical starting points as p21‐activated kinase 4 inhibitors. Rsc Advances. 2015;5:23202‐23209. [Google Scholar]
- 42. Kern DH, Morgan CR, Hildebrandzanki SU. In vitro pharmacodynamics of 1‐beta‐D‐arabinofuranosylcytosine: synergy of antitumor activity with cis‐diamminedichloroplatinum(II). Can Res. 1988;48:117‐121. [PubMed] [Google Scholar]
- 43. Zhang WJ, Li Y, Wei MN, et al. Synergistic antitumor activity of regorafenib and lapatinib in preclinical models of human colorectal cancer. Cancer Lett. 2017;386:100‐109. [DOI] [PubMed] [Google Scholar]
- 44. Gooz M. ADAM‐17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Richards FM, Tape CJ, Jodrell DI, Murphy G. Anti‐tumour effects of a specific anti‐ADAM17 antibody in an ovarian cancer model in vivo. PLoS ONE. 2012;7:e40597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mcgowan PM, Ryan BM, Hill AD, Mcdermott E, O'Higgins N, Duffy MJ. ADAM‐17 expression in breast cancer correlates with variables of tumor progression. Clin Cancer Res. 2007;13:2335. [DOI] [PubMed] [Google Scholar]
- 47. Baumgart A, Seidl S, Vlachou P, et al. ADAM17 regulates epidermal growth factor receptor expression through the activation of Notch1 in non‐small cell lung cancer. Can Res. 2010;70:5368‐5378. [DOI] [PubMed] [Google Scholar]
- 48. Blanchot‐Jossic F, Jarry A, Masson D, et al. Up‐regulated expression of ADAM17 in human colon carcinoma: co‐expression with EGFR in neoplastic and endothelial cells. J Pathol. 2005;207:156‐163. [DOI] [PubMed] [Google Scholar]
- 49. Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32‐43. [DOI] [PubMed] [Google Scholar]
- 50. Lu J, Ye X, Fan F, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged‐1. Cancer Cell. 2013;23:171‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suliman MA, Zhang Z, Na H, et al. Niclosamide inhibits colon cancer progression through downregulation of the Notch pathway and upregulation of the tumor suppressor miR‐200 family. Int J Mol Med. 2016;38:776‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Qiao L, Wong BCY. Role of Notch signaling in colorectal cancer. Carcinogenesis. 2009;30:1979‐1986. [DOI] [PubMed] [Google Scholar]
- 53. Meng RD, Shelton CC, Li YM, et al. Gamma‐Secretase inhibitors abrogate oxaliplatin‐induced activation of the Notch‐1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Can Res. 2009;69:573‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther. 2013;139:95‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim SL, Kim SH, Trang KTT, et al. Synergistic antitumor effect of 5‐fluorouracil in combination with parthenolide in human colorectal cancer. Cancer Lett. 2013;335:479‐486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials