

Abstract
Objectives
In our ongoing studies to develop ER targeting agents, we screened for dual‐acting molecules with a hypothesis that a single molecule can also target both ER positive and negative groups of breast cancer.
Materials and methods
1‐(2‐(4‐(Dibenzo[b,f]thiepin‐10‐yl)phenoxy)ethyl)piperidine (DTPEP) was synthesized and screened in both MCF‐7 (ER+ve) and MDA‐MB‐231 (ER‐ve) cells. Assays for analysis of cell cycle, ROS, apoptosis and MMP loss were carried out using flow cytometry. Its target was investigated using western blot, transactivation assay and RT‐PCR. In vivo efficacy of DTPEP was validated in LA‐7 syngeneic rat mammary tumour model.
Results
Here, we report identification of dual‐acting molecule DTPEP that downregualtes PI3K/Akt and PKCα expression, induces ROS and ROS‐dependent apoptosis, loss of mitochondrial membrane potential, induces expression of caspase indicative of both intrinsic and extrinsic apoptosis in MCF‐7 and MDA‐MB‐231 cells. In MCF‐7 cells, DTPEP downregulates ERα expression and activation. In MDA‐MB‐231 cells, primary cellular target of DTPEP is not clearly known, but it downregualtes PI3K/Akt and PKCα expression. In vivo study showed regression of LA‐7 syngeneic mammary tumour in SD rat.
Conclusions
We identified a new dual‐acting anti‐breast cancer molecules as a proof of concept which is capable of targeting both ER‐positive and ER‐negative breast cancer.
Abbreviation
- CCCP
carbonyl cyanide m‐chlorophenylhydrazone
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DCFH‐DA
2,7‐dichlorodihydrofluorescein diacetate
- E2
17β‐estradiol
- ER
oestrogen receptor
- JC‐1
5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolylcarbocyanine iodide
- MMP
mitochondrial membrane potential
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide
- NAC
N‐acetyl‐l‐cysteine
- PCNA
proliferating cell nuclear antigen
- PI
propidium iodide
- PKC
protein kinase c
- primary cells
primary breast cancer cells derived from ER‐ve breast adenocarcinoma tissue of breast cancer patient
- PR
progesterone receptor
1. INTRODUCTION
Breast cancer in the majority of subjects are hormone sensitive and promoted by oestrogens.1 The key role of oestrogens in ER‐positive hormone sensitive breast tumours is the core of developing drugs targeting ER through competition with endogenous oestrogens to block its tumour promoting action.2 Tamoxifen (TAM) is the first in class non‐steroidal anti‐oestrogen drug which is still used for ER‐positive breast cancer patients.3 In addition to acting on ER as an anti‐oestrogen, tamoxifen is also known to have some other off‐target effect due to which it shows some degree of efficacy against ER‐negative breast cancers.4, 5, 6, 7, 8 We have previously reported 1‐(2‐(4‐(Dibenzo[b,f]thiepin‐10‐yl)phenoxy)ethyl)piperidine (DTPEP) compound designed to target ER as anti‐cancer agent.9 Here, we are reporting detailed mechanism of lead compound DTPEP in both ER‐positive and ER‐negative breast cancer cells.
2. MATERIALS AND METHODS
2.1. Synthesis of DTPEP
The lead compound DTPEP and tamoxifen share structural similarity as shown in Figure 1A. DTPEP was synthesized, purified and characterized according to our previously reported method (supplementary data).9
Figure 1.
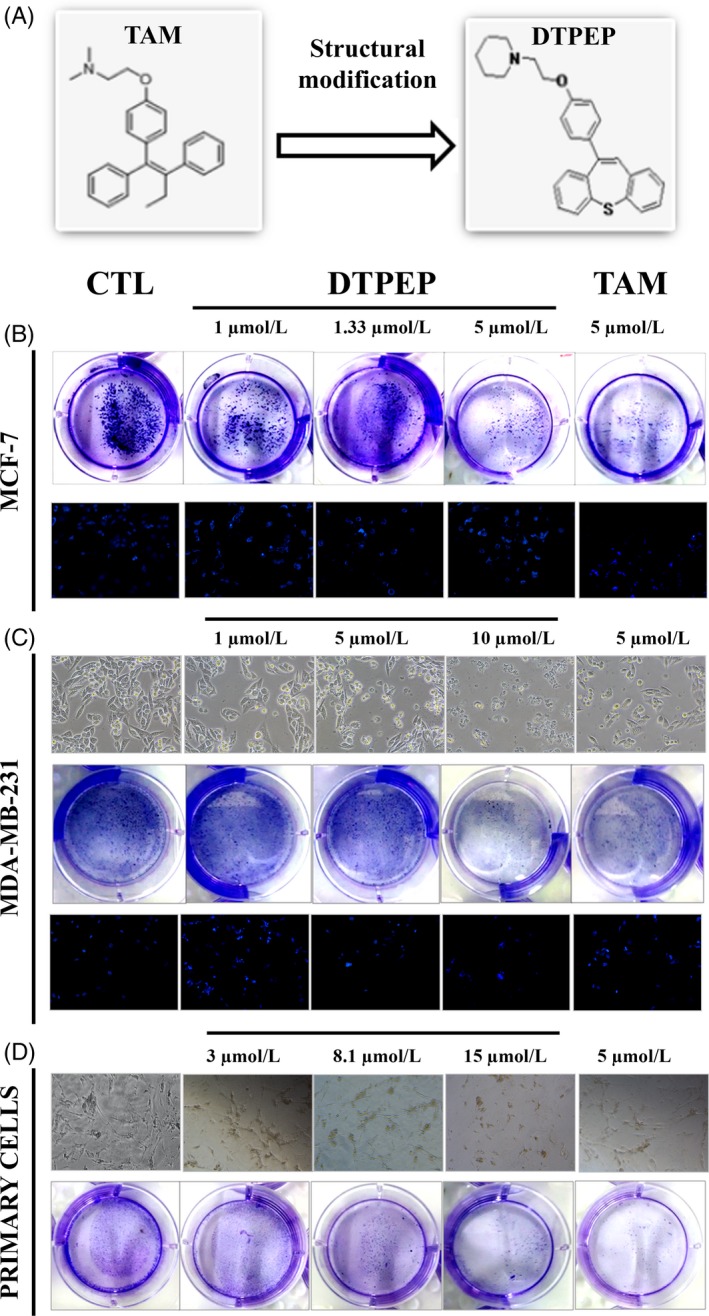
(A) Strucure of tamoxifen (TAM) and DTPEP. (B) Colony formation and DAPI staining of MCF‐7 cells after the treatment of DTPEP and TAM at various concentrations for 24 h. (C). White light microphotograph (10X maginfication), colony formation on 7th day post‐treatment withdrawal and DAPI staining (10X maginfication) of MDA‐MB‐231 cells after treatment of DTPEP and TAM at various concentrations for 24 h. (D). White light microphotograph of primary cells and colony formation on 7th day post‐treatment withdrawal in primary cells after treatment of DTPEP and TAM at various concentrations for 24 h
2.2. Plasmids
The 2xERE‐pS2‐bearing pGL3‐luc plasmid, ERα (pSG5‐mERα) plasmid and ERβ (pSG5‐hERβ) plasmid were kind gifts from Prof Malcolm G. Parker, Imperial Cancer Research Fund, London, UK.10 The pRL‐luc plasmid was procured from Promega (Wisconsin, Madison, USA).
2.3. Cells and cell culture condition
Breast cancer cell lines MCF‐7 (ER positive), MDA‐MB‐231 (ER negative), MCF‐10A (non‐tumourigenic epithelial cell line) and LA‐7 (rat mammary tumour cell line) were obtained from ATCC (Manassas, Virginia, USA). HEK‐293 (human embryonic kidney epithelial cell line) cells were obtained from institutional repository of CSIR‐CDRI. MCF‐7, MDA‐MB‐231 and HEK‐293 were maintained in DMEM. MCF‐10A was maintained in DMEM phenol red supplemented with 10% horse serum, 100 ng/mL cholera toxin, 20 ng/mL epidermal growth factor, 500 ng/mL hydrocortisone and 10 μg/mL insulin. LA‐7 (rat mammary cancer cell line) was maintained in DMEM phenol red supplemented with 10% FBS, 50 nmol/L hydrocortisone and 5 μg/mL bovine insulin.
2.4. Primary cell culture from breast adenocarcinoma tissues
The study was ethically approved by the Institutional Human Ethics Committee of King George's Medical University (6610/Ethics/R.Cell‐15) and CSIR‐CDRI (CDRI/IEC/2017/A5). Breast cancer patients were recruited at the Department of Surgery, King George's Medical University, Lucknow, India. Clinically and histologically confirmed ER‐negative high‐grade infiltrating ductal breast carcinoma tissue were only used for primary cell culture studies. Primary cell culture was carried out as per previously described method.11 In brief, tissues were collected in DMEM‐F12 containing antibiotic, minced and incubated with 1 mg/ml collagenase and DNase (2 mg/mL) in DMEM‐F12 for 2 hours at 37°C with periodic mixing, digested tissue was mechanically dissociated by repeated pipetting and resuspended in 10 mL of fresh DMEM‐F12. Suspension was cetrifuged and supernatant containing cells were separated from tissue clumps, washed twice with DMEM‐F12 containing 10% FBS, transferred into culture flasks and incubated at 37°C with 5% CO2.
2.5. MTT assay
MTT assay was carried out as previously described.12
2.6. Colony formation assay
Cancer cells were seeded at the rate of 5000 cells per well in six‐well culture plate and incubated overnight. Cells were treated with compound for 24 hours. Then, media along with compound were washed with PBS and incubated in fresh DMEM media for 7 days. At the end of incubation period, media were removed, cells were fixed with methanol, stained with 0.4% crystal violet and images were captured.
2.7. DAPI staining
Cells were seeded at a density of 10 000 cells per well in four‐well glass chambers slide and grown overnight. Then, cells were treated with compound for 24 hours, washed and fixed with 4% paraformaldehyde. Cells were stained with DAPI (0.5 mg/mL) for 30 minutes at 37°C, observed under fluorescent microscope and image of random fields from each group were captured (Leica, Wetzlar, Hesse, Germany) using blue filter.13
2.8. Cell cycle analysis
Cell cycle analysis was carried out as previously described.14 The percentage of DNA content at different phases of the cell cycle was analysed using FACS Calibur flow cytometer (Becton‐Dickinson, Franklin Lakes, NJ, USA) and analysed with CellQuest software (Becton‐Dickinson).
2.9. Annexin V‐FITC and PI staining
Assay was carried out as previously described using Annexin V‐FITC and PI kit as per manufacturer instructions (Sigma, St. Louis, MO, USA).15 Live, apoptotic and necrotic cell populations were differentiated using flow cytometer (Becton‐Dickinson).
2.10. Measurement of intracellular ROS
Cancer cells were plated in the density of 1 × 105 cells/well in six‐well culture plates for 24 hours. Cells were treated with compound for 24 hours and then cells were trypsinized, fixed in 1 mL methanol, stained with DCFH‐DA stain (2 μg/mL) for 30 minutes at 37°C followed by PBS washing twice. The stained cells were measured for ROS accumulation using flow cytometer (Becton‐Dickinson).
2.11. Measurement of mitochondrial membrane potential
MMP assay was carried out as previously described.16 The change in the MMP was estimated by calculating the ratio of fluorescence at 590 nm (green) and 530 nm (red).
2.12. Western blotting
Cells from various treatment groups were washed with ice‐cold PBS, lysed in RIPA buffer containing phosphatase and protease inhibitor cocktail (Sigma‐Aldrich). Lysates containing equal amount of protein were electrophoresed and transferred to PVDF membrane (Millipore, Bangalore, Karnataka, India), probed with appropriate primary antibodies. Blots were developed using ECL solution (Immobilon; Millipore, Billerica, MA, USA) and scanned with gel documentation system (ImageQuant LAS4000, GE, Piscataway, NJ, USA).17 Each experiment was repeated minimum three times. Quantitation of band intensity was carried out by densitometry using Quantity One 1‐D analysis software version 4.6.6 (Biorad, Hercules, California, USA).
2.13. RT‐PCR and quantitative real‐time PCR
Total RNA was extracted from the cells of various treatment groups using Trizol (Invitrogen, Carlsbad, CA, USA). cDNA synthesized with RevertAid H Minus Reverse Transcriptase cDNA synthesis kit (Fermentas, Burlington, Ontario, Canada) using 2 μg of total RNA. Quantitative PCR was performed for assessing the expression of selected genes using SYBR Green (Roche, Indianapolis, IN,). Relative gene expression levels were determined using quantitative real‐time PCR (LightCycler480; Roche) and fold change in expression of different genes were determined after normalizing with 18S.18 Experiments were repeated three times and data were expressed in mean fold change ±SE. The details of primers used are shown in supplementary Table S1.
2.14. Immunofluorescence staining
MCF‐7 cells were grown in four‐well chamber slides at the rate of 1 × 103 per well (Pocheon, Gyeonggi‐do, South Korea) and treated with vehicle and test compounds for 30 min with or without pretreatment of 10 nmol/L of estradiol (E2). Cells were then fixed in methanol and acetone in 1:1 ratio at 4°C, permeabilized with 0.1% triton X‐100, washed with PBS and blocked with 2% BSA. Furthermore, incubated with ERα antibody for overnight followed by 1‐hour incubation with fluorescence‐tagged secondary antibody, then counter‐staining done with DAPI for 5 minutes. Images were captured at 40X magnification with confocal microscope.17
2.15. In vivo efficacy study in LA‐7 derived syngenic rat mammary tumour
All experimental procedures were done according to standard protocols approved by Institutional Animal Ethics Committee (IAEC/2014/89). In brief, 6 × 106 LA‐7 cells were transplanted orthotopically into mammary fat pad of adult female Sprague Dawley (SD) rats. When tumours became measurable, animals were randomized and divided into four groups (n = 4). Two groups were orally administered with DTPEP (10 and 20 mg/kg body weight per day), one group orally administered with tamoxifen (20 mg/kg body weight per day) for 25 days and remaining group used as negative vehicle control. Tumour size and body weight were measured on every fifth day. Tumour volume was calculated using formula, V = [(Length) × (Width)2]/2.19 Finally, tumours and other organs were collected in RNA later as well as 10% neutral‐buffered formalin while lungs were collected in Bouin's solution.20 Percentage of tumour incidence was calculated as percentage of tumour‐bearing rats in a group divided by total rats in that group while percentage of tumour burden was calculated as percentage of total final weight of ex situ tumours of a group divided by total final weight of animals in that group.
2.16. Haematoxylin and eosin staining
Approximately 5.0 μm thick sections were prepared from mammary tumour tissues collected from different groups and stained with haematoxylin and eosin using standard method.21 Stained tissue section was mounted with coverslip, observed under microscope and randomly selected fields of each group were recorded.
2.17. Immunohistochemistry of ERα and PKCα
Mammary tumours tissue sections were deparaffinized, rehydrated in PBS, pre‐treated at 60°C for 30 minutes for antigen retrieval, blocked in 5.0% BSA for 2 hours and subsequently incubated with antibody ERα (MC‐20) and PKC α (C‐20) at 1:1000 dilution in 2.0% BSA in a humidified chamber at 4°C for overnight. Tissue sections were washed with PBS and incubated with SuperPicture polymer detection kit (Cat. no. 87‐8963; Invitrogen) containing horseradish peroxidase conjugated secondary antibody. Colour developed by using 3,3‐diaminobenzidine peroxidase substrate for 1 minute before counter‐staining with haematoxylin (Sigma‐Aldrich). Sections were observed under microscope and image was recorded from randomly selected fields of each group.22
2.18. Tunel assay
DeadEnd fluorometric TUNEL system (Promega; cat. no. G3250) was used for Tunel assay. DAPI‐containing mounting media were used for mounting with coverslip and samples were analysed using fluorescence confocal microscope.
2.19. Transient transfection and ER transactivation assay
For analysis of ERE‐mediated transcription, MCF‐7 cells were transfected with 100 ng of pERE‐Luc using Lipofectamine‐2000™ transfection reagent (Invitrogen). Then, 100 ng of ERα or ERβ expression plasmids was cotransfected with pERE‐Luc in ER‐negative MDA‐MB‐231 breast cancer cell line. In addition, 50 ng of pRL‐SV40‐luc was cotransfected for using it for normalization of transfection efficiencies. After 5 hours of transfection, cells were treated with vehicle, E2 and different concentrations of DTPEP. After 18 hours, cells were processed using lysis buffer and then proceeded for luciferase activity measurement using Dual Luciferase Assay System (Promega). The firefly luciferase intensity for every sample was normalized through transfection efficiency obtained from renilla luciferase activity.23 Each experiments were performed in triplicate of each test groups with three replicates of each experiment.
2.20. Statistical analysis
All the analysed results are expressed as mean ± SEM derived from at least three independent experiments with triplicates of each test group. Statistical significance was determined by ANOVA and Newmann‐Keul's test or by paired Student's t test. The levels of probability were noted and P values <.05 were considered statistically significant. P‐values were noted as, if significant P < .05 (*), P < .01 (**), P < .001 (***). Statistical analysis was carried out using Microsoft Excel and Prism software 5.0 (GraphPad Software, La Jolla, CA, USA).
3. RESULTS
3.1. DTPEP inhibits breast cancer cell proliferation
DTPEP exhibited significant anti‐cancer activity against various breast cancer cells (MCF‐7, MDA‐MB‐231, LA‐7, primary cells) as revealed by MTT assay (Supplementary Table S2). MCF‐7 cells were significantly inhibited even in the presence of 10 nmol/L E2 while DTPEP appeared safe towards MCF‐10A and HEK‐293 cells. DTPEP also significantly decreased the number of colony formation in MCF‐7, MDA‐MB‐231 and primary cells (Figure 1B‐D). In addition, clear morphological changes suggestive of cell proliferation inhibition was observed in both MDA‐MB‐231 and primary cells upon 24 hours treatment with DTPEP (Figure 1C,D). On nuclear staining with DAPI, MCF‐7 cells and MDA‐MB‐231 cells treated with various concentrations of DTPEP showed a significant induction of nuclear condensation and increase in apoptotic nuclei (Figure 1B,C).
3.2. DTPEP induces G0/G1 arrest, cellular ROS, apoptosis and altered MMP in breast cancer cells
DTPEP induced a significant arrest of MDA‐MB‐231 and primary cells in G0/G1 phase of cell cycle in a dose dependent manner (Figure 2A,C). This is similar to DTPEP‐induced cell cycle arrest that we previously reported in MCF‐7 cells.9 In addition, DTPEP significantly increased early as well late apoptosis in both MDA‐MB‐231 and primary breast cancer cells (Figure 2B,D). DTPEP induced a significant dose‐dependent increase of cellular ROS level in MCF‐7, MDA‐MB‐231 and primary cells (Figure 3A‐C). JC‐1 stained cells show a decrease in red fluorescence and increase in green fluorescence during mitochondrial membrane depolarization due to loss of mitochondrial membrane integrity. We observed higher green fluorescing cells after treatment of DTPEP for 24 hours in MCF‐7 cells, MDA‐MB‐231 and primary cells. This suggests that DTPEP induces a significant depolarization of mitochondria possibly associated with apoptosis (Figure 3D‐F).
Figure 2.
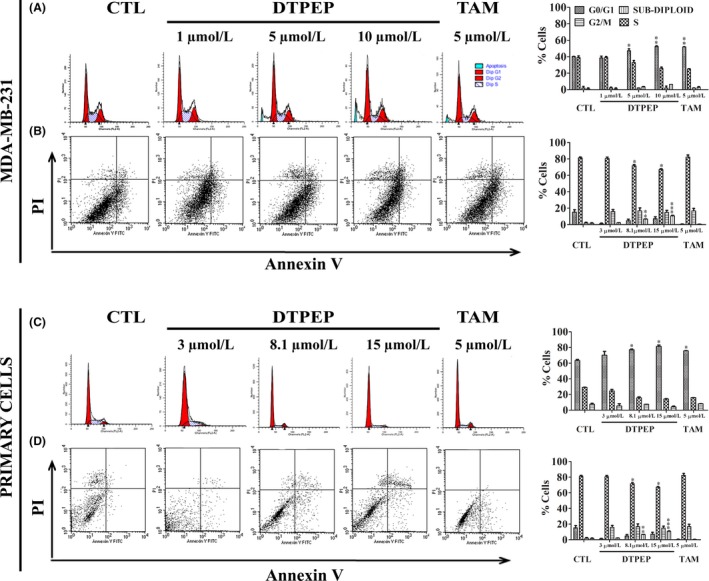
Effect of DTPEP and TAM in cell cycle and apoptosis of breast cancer cell, MDA‐MB‐231 (A, B) and primary cells (C, D). (A, C). For cell cycle PI‐stained cells, number and cell size were counted using flow cytometry. After gating out cell debris and aggregates, 5000‐10 000 events were collected for analysis of each sample. The percentage of cells in different phases of cell cycle was calculated based on their PI stained DNA content vs cell size. (B, D). The per cent cells undergoing apoptosis was determined using Annexin V‐FITC & PI double staining assay and flow cytometry (FACS Calibur, Becton‐Dickinson, San Jose, CA, USA). After gating out cell debris and aggregates, 10 000 events were collected for analysis of each sample. All values are expressed as mean with their standard errors (mean ± SEM, N = 3) derived from three independent cytometry assay and presented with histograms on corresponding right side panels. Statistical analysis of each parameter for the compound treated groups was compared with non‐treated groups using one‐way ANOVA (non‐parametric) with Newman‐Keuls post hoc test. The difference was considered statistically significant if *P < .05. **P < .01 and ***P < .001 vs control
Figure 3.
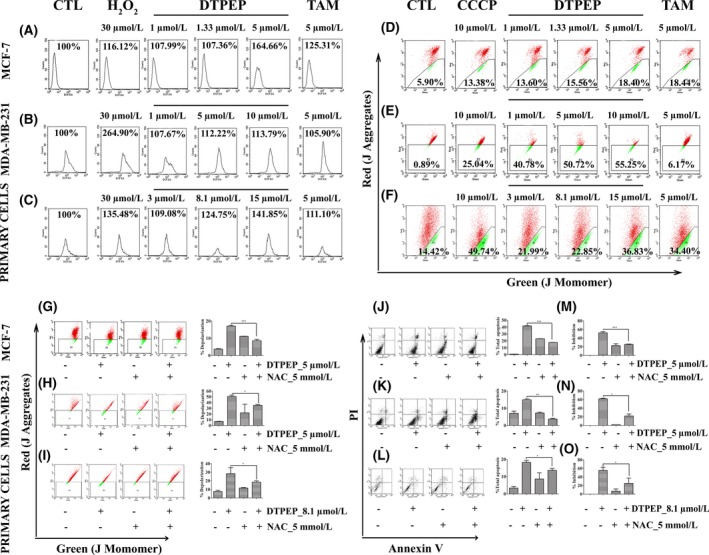
DTPEP induced ROS generation, mitochondrial depolarization and ROS‐dependent mitochondrial depolarization and apoptosis in breast cancer cells. (A‐C) DTPEP induced ROS generation in breast cancer cells, (A) MCF‐7 cells, (B) MDA‐MB‐231 and (C) primary cells. H2O2 was used as a positive control to induce cellular ROS. After gating out cell debris, 10 000 events were collected for analysis of each sample. All values of ROS are expressed as percentage of ROS generation derived from CellQuest software‐based analysis of acquired cytometric data. (D‐F). DTPEP induced mitochondrial depolarization in breast cancer cells, (D) MCF‐7 cells, (E) MDA‐MB‐231 and (F) primary cells. 10 μmol/L CCCP was used as positive control to induce mitochondrial depolarization and added 2 h before harvesting of cells for flow cytometric analysis. After gating out cell debris, 10 000 events were collected for analysis of each sample. Data of mitochondrial depolarization are expressed as % gated population of monomeric JC‐1 with green fluorescence indicative of low ΔΨm and aggregates JC‐1 with red fluorescence indicative of high ΔΨm derived from cytometry (FACS Calibur, Becton‐Dickinson, San Jose, CA, USA). (G‐I) DTPEP induced ROS‐dependent mitochondrial depolarization of breast cancer cells (G) MCF‐7 cells, (H) MDA‐MB‐231 and (I) primary cells. (J‐L). DTPEP induced ROS‐dependent apoptosis determined by Annexin V‐FITC & PI double staining assay using flow cytometry in breast cancer cells, (J) MCF‐7 cells, (K) MDA‐MB‐231 and (L) primary cells. After gating out cell debris, 10 000 events were collected for analysis of each sample. Data are expressed as % total apoptotic population by combining lower right (early apoptotic) and upper right (late apoptotic) quadrant data of flow cytogram (M‐O). DTPEP‐induced ROS‐dependent loss of percentage inhibition was determined with MTT assay of breast cancer cells (M) MCF‐7 cells, (N) MDA‐MB‐231 and (O) primary cells. Data are expressed as % cell inhibition compared to control. In all the assays, 5 mM of NAC was used as a standard ROS scavenger. Statistical analysis of each parameter of DTPEP treated in the presence of NAC groups was compared with DTPEP alone treated groups using one‐way ANOVA (non‐parametric) with Newman‐Keuls post hoc test. The difference was considered statistically significant if *P < .05, **P < .01 and ***P < .001 DTPEP in the presence of NAC vs DTPEP alone treated group
3.3. DTPEP‐induced loss of MMP and apoptosis are ROS dependent
It is known that a majority of chemotherapeutic agents induces cellular ROS which in turn is responsible for mitochondria‐mediated apoptosis.24 Cells treated with DTPEP for 24 hours in the presence of NAC, as ROS scavenger showed that mitochondrial depolarization caused by DTPEP was significantly decreased in MCF‐7, MDA‐MB‐231 and primary cells (Figure 3G‐I). Similarly, NAC also significantly decreased apoptosis induced by DTPEP in MCF‐7, MDA‐MB‐231 and primary cells (Figure 3J‐L). These suggest that DTPEP‐induced loss of mitochondrial membrane potential and apoptosis in breast cancer is ROS‐dependent. Additional experiment was performed using MTT assay in the presence of NAC, to evaluate the role of ROS in DTPEP‐induced loss of cell viability. We found that the percentage inhibition of breast cancer cells was significantly decreased by DTPEP in the presence of NAC as compared to the absence of NAC (Figure 3M‐O).
3.4. DTPEP induces caspase‐dependent apoptosis
Caspase 8 is a mediator of extrinsic pathway while caspase‐9 is mediator of intrinsic pathway of apoptosis. Breast cancer cells were treated with either of pan‐caspase inhibitor (z‐VAD‐FMK) or caspase 8 inhibitor (z‐LETD‐FMK) or caspase 9 inhibitor (z‐LEHD‐FMK) and apoptosis measured with annexin‐V‐FITC/PI double staining assay. Results showed a significant decrease in apoptosis irrespective of caspase inhibitors used in all types of breast cancer cells (Figure 4A,B,D,E,I,J,L,M,Q,R). This clearly indicates that DTPEP depends on both extrinsic and intrinsic pathway of apoptosis. MTT assay with 50 μmol/L z‐VAD‐FMK, 20 μmol/L z‐LETD‐FMK and 20 μmol/L z‐LEHD‐FMK also showed a significant decrease loss of viability in the presence of caspase inhibitors in breast cancer cells (Figure 4C,F,K,N,S).
Figure 4.
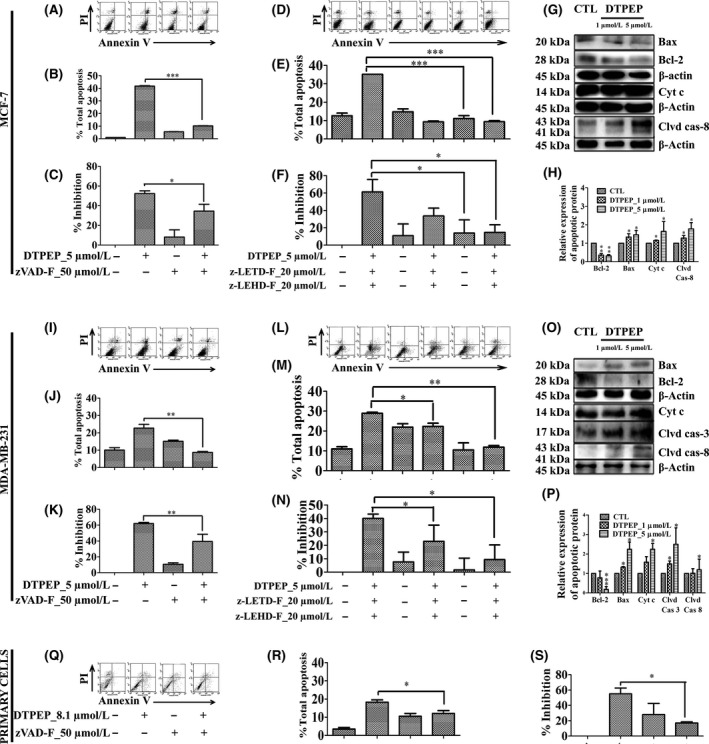
DTPEP‐induced caspase‐dependent apoptosis determined by Annexin V‐FITC & PI double staining assay using flow cytometry and loss of percentage inhibition was determined with MTT assay of breast cancer cells (A‐F) MCF‐7 cells, (I‐N) MDA‐MB‐231 and (Q‐S) primary cells. Here, 50 μmol/L of z‐VAD‐FMK (pan‐caspase inhibitor), 20 μmol/L of z‐LETD‐FMK (caspase‐8 inhibitor) and 20 μmol/L of z‐LEHD‐FMK (caspase‐9 inhibitor) were used. Statistical analysis of each parameter of DTPEP treated in the presence of z‐VAD‐FMK, z‐LETD‐FMK and z‐LEHD‐FMK groups was compared with DTPEP alone treated groups using one‐way ANOVA (non‐parametric) with Newman‐Keuls post hoc test. The difference was considered statistically significant if *P < .05, **P < .01, and ***P < .001 DTPEP in the presence of z‐VAD‐FMK, z‐LETD‐FMK and z‐LEHD‐FMK vs DTPEP alone treated group. DTPEP induced mediator of both extrinsic and intrinsic apoptosis pathway in breast cancer cells (G,H) MCF‐7 cells, and (O,P) MDA‐MB‐231. Each experiment was repeated three times and quantitation of band intensity was performed by densitometry (H & P) using Quantity One® software (v.4.5.1). Statistical analysis of each parameter for the compound treated groups was compared with non‐treated groups using one‐way ANOVA (non‐parametric) with Newman‐Keuls post hoc test. The difference was considered statistically significant if *P < .05. **P < .01 and ***P < .001 vs control. Primary antibodies used Bax (cat. 2772, cst), Bcl‐2 (cat. 2876, cst), Cleaved Caspase‐8 (cat. 9496, cst), Cleaved Caspase‐3 (cat. 9661, cst), Cytochrome c (cat. 4272, cst) and β‐actin (cat. 4970, cst)
Cleaved caspase‐8 is a characteristic of extrinsic apoptotic pathway, whereas alterations in Bax, Bcl‐2, cytochrome c and cleavage of caspase‐3 are indicator of activation of intrinsic apoptotic pathway. We found that DTPEP treatment significantly increased cleaved caspase‐8, pro‐caspase Bax, cytochrome c and significantly decreased anti‐apoptotic Bcl‐2 in both MCF‐7 (Figure 4G,H) and MDA‐MB‐231 cells along with increased cleaved caspase‐3 (Figure 4O,P) in a dose‐dependent manner. Overall, these results further support that DTPEP induces both extrinsic and intrinsic apoptosis pathway in breast cancer cells.
3.5. DTPEP inhibits exogenous E2‐induced cell proliferation, suppressed ER pathway genes/proteins and downregulates ERα and ERβ upregulates in MCF‐7 cells
DTPEP was designed for targeting ER based on tamoxifen structure (Figure 1A). We observed a significant reduction in E2‐induced proliferation of MCF‐7 cells by DTPEP in a concentration‐dependent manner suggesting that DTPEP competes with E2 (Figure 5B). Furthermore, DTPEP also suppressed expression of E2‐responsive genes. Greb1 (growth regulation by oestrogen in breast cancer 1), Ctsd (Cathepsin D), Cxl12 (Chemokine (C‐X‐C motif) ligand 12), Bmp (Bone morphogenetic protein), Rbbp8 (retinoblastoma‐binding protein 8), Igfbp4 (insulin‐like growth factor binding protein 4) and Abca3 (ATP‐binding cassette subfamily A) and upregulated Bmp (bone morphogenetic protein) (Figure 5H). DTPEP also decreased PR (progesterone receptor) and PCNA (proliferating cell nuclear antigen) expression (Figure 5F,G). DTPEP enhanced expression of cyclin‐dependent kinase inhibitor p21 protein (Figure 5F,G). Furthermore, DTPEP significantly decreased overall expression of ERα in MCF‐7 as compared to untreated MCF‐7 cells (Figure 5A). ERα expression was decreased by DTPEP even in the presence of E2 treatment as compared to E2 alone treated MCF‐7 cells (Figure 5A). DTPEP caused increase in level of ERβ both at transcript and protein level (Figure 5C‐E). DTPEP also decreased expression of phospho‐ER α (Ser118) which is important for ER signalling as it can direct recruitment of promoter complexes leading to gene‐specific transcription25 (Figure 5D,E). These data suggest that DTPEP induces dose‐dependent decrease in expression of ERα along with concomitant increase in expression of ERβ which correlates with DTPEP‐induced inhibition of MCF‐7 proliferation. DTPEP also significantly decreased ERE‐mediated transcription in concentration‐dependent manner as compared to control (Figure 5I). Decrease in ERE‐mediated transcription was significant even at 1 μmol/L in DTPEP + E2 treated group in comparison to E2 (P < .001) indicating that the compound acts by antagonizing E2 by stimulating transcription via classical (ERE‐mediated) pathway (Figure 5I). Overall, these data suggest that DTPEP action on MCF‐7 cell is ER‐dependent. To further assess specific involvement of ERα or ERβ, their expression plasmids were cotransfected with pERE‐Luc plasmids in ER‐negative MDA‐MB‐231 cells. Here, DTPEP caused a significant decrease in ERα‐mediated ERE promoter activity both in the presence and absence of E2 (Figure 5J). In case of E2‐treated group, the decrease observed was significant even at 1 μmol/L (P < .01) in comparison to control and in comparison to E2 (P < .001). However, DTPEP in the presence of E2‐augmented ERβ‐mediated ERE transactivation in a concentration‐dependent manner (P > .05‐.001) (Figure 5J). Thus, the results suggested that DTPEP acts through modulation of both ERα‐ and ERβ‐mediated classical oestrogen signalling pathways in breast cancer cells.
Figure 5.
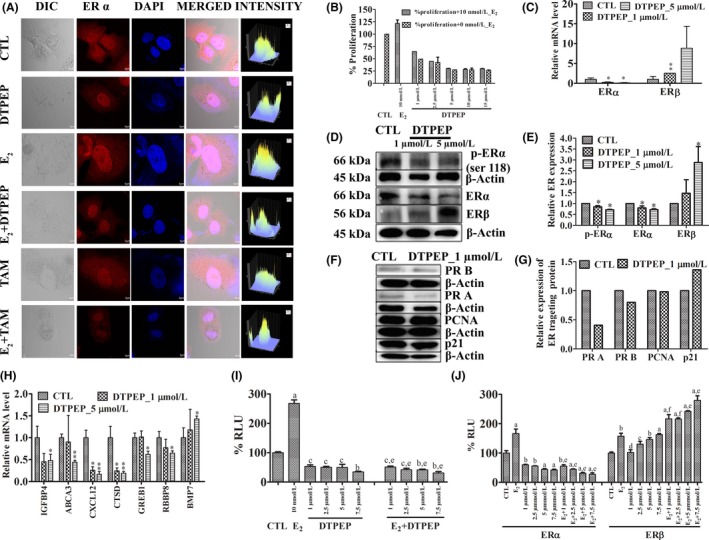
(A) DTPEP and tamoxifen downregulates ERα protein expression in MCF‐7 cells irrespective of E2 presence. ERα was detected with fluorophore‐tagged secondary antibody and counter‐staining done with DAPI. Microphotograph, 40× magnification were acquired with confocal microscope (FLUOVIEW FV1200 Multi Photon Laser Scanning Microscope,). Immunofluorescence correlating with ERα protein expression was quantitated by using software FV10‐ASW Ver.4.1,FV1200, Olympus showed on the right column. (B) DTPEP inhibits E2‐induced proliferation of MCF‐7 cells. MCF‐7 cells were treated in different concentration of DTPEP with or without 10 nmol/L E2 in phenol red free DMEM media with 0.5% of charcoal striped FBS for 24 h and results were determined by MTT assay. DTPEP on downregulates ERα and upregulates ERβ in MCF‐7 cells. (C) Relative mRNA level of ERα and ERβ. (D) ER status of MCF‐7 cells with western blot, (E) Densitometry of western blot of ER status. Effect of DTPEP on ER‐targeting/responsive/dependent protein (F) evaluated by western blot, its densitometry (G) and genes (H) expression determined by real‐time PCR. Statistical analysis of each parameter for the compound treated groups was compared with non‐treated groups using one‐way ANOVA (non‐parametric) with Newman‐Keuls post hoc test. The difference was considered statistically significant if *P < .05 and **P < .01 vs control. DTPEP modulates classical (ERE‐mediated) transcriptional activation. (I) Transcriptional activation of the ERE promoter by total ER in MCF‐7 and (J) by transiently transfected ERα and ERβ in MDA‐MB231 in response to compound either alone or in the presence of 10 nmol/L E2 for agonistic and antagonistic activity. MCF‐7 cells were transfected with ERE‐luc reporter plasmid and MDA‐MB‐231 cells were cotransfected with ERα or ERβ expression vector along with pERE‐luciferase reporter plasmid and incubated with various concentrations of compound for 24 h. Renilla luciferase pRL‐luc plasmid was used for internal control. Results are described as % of normalized relative luciferase unit (RLU). Results are expressed as mean ± SEM, n = 3. P values are a—P < .001, b—P < .01, c—P < .05 and d—P > .05 vs control and e—P < .001, f—P < .01, g—P < .05 and h—P > .05 vs E2 . Primary antibodies used Phospho‐Oestrogen Receptor α (Ser118) (cat. 251, cst), p21 (cat. 2947, cst), ERα (MC‐20) (cat.sc‐542) and ERβ (cat.sc‐8974), PR, (cat.sc‐528), PCNA (cat.sc‐53407) and β‐actin (cat. 4970, cst)
3.6. DTPEP downregulates PKCα and PI3K/Akt survival pathway in breast cancer cells
Furthermore, DTPEP significantly decreased total apoptosis in the presence of E2 as compared to in the absence of E2 in MCF‐7 cells, but there is no significant decrease in MDA‐MB‐231 cells (Figure 6A,B). Thus, it suggests that DTPEP‐induced apotosis in ER‐positive MCF‐7 cells can be partly countered by E2, but DTPEP‐induced apotosis in ER‐negative MDA‐MB‐231 cells indicate that it may interact with cellular target other than ER. Previous studies report that tamoxifen downregulates PKCα which is considered as its non‐ER–mediated action.5, 26, 27, 28, 29 DTPEP also decreased PKCα in time‐dependent manner in both ER‐positive and ER‐negative cell lines (Figure 6C,D,G,H). PKCα inhibition by DTPEP in ER‐negative MDA‐MB‐231 cells is significantly higher than that of tamoxifen. Thus, DTPEP can target PKCα irrespective of the absence or presence of ER. In addition, DTPEP decreased phosphorylated PI3K and activation of Akt in both MCF‐7 and MDA‐MB‐231 cell lines (Figure 6E,F,I,J).
Figure 6.
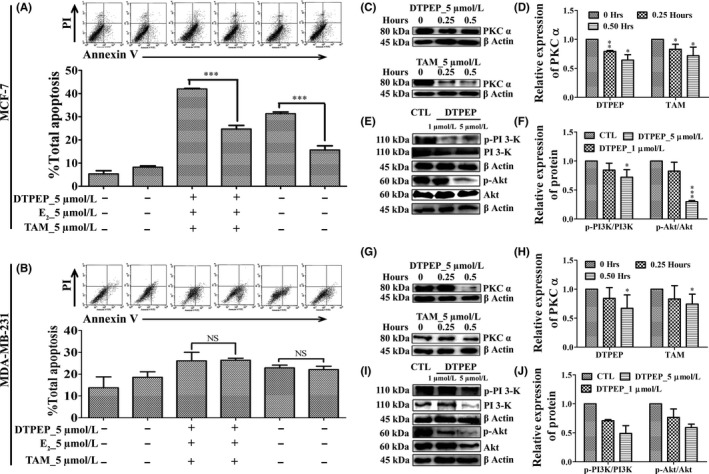
Effect of DTPEP and TAM on non‐ER pathway of apoptosis of breast cancer cell (A) MCF‐7 cells and (B) MDA‐MB‐231 cell. Statistical analysis of each parameter for the DTPEP and tamoxifen treated in the presence of E2 groups was compared with DTPEP and tamoxifen alone treated groups using one‐way ANOVA (non‐parametric) with Newman‐Keuls post hoc test. The difference was considered statistically significant if *P < .05, **P < .01 l and ***P < .001 DTPEP and tamoxifen in the presence of E2 vs DTPEP and tamoxifen alone treated group. NS = not significant. DTPEP activates non‐ER pathway PKCα like TAM in breast cancer cells irrespective of their ER status (C, D) MCF‐7 cells and (G, H) MDA‐MB‐231 cell and cells were treated for 0, 0.25 and 0.5 h. DTPEP inhibited PI3‐K/Akt pathway in breast cancer cells (E, F) MCF‐7 cells and (I, J) MDA‐MB‐231 cell. Statistical analysis of each parameter for the compound treated groups was compared with non‐treated groups using one‐way ANOVA (non‐parametric) with Newman‐Keuls post hoc test. The difference was considered statistically significant if *P < .05. **P < .01 and ***P < .001 vs control. Primary antibodies used PKC α (C‐20) (cat.sc‐208), PI‐3kinase p110β (cat.sc‐602), p‐PI3kinase p110γ Tyr 485 (cat.sc‐130211), Akt (Pan) (cat. 4685, cst), Phospho‐Akt (Ser473) (cat. 4051, cst), and β‐actin (cat. 4970, cst)
3.7. DTPEP inhibits in vivo on tumour growth in LA‐7 syngenic mammary tumour model
In syngenic mammary tumour model study, body weight of animals treated with DTPEP showed normal gain suggesting lack of any apparent acute adverse effects (Figure 7G,H). Major alteration in gross body weight is an important preliminary indication of toxicity or side effects of administered agent.30, 31 In addition, there was no significant changes in mean absolute and relative total weight of major vital organs measured at the end of trial (Figure 7E,F). Interestingly, DTPEP caused a significant decrease in mean in situ tumour volume (Figure 7D) and mean ex situ tumour weight measured at the end of the trial (Figure 7B,C). Tumour incidence and tumour burden were found decrease upon treatment with DTPEP and comparable with tamoxifen (Figure 7J,K). All the animals showed single tumour initially and we did not observe in any change in tumour multiplicity in any of the group throughout the study period. In addition, DTPEP also significantly decreased the number of metastatic nodule in lungs of the treated group in a dose‐dependent manner as compared to control group (Figure 7A,I) suggesting that DTPEP may have metastasis preventing effect as well. Furthermore, H&E‐stained tumour sections of DTPEP‐treated group presented large areas of stroma with depleted malignant epithelial cells unlike the vehicle‐treated group (Figure 8). Epithelial cells showed reduced nuclear pleomorphy, reduced nucleus to cell ratio with increased cellular necrosis and apoptosis. DTPEP treatment significantly increased fluorescence indicative of fragmented DNA in mammary tumour as compared to negative control group (Figure 8). DTPEP treatment also significantly reduced expression of ERα and PKCα protein in tumour tissue in comparison to control group (Figure 8). These observations overall suggest reversal of cellular changes in tumour tissue indicative of favourable prognosis.
Figure 7.
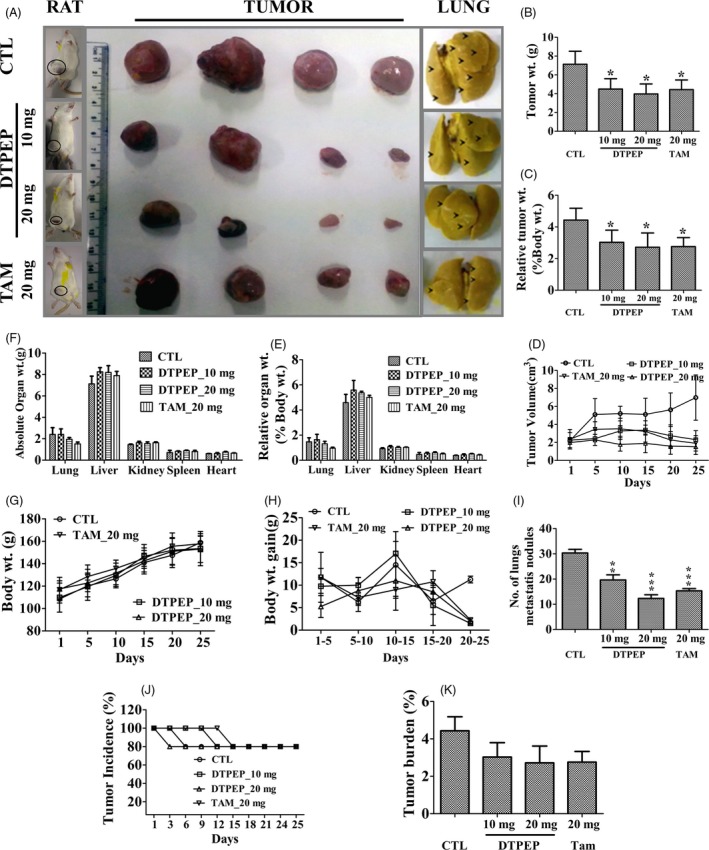
(A) Pictorial representation of tumour size ex situ and in situ LA‐7 rat syngenic mammary tumour model at 25 days of oral administration of vehicle (n = 4), DTPEP (10 mg/kg, n = 4 and 20 mg/kg, n = 4) and TAM (20 mg/kg, n = 4). (A, I). DTPEP reduces metastatic nodules in lung of LA‐7 rat mammary tumour model. (B). DTPEP induces loss of ex situ tumour weight, (C). relative tumour weight and (D) in situ tumour volume. Oral administration of DTPEP does not disrupt absolute organ weight (E), relative organ weight (F), total body weight (G) and total body weight gain (H). Statistical analysis of each parameter for the compound‐treated groups was compared with non‐treated groups using one‐way ANOVA (non‐parametric) with Newman‐Keuls post hoc test. The difference was considered statistically significant if *P < .05. **P < .01 and ***P < .001 vs control
Figure 8.
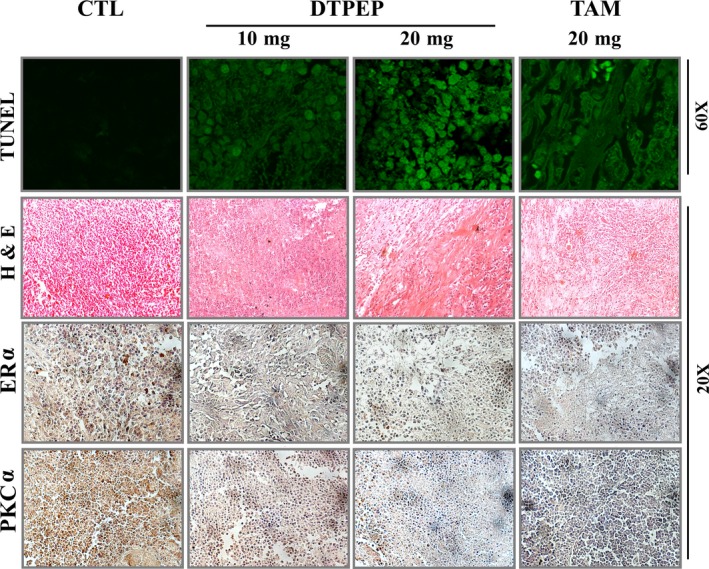
DTPEP and TAM induces apoptosis in mammary tumour tissue of LA‐7 rat mammary tumour model. The fragmented DNA indicative of apoptotic cells were visualized with TUNEL stain using 60× magnification of confocal microscope (FV1200 Multi Photon Laser Scanning Microscope; Olympus, Tokyo, Japan). DTPEP and tamoxifen reverses aggressively proliferating malignant cellular histomorphology of rat mammary tumour (H & E). DTPEP and tamoxifen downregulates expression of ERα and PKCα in rat mammary tumour (IHC). Images were captured at 20× magnification of CKX41 Trinocular with cooled CCD camera Model Imaging MP5.0‐RTV‐CLR‐10‐c from Olympus
4. DISCUSSION
Standard drugs like tamoxifen are beneficial against ER‐positive breast cancers.32, 33, 34 ER‐negative breast cancers are very aggressive tumours, lack safe standard therapeutics and result in higher patient mortality. Some report suggests activity of tamoxifen against ER‐negative breast cancer, possibly through some ER‐independent off‐target effect.5, 35 Some groups have attempted collective targeting of ER and other targets in breast cancer, mostly through hybrid molecules.36, 37, 38, 39 In pursuit of new drug discovery, a series of constrained tricyclic compounds of substituted dibenzo[b,f]thiepine and dibenzo[b,f]oxepines that were structurally analogous to tamoxifen were synthesized. DTPEP is appeared to be the most potent compound in the series equally acting against ER‐positive breast cancer cell lines (MCF‐7 with IC50 of 1.33 μmol/L) and ER‐negative breast cancer cell line (MDA‐MB‐231 with IC50 of 5 μmol/L; ER negative primary cells with IC50 of 8.1 μmol/L). It is devoid of any cytotoxic side effect on non‐cancer cells, HEK‐293 and MCF‐10A cells up to 50 μmol/L concentration.
Cell division and progress of cell cycle are most critical for unrestrained cancer cell proliferation and disruption of cell cycle is major end goal of effective anti‐cancer agent.40 DTPEP restricted the cell cycle at G0/G1 phase of breast cancer cells in dose‐dependent manner (Figure 2A,C). DTPEP induced apoptosis in MCF‐7, MDA‐MB‐231 and primary cells in caspase‐dependent manner. Selective caspase inhibition studies showed that DTPEP induces both extrinsic and intrinsic apoptosis. Several compounds like indirubin derivative (8‐Rha‐β),41 quinoline derivative (PQ1),42 sulphamoylated 2‐methoxyestradiol analogues,43 myricetin,44 indole‐coumarin‐thiadiazole hybrids45 are reported to induce extrinsic and intrinsic apoptosis. These dual‐acting compounds offer possibility of more comprehensive attack on cancer cells.
Our results also showed that DTPEP increased ROS generation in cancer cells in a dose‐dependent manner. Tamoxifen and several other chemotherapeutic drugs are well known to induce ROS that subsequently induces apoptosis and regression of cancer. Hence, DTPEP‐induced high level of ROS should possibly responsible for apoptosis in treated breast cancer cells. We know that ROS generation is critical for loss of mitochondrial membrane integrity and apoptosis, we observed the same upon DTPEP treatment. ROS‐induced mitochondrial membrane damage leads to either or both activation of proapoptotic factors and inhibition of anti‐apoptotic factors.46, 47, 48 Our data suggest that DTPEP‐enhanced expression of level of cytochrome c, bax and decreased bcl‐2 proteins in both MCF‐7 and MDA‐MB‐231 cells. Thus, we can conclude that DTPEP induces breast cancer cell death through ROS‐dependent apoptosis through both extrinsic and intrinsic pathway.
ERα plays a crucial role in the progression of breast cancer49 being the receptor for mitogenic growth promoter, oestrogen.50 Interruption of oestrogenic activity by targeting its receptor results desired anti‐cancer effects.51 DTPEP inhibited growth of ER‐positive breast cancer cell both in the absence and presence of exogenous E2. We assume that in the absence of exogenous E2, the compound exerted its growth inhibitory activity by neutralizing endogenous E2 and also possibly through some other non‐ER pathway. Due to its action through non‐ER pathway, DTPEP appears active against ER‐negative breast cancer cells. In ER‐positive MCF‐7 cells, DTPEP decreased expression of ERα with concomitant increase in expression of ERβ at both transcript and protein level. The increased ERβ expression favours formation of ERα‐ERβ heterodimer thereby suppressing ERα homodimers responsible promotion of breast cancer.52 ER regulates gene expression via classical pathway through ERE and non‐classical pathway through AP‐1 sites. Here, DTPEP decreased E2‐induced ERα‐ERE‐mediated transcriptional activation and at the same time increased the ERβ‐ERE‐mediated transactivation. In addition, DTPEP also altered ER‐responsive transcripts and proteins in treated MCF‐7 cells supporting DTPEP acts on ER. Oestrogen and ER‐dependent signalling pathway have been shown to activate Akt and its downstream cascade.53 DTPEP inhibits PI3K/Akt signalling through downregulation of phosphorylation of p‐PI3K at tyr 485 and p‐Akt at ser 473 in both MCF‐7 and MDA‐MB‐231 cell. Akt may be activated in cancer cells through various factors including oestrogen. Few reports suggest Akt activation by oestrogen may not be inhibited ER antagonists action.54 Here also, we believe that the non‐ER–dependent action of DTPEP must be responsible for inhibition of Akt in ER‐negative breast cancer cells. Tamoxifen is reported to have non‐ER–dependent action in cancer cells via inhibition of PKC.4, 5, 6, 7, 8 PKC play a crucial role in the signal transduction that influences cell growth and transformation in cancer cells.55 DTPEP also significantly downregulates PKCα protein expression in time‐dependent manner in both MCF‐7 and MDA‐MB‐231 cells. Thus, we assume that this could partly explain its anti‐cancer action in ER‐negative breast cancer cells, including MDA‐MB‐231 cell line and ER‐negative primary breast cancer cells derived from patient tumour. However, further investigation is warranted to understand how cellular targeting of PI3‐K/Akt and PKCα is achieved in ER‐indendepent anti‐cancer action of DTPEP.
Animal tumour models are better representation of complex biological test systems and regarded essential for preclinical investigation of anti‐cancer therapeutics. Our LA‐7 syngenic rat mammary tumour model studies clearly established in vivo efficacy of DTPEP with a significant reduction of orthotopic mammary tumour weight and volume. This was further supported by histological changes at tumour tissue level indicative of anti‐cancer effect of DTPEP. Furthermore, our animal model studies also validated in vitro observation confirming induction of apoptosis in tumour tissue by DTPEP. The observed regression of mammary tumour correlated with downregulation of ERα and PKCα expression in tumour tissue. Additionally, DTPEP treatment did not resulted in gross alteration body weight as well as major vital organs suggesting general safety of DTPEP. Thus, in vivo studies convincingly established that DTPEP induces significant tumour regression through downregulation both ERα and non‐ERα pathways with concomitant apoptosis. Overall, these data suggested that DTPEP shows strong anti‐breast cancer activity both in vitro and in vivo with considerable safety. DTPEP offers newer possibilities of dual targeting of ER‐positive and ‐negative breast cancer through involvement of both intrinsic and extrinsic apoptosis pathway.
ACKNOWLEDGEMENTS
A. A., M. I. A. and S. J. are thankful to CSIR, New Delhi and P. P. is thankful to ICMR, New Delhi, India, for financial assistance. Authors acknowledge Mr. A. L. Vishwakarma for flow cytometric analysis. This is CDRI communication no. 9700.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
Arun A, Ansari MI, Popli P, et al. New piperidine derivative DTPEP acts as dual‐acting anti‐breast cancer agent by targeting ERα and downregulating PI3‐K/Akt‐PKCα leading to caspase‐dependent apoptosis. Cell Prolif. 2018;51:e12501 10.1111/cpr.12501
Funding information
Council of Scientific and Industrial Research (CSIR), Indian Council of Medical Research (ICMR), Government of India
REFERENCES
- 1. Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol. 2013;75:225‐240. [DOI] [PubMed] [Google Scholar]
- 2. Baumann CK, Castiglione‐Gertsch M. Estrogen receptor modulators and down regulators: optimal use in postmenopausal women with breast cancer. Drugs. 2007;67:2335‐2353. [DOI] [PubMed] [Google Scholar]
- 3. Maximov PY, McDaniel RE, Jordan VC. Tamoxifen: Pioneering Medicine in Breast Cancer. Basel: Springer; 2013. [Google Scholar]
- 4. Anzai Y, Holinka CF, Kuramoto H, Gurpide E. Stimulatory effects of 4‐hydroxytamoxifen on proliferation of human endometrial adenocarcinoma cells (Ishikawa line). Cancer Res. 1989;49:2362‐2365. [PubMed] [Google Scholar]
- 5. Gundimeda U, Chen Z‐H, Gopalakrishna R. Tamoxifen modulates protein kinase C via oxidative stress in estrogen receptor‐negative breast cancer cells. J Biol Chem. 1996;271:13504‐13514. [DOI] [PubMed] [Google Scholar]
- 6. Lam H‐YP. Tamoxifen is a calmodulin antagonist in the activation of cAMP phosphodiesterase. Biochem Biophys Res Commun. 1984;118:27‐32. [DOI] [PubMed] [Google Scholar]
- 7. Sudo K, Monsma FJ Jr, Katzenellenbogen BS. Antiestrogen‐binding sites distinct from the estrogen receptor: subcellular localization, ligand specificity, and distribution in tissues of the rat. Endocrinology. 1983;112:425‐434. [DOI] [PubMed] [Google Scholar]
- 8. Watts C, Murphy L, Sutherland R. Microsomal binding sites for nonsteroidal anti‐estrogens in MCF 7 human mammary carcinoma cells. Demonstration of high affinity and narrow specificity for basic ether derivatives of triphenylethylene. J Biol Chem. 1984;259:4223‐4229. [PubMed] [Google Scholar]
- 9. Ansari MI, Hussain MK, Arun A, et al. Synthesis of targeted dibenzo [b, f] thiepines and dibenzo [b, f] oxepines as potential lead molecules with promising anti‐breast cancer activity. Eur J Med Chem. 2015;99:113‐124. [DOI] [PubMed] [Google Scholar]
- 10. Valentine JE, Kalkhoven E, White R, et al. Mutations in the estrogen receptor ligand binding domain discriminate between hormone‐dependent transactivation and transrepression. J Biol Chem. 2000;275:25322‐25329. [DOI] [PubMed] [Google Scholar]
- 11. Besch GJ, Wolberg WH, Gilchrist KW, Voelkel JG, Gould MN. A comparison of methods for the production of monodispersed cell suspensions from human primary breast carcinomas. Breast Cancer Res Treat. 1983;3:15‐22. [DOI] [PubMed] [Google Scholar]
- 12. Ansari MI, Arun A, Hussain MK, et al. Discovery of 3, 4, 6‐Triaryl‐2‐pyridones as potential anticancer agents that promote ROS‐independent mitochondrial‐mediated apoptosis in human breast carcinoma cells. ChemistrySelect. 2016;1:4255‐4264. [Google Scholar]
- 13. Wang Y, Yu H, Zhang J, et al. Hesperidin inhibits HeLa cell proliferation through apoptosis mediated by endoplasmic reticulum stress pathways and cell cycle arrest. BMC Cancer. 2015;15:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sashidhara KV, Singh LR, Choudhary D, et al. Design, synthesis and in vitro evaluation of coumarin–imidazo [1, 2‐a] pyridine derivatives against cancer induced osteoporosis. RSC Adv. 2016;6:80037‐80048. [Google Scholar]
- 15. Chauhan K, Arun A, Singh S, et al. Bivalent approach for homodimeric estradiol based ligand: synthesis and evaluation for targeted theranosis of ER (+) breast carcinomas. Bioconjug Chem. 2016;27:961‐972. [DOI] [PubMed] [Google Scholar]
- 16. Arun A, Patel OPS, Saini D, et al. Anti‐colon cancer activity of Murraya koenigii leaves is due to constituent murrayazoline and O‐methylmurrayamine A induced mTOR/AKT downregulation and mitochondrial apoptosis. Biomed Pharmacother. 2017;93:510‐521. [DOI] [PubMed] [Google Scholar]
- 17. Hamidullah Saini KS, Ajay A, et al. Triazole analog 1‐(1‐benzyl‐5‐(4‐chlorophenyl)‐1H‐1,2,3‐triazol‐4‐yl)‐2‐(4‐bromophenylamino)‐1‐(4 ‐chlorophenyl)ethanol induces reactive oxygen species and autophagy‐dependent apoptosis in both in vitro and in vivo breast cancer models. Int J Biochem Cell Biol. 2015;65:275‐287. [DOI] [PubMed] [Google Scholar]
- 18. Lee M‐Y, Marina M, King JL, et al. Differential expression of centrosome regulators in Her2 + breast cancer cells versus non‐tumorigenic MCF10A cells. Cell Div. 2014;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Upadhyaya K, Hamidullah Singh K, et al. Identification of gallic acid based glycoconjugates as a novel tubulin polymerization inhibitors. Org Biomol Chem. 2016;14:1338‐1358. [DOI] [PubMed] [Google Scholar]
- 20. Matsuda Y, Fujii T, Suzuki T, et al. Comparison of fixation methods for preservation of morphology, RNAs, and proteins from paraffin‐embedded human cancer cell‐implanted mouse models. J Histochem Cytochem. 2011;59:68‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cardiff RD, Miller CH, Munn RJ. Manual hematoxylin and eosin staining of mouse tissue sections. Cold Spring Harb Protoc. 2014;6:655‐658. [DOI] [PubMed] [Google Scholar]
- 22. Saini KS, Hamidullah Ashraf R, et al. New orally active DNA minor groove binding small molecule CT‐1 acts against breast cancer by targeting tumor DNA damage leading to p53‐dependent apoptosis. Mol Carcinog. 2017;56:1266‐1280. [DOI] [PubMed] [Google Scholar]
- 23. Blesson CS, Awasthi S, Kharkwal G, et al. Modulation of estrogen receptor transactivation and estrogen‐induced gene expression by ormeloxifene—A triphenylethylene derivative. Steroids. 2006;71:993‐1000. [DOI] [PubMed] [Google Scholar]
- 24. Simon H‐U, Haj‐Yehia A, Levi‐Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415‐418. [DOI] [PubMed] [Google Scholar]
- 25. Duplessis TT, Williams CC, Hill SM, et al. Phosphorylation of estrogen receptor α at serine 118 directs recruitment of promoter complexes and gene‐specific transcription. Endocrinology. 2011;152:2517‐2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Brian CA, Housey GM, Weinstein IB. Specific and direct binding of protein kinase C to an immobilized tamoxifen analogue. Can Res. 1988;48:3626‐3629. [PubMed] [Google Scholar]
- 27. O'Brian CA, Liskamp RM, Solomon DH, et al. Inhibition of protein kinase C by tamoxifen. Cancer Res. 1985;45:2462‐2465. [PubMed] [Google Scholar]
- 28. Boyan B, Sylvia V, Frambach T, et al. Estrogen‐dependent rapid activation of protein kinase C in estrogen receptor‐positive MCF‐7 breast cancer cells and estrogen receptor‐negative HCC38 cells is membrane‐mediated and inhibited by tamoxifen. Endocrinology. 2003;144:1812‐1824. [DOI] [PubMed] [Google Scholar]
- 29. Matsuoka H, Tsubaki M, Yamazoe Y, et al. Tamoxifen inhibits tumor cell invasion and metastasis in mouse melanoma through suppression of PKC/MEK/ERK and PKC/PI3K/Akt pathways. Exp Cell Res. 2009;315:2022‐2032. [DOI] [PubMed] [Google Scholar]
- 30. Raza M, Al‐Shabanah O, El‐Hadiyah T, et al. Effect of prolonged vigabatrin treatment on hematological and biochemical parameters in plasma, liver and kidney of Swiss albino mice. Sci Pharm. 2002;702:135‐145. [Google Scholar]
- 31. Teo S, Stirling D, Thomas S, et al. A 90‐day oral gavage toxicity study of D‐methylphenidate and D,L‐methylphenidate in Sprague‐Dawley rats. Toxicology. 2002;179:183‐196. [DOI] [PubMed] [Google Scholar]
- 32. Delozier T, Julien J‐P, Juret P, et al. Adjuvant tamoxifen in postmenopausal breast cancer: preliminary results of a randomized trial. Breast Cancer Res Treat. 1986;7:105‐109. [DOI] [PubMed] [Google Scholar]
- 33. Stewart HJ, Prescott RJ, Forrest APM. Scottish adjuvant tamoxifen trial: a randomized study updated to 15 years. J Natl Cancer Inst. 2001;93:456‐462. [DOI] [PubMed] [Google Scholar]
- 34. Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P‐1 study. J Natl Cancer Inst. 2005;97:1652‐1662. [DOI] [PubMed] [Google Scholar]
- 35. Manna S, Holz MK. Tamoxifen action in ER‐negative breast cancer. Sign Transduct Insights. 2016;5:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boulay A, Rudloff J, Ye J, et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005;11:5319‐5328. [DOI] [PubMed] [Google Scholar]
- 37. O'Boyle NM, Pollock JK, Carr M, et al. Β‐lactam estrogen receptor antagonists and a dual‐targeting estrogen receptor/tubulin ligand. J Med Chem. 2014;57:9370‐9382. [DOI] [PubMed] [Google Scholar]
- 38. Tang C, Li C, Zhang S, et al. Novel bioactive hybrid compound dual targeting estrogen receptor and histone deacetylase for the treatment of breast cancer. J Med Chem. 2015;58:4550‐4572. [DOI] [PubMed] [Google Scholar]
- 39. Luo G, Li X, Zhang G, et al. Novel SERMs based on 3‐aryl‐4‐aryloxy‐2H‐chromen‐2‐one skeleton‐A possible way to dual ERα/VEGFR‐2 ligands for treatment of breast cancer. Eur J Med Chem. 2017;140:252‐273. [DOI] [PubMed] [Google Scholar]
- 40. Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berger A, Quast S‐A, Plötz M, et al. Sensitization of melanoma cells for death ligand‐induced apoptosis by an indirubin derivative‐enhancement of both extrinsic and intrinsic apoptosis pathways. Biochem Pharmacol. 2011;81:71‐81. [DOI] [PubMed] [Google Scholar]
- 42. Ding Y, Nguyen TA. PQ1, a quinoline derivative, induces apoptosis in T47D breast cancer cells through activation of caspase‐8 and caspase‐9. Apoptosis. 2013;18:1071‐1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Visagie M, Theron A, Mqoco T, et al. Sulphamoylated 2‐methoxyestradiol analogues induce apoptosis in adenocarcinoma cell lines. PLoS ONE. 2013;8:e71935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang H, Chen AY, Ye X, et al. Myricetin inhibits proliferation of cisplatin‐resistant cancer cells through a p53‐dependent apoptotic pathway. Int J Oncol. 2015;47:1494‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kamath PR, Sunil D, Joseph MM, et al. Indole‐coumarin‐thiadiazole hybrids: an appraisal of their MCF‐7 cell growth inhibition, apoptotic, antimetastatic and computational Bcl‐2 binding potential. Eur J Med Chem 2017;136:442‐451. [DOI] [PubMed] [Google Scholar]
- 46. Puthalakath H, Huang DC, O'Reilly LA, et al. The proapoptotic activity of the Bcl‐2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287‐296. [DOI] [PubMed] [Google Scholar]
- 47. Harada H, Quearry B, Ruiz‐Vela A, et al. Survival factor‐induced extracellular signal‐regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci U S A. 2004;101:15313‐15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gomez‐Bougie P, Bataille R, Amiot M. Endogenous association of Bim BH3‐only protein with Mcl‐1, Bcl‐xL and Bcl‐2 on mitochondria in human B cells. Eur J Immunol. 2005;35:971‐976. [DOI] [PubMed] [Google Scholar]
- 49. Dickson RB, Lippman ME. Growth factors in breast cancer. Endocr Rev. 1995;16:559‐589. [DOI] [PubMed] [Google Scholar]
- 50. Bai Y, Giguère V. Isoform‐selective interactions between estrogen receptors and steroid receptor coactivators promoted by estradiol and ErbB‐2 signaling in living cells. Mol Endocrinol. 2003;17:589‐599. [DOI] [PubMed] [Google Scholar]
- 51. Macgregor JI, Jordan VC. Basic guide to the mechanisms of antiestrogen action. Pharmacol Rev. 1998;50:151‐196. [PubMed] [Google Scholar]
- 52. Fox EM, Davis RJ, Shupnik MA. ERβ in breast cancer—onlooker, passive player, or active protector? Steroids. 2008;73:1039‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ahmad S, Singh N, Glazer RI. Role of AKT1 in 17β‐estradiol‐and insulin‐like growth factor I (IGF‐I)‐dependent proliferation and prevention of apoptosis in MCF‐7 breast carcinoma cells. Biochem Pharmacol. 1999;58:425‐430. [DOI] [PubMed] [Google Scholar]
- 54. Tsai E‐M, Wang S‐C, Lee J‐N, Hung M‐C. Akt activation by estrogen in estrogen receptor‐negative breast cancer cells. Cancer Res. 2001;61:8390‐8392. [PubMed] [Google Scholar]
- 55. Jaken S. Protein kinase C and tumor promoters. Curr Opin Cell Biol. 1990;2:192‐197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials